
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu–Mg synergy enhanced synthesis of methyl formate over noble metal-free heterogeneous catalyst systems†
Jyotishman
Kaishyop
 ab,
Arpan
Mukherjee
a,
Abhay Giri
Goswami
c,
Tuhin Suvra
Khan
bd and
Ankur
Bordoloi
*ab
ab,
Arpan
Mukherjee
a,
Abhay Giri
Goswami
c,
Tuhin Suvra
Khan
bd and
Ankur
Bordoloi
*ab
aNanocatalyst Area, Light Stock Processing Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, Uttarakhand, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, 201002, India
cDepartment of Chemistry, IIT Gandhinagar, Palaj, Gujarat 382355, India
dClimate Change and Data Science Division, CSIR-Indian Institute of Petroleum, Dehradun 248005, Uttarakhand, India
First published on 11th November 2024
Abstract
To address the challenges associated with storage and transportation of hydrogen, Liquid Organic Hydrogen Carriers (LOHCs) like methyl formate (MF) offer a promising and sustainable solution for storing hydrogen (8.4% wt) under ambient conditions. The conversion of anthropogenic CO2 to MF in the presence of methanol, under base-free conditions, is attracting significant research attention, as it not only helps to limit the atmospheric CO2 but also produces MF as an efficient LOHC. In this study, a supported Cu nanocatalyst was synthesized, demonstrating notable activity for CO2 hydrogenation to MF, achieving a CO2 conversion of 14.01%, a turnover frequency (TOF) of 186 h−1, and selectivity greater than 99%. These results represent the highest performance reported among noble-metal-free catalyst systems. The Cu–Mg synergy enhanced catalytic activity was thoroughly investigated in the quest for improved activity. Comprehensive characterization studies were conducted to elucidate the structure–activity relationship, and the catalyst exhibited consistent performance in recycling and long-term experiments. Additionally, a plausible reaction mechanism for the catalytic process has been proposed.
Sustainability spotlightThe sustainable conversion of CO2 to valuable chemicals like methyl formate (MF) through hydrogenation represents a promising approach to address both carbon capture and utilization. This process not only reduces the levels of CO2 in the atmosphere, mitigating its environmental impact, but also provides an efficient method for producing MF, a compound with significant industrial applications, including as a Liquid Organic Hydrogen Carrier (LOHC). The use of non-noble metal catalysts, such as copper supported on MgO–ZrO2, offers an environmentally friendly and cost-effective solution, aligning with green chemistry principles. By optimizing catalytic activity and selectivity, researchers are advancing the potential of this technology to contribute to a circular carbon economy, where CO2 is transformed from a waste product into a valuable resource. This study focuses on sustainable CO2 hydrogenation to MF which is crucial for developing energy-efficient and scalable processes that support global efforts toward carbon neutrality. |
Introduction
The skyrocketing demand for energy in the world is currently met through the combustion of fossil fuels, leading to the uncontrolled release of anthropogenic CO2 into the atmosphere and the rapid depletion of these resources from the earth's crust. The dramatic rise in atmospheric CO2 levels has resulted in severe examples of worsening global climate and causes environmental crises.1 Consequently, the global effort focuses on adopting renewable energy sources to replace fossil fuels and reduce human-caused CO2 emissions. Hydrogen is viewed as a long-term substitute for fossil fuels as it is an industrial fuel with high energy density potentially produced from renewable sources through methods like electrochemical water splitting.2 In terms of revenue, with a compound annual growth rate (CAGR) of 7.8% from 2023, $410.6 billion of the global hydrogen market is anticipated to reach by 2030. However, storing and transporting hydrogen is a significant challenge because it is highly flammable and needs to be stored at high pressure and cryogenic temperature. Liquid organic hydrogen carriers (LOHCs), which can be reversibly hydrogenated and dehydrogenated to release H2, are an up-and-coming alternative to storing hydrogen under ambient conditions and transporting large quantities of hydrogen over long distances. Currently, methanol and ammonia are commonly used as liquid organic hydrogen carriers due to the higher capacity of hydrogen; however, these two are classified as toxic and flammable. Arenas, also known as aromatic compounds like benzene, toluene, naphthalene, xylene, phenol, cresols, etc., a class of LOHCs, offer notable hydrogen density (5.8–7.3 wt%) and are easy to handle; nevertheless, they are less available and pose toxicity issues. Formic acid (FA) can be directly obtained from CO2 and has favourable thermodynamic properties, allowing for easy dehydrogenation under ambient conditions; however, it has low hydrogen content (<5 wt%) and is corrosive. Therefore, there is significant interest in developing new practical hydrogen vectors that address these limitations. Methyl formate (MF) has garnered attention due to its hydrogen storage capacity (8.4 wt%), which falls between that of MeOH (12.1 wt%) and FA (4.4 wt%) and is comparable to that of other LOHCs.3,4 In particular, MF's volumetric energy density is equal to 1200 bar of compressed hydrogen. Except for FA, MF's dehydrogenation is noticeably more thermodynamically preferred than other hydrogen carriers. Moreover, MF is categorized as a crucial intermediary for synthesising formic acid, antileukemic agents and other fine chemicals. In addition, MF has many other industrial uses, such as a blowing agent for foams and as an agricultural fumigant for tobacco, dried fruits, and cereals.Currently, MF is industrially produced through the carbonylation of methanol, typically using a base catalyst such as sodium methoxide.5 This process is extremely toxic due to the use of CO as a raw material and the highly explosive nature of sodium methoxide. Therefore, the catalytic hydrogenation of CO2 to MF holds more significant potential for the preparation of MF, offering dual benefits, such as reducing atmospheric CO2 and providing a greener method for producing MF as a potential liquid organic hydrogen carrier. Table 1 lists a few homogeneous catalysts and their processes reported recently for the synthesis of MF through CO2 hydrogenation.
Despite the exceptional performance of homogeneous catalysts, the separation and recycling of metal complexes, isolation of the desired product from the base, and the use of expensive and air-sensitive ligands pose significant barriers to the practical application of these applied homogeneous catalysts. In contrast, heterogeneous catalysts offer easier separation, handling, and recyclability, making them particularly suitable for continuous operations. Consequently, significant research has focused on designing and developing heterogeneous catalysts for the synthesis of MF through CO2 hydrogenation (Fig. 1). Historically, Krocher et al. reported a RuCl2[PMe2(CH2)2Si(OEt)3]3 catalyst supported over a silica hybrid gel, which was highly effective for the hydrogenation of CO2 to methyl formate in the presence of NEt3 as a base additive.7 Furthermore, methyl formate (MF) was synthesized efficiently by hydrogenating scCO2 in methanol solvent over a ruthenium catalyst with a TOF of 55 h−1 at 80 °C.10 Later, Tsang and his group showcased that Pd/Cu/ZnO and Cu/ZnO catalysts were capable of hydrogenating CO2 into MF under base-free conditions, although the catalytic activity was poor, and the CO by-product was produced.11 They demonstrated a one-step CO2 hydrogenation to MF over a Pd/Cu/ZnO catalyst with excessive methanol in hydrogen.12 Furthermore, Xin and colleagues reported the photocatalytic reduction of CO2 to MF using ZnS and CuO/TiO2 as catalysts.13
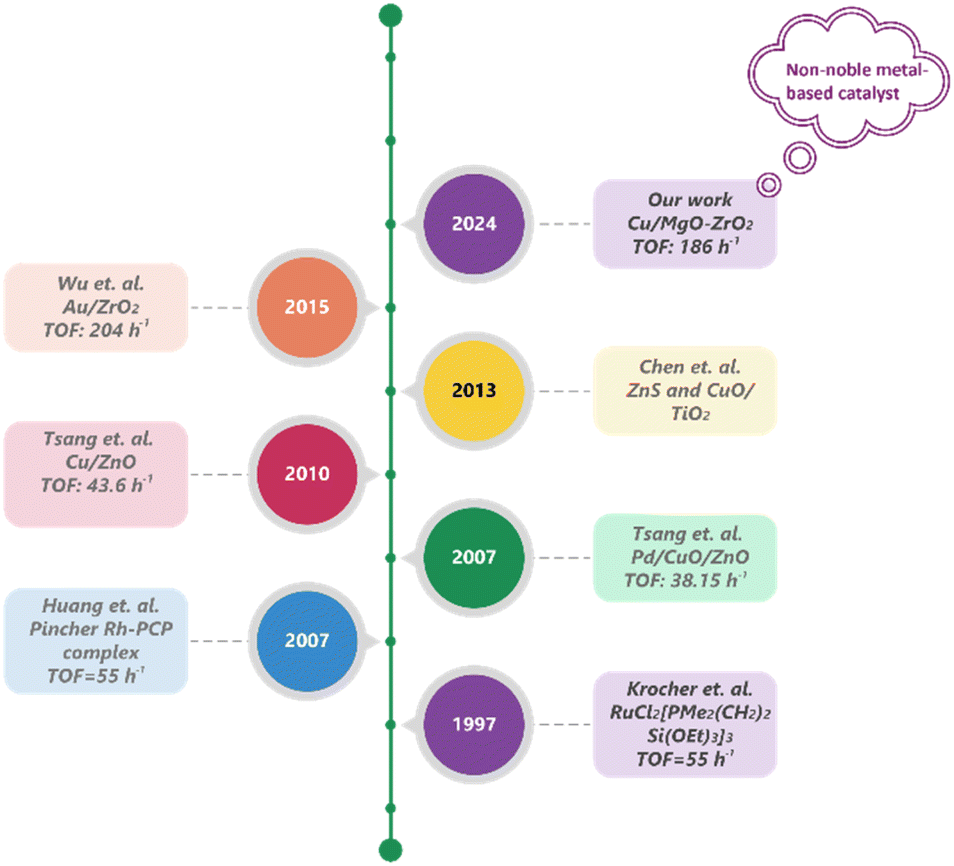 | ||
| Fig. 1 Periodic development of heterogeneous catalysts for the hydrogenation of CO2 to methyl formate (MF). | ||
Recently, Congyi Wu and colleagues proposed an Au/ZrO2 catalyst for the hydrogenation of CO2 to MF, achieving a TOF of over 200 h−1 at 140 °C and 16 MPa total pressure. They explained that ZrO2 is a more suitable support for this reaction compared to CeO2 and TiO2 due to its amphoteric nature. The basic sites support the adsorption and activation of CO2, while the acidic sites are responsible for the desorption of the generated formic acid.14 In the absence of a base, it was found that Cu catalysts are highly active for the hydrogenation of CO2 to formic acid and methanol.15–18 MF can be produced by the esterification process between formic acid and methanol.14 MgO serves a dual function in the hydrogenation of CO2 to MF. It enhances the basicity of the catalyst surface, promoting CO2 adsorption and activation19 while also facilitating the esterification between methanol (MeOH) and formic acid (HCOOH) to form MF by increasing the nucleophilicity of the alcohol. Specifically, MgO facilitates the deprotonation of methanol, leading to the production of methoxide ions forming stronger nucleophiles than neutral methanol.20 The enhanced nucleophilicity allows the methoxide ion to more effectively attack the electrophilic carbonyl carbon of formic acid. The carbonyl carbon, being partially positively charged due to the polarization of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, becomes a target for nucleophilic attack. This process forms a tetrahedral intermediate, which is crucial for the subsequent conversion into ester products, particularly methyl formate. Therefore, it can be expected that hydrogenating CO2 in the presence of methanol can synthesize MF utilizing Cu as the catalyst supported over MgO and ZrO2, without the need for any base additives.
O bond, becomes a target for nucleophilic attack. This process forms a tetrahedral intermediate, which is crucial for the subsequent conversion into ester products, particularly methyl formate. Therefore, it can be expected that hydrogenating CO2 in the presence of methanol can synthesize MF utilizing Cu as the catalyst supported over MgO and ZrO2, without the need for any base additives.
Aligning with this, a Cu catalyst was supported over MgO–ZrO2 for the CO2 hydrogenation to MF with a conversion of 14.01% with a selectivity of more than 99% in an aqueous medium. In two hours and under industrially relevant conditions (T = 393 K and P = 10 MPa), a maximum turnover number (TON) of 372 was achieved for MF. Furthermore, a detailed explanation of the impact of MgO doping in improving the MF yield was provided. The catalyst is incredibly durable and has consistently performed well in ongoing recycling testing.
Experimental
Preparation of catalysts (xCu/yMgO–ZrO2)
The catalyst was synthesized using the template assisted modified sol–gel method,21 as illustrated in Part A of Scheme 1. In a typical synthesis process, 1 g of P123 was dissolved in 20 mL of deionized water, serving as the structure-directing agent, and stirred vigorously. A stoichiometric amount of magnesium nitrate hexahydrate was then added to this solution, which was labelled solution A. Separately, a specified amount of zirconium n-isopropoxide and acetic acid were added to anhydrous ethanol to produce solution B. Solutions A and B were then combined and stirred for 30 minutes. After stirring for an hour, a small amount of deionized water was added to the mixture. After that, the mixture was kept for ten hours at 80 °C, producing a lumpy, thick white gel, which was refluxed in a pH ≈ 11 NaOH solution, for 24 h. The solution was then filtered and washed multiple times with deionised water to remove Na+. The obtained products were dried for 2 h, followed by calcination at 700 °C for two hours, yielding white MgO–ZrO2 powders. In the subsequent step (Part B, Scheme 1), the prepared support powders were mixed with varying amounts of Cu(NO3)2·3H2O (Sigma-Aldrich) dissolved in a specified amount of deionized water. The mixtures were stirred for 4 hours, followed by drying and calcination at 400 °C for 4 hours. The resulting powders were the catalysts with different Cu contents. The catalysts were designated as xC–yMZ, where x and y represent the weight percentages of Cu and MgO, respectively.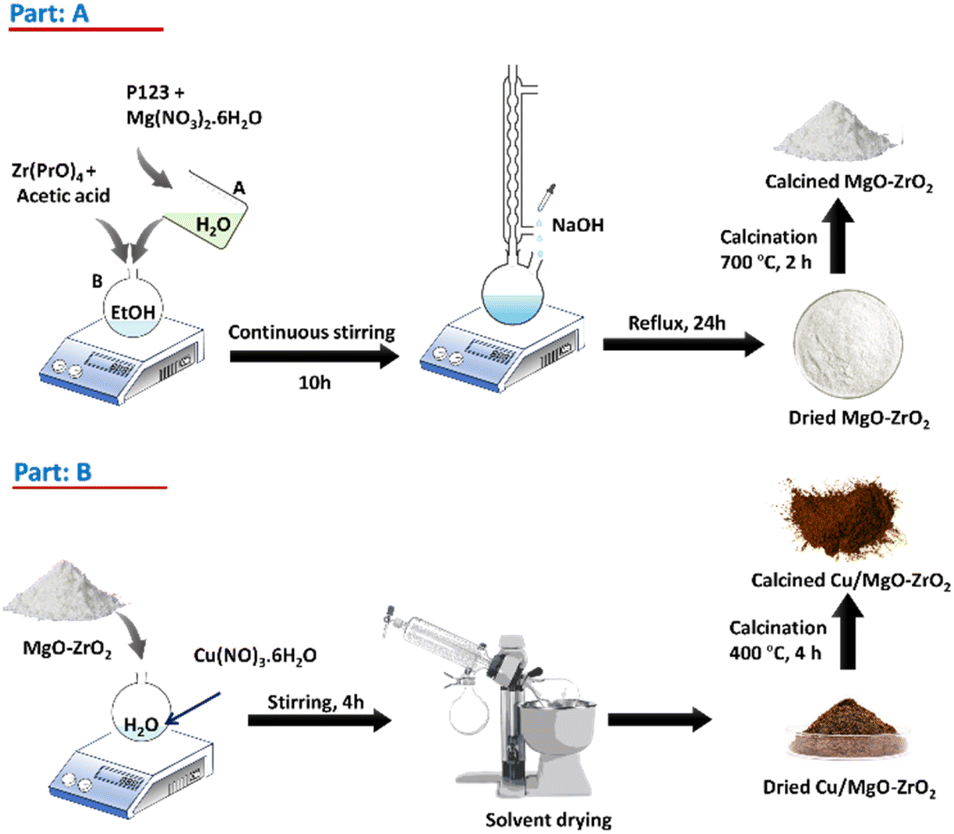 | ||
| Scheme 1 Synthesis scheme of Part A: MgO–ZrO2 support and Part B: Cu/MgO–ZrO2 catalysts. | ||
Characterisation of catalysts
The prepared catalysts were characterized using advanced techniques, including XRD, N2 physisorption analysis, H2-TPR, CO2-TPD, FE-SEM, HR-TEM, XPS, and TGA. XRD characterization was done with a PROTO AXRD® benchtop powder diffractometer with a radiation source of Cu-Kα. Diffraction patterns were recorded with a step size of 0.02° in the region 10°–80°. Recorded peaks were evaluated using the JCPDS (electronic database system). The specific surface area and porosity of the calcined samples were analyzed using a Micromeritics Tristar 3000 setup using the BET equation. Moreover, the basic strength of copper catalysts was investigated in a fixed bed column with a CO2-Temperature Programmed Desorption (TPD) analyzer (Micromeritics Autochem 2020). H2-Temperature Programmed Reduction (TPR) was performed to elucidate the reducibility of the catalysts on a Micromeritics Autochem 2020 instrument. The M/s FEI Quanta 200F instrument was used for Field Emission-Scanning Electron Microscopy (FE-SEM) analysis with an X-ray source of tungsten filament doped with LaB6. Signals were detected with an ETD detector in a high vacuum. High Resolution Transmission Electron Microscopy (HR-TEM) analysis was done using a JEOL JEM 2100 microscope. X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Scientific NEXSA XPS spectrometer that featured a monochromatic Al Kα X-ray source operating at 15 kV. This analysis was carried out under conditions of ultrahigh vacuum ranging from 10−8 to 10−9 mbar. The reference for calibrating the binding energies of individual elements was established by utilizing the binding energy peak of C 1s at 284.8 eV. Thermogravimetry analysis (TGA) was carried out using a PerkinElmer 8000 thermogravimetric analyzer. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) analysis was carried out for determining elemental composition of the catalyst, which was performed on a Thermo Scientific iCAPTM RQ instrument.General method for CO2 hydrogenation to methyl formate
The CO2 hydrogenation reaction was conducted in a high-pressure batch reactor (25 mL, Parr Instrument Company) equipped with a mechanical stirrer. The autoclave reactor was charged with 2 mL of methanol and 0.08 g of catalyst, and then it was closed. After sealing, the reactor was flushed with N2 twice to remove air, followed by pressurization to a final total pressure of CO2/H2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, p/p) varying from 6 to 12 MPa at room temperature. The reactor was then brought to the desired reaction temperature, and stirring was started at a speed of 400 rpm, which was considered as the starting of the reaction. After the completion of the reaction, the reactor was depressurized at room temperature, the vessel was opened, and the reaction solution was recovered by centrifugation.
:1, p/p) varying from 6 to 12 MPa at room temperature. The reactor was then brought to the desired reaction temperature, and stirring was started at a speed of 400 rpm, which was considered as the starting of the reaction. After the completion of the reaction, the reactor was depressurized at room temperature, the vessel was opened, and the reaction solution was recovered by centrifugation.
Analysis of products
The gaseous products formed in the reaction were analyzed using an Agilent 7890B GC with a thermal conductivity detector and a flame ionization detector. The liquid products were analyzed by TLC and 1H NMR. 1H NMR of the reaction mixture was carried out on a 500 MHz Bruker Avance III spectrometer equipped with a 5 mm broadband fluorine observe (BBFO) probe. 10% w/v of the product mixture was prepared in CDCl3 for all the measurements. All the experiments were carried out with a 1H π/2 pulse of 12.9 μs. Sixteen transients were co-added, and the spectrum was processed with lb = 0.03 Hz, exponentially multiplied, Fourier transformed, phase-corrected and referenced to TMS. 5 s relaxation delays were taken between successive measurements. The concentration of methyl formate was determined by plotting a standard calibration curve using a graphical method. The injection volume of the sample was 10 μL, which was used in a standard calibration curve with different concentrations of methyl formate. Five solutions of methyl formate and methanol with different concentrations were made for the calibration curve. The CO2 conversion, selectivity, TON and TOF were determined with the following formulae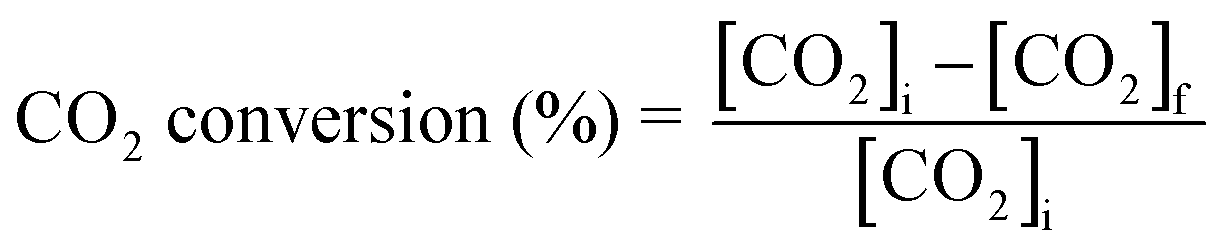 | (1) |
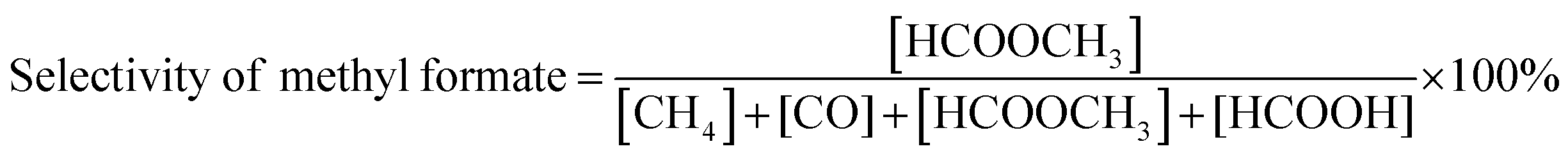 | (2) |
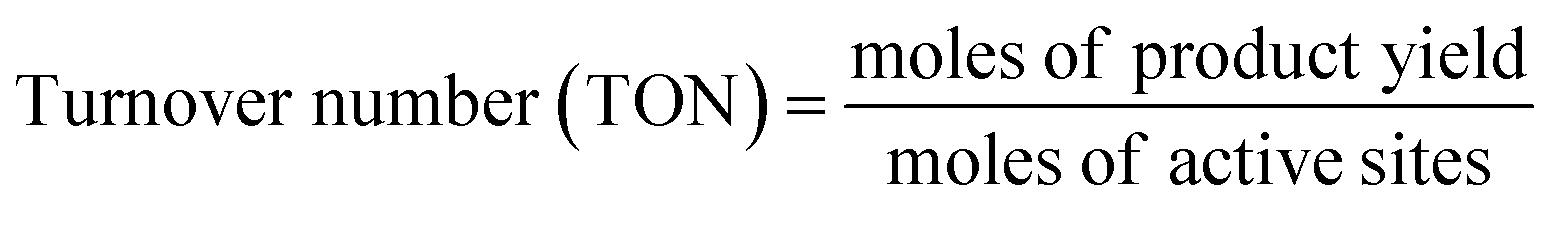 | (3) |
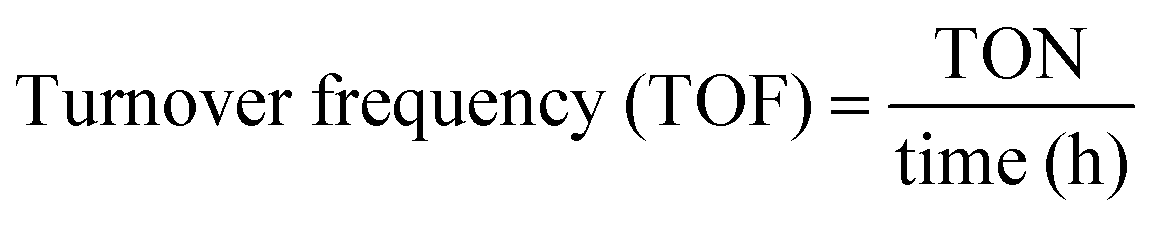 | (4) |
Results and discussion
Catalyst performance
The activity of Cu catalysts supported over MgO–ZrO2 for the hydrogenation of CO2 to MF in the presence of MeOH was systematically studied in a 25 mL batch reactor. Prior to any reaction, the catalyst was reduced in the dynamic flow of H2 and N2 (H2:N2 = 1:2) at 450 °C for 2 h.
The catalytic performance of xC-yMZ catalysts was studied to evaluate the effect of Cu loading, as shown in Fig. 2(a). The 20MZ support alone exhibited no catalytic activity in the conversion of CO2 to MF. However, when 2% wt Cu was added to the MgO–ZrO2 support (2C-20MZ), the conversion rate of CO2 to MF reached 10.31%, with selectivity for MF exceeding 99%. This result confirmed that metallic copper is crucial for the hydrogenation of CO2 to MF by facilitating the dissociation of H2 molecules.22 With the increase in the Cu loading up to 4% wt (4C-20MZ), a notable improvement in the catalyst activity was observed with a CO2 conversion of 13.52% and selectivity > 99%. This enhancement can be attributed to the greater number of active sites for H2 spillover resulting from the increased Cu loading. The highest catalytic activity was observed with 5% wt loading of Cu (5C-20MZ), achieving a maximum CO2 conversion of 14.01% and the selectivity exceeding 99%. However, upon increasing the Cu loading to 6 wt% (6C-20MZ), a slight decline in catalytic performance was observed, with CO2 conversion dropping to 13.77%. This reduction can be attributed to a decrease in active sites caused by the agglomeration of Cu particles, as indicated by the larger particle size observed in the TEM analysis. Furthermore, the increase in Cu loading up to 8 (8C-20MZ) and 10% wt (10C-20MZ) resulted in a gradual decline in CO2 conversion to 13.27% and 12.63%, respectively, while maintaining a selectivity of over 99%. As mentioned earlier, this decrease can be attributed to the sintering of Cu atoms, which reduced the number of active sites available for H2 spillover.
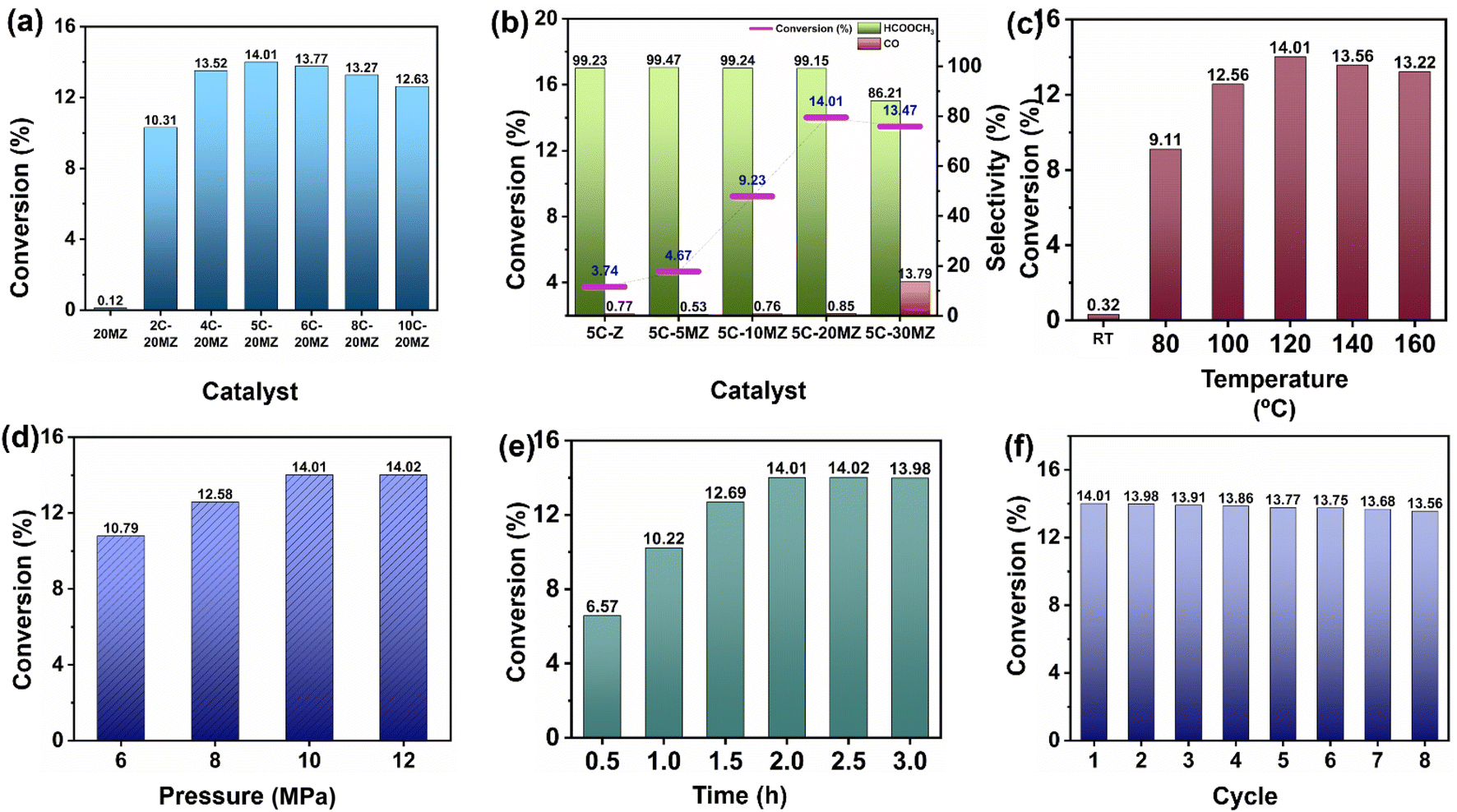 | ||
| Fig. 2 (a) Impact of Cu loading on CO2 conversion (reaction conditions: catalyst: 80 mg, pressure: 10 MPa, temperature: 120 °C, and time: 2 h), (b) impact of MgO loading on CO2 conversion (reaction conditions: catalyst: 80 mg, pressure: 10 MPa, temperature: 120 °C, and time: 2 h), (c) effect of temperature (reaction conditions: catalyst: 80 mg, pressure: 10 MPa, and time: 2 h), (d) effect of pressure (reaction conditions: catalyst: 80 mg, temperature: 120 °C, and time: 2 h), (e) effect of time (reaction conditions: catalyst: 80 mg, pressure: 10 MPa, and temperature: 120 °C), and (f) recyclability (reaction conditions: catalyst: 80 mg, pressure: 10 MPa, temperature: 120 °C, and time: 2 h). | ||
Surface basicities of the catalysts are essential for CO2 hydrogenation by enabling acid–base interactions between acidic CO2 and the catalysts' basic sites. To study the impact of MgO on the catalytic performance for the hydrogenation of CO2 to MF, a series of 5% wt. Cu catalysts were synthesized by varying the loading of MgO from 5 to 30% wt. Fig. 2(b) illustrates the effect of MgO on the catalysts for the hydrogenation of CO2 to MF. In the absence of MgO, the 5C-Z catalyst demonstrated a low CO2 conversion of just 3.74%. A slight improvement was observed in catalytic activity with a CO2 conversion of 4.57%, while 5% MgO (5C-5MZ) was incorporated in the catalyst. Upon the addition of 10% MgO (5C-10MZ), a dramatic increase in CO2 conversion was observed, with the conversion rate soaring to 9.23%. This nearly threefold improvement compared to the 5C-Z catalyst underscores the significant role that MgO plays in enhancing the catalytic performance. The presence of MgO likely increases the surface basicity, which is crucial for CO2 adsorption and activation, thereby facilitating the hydrogenation process. A further increase in the MgO loading to 20% (5C-20MZ) resulted in an even higher CO2 conversion rate of 14.01%, indicating that the optimal MgO content significantly boosts the catalytic activity. However, when the MgO content was increased to 30% (5C-30MZ), a negative impact on the catalyst activity was observed. The CO2 conversion rate decreased to 13.47%, and the selectivity for MF lowered to 86.21%, while the production of CO as a competitive by-product indicated a decline in the overall catalytic efficiency. This decrease in performance with excessive MgO can be attributed to the overly strong acid–base interactions between CO2 and the catalyst surface, which hinder the desorption of reaction products. Additionally, excessive basicity may promote the reverse water–gas shift reaction (RWGS), an unfavourable side reaction during CO2 hydrogenation that produces CO and water, further reducing the efficiency of MF production.
In addition to the Cu and MgO loading, the catalytic activity of 5C-20MZ for CO2 hydrogenation to MF is significantly influenced by various reaction parameters, including the temperature, pressure, and reaction time. To thoroughly understand the effect of temperature on the catalytic performance, a study was conducted over a temperature range from 80 °C to 160 °C while applying a total CO2:H2 (1:1) pressure of 10 MPa. The results of this study are illustrated in Fig. 2(c). At room temperature, the catalyst showed no significant CO2 conversion, indicating that the reaction requires an elevated temperature to proceed. When the temperature was increased to 80 °C, a noticeable improvement in catalytic activity was observed, achieving a CO2 conversion rate of 9.11% with the MF selectivity exceeding 99%. A further increase of the temperature to 100 °C led to an even higher CO2 conversion of 12.56% while still maintaining high selectivity for MF. This trend of increasing catalytic activity with temperature continued, reaching its peak at 120 °C. At this optimal temperature, the catalyst exhibited the highest CO2 conversion rate of 14.01%, with MF selectivity remaining above 99%. This suggests that 120 °C is the most favourable temperature for this catalytic system under the given conditions. However, when the temperature was further increased to 140 °C and 160 °C, no significant improvement in catalytic activity was observed. The CO2 conversion rates slightly decreased to 13.56% and 13.22%, respectively. This decline can be attributed to the exergonic nature of the CO2 hydrogenation reaction to MF. As the temperature increased, the equilibrium constant for the reaction decreased, resulting in lower CO2 conversion despite the availability of higher thermal energy. This phenomenon is consistent with the principles of chemical thermodynamics, where higher temperatures can shift the equilibrium of an exergonic reaction towards the reactants.
Similarly, the impact of total CO2 (1:1) pressure on the hydrogenation reaction for the conversion of CO2 to MF was examined methodically. Fig. 2(d) showcases the CO2 conversion to MF at different pressures, spanning from 6 to 12 MPa while maintaining a constant temperature of 120 °C. A significant enhancement in CO2 conversion was noted, increasing from 10.79% to 12.58% as the total pressure increased from 6 MPa to 8 MPa. This upward trend continued, with the CO2 conversion reaching 14.01% when the pressure was further increased to 10 MPa. This demonstrates a clear linear relationship between conversion and pressure, which aligns with Le Chatelier's principle. According to this principle, increasing the pressure in a chemical reaction favours the formation of products by shifting the equilibrium toward the right. In addition to this, higher CO2 pressures lead to improved adsorption efficiency on catalytic surfaces, resulting in a higher catalytic activity for CO2 conversion. However, no significant increase in conversion was observed when the total pressure was increased beyond 10 MPa. This phenomenon can be attributed to several factors. One possibility is catalyst deactivation, which can occur due to over-acidification of the reaction medium. As the pressure increases, the concentration of reactants might reach a saturation point, limiting further improvement in the conversion efficiency. Additionally, the reaction dynamics might change at higher pressures, leading to potential side reactions or reduced efficiency in the desired hydrogenation process.
Among the various parameters influencing the catalytic activity of 5C-20MZ for CO2 hydrogenation to MF, the reaction time was carefully examined to understand its effect on conversion rates. The results highlighted a clear correlation between the reaction time and the efficiency of CO2 conversion (Fig. 2(e)). The experimental data demonstrated that as the reaction time increased, there was a progressive enhancement in the conversion of CO2 to MF. Specifically, at a reaction time of 0.5 hours, the conversion rate was 6.57%. This rate improved to 10.22% at 1 hour and further to 12.69% at 1.5 hours. The maximum conversion rate of 14.01% was achieved at a reaction time of 2 hours, and this rate remained constant even when the reaction time was extended to 2.5 and 3 hours.
This indicates that the catalytic activity reached a saturation point at 2 hours, beyond which no significant increase in conversion was observed. The consistency in conversion beyond 2 h suggests that the reaction equilibrium was achieved, and the active sites on the catalyst were fully utilized within this time frame. Additionally, the constant selectivity for MF above 99% throughout these durations underscores the catalyst's efficiency and stability in promoting the desired reaction without significant side reactions.
The recyclability of the 5C-20MZ catalyst for CO2 hydrogenation to MF was extensively evaluated, showcasing its outstanding stability and consistent catalytic performance across multiple cycles, as depicted in Fig. 2(f). During the first cycle, the 5C-20MZ catalyst achieved a notable CO2 conversion rate of 14.01% with a selectivity of over 99%, highlighting its high efficiency in facilitating the targeted reaction. After completing the first cycle, the catalyst was recovered and reused in subsequent cycles to thoroughly assess its durability and robustness. Remarkably, even after several cycles, the 5%Cu/MgO–ZrO2 catalyst continued to demonstrate high catalytic activity. The CO2 conversion rate was slightly reduced; however, it remained high at 13.56% in the eighth cycle while maintaining an MF selectivity of more than 99%. This minimal decrease in catalytic performance over multiple cycles indicates the exceptional recyclability and stability of the 5%Cu/MgO–ZrO2 catalyst.
Plausible mechanism
The plausible mechanism of CO2 hydrogenation to methyl formate in the presence of methanol over the Cu/MgO–ZrO2 catalyst involves a multi-step process where CO2 is converted into intermediate products before forming methyl formate, as illustrated in Scheme 2. Initially, CO2 molecules adsorb onto the active sites of the Cu/MgO–ZrO2 catalyst. The presence of ZrO2 enhances the adsorption of CO2 due to its unique properties, which include a high surface area and the ability to interact synergistically with copper. This adsorption leads to the formation of formate or carboxylate species, which are crucial intermediates in the reaction. Besides this, Cu facilitates the H2 spillover process. Once the CO2 is adsorbed, it undergoes hydrogenation to form formates, where the hydrogen reacts with the adsorbed CO2 to produce intermediates such as formic acid. In the next phase, methanol acts as both a solvent and a reactant, promoting the esterification reaction between the formed formate and itself. During this esterification process, the formate reacts with methanol to produce methyl formate and water. MgO in the catalysts plays a vital role in the esterification process by improving the nucleophilicity of the methanol. In the presence of MgO, methanol can more readily attack the electrophilic carbonyl carbon of the formic acid resulting in the formation of intermediates that ultimately yield the desired ester product.23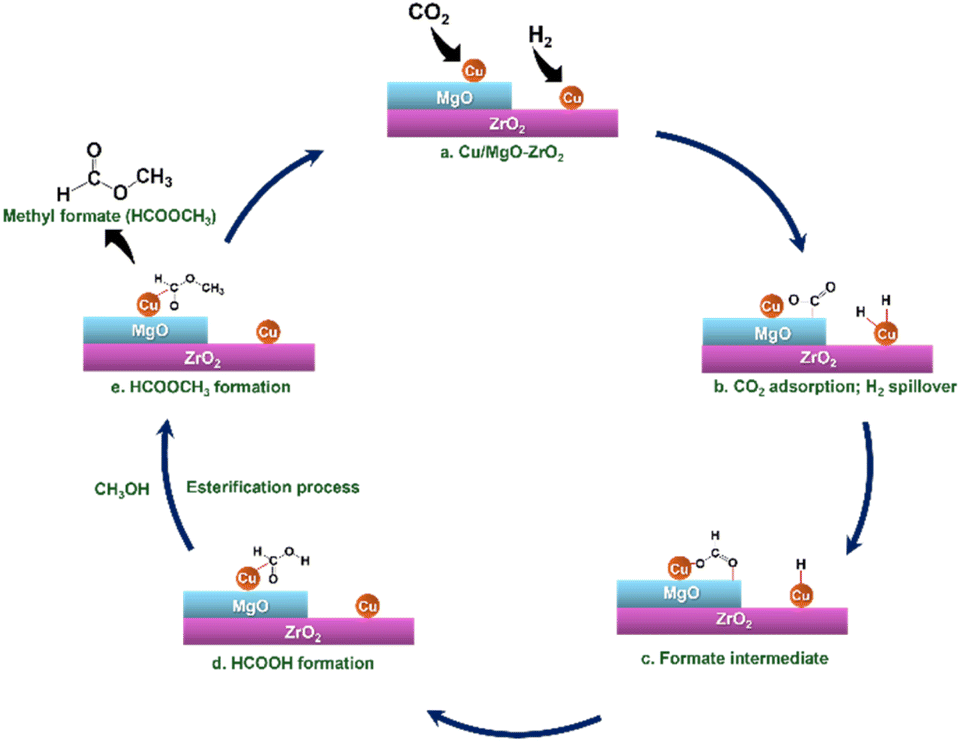 | ||
| Scheme 2 Plausible mechanism for the hydrogenation of CO2 to MF in the presence of MeOH over the Cu/MgO–ZrO2 catalyst. | ||
Basic characterization
The powder X-ray diffraction (XRD) analysis was performed to investigate the structural properties of the 20MZ support and the 5C-20MZ catalyst, and the corresponding diffractogram is illustrated in Fig. 3(a). For both the freshly synthesized 20MZ support and the 5C-20MZ catalyst, several distinct diffraction peaks were observed, specifically at 2θ angles of 30.43°, 35.38°, 50.41°, 60.23°, and 74.59°. These peaks correspond to the (101), (110), (112), (211), and (220) lattice planes, respectively, which are characteristic of a tetragonal ZrO2 crystalline phase, in excellent agreement with the standard data from the JCPDS card no. 81-1455.24 Moreover, a slight, nevertheless noticeable peak at 29.20° was detected, which is indexed to diffractions from the (−111) lattice plane of the monoclinic phase, aligning with the JCPDS PDF card no. 37-1484.25,26 This phase transition is attributed to the calcination of the catalyst at 700 °C, as calcination temperatures exceeding 400 °C result in the transformation of zirconia from the tetragonal to the monoclinic phase.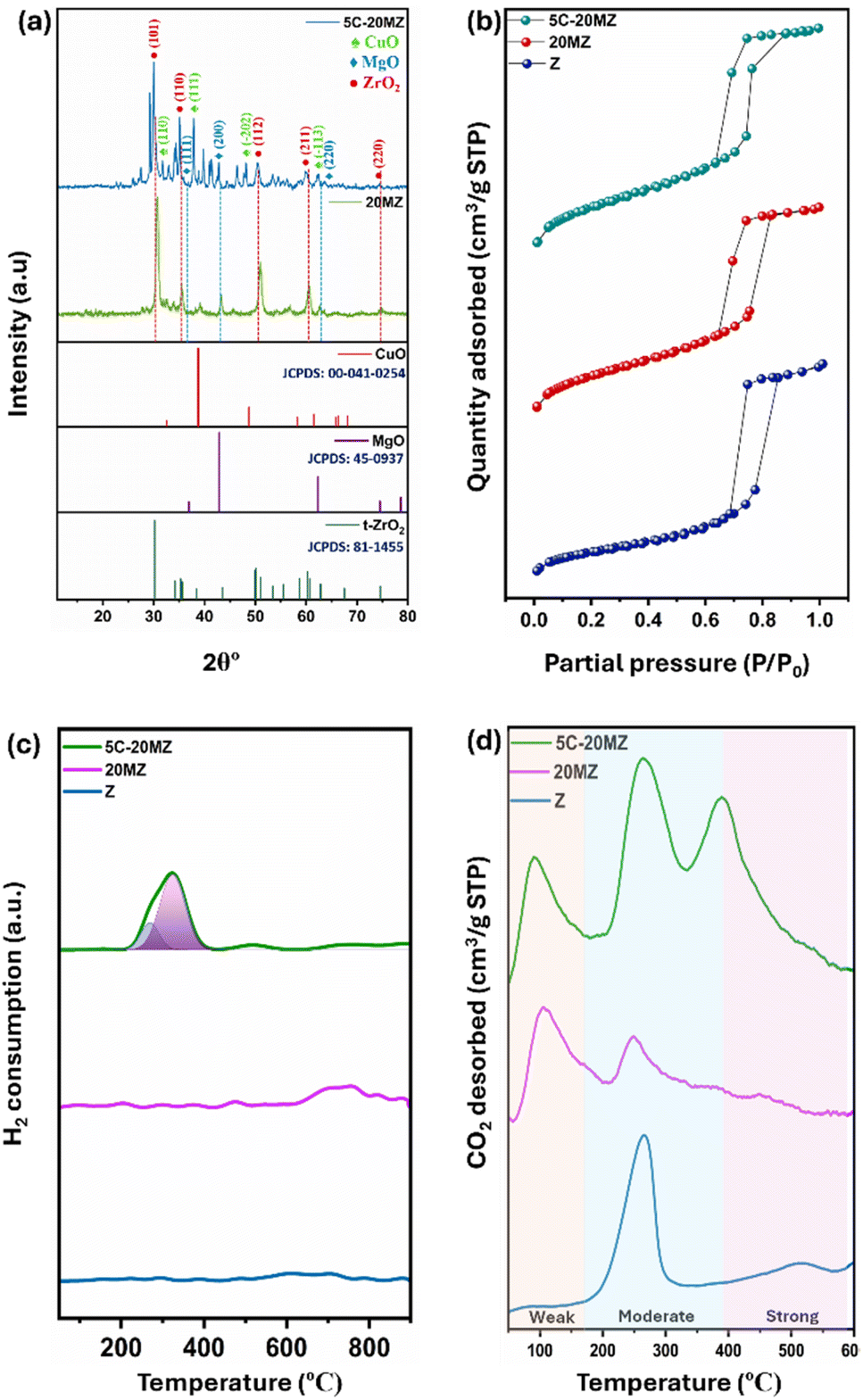 | ||
| Fig. 3 (a) XRD diffractograms of 20MZ and 5C-20MZ, (b) N2 physisorption isotherms of Z, 20MZ, 5C-20MZ, (c) H2-TPR profiles of Z, 20MZ, and 5C-20MZ, and (d) CO2-TPD profiles of Z, 20MZ, and 5C-20MZ. | ||
The occurrence of MgO was also confirmed by characteristic diffraction peaks at 2θ angles of 36.9°, 42.9°, and 62.3°. These peaks correspond to the (111), (200), and (220) planes of MgO, respectively, which is consistent with the JCPDS PDF card no. 45-0937.27,28 This confirms the successful incorporation of MgO into the support structure, which plays a crucial role in the overall catalytic performance of the material.
Furthermore, the characteristic diffraction peaks of CuO were identified at 2θ angles of 33.9°, 37.8°, 49.4°, and 61.7°. These peaks are associated with the (110), (111), (−202), and (−113) crystal planes, respectively, and correspond closely with the standard JCPDS card no. 00-041-0254.29
N2 physisorption analysis was performed to thoroughly investigate the porosity and surface area of the synthesized catalyst, and the isotherms generated from this analysis are presented in Fig. 3(b). The N2 adsorption–desorption isotherms for all the catalysts exhibited a type IV pattern, with an H4 hysteresis loop, indicating the presence of well-defined mesopores predominantly within a narrow pore size ranging between 9 and 12 nm. The ZrO2 support, denoted as Z, demonstrated a substantial BET surface area of 78 m2 g−1 alongside an average pore size of 11.8 nm. However, when MgO was incorporated into the ZrO2 matrix, a noticeable decrease in the BET surface area to 66 m2 g−1 was observed. This reduction can be attributed to pore blocking, where the introduction of MgO likely leads to partial obstruction of the pores on the catalyst surface. Furthermore, the addition of Cu nanoparticles resulted in an even further reduction of the surface area, bringing it down to 61 m2 g−1. The presence of Cu nanoparticles could potentially block more channels within the catalyst, resulting in an additional decrease in the surface area.
The H2-TPR analysis was carried out to examine the reducibility of the catalysts, with the TPR profiles for the Z, 20MZ, and 5C-20MZ catalysts presented in Fig. 3(c). The profiles for Z and 20MZ did not display any significant peaks as both MgO and ZrO2 are known to be irreducible supports. However, with the introduction of Cu nanoparticles, a distinct peak emerged at around 250 °C, indicating the reduction of highly dispersed CuO nanoparticles supported on MgO–ZrO2. Additionally, the small and broad peaks observed beyond 400 °C correspond to the reduction of larger, more agglomerated CuO particles, which are present in minimal amounts. The H2-TPR profile suggests that all the reducible sites in the catalysts are located in the low-temperature region, which implies that the CuO nanoparticles are well dispersed on the ZrO2 surface, which is beneficial for the catalytic activity since reduced copper sites are often the active centres for hydrogenation reactions.30
The temperature-programmed desorption of CO2 (CO2-TPD) was performed to gain insights into the surface basicity of the catalysts, and the desorption profiles are illustrated in Fig. 3(d). The analysis categorizes the basic sites into weak, moderate, and strong, with specific temperature ranges corresponding to each category. A more detailed quantification of these sites is outlined in Table S2.† The amount of CO2 desorbed revealed that the basicity of the catalysts increases when MgO is incorporated into the ZrO2 matrix, as MgO introduces a greater number of surface basic sites on the ZrO2 support. In terms of temperature ranges, desorption between 90 and 180 °C is associated with weak basic sites, which typically contribute to physical adsorption rather than chemical activation of CO2. While these sites play a role in initial CO2 capture, they are not as effective in catalyzing subsequent reactions. However, desorption between 180 and 400 °C is linked to moderate basic sites, which are more active for CO2 adsorption and activation for further reaction.31 Desorption beyond 400 °C corresponds to strong basic sites, which are the most effective in holding and activating CO2 molecules for hydrogenation.32 The CO2-TPD profiles indicate that pure ZrO2 primarily exhibits a peak in the moderate basic site region, suggesting that ZrO2 alone offers a limited but significant capability for CO2 activation. However, when MgO is incorporated, the catalyst surface gains weak basic sites in addition to those moderate sites, enhancing the overall basicity of the catalysts and thereby broadening the range of CO2 adsorption capabilities. Furthermore, the addition of Cu to the MgO–ZrO2 catalyst results in the appearance of a third desorption peak at around 400 °C. This peak signifies the presence of strong basic sites, which were not prominent in the MgO–ZrO2 support alone. The presence of these strong basic sites, introduced by Cu, further elevates the catalyst's basicity, making it even more effective in CO2 adsorption and subsequent hydrogenation reactions to MF.
Field emission scanning electron microscopy (FE-SEM) was conducted on the best-performing catalyst, 5C-20MZ, to examine the morphology of the synthesized material. As illustrated in Fig. 4(a)–(c), the 5C-20MZ catalyst exhibited an irregular lamellar structure with a smooth, clean surface. The energy-dispersive X-ray spectroscopy (EDS) spectra of the 5C-20MZ nanoparticles, shown in Fig. 4(d), confirmed the presence of Cu, Mg, Zr, and O in the nano-mixed oxide. Additionally, Fig. 4(e)–(h) present the EDS mapping, where the distribution of Cu, Mg, Zr, and O elements is clearly visible. This confirms that Cu is evenly dispersed throughout the MgO–ZrO2 particles, a finding further corroborated by TEM-EDS analysis.
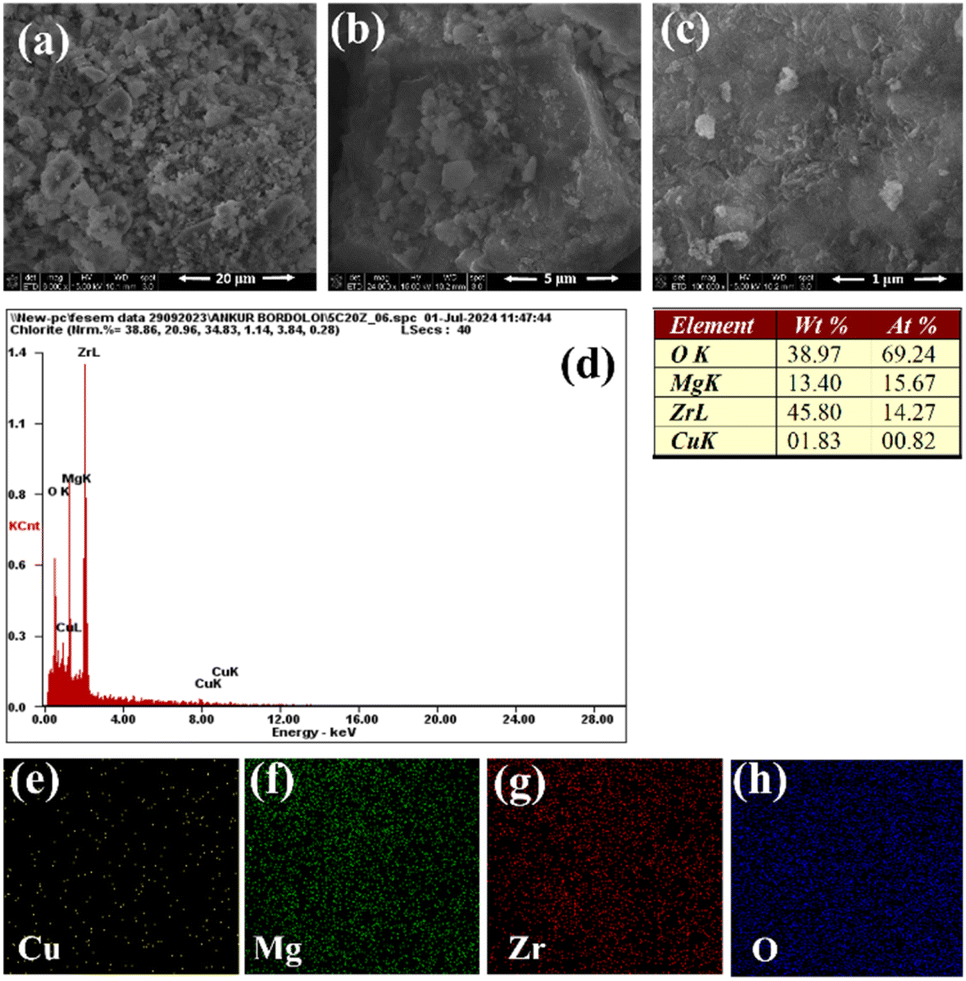 | ||
| Fig. 4 (a–c) FE-SEM images, (d) EDX spectrum, and (e–h) elemental mapping of 5C-20MZ. | ||
Transmission electron microscopy (TEM) analysis was carried out to study the surface morphology and determine the particle size. As seen from Fig. 5(a) and (b), the uniform elongated platelets and star-like structures were composed of short nanorods with a diameter of approximately 5 nm and a length of approximately 10 nm. This confirmed that the nanocrystals composed of star-like ZrO2 nanostructures were anisotropic. Also, the TEM images depicted the mono-dispersivity and spherical shape of the Cu nanoparticles on the surface. HR-TEM analysis showed that the Cu nanoparticles possess a size of 9.8 nm and exhibited an interplaner d-spacing value of 0.203 nm, which indicated the presence of the Cu (111) plane, as shown in Fig. 5(c) and (d). The selective area electron diffractogram (SAED) showed that the catalyst is highly crystalline in nature (Fig. 5(e)). The EDS analysis showed the composition of all the elements on the surface (Fig. 5(g)). Fig. 5(h)–(k) demonstrate the elemental composition, which shows that Cu particles are highly dispersed over the catalyst's surface.
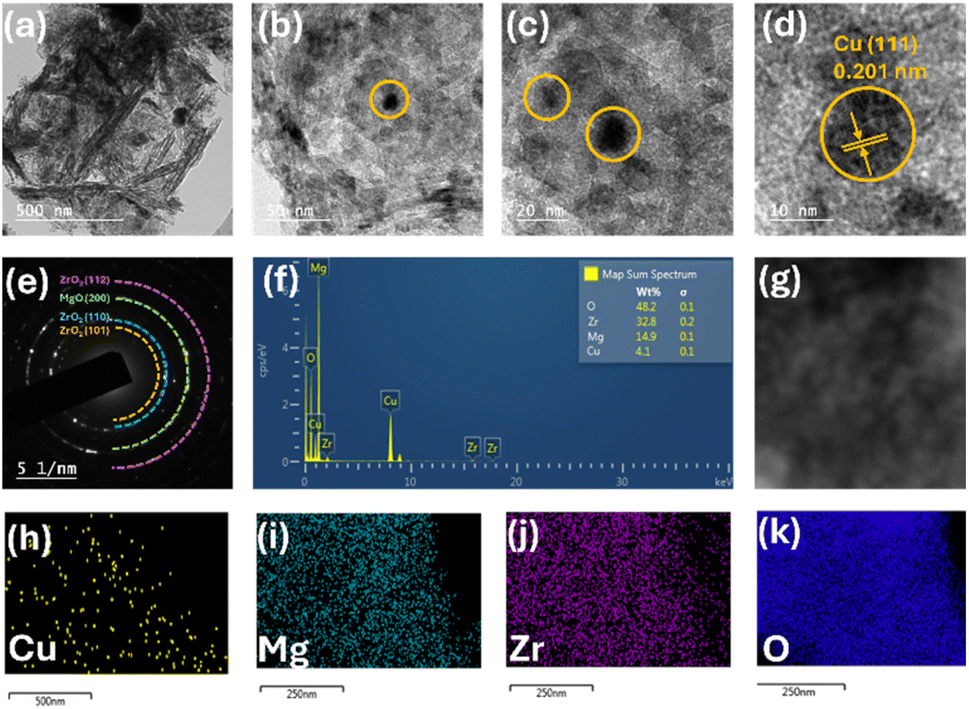 | ||
| Fig. 5 (a and b) TEM images of 5C-20MZ, (c and d) HR-TEM images of 5C-20MZ, (e) SAED pattern, (f) EDX elemental composition, (g) electron microscopy image and (h–k) elemental mapping. | ||
X-ray photoelectron spectroscopy was used to analyze the oxidation states of the elements present in the reduced 5C-20MZ catalyst. The fitted spectra of the Cu2p3/2 region featured two distinct peaks at 932.48 eV and 934.68 eV, indicating the presence of metallic Cu0/Cu1+ and Cu2+, respectively (Fig. 6(a)).33,34 The existence of Cu in +1 and +2 oxidation states may be attributed to the oxidation of surface Cu during sample handling, as Cu is an easily oxidized material at room temperature. Furthermore, Cu LMM analysis revealed that the ratio between Cu0 and Cu1+ was 2, as shown in Fig. 6(b). The peak at 940.18 eV is the satellite peak of Cu, which suggested a significant strong metal–support interaction (SMSI) between Cu and MgO–ZrO2 of the catalyst. Fig. 6(c) shows a single peak at 1303.3 eV, assigned to the +2-valence state of the MgO–ZrO2 catalyst. Fig. 6(d) depicts the Zr3d core level spectra showing two peaks at a binding energy of 181.18 eV in the Zr3d5/2 region and 183.58 eV in the Zr3d3/2 region, representing the Zr4+ oxidation state.35–37 As illustrated in Fig. 6(e), the oxygen 1s peak at 530.1 eV corresponds to the surface lattice oxygen, the peak at 531.3 eV corresponds to the oxygen deficiency on the surface, and the peak at 533.4 eV resulted due to the surface hydroxyl (–OH) groups.38
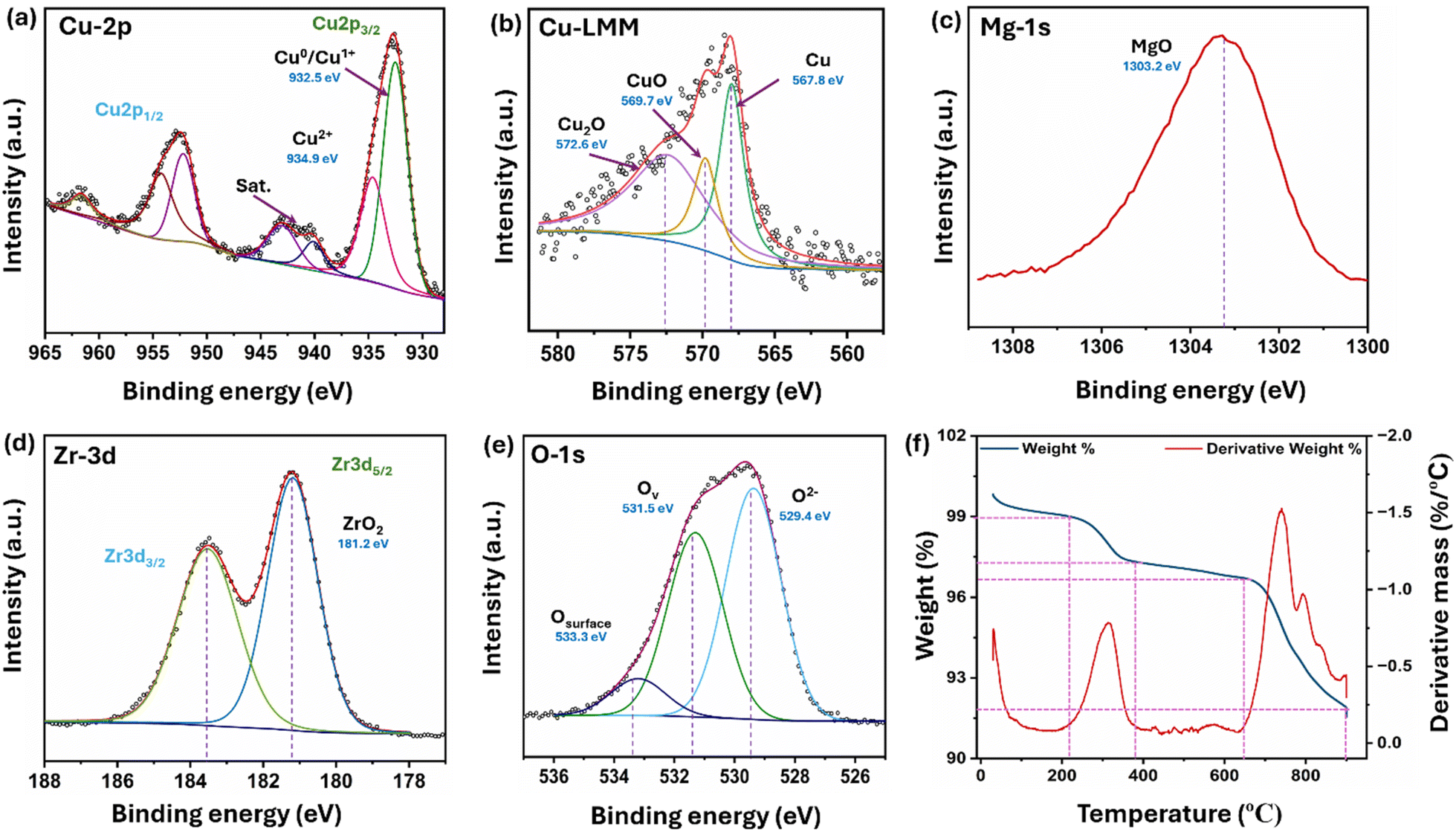 | ||
| Fig. 6 XPS patterns for (a) Cu 2p, (b) Cu LMM, (c) Mg 1s (d) Zr 3d, and (e) O 1s; (f) TGA profile of 5C-20MZ. | ||
The thermal stability of the 5C-20MZ catalyst was thoroughly investigated using thermogravimetry analysis (TGA) and differential thermal analysis (DTA). The specific focus of this investigation was on CuO/MgO–ZrO2, and the resulting thermogram is illustrated in Fig. 6(f). The temperature range for these analyses spanned from 30 °C to 850 °C, with a controlled heating rate of 5 °C per minute. The thermogravimetry (TG) profile showed two distinct phases of weight loss corresponding to the exothermic peaks in the DTA profile. The first weight loss occurred between 212 °C and 387 °C, resulting in a 1.4% reduction in weight. This phase likely represents the loss of adsorbed water or the decomposition of surface hydroxyl groups. In the second phase of weight loss, a minimal reduction in weight (4.6%) was observed between 644 °C and 850 °C. This stage is associated with the more substantial breakdown of the remaining hydroxide structure, leading to the formation of the CuO/MgO–ZrO2 phase.
The results of the analysis are further supported by DTA analysis, where the thermal decomposition of the hydroxide involves two major exothermic peaks, as observed in the differential thermal analysis curve. These peaks indicate significant thermal events, specifically related to the breakdown of the hydroxide structure. The first exothermic peak occurs at around 304 °C, while the second peak is observed at 750 °C. These peaks suggest that the leftover hydroxide maintains its thermal stability up to nearly 644 °C, beyond which decomposition begins. This observation suggests that the catalyst is highly stable up to 640 °C, beyond which there is a significant loss of 6% weight, possibly due to the decomposition of hydroxides left over the surface, as mentioned above.
Product analysis
The reaction mixture was analyzed by employing a 500 MHz 1H-NMR spectrometer using CDCl3 as a solvent and TMS as an internal reference. Methanol and methyl formate both contain methyl (–CH3) groups, but their chemical environments differ due to the presence of electronegative oxygen atoms in the –COOCH3 group of methyl formate. This difference in the chemical environment causes the protons in the –COOCH3 group to experience a deshielding effect, making them resonate at different chemical shift (ppm) values in the 1H-NMR spectrum compared to the protons in the CH3 group of methanol.In the recorded 1H-NMR spectrum (Fig. 7), the methyl group in methanol typically appears as a sharp singlet at around 3.32 ppm. On the other hand, the –COOCH3 group in methyl formate resonates in a broader range, typically between 3.4 and 4.8 ppm. The precise chemical shift of the –COOCH3 group can vary depending on the specific structure and conditions, and in this case it appears at 3.69 ppm. The integration of these peaks provides quantitative information about the relative amounts of methanol and methyl formate in the mixture. The hydroxyl (–OH) proton in methanol does not show up in the 1H-NMR spectrum because of deuterium exchange with the solvent CDCl3. This means that only the signals corresponding to the –CH3 groups of methanol and methyl formate are observed.
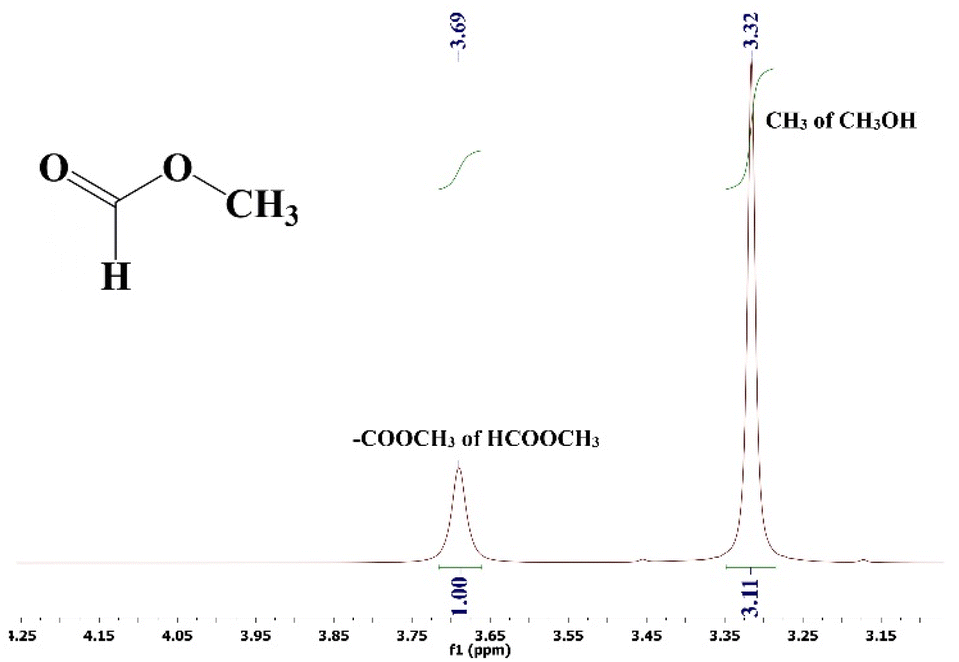 | ||
| Fig. 7 1H-NMR spectra of the isolated reaction mixture in CDCl3. | ||
Conclusions
In conclusion, supported Cu nanocatalysts were reported as non-noble catalysts for the synthesis of MF through CO2 hydrogenation in the presence of methanol. The introduction of MgO in the catalysts played a dual role by increasing the catalyst surface's basicity for CO2 adsorption and activation and facilitating the esterification between MeOH and HCOOH to produce MF. Among all the catalysts, 5Cu/20MgO–ZrO2 with optimum loading of Cu and MgO exhibited very high activity with 14.01% CO2 conversion and more than 99% selectivity for the MF under optimum reaction conditions without any basic additive. The effectiveness of the catalysts was largely influenced by the properties of the support and the size of the Cu nanoparticles. The turnover frequency (TOF) of the 5Cu/20MgO–ZrO2 catalyst was measured to exceed 185 h−1 at 120 °C and 10 MPa. Therefore, the catalytic process was likely to have progressed by the formation of a formic acid intermediate, followed by the esterification of formic acid with methanol to yield MF.Data availability
Data will be made available on suitable request.Author contributions
Jyotishman Kaishyop was responsible for the work plan, catalyst synthesis and characterization, as well as catalyst evaluation, and drafting of the original manuscript. Arpan Mukherjee handled data curation and contributed to the original draft writing. Abhay Giri Goswami was involved in the synthesis and characterization of catalysts. Tuhin Suvra Khan helped with formal analysis, as well as with reviewing and editing of the manuscript. Ankur Bordoloi conceptualized and oversaw the project, secured the funding, and participated in the review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JK expresses gratitude to the DST-Inspire Fellowship, New Delhi, India, for the fellowship support. A. B. sincerely acknowledges CSIR-India (HCP-0048) for the grant. The authors extend their thanks to the Director of CSIR-IIP, Dehradun, for his valuable encouragement. Additionally, they appreciate the Analytical Science Division of CSIR-IIP for providing analytical services.Notes and references
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- T. T. Le, P. Sharma, B. J. Bora, V. D. Tran, T. H. Truong, H. C. Le and P. Q. P. Nguyen, Int. J. Hydrogen Energy, 2024, 54, 791–816 CrossRef CAS.
- R. Sang, Z. Wei, Y. Hu, E. Alberico, D. Wei, X. Tian, P. Ryabchuk, A. Spannenberg, R. Razzaq, R. Jackstell, J. Massa, P. Sponholz, H. Jiao, H. Junge and M. Beller, Nat. Catal., 2023, 6(6), 543–550 CrossRef CAS.
- J. Kaishyop, T. S. Khan, S. Panda, P. R. Chandewar, D. Shee, T. C. R. Rocha, F. C. Vicentin and A. Bordoloi, Green Chem., 2023, 25, 7729–7742 RSC.
- C. Adami, M. Slany, J. Karl, G. Kaibel, M. Schäfer, P. Zehner and M. Röper, Method for producing methyl formate, US Pat., US7053239B2, 2003, vol. 8 Search PubMed.
- O. Kröcher, R. A. Köppel and A. Baiker, Chem. Commun., 1997, 453–454 RSC.
- K. W. Huang, J. H. Han, C. B. Musgrave and E. Fujita, Organometallics, 2007, 26, 508–513 CrossRef CAS.
- K. M. Kerry Yu and S. C. Tsang, Catal. Lett., 2011, 141, 259–265 CrossRef CAS.
- K. M. K. Yu, C. M. Y. Yeung and C. T. Shik, J. Am. Chem. Soc., 2007, 129, 6360–6361 CrossRef CAS.
- J. Chen, F. Xin, S. Qin and X. Yin, Chem. Eng. J., 2013, 230, 506–512 CrossRef CAS.
- C. Wu, Z. Zhang, Q. Zhu, H. Han, Y. Yang and B. Han, Green Chem., 2015, 17, 1467–1472 RSC.
- J. Kaishyop, J. Gahtori, S. Dalakoti, M. J. Gazi, T. S. Khan and A. Bordoloi, J. Mater. Chem. A, 2024, 12, 8457–8473 RSC.
- C. L. Chiang, K. S. Lin and H. W. Chuang, J. Clean. Prod., 2018, 172, 1957–1977 CrossRef CAS.
- J. Gahtori, J. Kaishyop, G. Singh, T. S. Khan, F. C. Vicentin, T. C. R. Rocha and A. Bordoloi, Chem. Commun., 2023, 59, 12475–12478 RSC.
- V. K. Shrivastaw, J. Kaishyop, T. S. Khan, D. Khurana, G. Singh, S. Paul, B. Chowdhury and A. Bordoloi, ChemCatChem, 2024, e202400534 CrossRef CAS.
- S. K. Sharma, T. S. Khan, R. K. Singha, B. Paul, M. K. Poddar, T. Sasaki, A. Bordoloi, C. Samanta, S. Gupta and R. Bal, Appl. Catal., A, 2021, 623, 118239 CrossRef CAS.
- C. Chizallet, M. L. Bailly, G. Costentin, H. Lauron-Pernot, J. M. Krafft, P. Bazin, J. Saussey and M. Che, Catal. Today, 2006, 116, 196–205 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Lkariya and R. Noyori, J. Chem. Soc., Chem. Commun., 1995, 707–708 RSC.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS.
- M. Yadav, J. C. Linehan, A. J. Karkamkar, E. Van Der Eide and D. J. Heldebrant, Inorg. Chem., 2014, 53, 9849–9854 CrossRef CAS.
- X. Huang, Y. Ji, T. Wei, L. Jia, D. Yan and J. Li, Curr. Res. Green Sustain. Chem., 2021, 4, 100183 CrossRef CAS.
- X. Xin, P. Gao and S. Li, Catal. Sci. Technol., 2024, 14(18), 5439–5449 RSC.
- N. Zhong, L. Z. Cheong and X. Xu, Eur. J. Lipid Sci. Technol., 2014, 116, 97–107 CrossRef CAS.
- N. C. Horti, M. D. Kamatagi, S. K. Nataraj, M. N. Wari and S. R. Inamdar, Nano Express, 2020, 1, 010022 CrossRef.
- R. C. Garvie and M. V. Swain, J. Mater. Sci., 1985, 20, 1193–1200 CrossRef CAS.
- D. Tan, G. Lin, Y. Liu, Y. Teng, Y. Zhuang, B. Zhu, Q. Zhao and J. Qiu, J. Nanopart. Res., 2011, 13, 1183–1190 CrossRef CAS.
- T. Somanathan, V. M. Krishna, V. Saravanan, R. Kumar and R. Kumar, J. Nanosci. Nanotechnol., 2016, 16, 9421–9431 CrossRef CAS.
- K. D. Salman, H. H. Abbas and H. A. Aljawad, J. Phys. Conf., 2021, 1973, 012104 CrossRef CAS.
- I. Khaldari, M. R. Naghavi and E. Motamedi, RSC Adv., 2021, 11, 3346–3353 RSC.
- C. Zhong, X. Guo, D. Mao, S. Wang, G. Wu and G. Lu, RSC Adv., 2015, 5, 52958–52965 RSC.
- A. S. Al-Fatesh, Y. Arafat, H. Atia, A. A. Ibrahim, Q. L. M. Ha, M. Schneider, M. M. Pohl and A. H. Fakeeha, J. CO2 Util., 2017, 21, 395–404 CrossRef CAS.
- D. Goma, J. J. Delgado, L. Lefferts, J. Faria, J. J. Calvino and M. Á. Cauqui, Nanomaterials, 2019, 9, 1582 CrossRef CAS PubMed.
- J. Kaishyop, J. Gahtori, S. Dalakoti, M. J. Gazi, T. S. Khan and A. Bordoloi, J. Mater. Chem. A, 2024, 12, 8457–8473 RSC.
- C. E. Dubé, B. Workie, S. P. Kounaves, A. Robbat, M. L. Aksub and G. Davies, J. Electrochem. Soc., 1995, 142, 3357–3365 CrossRef.
- N. Li, Y. Tong, D. Yi, X. Cui and X. Zhang, J. Alloys Compd., 2021, 868, 158771–158771 CrossRef CAS.
- M. A. Aramendía, V. Boráu, C. Jiménez, A. Marinas, J. M. Marinas, J. A. Navío, J. R. Ruiz and F. J. Urbano, Colloids Surf. A Physicochem. Eng. Asp., 2004, 234, 17–25 CrossRef.
- A. Galtayries, R. Sporken, J. Riga, G. Blanchard and R. Caudano, J. Electron. Spectrosc. Relat. Phenom., 1998, 88–91, 951–956 CrossRef CAS.
- Z. Wang, R. Lin, Y. Huo, H. Li and L. Wang, Adv. Funct. Mater., 2022, 32, 2109503 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00478g |
| This journal is © The Royal Society of Chemistry 2024 |