
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-assisted (de)lithiation to enhance photoelectrochemical storage in quasi-solid-state Li-ion batteries†
Xin
Mi
ab,
Jun
Pan
 a,
Menglin
Duan
ab,
Fuqiang
Huang
*ac and
Peng
Qin
*ab
a,
Menglin
Duan
ab,
Fuqiang
Huang
*ac and
Peng
Qin
*ab
aState Key Laboratory of High-Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Science, Shanghai 200050, China. E-mail: qinpeng@mail.sic.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Science, Beijing 100049, China
cState Key Lab of Metal Matrix Composites, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: huangfq@sjtu.edu.cn
First published on 29th October 2024
Abstract
The development of photo-assisted rechargeable batteries is an attractive approach to realize the conversion and storage of solar energy in a single device, but designing bifunctional electrodes and improving their safety are challenging. Herein, we chose CsPbBr3 perovskites as the photo-responsive unit and lead phytate as the ion storage unit for the construction of a dual-functional cathode. The designed photo-cathode possessed enhanced photon responsiveness to benefit solar conversion and reversible redox centers for direct energy storage. Moreover, considering the safety of photo-assisted Li-ion batteries (PA-LIBs), a polymer-based electrolyte was used instead of a traditional liquid-based electrolyte to further restrict leakage and decomposition issues. During the photo-assisted charging and discharging processes (AM 1.5 G, 100 mW cm−2), the specific capacity of the assembled quasi-solid-state PA-LIB increased by 11.4% and 64.8%, respectively, compared with conventional electric charging and discharging, thus providing an efficient and sustainable strategy to realize solar-driven electrochemical energy storage.
Sustainability spotlightSunlight is the most abundant source for providing clean energy. Significant progress has been made in efficiently harvesting solar energy using solar cells for the production of electricity; however, the generated electricity cannot be continuously stored. Photo-assisted Li-ion batteries (PA-LIBs) are recognized as a promising technology, which allows the simultaneous conversion and storage of solar energy in a two-electrode system. Herein, we demonstrate a dual function photocathode to fully utilize sunlight. In addition, considering the safety of light-assisted lithium-ion batteries, we introduce a polymer-based electrolyte. The aim of this work is in line with the following UN Sustainable Development Goals: responsible consumption and production (SDG 12); industry, innovation and infrastructure (SDG 9) and climate action (SDG 13). |
Introduction
Sunlight is the most abundant source for providing clean energy.1 Significant progress has been made in efficiently harvesting solar energy using solar cells for the production of electricity; however, the generated electricity cannot be continuously stored. In this case, photo-assisted Li-ion batteries (PA-LIBs) are recognized as a promising technology, allowing the simultaneous conversion and storage of solar energy in a two-electrode system.2 Photogenerated electrons and holes participate in redox reactions of lithium ions during the charging and discharging process, increasing the specific capacity and energy density of electrochemical reactions.3,4 Unfortunately, PA-LIBs reported thus far usually suffer from high cost, low solar utilization efficiency due to the mismatch between fast charge recombination and slow Li-ion storage kinetics, lack of in-depth understanding of the design strategy of bifunctional electrodes, and safety issues due to liquid electrolytes.5 Moreover, designing efficient bifunctional photoelectrodes is challenging but critical for the further development of PA-LIBs.6According to previous reports, a photoelectrode can be composed of (i) a single material with photo-activity and redox-activity,7–11 for example, LiMn2O4,9 WO3,7 and Cs3Bi2I9 perovskite nanosheets.11 The use of γ-LiV2O5 as a photocathode material for PA-LIBs without any additional additives has been reported.8 The photo-assisted fast charging mode enables the battery to be charged to 185 mA h g−1 in 5 min at a current density of 2 A g−1, showing a 270% increase in capacity compared to that under dark conditions. Volder et al. synthesized copper-hexahydroxybenzene as a bifunctional material, which was further mixed with reduced graphene oxide as a conductive additive and charge transfer medium. After exposure to light, the specific capacity of the battery increased from 107 mA h g−1 to 126 mA h g−1 at 200 mA g−1 and from 79 mA h g−1 to 97 mA h g−1 at 2000 mA g−1 under 1 sun illumination. However, the mismatch between fast charge recombination and slow Li-ion storage kinetics in a single material leads to significant energy loss.12 A photoelectrode can also be composed of (ii) two (or more) components with either photo-activity or redox-activity, through physical mixing or in situ growth.13–25 Zaghib et al. created a bifunctional photoelectrode by physically mixing the photoactive N719 dye with a lithium-storable LiFePO4 (LFP) unit.19 They demonstrated that electron–hole pairs were generated in the N719 dye after light illumination, the holes participated in the delithiation of LiFePO4 at the cathode and the electrons were reduced by oxygen at the anode to form an SEI. The discharge capacity was twice the theoretical value in light-assisted charging, but the photoconversion efficiency was low due to the fast charge recombination between the two units. For better charge extraction, Yang et al. constructed a core–shell heterojunction nanorod array (Ni/CdS@Ni3S2) via the hydrothermal method as a PA-LIB photoelectric electrode.25 The specific capacity of this PA-LIB increased during the charging and discharging cycles by 13.7% and 15.9%, respectively. Compared to in situ growth, physical mixing has advantages such as not requiring tedious multi-step synthesis processes and a wide material selection range. However, the selection of appropriate photo-responsive and ion storage units with matched energy levels and charge/ion transfer kinetics to realize efficient interface carrier separation/transportation and high light utilization efficiency is still a significant challenge.
Herein, a bifunctional photocathode was constructed by physically mixing the photo-responsive CsPbBr3 perovskite unit and the ion storage lead phytate (PbPA) unit. The CsPbBr3 unit is responsible for the photoresponse through the generation of electron–hole pairs under light illumination, while the PbPA unit is responsible for lithium ion storage through the adsorption/desorption mechanism during the discharge/charge process. During the light-assisted discharge process, the photogenerated electrons move from the conduction band of CsPbBr3 to PbPA, together with electron filling from the external circuit to the valence band of CsPbBr3, increasing the discharge voltage and capacity. During the following light-assisted charge process, due to the high electron density in PbPA, the electrons move from PbPA to the valence band of CsPbBr3 to combine with the holes, accompanied by the transfer of photogenerated electrons from the conduction band of CsPbBr3 to the Li anode through the external circuit, resulting in a reduction in the charging voltage. Moreover, considering the safety of PA-LIBs, a polymer-based electrolyte was used instead of the traditional liquid-based electrolyte to further restrict the electrolyte leakage and decomposition issues. During the photo-assisted charging/discharging processes (AM 1.5 G, 100 mW cm−2), the specific capacity of the assembled quasi-solid-state PA-LIB increased by 11.4% and 64.8%, respectively, compared with conventional electric charging/discharging.
Results
Basic characterization of the two units
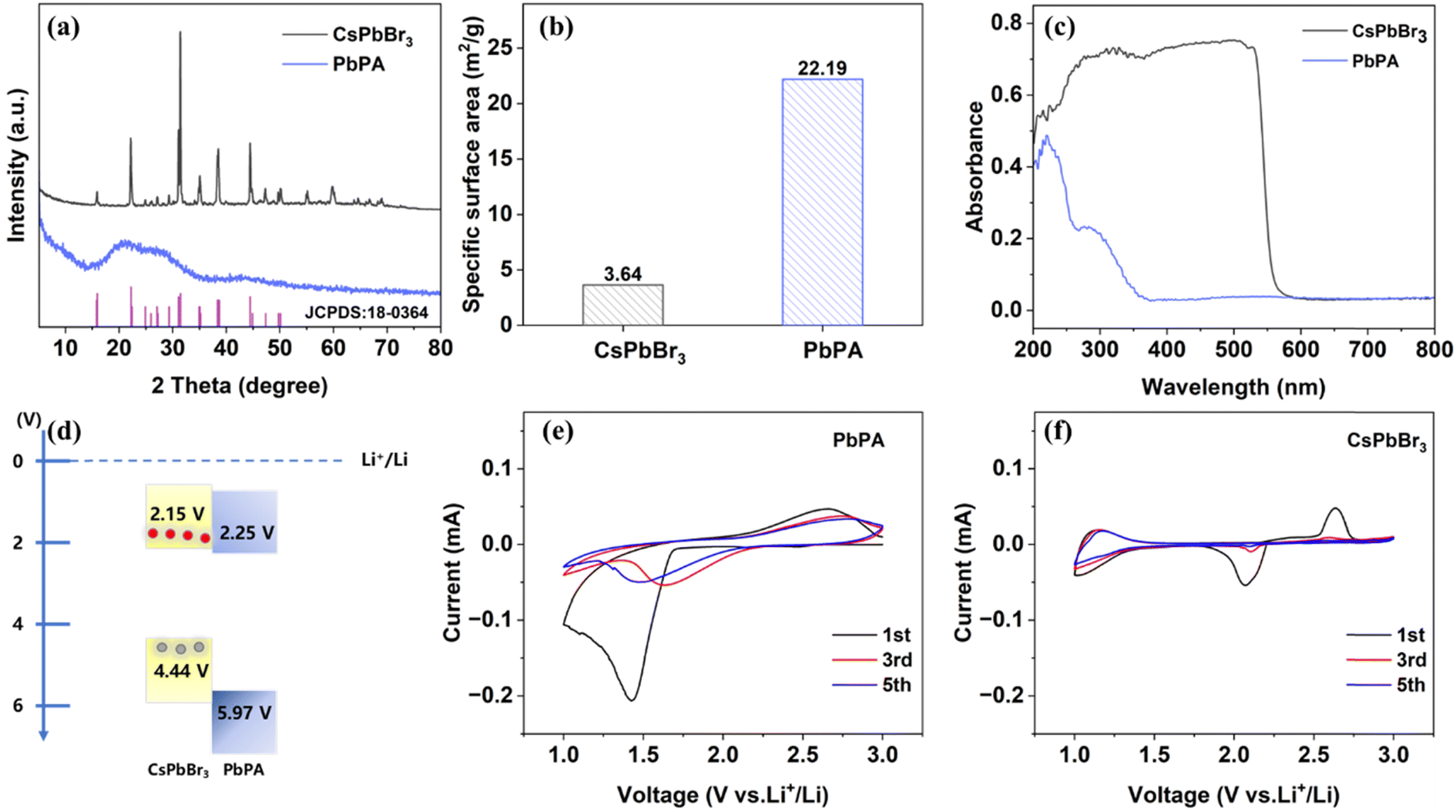
The photocathode consisted of CsPbBr3 perovskite as the photo-responsive unit and PbPA as the ion storage unit. The CsPbBr3 powder was synthesized by dropping hydrohalic acid into a DMSO solution containing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of CsBr and PbBr2. The orange precipitate was immediately observed under continuous stirring. The three-dimensional PbPA was prepared by coordinating lead ions with phytic acid. The phosphoric acid groups dispersed on the spatially twisted six-membered carbon ring on the surface are responsible for lithium-ion storage through the adsorption/desorption mechanism. The crystalline phase of the prepared CsPbBr3 and PbPA was investigated by X-ray diffraction (XRD). As shown in Fig. 1a, the diffraction peaks at 15.2°, 21.5°, 30.4°, 30.7°, and 34.2° are attributed to the (100), (110), (002), (200), and (210) crystal planes of CsPbBr3 (JCPDS # 18-0364),26,27 respectively. PbPA possesses amorphous features, and thus no crystalline peaks were observed.28 According to scanning electron microscopy (SEM), spherical particles with a diameter of 50–200 nm were observed for PbPA (ESI Fig. S1a†) and the crystal size of CsPbBr3 was found to be about 10–50 μm (ESI Fig. S1b†). A schematic diagram of the molecular structure and optical image of PbPA and CsPbBr3 are shown in ESI Fig. S2 and S3,† respectively.
:1 molar ratio of CsBr and PbBr2. The orange precipitate was immediately observed under continuous stirring. The three-dimensional PbPA was prepared by coordinating lead ions with phytic acid. The phosphoric acid groups dispersed on the spatially twisted six-membered carbon ring on the surface are responsible for lithium-ion storage through the adsorption/desorption mechanism. The crystalline phase of the prepared CsPbBr3 and PbPA was investigated by X-ray diffraction (XRD). As shown in Fig. 1a, the diffraction peaks at 15.2°, 21.5°, 30.4°, 30.7°, and 34.2° are attributed to the (100), (110), (002), (200), and (210) crystal planes of CsPbBr3 (JCPDS # 18-0364),26,27 respectively. PbPA possesses amorphous features, and thus no crystalline peaks were observed.28 According to scanning electron microscopy (SEM), spherical particles with a diameter of 50–200 nm were observed for PbPA (ESI Fig. S1a†) and the crystal size of CsPbBr3 was found to be about 10–50 μm (ESI Fig. S1b†). A schematic diagram of the molecular structure and optical image of PbPA and CsPbBr3 are shown in ESI Fig. S2 and S3,† respectively.
 | ||
| Fig. 1 Physical characterization. (a) XRD patterns. (b) Specific surface area. (c) UV-vis absorption spectra of PbPA and CsPbBr3. (d) Band diagram of the two units. (e and f) CV curves at 0.2 mV s−1 for the first, third and fifth cycle of PbPA and CsPbBr3. | ||
The specific surface area of CsPbBr3 and PbPA was calculated to be 3.64 m2 g−1 and 22.19 m2 g−1, respectively (Fig. 1b). The UV-vis absorption spectra of PbPA and CsPbBr3 are shown in Fig. 1c. As the photo-responsive unit, CsPbBr3 showed a sharp absorption edge at around 552 nm. As the ion storage unit, the absorption of PbPA was negligible in the visible region. After physically mixing CsPbBr3 with PbPA, the absorption edge showed a slight blue-shift to 548 nm. According to the Tauc equation, the band gaps of CsPbBr3 and PbPA were calculated to be 2.29 eV and 3.72 eV, respectively (ESI Fig. S5†). Furthermore, the Mott–Schottky plots were recorded at 1 kHz to illustrate the positions of the band structure (ESI Fig. S6†).29 The measurement was conducted in a three-electrode system under an argon gas environment, with Li foil serving as the counter electrode and reference electrode. The plots of CsPbBr3 and PbPA display positive slopes, suggesting their n-type semiconductor nature.30 The flat band potential of CsPbBr3 and PbPA was measured to be 2.25 V and 2.35 V versus Li+/Li, respectively, which is more positive by ∼0.1 V than the conduction band minimum (CBM). Thus, the corresponding CBM of CsPbBr3 and PbPA was estimated to be 2.15 V and 2.25 V versus Li+/Li, respectively. Based on the CBM and band gap, the valence band maximum (VBM) of CsPbBr3 and PbPA was determined to be 4.44 V and 5.97 V versus Li+/Li, respectively. The overall energy diagram is shown in Fig. 1d. Due to its excellent energy level arrangement, under illumination, the photogenerated electrons in CsPbBr3 could transfer to PbPA, achieving efficient carrier separation at the PbPA/CsPbBr3 interface.
Cyclic voltammetry (CV) was performed to further check the ion storage properties of PbPA and CsPbBr3. Fig. 1e and f show the CV curves of PbPA and CsPbBr3 during the first, third, and fifth cycles over 1.0–3.0 V at a scan rate of 0.2 mV s−1, respectively. The broad peaks located at around 1.4/2.6 V for PbPA and 2.1/2.7 V for CsPbBr3 in the first cycle can be assigned to the formation of a cathode electrolyte interface (CEI) film and/or irreversible capacity loss. In the case of PbPA, the pair of redox peaks at 1.65/2.75 V is attributed to the adsorption/desorption of lithium ions in the lead phytate framework, respectively. The area of the reduction peak is larger than that of the oxidation peak, indicating that some of the adsorbed lithium ions could not be extracted. In the second cycle, the redox peaks tended to overlap, showing the good reversibility of the adsorption–desorption process. Unlike PbPA, CsPbBr3 showed a redox peak only in the first cycle, indicating that after the initial pre-lithiation, the stabilized CsPbBr3 crystals did not contribute to the capacity during the following electrochemical process. According to the literature, the intercalation of lithium ions in the CsPbBr3 lattice does not influence the energy band gap.31
Electrochemical characterization of the quasi-solid-state lithium-ion batteries
To further determine the electrochemical performance, we compared three different conditions with the mass ratios of PbPA:CsPbBr3 = 4:1, 6:1, and 8:1. The photocathode was prepared by mixing PbPA/CsPbBr3, acetylene black, and PVDF in a weight ratio of 7:2:1, which together with a lithium metal anode and three-dimensional PVDF–HFP gel polymer-based electrolyte,32 constituted a quasi-solid-state Li-ion battery. The polymer-based electrolyte was used instead of the traditional liquid electrolyte to avoid the electrolyte leakage and decomposition issues, which showed an electrochemical window of 4.95 V and the lithium-ion diffusion coefficient of 0.444 cm2 s−1 (ESI Fig. S7 and S8†).
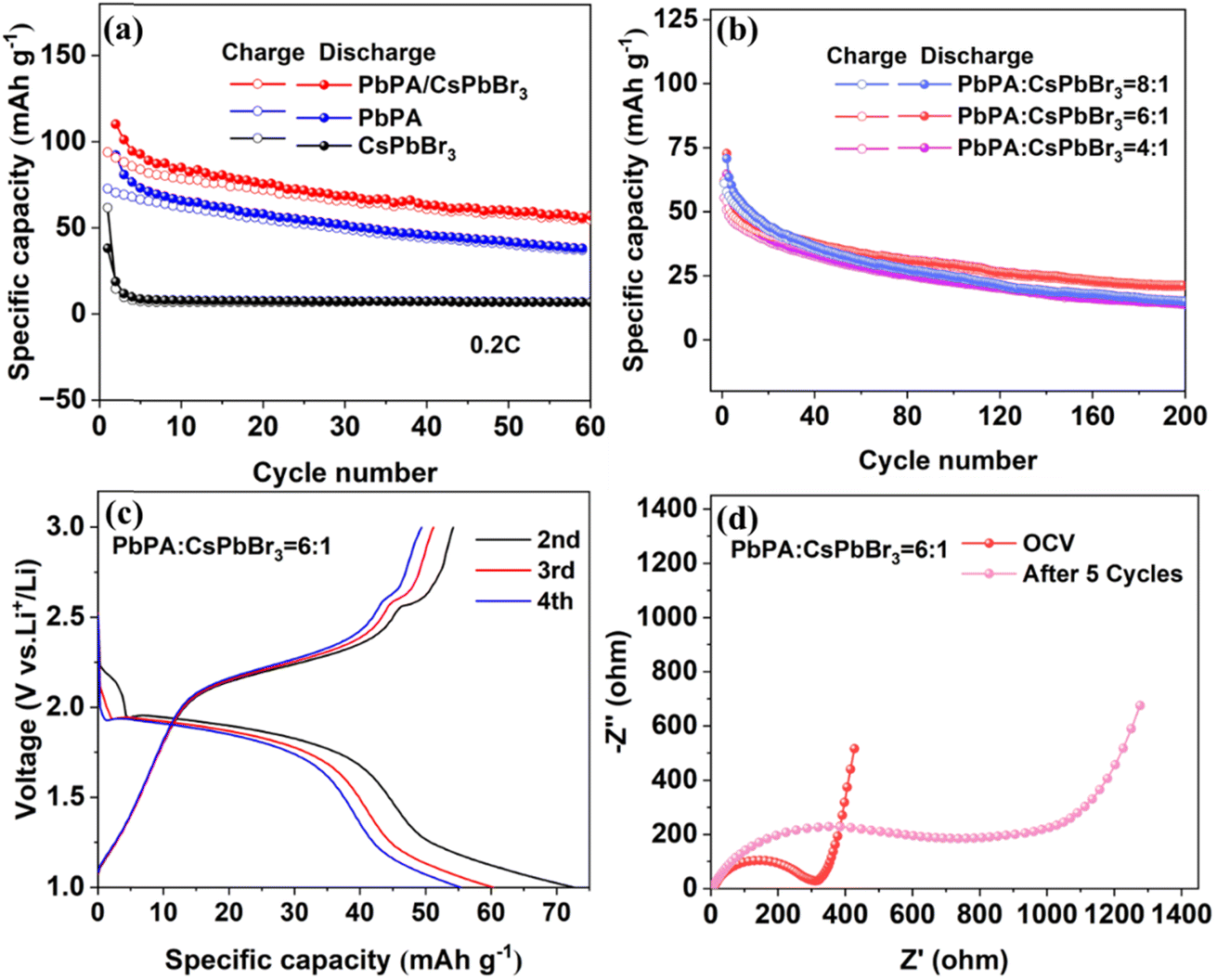
Fig. 2a shows the cycling performance of the PbPA:CsPbBr3 = 6:1 battery at a current density of 0.2C (1C = 100 mA h g−1) in the range of 1.0 to 3.0 V without light illumination. CsPbBr3 and PbPA alone were also tested under the same conditions for comparison. The PbPA battery showed an initial discharge capacity of 92.2 mA h g−1, maintaining 38.5 mA h g−1 after 60 cycles. Under the same conditions, the capacity of the CsPbBr3 battery was negligible, except for the initial irreversible Li insertion, which is consistent with the CV curves. After physical mixing, the PbPA:CsPbBr3 = 6:1 battery showed an initial capacity of 72.7 mA h g−1, maintaining 33.8 mA h g−1 after 60 cycles, which was slightly lower than that of PbPA due to the reduction of the ion storage unit. To check the effect of the ratio of the two units, the batteries based on PbPA:CsPbBr3 = 4:1 and 8:1 were also tested for comparison. As shown in Fig. 2b, with an increase in the proportion of PbPA, a slightly higher capacity that the initial cycles but worse stability was observed for the battery based on PbPA:CsPbBr3 = 8:1. Alternatively, a decrease in the proportion of PbPA led to a reduction in both the capacity and stability. Based on this, the PbPA:CsPbBr3 = 6:1 battery was chosen for our further studies. Fig. 2c shows the charge/discharge curves of the PbPA:CsPbBr3 = 6:1 battery from the 2nd to 4th cycles, where the two long platforms signify the adsorption/desorption lithium reaction. The PbPA:CsPbBr3 = 6:1 battery exhibited an initial coulombic efficiency of 19.01% (ESI Fig. S9†). Its low coulombic efficiency is related to the irreversible electrochemical reaction in the two units, the formation of a CEI film, and the asymmetric adsorption/desorption of lithium ions. Thus, the first cycle discharge capacity was not drawn in the cycling performance pattern to facilitate a comparison of the data. Furthermore, electrochemical impedance spectroscopy (EIS) demonstrated that after five cycles, the interfacial resistance increased from 310 Ω to 790 Ω, signifying the formation of a CEI film (Fig. 2d).
 | ||
| Fig. 2 Electrochemical characterization. (a) Cycling performance of the batteries based on PbPA, CsPbBr3, and PbPA:CsPbBr3 = 6:1 at 0.2C (1C = 100 mA h g−1). (b) Cycling performance of the batteries based on PbPA:CsPbBr3 = 4:1, 6:1, and 8:1 at 0.2C. (c) Charge/discharge profiles of the PbPA:CsPbBr3 = 6:1 battery. (d) EIS spectra of the pristine PbPA:CsPbBr3 = 6:1 battery and that after working for five cycles. | ||
Photo-electrochemical performance of the quasi-solid-state PA-LIB
For the preparation of the PA-LIB, the uncured GPE was directly coated on the cathode film, which was then torn off from the aluminum foil to form a self-supporting photocathode. The coin-cell battery was perforated with a hole with a diameter of 6 mm and sealed with a single-sided conductive plastic sheet (covered with graphene nano-silver wires) and UV-curable adhesive to ensure the transmission of light (ESI Fig. S11†). The interior of the homemade PA-LIB consisted of nickel foam, lithium foil, gel polymer electrolyte, and the PbPA/CsPbBr3 electrode (Fig. 3a). The PA-LIB was firstly cycled in the dark for stabilization, and then cycled under light (Xe lamp (AM 1.5 G, 100 mW cm−2)) and dark alternately.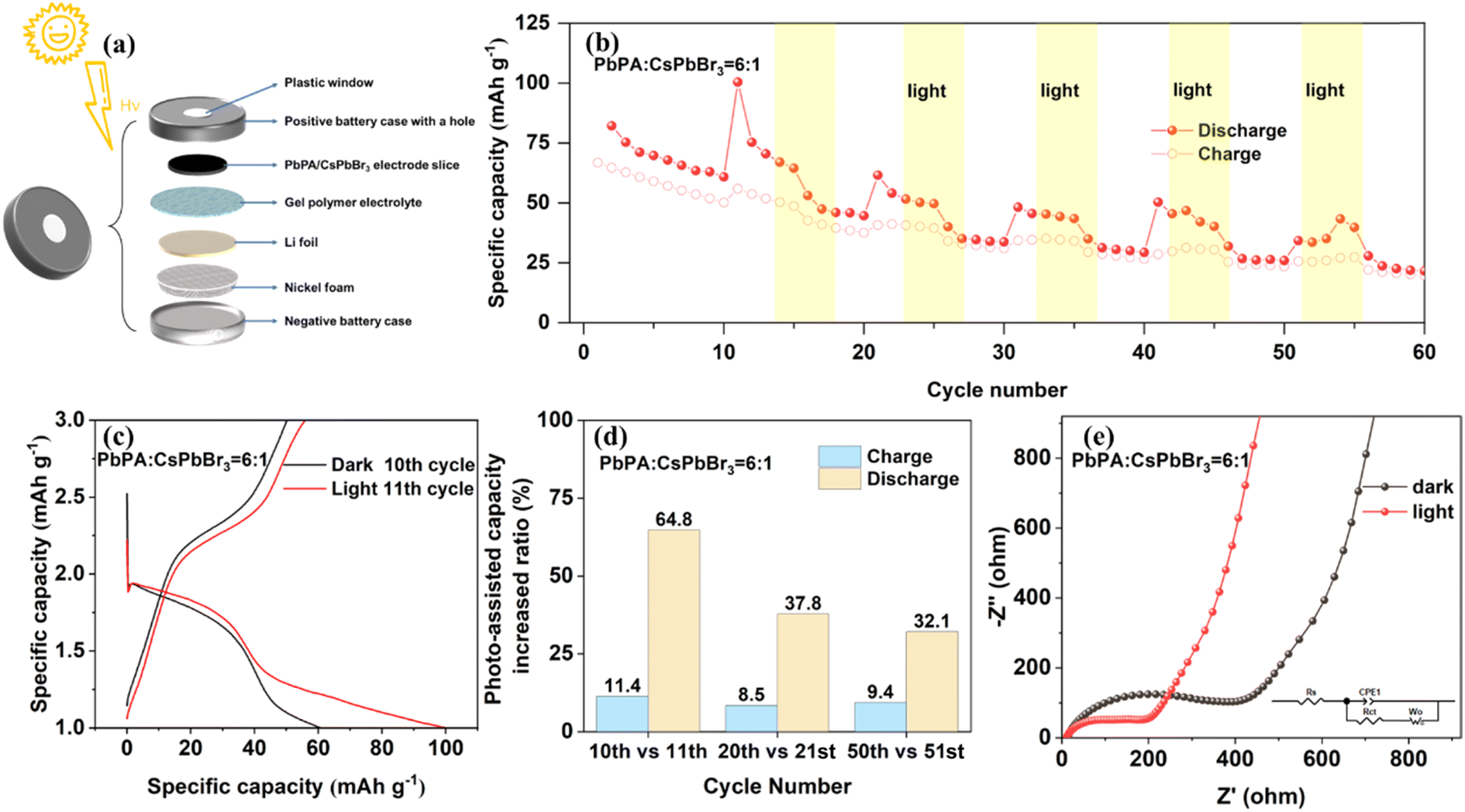 | ||
| Fig. 3 Photo-electrochemical performance of the quasi-solid-state PA-LIB based on PbPA:CsPbBr3 = 6:1 working under dark and Xe lamp (100 mW cm−2) conditions. (a) Schematic of PA-LIBs. (b) Photo-electrochemical performance of the PbPA:CsPbBr3 = 6:1 PA-LIB at 0.2C. (c) Comparison of the charge/discharge curves of the 10th cycle in the dark and 11th cycle under light of the PbPA:CsPbBr3 = 6:1 PA-LIB at 0.2C. (d) Photo-assisted capacity increasing ratio at different cycle numbers. (e) EIS spectra with and without light. | ||
As shown in Fig. 3b, the PA-LIB based on PbPA:CsPbBr3 = 6:1 showed a significantly enhanced capacity under light illumination compared to that under dark conditions. This increase in capacity was sustained under light and disappeared when the lamp was turned off. Specifically, on comparing the 10th cycle in the dark and the 11th cycle under light, the discharge capacity increased from 60.9 to 100.4 mA h g−1 with an increase rate of 64.8%, and the charge capacity increased from 50.2 to 55.9 mA h g−1 with an increase rate of 11.4%, indicating the effective conversion and storage of solar energy into electrochemical energy (Fig. 3c). When CsPbBr3 was used alone, a notable increase in the device capacity was only observed in the first illumination cycle, consistent with the earlier conclusion that after the initial pre-lithiation, CsPbBr3 did not participate in the following electrochemical process (ESI Fig. S12†). Under the light-assisted conditions, the light reduced the charging platform, improved the discharge platform, and effectively extended the voltage platform. This significantly improved the energy efficiency, allowing a higher voltage discharge through lower voltage charging. After reducing the current density from 0.2C to 0.08C, the charge platform decreased by 0.25 V and the discharge platform increased by 0.5 V, corresponding to an extra improvement in energy efficiency (ESI Fig. S10†). The increased ratio between different cycles was also calculated, as shown in Fig. 3d. As a quasi-solid-state battery, the PA-LIB showed good photo-responsive activity with an increase in the charge and discharge rate by 9.4% and 32.1%, respectively, after 50 cycles.25 We performed EIS fitting to further understand the kinetics (Fig. 3e). The fitted circuit element diagram is shown in the inset of Fig. 3e, where Rs represents the electrolyte resistance, which was almost unchanged before (13 Ω) and after light illumination (11 Ω). According to the fitting results, the charge transfer resistance (Rct) decreased from 400 to 200 Ω after light illumination. These results demonstrate that the solar energy was effectively converted to chemical energy during both the charge and discharge processes.
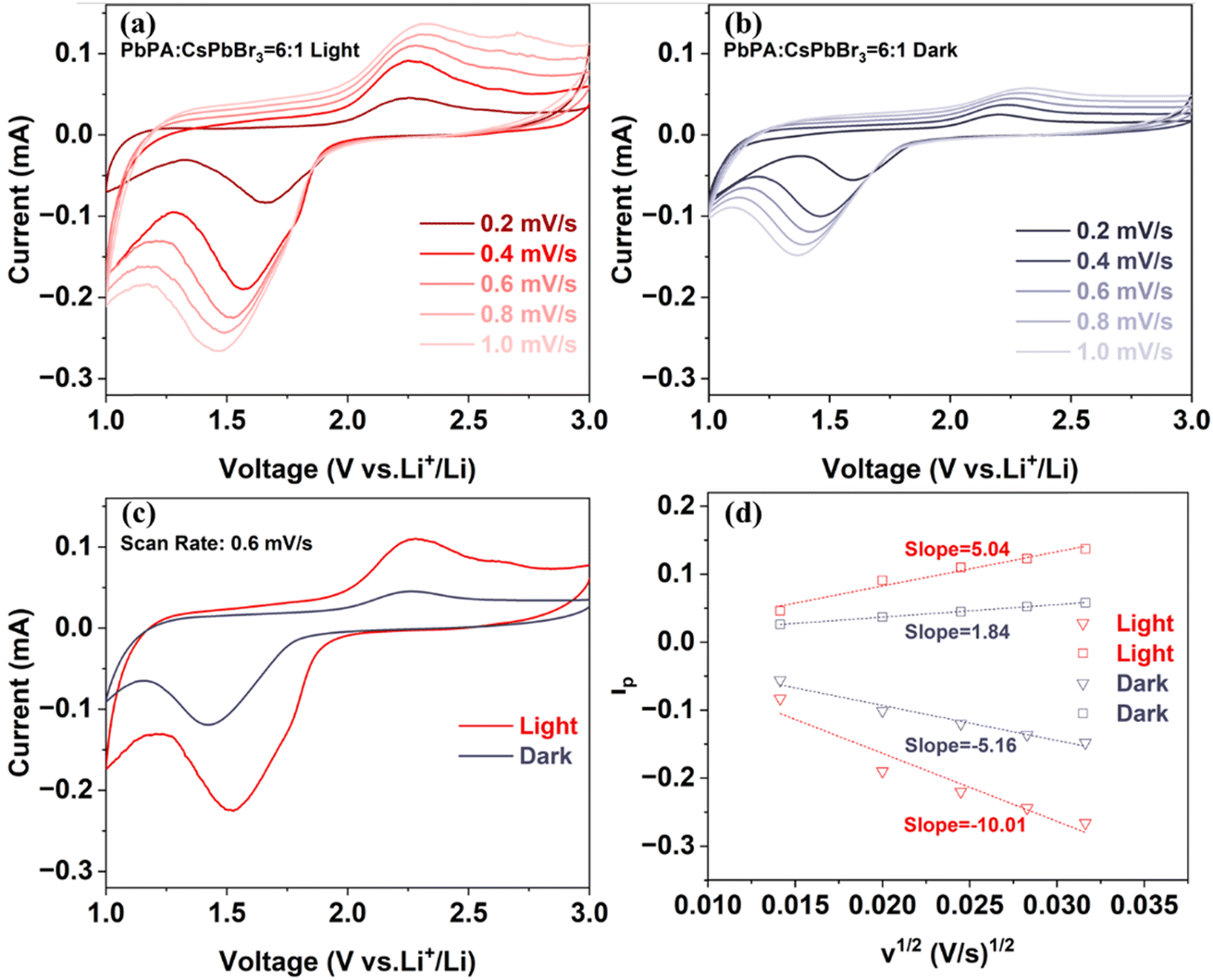
Cyclic voltammetry (CV) measurements were further performed at various scanning rates under both light and dark conditions to better understand the effect of light on the electrochemical reactions. As shown in Fig. 4a and b, at a scan rate of 0.2 mV s−1, the CV curves measured under both light and dark conditions showed two redox peaks associated with the adsorption and desorption of lithium ions on the PbPA framework, respectively. With an increase in the scan rate, the reduction peak shifted to the lower potential direction, and the oxidation peak shifted to the higher potential direction. The reversibility of the redox reaction was worse with an increase in the scan rate. Fig. 4c shows the CV curves under dark and light at 0.6 mV s−1 for comparison. Under light illumination, the CV curve area increased by 123%, indicating the potential for an enhancement in capacity. Moreover, the decreased oxidation–reduction potential gap means that the charge/discharge behavior of lithium ions was effectively improved under light-assisted conditions.
 | ||
| Fig. 4 Electrochemistry profiles of the quasi-solid-state PA-LIB based on PbPA:CsPbBr3 = 6:1 working under different conditions. CV curves at different scan rates (0.2–1.0 mV s−1) measured under light illumination (a) and in the dark (b). (c) CV curves at a scan rate of 0.6 mV s−1 with and without light. (d) Diffusion constant analysis under dark and light conditions. | ||
The peak current of CVs can be related to the diffusion constant (D) of lithium ions as follows:
| ip = 0.4463F(F/RT)1/2ACD1/2ν1/2 = ND1/2ν1/2. |
Considering that the electrode area is not influenced by light, then N = 0.4463F(F/RT)1/2AC can be regarded as a constant value for dark and illuminated conditions. Hence, according to the following relation, we can estimate the diffusion constant from the slope of ipvs. ν1/2, as shown in Fig. 4d. The enhancement in the calculated diffusion constant was 94% and 173% for the cathodic and anodic peak under 1 sun illumination compared to that in dark conditions, respectively, indicating that light is beneficial for the diffusion of lithium ions in the battery.
The working mechanism of the quasi-solid-state PA-LIB
To gain a deeper understanding of the working mechanism of the dual-functional electrode under illumination, ex situ X-ray photoelectron spectroscopy (XPS) was performed for the photocathode based on PbPA:CsPbBr3 = 6:1 after discharging to 1.0 V (D 1.0 V) and charging to 3.0 V (C 3.0 V) (Fig. 5a). In the absence of light, the peaks of Pb 4f7/2 and Pb 4f5/2 shifted from 140.2 eV and 145.1 eV in the open circuit voltage state (OCV) to 139.1 eV and 144.0 eV after discharging to 1.0 V, respectively. With the adsorption of lithium ions, the electron density around PbPA/CsPbBr3 increased, leading to a shift in the binding energy to the lower energy direction.33 Subsequently, with the extraction of lithium ions during the next charging process, the binding energy shifted back to a high energy (139.4 eV and 144.3 eV). However, due to the irreversible electrochemical reaction and the formation of a CEI film during the first cycle, it could not move back exactly to the OCV state. A similar phenomenon was also observed for the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of P 2p located at 134.9 eV, which was gradually reduced after discharging to 1.0 V and recovered when charged to 3.0 V, indicating that the lithium ions were mainly stored on the surface of PbPA through the –PO3Pb group and the adsorption/desorption of lithium ions were basically reversible. Under the light-assisted conditions, the involvement of photo-generated electron–hole pairs led to a further improvement in reduction for both Pb 4f (138.5 eV and 143.4 eV) and P 2p compared to the regular discharging, suggesting that more lithium ions could be stored in the PbPA/CsPbBr3 electrode. After photo-assisted charging to 3.0 V, the Pb 4f7/2 and Pb 4f5/2 peaks shifted back to 139.5 eV and 144.4 eV, respectively, which is closer to the OCV state compared to the regular charge process. Obviously, the photo-generated carriers promoted both the reduction and oxidation reactions to drive the discharge/charge processes more completely. Meanwhile, the binding energy changes were more obvious in the photo-assisted discharge process than the charge process, indicating that the effect of light is more significant for discharging, which is consistent with the improvement in specific capacity.
O bond of P 2p located at 134.9 eV, which was gradually reduced after discharging to 1.0 V and recovered when charged to 3.0 V, indicating that the lithium ions were mainly stored on the surface of PbPA through the –PO3Pb group and the adsorption/desorption of lithium ions were basically reversible. Under the light-assisted conditions, the involvement of photo-generated electron–hole pairs led to a further improvement in reduction for both Pb 4f (138.5 eV and 143.4 eV) and P 2p compared to the regular discharging, suggesting that more lithium ions could be stored in the PbPA/CsPbBr3 electrode. After photo-assisted charging to 3.0 V, the Pb 4f7/2 and Pb 4f5/2 peaks shifted back to 139.5 eV and 144.4 eV, respectively, which is closer to the OCV state compared to the regular charge process. Obviously, the photo-generated carriers promoted both the reduction and oxidation reactions to drive the discharge/charge processes more completely. Meanwhile, the binding energy changes were more obvious in the photo-assisted discharge process than the charge process, indicating that the effect of light is more significant for discharging, which is consistent with the improvement in specific capacity.
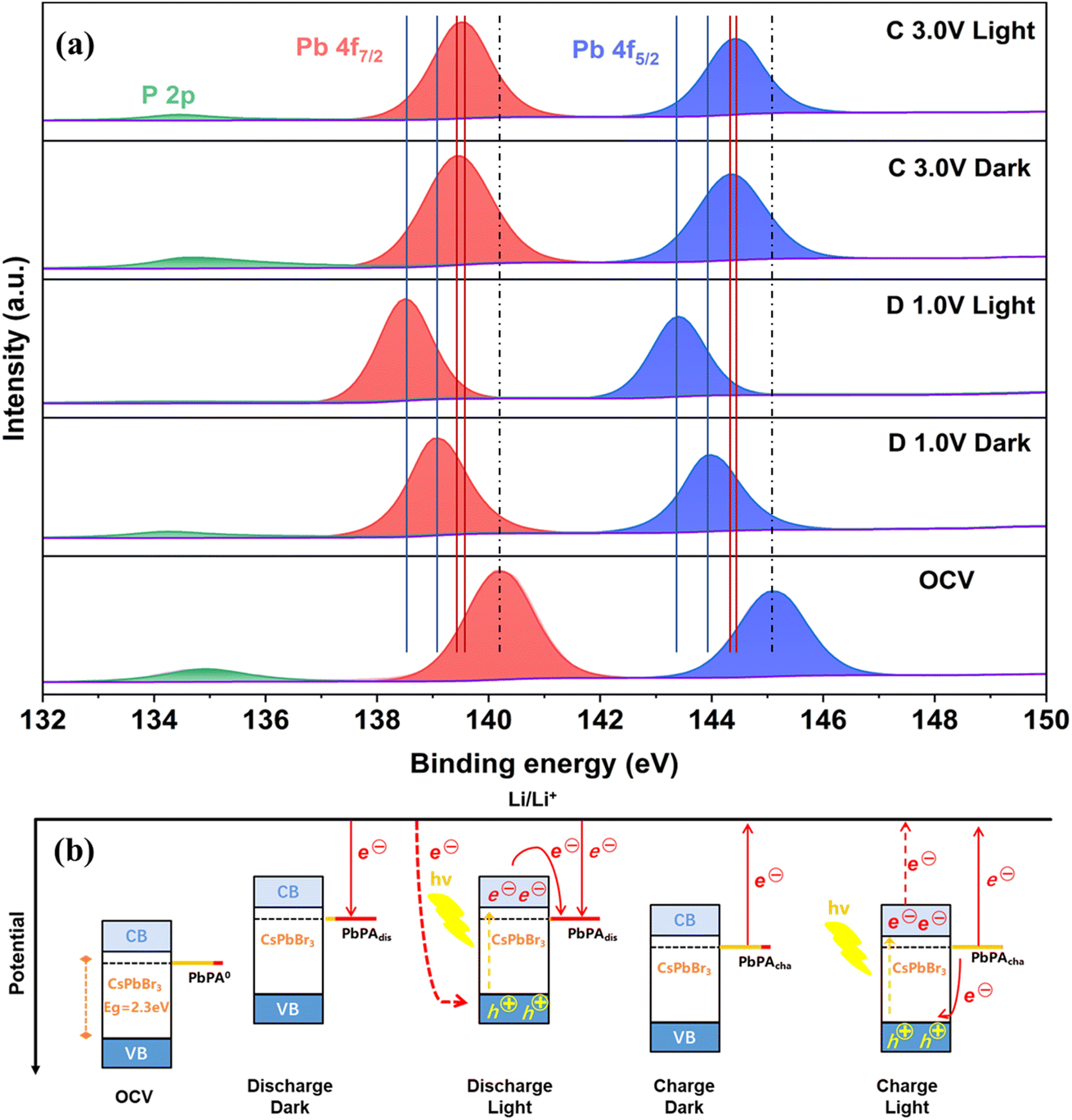 | ||
| Fig. 5 Photo-assisted working mechanism. (a) XPS Pb 4f and P 2p spectra of the PbPA:CsPbBr3 = 6:1 cathode under different conditions. (b) Energy alignment and the carrier flow in the system. | ||
Fig. 5b shows the energy levels of the battery with and without light illumination. The OCV state displays the energy level and potential distribution of the CsPbBr3 and PbPA units under the open circuit voltage state. Lithium-ion migration and adsorption on the PbPA surface occur simultaneously with the injection of electrons from the external circuit for the conventional discharge process. During the photo-assisted discharge process, the photo-responsive CsPbBr3 unit is excited, accompanied by the formation of electron–hole pairs. The photo-generated electrons move from the CBM of CsPbBr3 to PbPA0, and the photo-generated holes are combined with the electrons from the external circuit. At the same capacity, the voltage of photo-assisted discharge is higher than that of the conventional discharge. During the following charge process, the lithium ions desorb from the PbPA surface, accompanied by the electrons moving from the photoelectrode to the lithium metal electrode. In the presence of light, the electrons move from PbPAdis to the VBM of CsPbBr3 to combine with the photo-generated holes, and the photo-generated electrons transfer from the CB of CsPbBr3 to the lithium metal electrode through the external circuit, resulting in a decrease in the charging voltage. According to these results, during the light-assisted discharge/charge processes, the photo-generated carriers promote the lithium redox reactions, leading to a higher energy output.
Conclusion
We developed a bifunctional photocathode by physically mixing the photo-responsive CsPbBr3 perovskite unit and the ion storage PbPA unit for quasi-solid-state photo-assisted Li-ion batteries. The photo-excited carriers could be effectively separated at the PbPA/CsPbBr3 interface and stored as chemical energy via reversible de/lithiation reactions. In the presence of light, the photo-generated carriers promoted the lithium redox reactions, leading to a higher energy output. During the photo-assisted charge and discharge processes (AM 1.5 G, 100 mW cm−2), the discharge capacity in the first cycle increased from 60.9 mA h g−1 to 100.4 mA h g−1, and the charge capacity increased from 50.2 mA h g−1 to 55.9 mA h g−1. Consequently, the specific capacity of the assembled quasi-solid-state PA-LIB was enhanced by 11.4% and 64.8%, respectively, compared with conventional electric charging/discharging. As the first quasi-solid-state photo-assisted Li-ion battery, our work demonstrates that the selection of appropriate photo-responsive and ion storage units with matching energy levels and charge/ion transfer kinetics can result in efficient interface carrier separation/transportation and high light utilization, providing a low cost and convenient way for the further development of bifunctional electrodes.Experimental
Synthesis of CsPbBr3
0.5 M PbBr2 (Alfa Aesar) and CsBr (Alfa Aesar) were dissolved in DMSO (Damas-Beta), and the solution was stirred at 90 °C for 4 h. After cooling to room temperature, hydrobromic acid (HBr, Sigma-Aldrich) was added to the solution and an orange precipitate was immediately observed. The precipitate was filtered and washed with ethanol three times, and finally dried at 60 °C overnight for further application.Synthesis of PbPA
15 mL of a 0.1 M of phytic acid (Macklin) in water was placed on a magnetic stirrer, and 150 mL of a 0.06 M Pb(NO3)2 (Damas-Beta) aqueous solution was dripped slowly into the above-mentioned solution at room temperature. A white precipitate appeared immediately, and the solution was stirred at room temperature for another 2 h. The precipitate was filtered and washed with water and ethanol, and finally dried at 60 °C overnight for further use.Synthesis of PbPA/CsPbBr3
The PbPA and CsPbBr3 powder were added to a mortar according to the mass ratio, and then manually ground for 1000 cycles.Preparation of gel polymer-based electrolyte (GPE)
To prepare GPE, poly(vinylidene fluoride-co-hexafluoropropylene) (P(VDF–HFP)) (1.0 g, Aladdin), lithium bis(trifluoromethylsulphonyl)imide (LiTFSI) (0.45 g, Alfa Aesar) and PC (1 mL, Sinopharm) were dissolved in acetone (10 mL, Sinopharm) under continuous stirring for 1 h at 60 °C, and subsequently for 8 h at room temperature. The centrifugal film was coated with a film scraper at a thickness of 1.0 mm and dried at 60 °C to obtain a PSE with a thickness of ∼100 μm.Fabrication of the quasi-solid-state PA-LIBs
The cathode was made by coating a slurry with the weight ratio of PbPA/CsPbBr3, conductive carbon black (Super P) and PVDF of 8:1:1 on aluminum foil with a thickness of 200 μm. After vacuum drying at 100 °C for 12 h, the cathode was coated with a layer of polymer electrolyte, dried at 60 °C, and then cut into a disc with a diameter of 12 mm. The self-supporting photocathode was obtained by stripping the aluminum foil with an active mass loading of 1.0–1.5 mg cm−2, and then transferred to a conductive polyacrylonitrile substrate. Finally, CR2032-type coin cells were assembled in an argon-filled glove box (MB-Labstar, 1200/780, H2O < 0.5 ppm; O2 < 0.5 ppm) with the GPE and Li metal anode.
Characterization
Powder XRD patterns were recorded on an X-ray diffractometer (Bruker D8 Advance, Germany), employing monochromatic Cu Kα as the radiation source. SEM images and energy dispersive X-ray spectra were recorded on a field-emission scanning electron microscope (HITACHI S4800, Japan). XPS spectra were obtained on an X-ray photo-electron spectrometer (Thermo Scientific, ESCALAB 250, USA). N2 absorption isotherms were measured on a physisorption analyzer (Micromeritics ASAP 2020 HD88, USA) at 77 K. Absorption spectra were recorded on a UV absorption spectrometer (Hitachi U-4100, Japan).Cyclic voltammograms (CVs) were recorded using an electrochemical workstation (CHI760E, China) at room temperature. EIS spectra were recorded on an electrochemical workstation (Autolab PGSTAT 302N, Switzerland) from 1 MHz to 0.1 Hz. Photo-assisted charging and discharging were performed on a LAND CT-2001A (China) combined with the solar simulator under the light intensity of 100 mW cm−2.
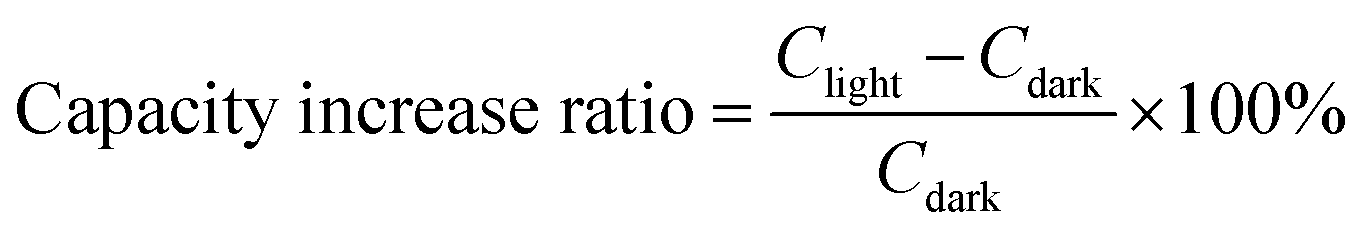
Data availability
The authors declare that the data supporting the findings of this study are available within this article and the ESI files.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 51872315), and the Science Foundation of the Chinese Academy of Sciences.References
- G. Hodes, J. Manassen and D. Cahen, Nature, 1976, 261, 403–404 CrossRef CAS.
- M. B. Hayat, D. Ali, K. C. Monyake, L. Alagha and N. Ahmed, Int. J. Energy Res., 2019, 43, 1049–1067 CrossRef.
- P. Zhang, M. Cai, Y. Wei, J. Zhang, K. Li, S. R. P. Silva, G. Shao and P. Zhang, Adv. Sci., 2024, 11(30), e2402448 CrossRef PubMed.
- N. Yan and X. Gao, Energy Environ. Mater., 2021, 5, 439–451 CrossRef.
- B. D. Boruah, Energy Storage Mater., 2021, 34, 53–75 CrossRef.
- A. D. Salunke, S. Chamola, A. Mathieson, B. D. Boruah, M. de Volder and S. Ahmad, ACS Appl. Energy Mater., 2022, 5, 7891–7912 CrossRef CAS.
- T. Kim, I. Mahdi, H. Choi, S. H. Lee and Y. Park, Energy Technol., 2022, 10, 2200891 CrossRef CAS.
- J. Wang, Y. Wang, C. Zhu and B. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 4071–4078 CrossRef CAS PubMed.
- A. Lee, M. Voros, W. M. Dose, J. Niklas, O. Poluektov, R. D. Schaller, H. Iddir, V. A. Maroni, E. Lee, B. Ingram, L. A. Curtiss and C. S. Johnson, Nat. Commun., 2019, 10, 4946 CrossRef PubMed.
- S. Ahmad, C. George, D. J. Beesley, J. J. Baumberg and M. De Volder, Nano Lett., 2018, 18, 1856–1862 CrossRef CAS PubMed.
- N. Tewari, S. B. Shivarudraiah and J. E. Halpert, Nano Lett., 2021, 21, 5578–5585 CrossRef CAS PubMed.
- Y.-X. Tan, X. Zhang, J. Lin and Y. Wang, Energy Environ. Sci., 2023, 16, 2432–2447 RSC.
- Q. Li, N. Li, M. Ishida and H. Zhou, J. Mater. Chem. A, 2015, 3, 20903–20907 RSC.
- X. Liu, H. Andersen, Y. Lu, B. Wen, I. P. Parkin, M. De Volder and B. D. Boruah, ACS Appl. Mater. Interfaces, 2023, 15, 6963–6969 CrossRef CAS PubMed.
- A. Kumar, R. Hammad, M. Pahuja, R. Arenal, K. Ghosh, S. Ghosh and T. N. Narayanan, Small, 2023, 19, e2303319 CrossRef PubMed.
- C. Xu, X. Zhang, L. Duan, X. Zhang, X. Li and W. Lu, Nanoscale, 2020, 12, 530–537 RSC.
- A. Kumar, P. Thakur, R. Sharma, A. B. Puthirath, P. M. Ajayan and T. N. Narayanan, Small, 2021, 17, e2105029 CrossRef PubMed.
- Z. Wang, H. C. Chiu, A. Paolella, K. Zaghib and G. P. Demopoulos, ChemSusChem, 2019, 12, 2220–2230 CrossRef CAS PubMed.
- A. Paolella, C. Faure, G. Bertoni, S. Marras, A. Guerfi, A. Darwiche, P. Hovington, B. Commarieu, Z. Wang, M. Prato, M. Colombo, S. Monaco, W. Zhu, Z. Feng, A. Vijh, C. George, G. P. Demopoulos, M. Armand and K. Zaghib, Nat. Commun., 2017, 8, 14643 CrossRef CAS PubMed.
- N. Chamidah, S. Tsuchida, T. Yaji, A. Irizawa, C. Zhong, K.-i. Okazaki and Y. Orikasa, Electrochem. Commun., 2023, 149, 107459 CrossRef CAS.
- B. D. Boruah, B. Wen and M. De Volder, Nano Lett., 2021, 21, 3527–3532 CrossRef CAS PubMed.
- A. Khataee, J. Azevedo, P. Dias, D. Ivanou, E. Dražević, A. Bentien and A. Mendes, Nano Energy, 2019, 62, 832–843 CrossRef CAS.
- M. X. Tran, G. Liu, R. E. A. Ardhi, S.-W. Lee and J. K. Lee, Energy Storage Mater., 2024, 65, 103145 CrossRef.
- Q. Zhang, M. Wei, Q. Dong, Q. Gao, X. Cai, S. Zhang, T. Yuan, F. Peng, Y. Fang and S. Yang, J. Energy Chem., 2023, 79, 83–91 CrossRef CAS.
- Q. Dong, M. Wei, Q. Zhang, L. Xiao, X. Cai, S. Zhang, Q. Gao, Y. Fang, F. Peng and S. Yang, Chem. Eng. J., 2023, 459, 141542 CrossRef CAS.
- A. Kostopoulou, D. Vernardou, D. Makri, K. Brintakis, K. Savva and E. Stratakis, J. Power Sources Adv., 2020, 3, 100015 CrossRef.
- S. Liu, K. Zhang, L. Tan, S. Qi, G. Liu, J. Chen and Y. Lou, Electrochim. Acta, 2021, 367, 137352 CrossRef CAS.
- W.-J. Dong, J.-B. Le, Y. Jin, G.-Q. Zhang, B. Ye, P. Qin and F.-Q. Huang, Rare Met., 2022, 42, 122–133 CrossRef.
- G. Y. Qiao, D. Guan, S. Yuan, H. Rao, X. Chen, J. A. Wang, J. S. Qin, J. J. Xu and J. Yu, J. Am. Chem. Soc., 2021, 143, 14253–14260 CrossRef CAS PubMed.
- Z. Zhu, X. Shi, G. Fan, F. Li and J. Chen, Angew Chem. Int. Ed. Engl., 2019, 58, 19021–19026 CrossRef CAS PubMed.
- Q. Jiang, M. Chen, J. Li, M. Wang, X. Zeng, T. Besara, J. Lu, Y. Xin, X. Shan, B. Pan, C. Wang, S. Lin, T. Siegrist, Q. Xiao and Z. Yu, ACS Nano, 2017, 11, 1073–1079 CrossRef CAS PubMed.
- J. Pan, H. Peng, Y. Yan, Y. Bai, J. Yang, N. Wang, S. Dou and F. Huang, Energy Storage Mater., 2021, 43, 165–171 CrossRef.
- J. Pan, K. Yuan, X. Mi, Y. Lu, Y. Yu, J. Yang, S. Dou and P. Qin, ACS Nano, 2023, 17, 21360–21368 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00494a |
| This journal is © The Royal Society of Chemistry 2024 |