Aqueous-mediated DABCO and DABCO-ionic liquid catalysed synthesis of 3-acetylcoumarins: exploration by kinetic, electrochemical and spectroscopic studies
Received
12th August 2024
, Accepted 23rd October 2024
First published on 7th November 2024
Abstract
A more efficient, green and user-friendly approach for the synthesis of a combinatorial library of 3-acetylcoumarins from easily available ethylacetoacetate (EAA) and o-hydroxyaldehydes in water at room temperature was developed with excellent yields by modifying novel methods. The experimental method is facile and more economical than traditional methods, requiring no further product purification. Furthermore, the experimental conditions were optimized at the gram scale. The catalyst was reusable, resulting in excellent yields in a shorter time. Additionally, the by-product ethanol formed in the reaction was distilled out. Interestingly, the use of DABCO, DABCO-ionic liquids and DABCO salts as catalysts yielded excellent results (95–98%) in a shorter time. All products were identified by comparing their physical and spectroscopic data. The versatility of the developed green approach encouraged us to investigate the kinetics of the slowest step in the reaction and arrive at a rate law for the reaction. The kinetic and thermodynamic experimental results prompted us to undertake spectroscopic studies, including IR and 1H NMR, which demonstrated that the catalyst initiated the mechanism. Additionally, cyclic voltammetry (CV), a key electroanalytical tool, showed both the spontaneity of the reaction and the tautomerism of EAA.
Sustainability spotlight
Knoevenagel condensation is commonly used for the synthesis of 3-acetylcoumarins, where piperidine is employed as a base. However, this base is removed through an excess alcohol washing, during which a significant amount of soluble product may be lost, resulting in a reduced yield. Second, piperidine is toxic upon skin contact or inhalation, potentially causing severe skin burns and eye damage. Our focus is on developing a protocol to accelerate 3-acetylcoumarins synthesis using a green method. In this context, we envisioned the use of highly efficient catalysts (DABCO and DABCO-ionic liquid) that could be easily recovered and reused. The by-product is ethanol, which can be distilled under reduced pressure and reused when necessary. Remarkably, we achieved this reaction at room temperatures in very short time periods, and we verified the process using kinetic, electrochemical, and spectroscopic studies. So far, spectroscopic techniques have shown the tautomerism of EAA using various methods. Curiosity regarding EAA tautomerism and also the arrested form of tautomerism made us propose the use of the electrochemical technique CV, which is still reliable and not time consuming. The isolated products were more than 95% pure (no purification required). Our work emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7) and industry, innovation and infrastructure (SDG 9).
|
Introduction
Half of all known organic compounds are heterocycles, making this class of chemicals extremely significant. Indeed, the majority of vitamins, optoelectronics, pharmaceuticals, and several other natural goods are heterocycles. Benzopyrones represent an important class of heterocyclic compounds, of which benzo-α-pyrones, commonly called coumarins, are significant and involve the fusion of a pyrone ring with a benzene nucleus.1 Vogel2 first isolated coumarin from tonka beans. Although coumarin is a naturally active substance, its activity can be enhanced by combining it with various chemical moieties. Conventional methods for producing the coumarin nucleus include the Pechmann,3 Knoevenagel,4 Perkin,5 Reformatsky,6 and Wittig7 condensation processes. Ongoing attempts have been made towards novel, practical and adaptable methods for synthesizing the coumarin nucleus with clear structural characteristics. Several modifications, including variations in the catalyst and reaction conditions, have been introduced to improve the efficacy of these classical reactions.8 In recent years, versatile coumarin synthesis has been achieved through organopalladium intermediates.9 Additionally, coumarins and their derivatives have been utilized as key components in a wide range of applications, including laser dyes,10 fluorescent metal ion sensors,11 solution dynamic probes,12 and, more recently, organic sensitizers in high-efficiency dye-sensitized solar cells.13
For instance, derivatives of 3-acetylcoumarins have found numerous useful applications, including as fluorescence brighteners,14 inhibitors of Zn corrosion in acidic media,15 and in biological applications due to their various activities, such as antibacterial,16 EGFR inhibition,17 anti-breast cancer,18 α-amylase inhibition,19 antimycobacterial,20 anti-inflammatory and analgesic,21,22 and fluorescence dye properties.23 According to previous studies, 3-acetylcoumarins have been synthesized using various catalysts,24–30 with the various protocols having distinct advantages and disadvantages. Ultimately, chemists usually aim for new approaches that can offer more merits and less demerits, such as a workable procedure, high purity of product, good yield, and no effects on human health or the environment. Undoubtedly, there are a few advantages in the reported works, but there are more disadvantages too in terms of the usage of organic solvents for the reaction, excess usage of solvents for washing, usage of certain hazardous catalysts in the reactions that can cause health issues, and economic concerns about the more costlier catalyst and reagents used. However, many eco-friendly DABCO-based organocatalysts have been screened in organic synthesis, and these catalysts were found to show good to excellent performance in many organic reactions.31
Many methods have been reported, among which the Knoevenagel condensation is commonly used for the synthesis of 3-acetylcoumarins; however, piperidine is used as a base, which is removed by excess alcohol washing, which may also lead to the loss of soluble product and a reduction in the % yield. Other reported methods have similar disadvantages with product isolation and purification. Also, many reports have discussed the organic reaction mechanism via quantitative approaches. In recent years, qualitative approaches, such as electrochemical and spectroscopic methodologies, have yielded more authentic and impressive information about organic reaction mechanisms involving electron transfer. Such electron transfer can tune the thermodynamic or kinetic selectivity to form organic reactive intermediates. These intermediates can aid understanding the feasibility and kinetics of electron transfer, which clearly depends on the concentration of the reactant/catalyst.
Moreover, electroanalytical techniques offer a variety of ways to learn about the basic interactions, lifetimes, and reactivities that underlie redox reactions.32 Nevertheless, the organic synthetic community has not embraced these tools extensively, with the majority of these techniques still being within the domain of analytical and physical chemists. This perspective seeks to provide an overview of the essential approaches for a synthetic chemist audience, with recommendations on how to get useful information on mechanistic issues. The information summarized herein will exemplify electrochemistry as a tool to choose the perfect catalyst, and for the identification and quantification of organic intermediate, kinetics, etc. A deeper comprehension of organic processes will be possible with knowledge of how these factors impact a mechanism, and could facilitate the creation of innovative synthetic strategies. Herein, we have come up with a user-friendly, green approach for the synthesis of 3-acetylcoumarins; in which water acts as a green solvent and DABCO-based green base and catalyst. A detailed investigation was carried out for the formation of 3-acetyl coumarin and also its mechanism was determined using kinetic, thermodynamic, cyclic voltammetry, and spectroscopic approaches.
Results and discussion
Optimization study
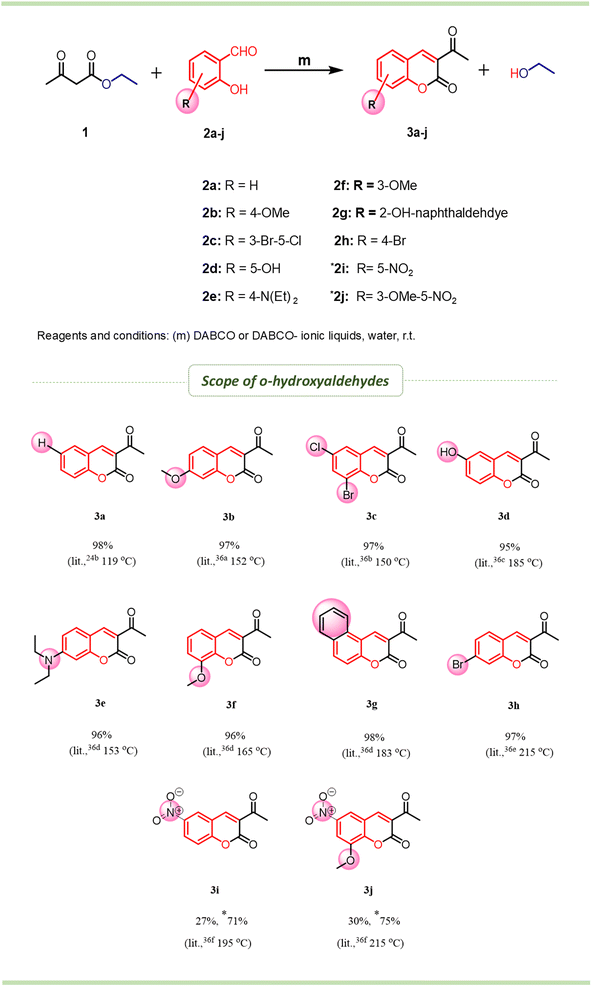
A green method involving the condensation of EAA (1) with o-hydroxyaldehydes (2) took place smoothly in the presence of diazabicyclo [2, 2, 2] octane (DABCO) as a base in water at room temperature, which resulted in the formation of 3-acetylcoumarins (3) in excellent yield (Scheme 2). As a model reaction, the condensation of 1 with 2a to generate 3-acetylcoumarin (3a) was investigated (Scheme 1) using various bases to optimize the yield, and the obtained results are presented in Table 1. The reaction was relatively fast (∼10–15 min) when 1 equiv. of DABCO was employed stoichiometrically. The other tested bases, like DBU (Table 1, entry 4) and DMAP (Table 1, entry 5), also gave good yields of the desired product 3a. In contrast, MeONa (Table 1, entry 1), EtONa (Table 1, entry 2), and NaOAc (Table 1, entry 3) gave poor yields and needed a longer time. Thus, it is clear from the aforementioned experiments that the best yield of 3-acetylcoumarin 3a could be obtained by employing DABCO as a green catalyst and water as a solvent.
 |
| | Scheme 1 Screening of different bases/catalysts* in the model reaction between EAA 1 and salicylaldehyde 2a. | |
 |
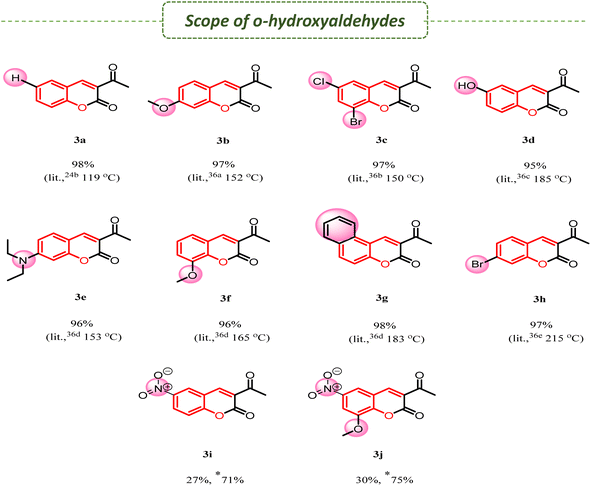
| | Scheme 2 Scope of the reaction of o-hydroxyaldehydes 2a–h with EAA 1 employing different catalysts. % Yield taken in the gram scale and melting point in oC. *Heating (65–70 °C), **literature value.24b,36a–f | |
Table 1 Optimization of the synthesis of 3-acetylcoumarin using various bases (Scheme 1)
| Entry |
Bases/catalysts |
Time |
Yield% |
| 1 |
MeONa |
2–3 h |
<60 |
| 2 |
EtONa |
2–3 h |
<60 |
| 3 |
NaOAc |
2–3 days |
<50 |
| 4 |
DBU |
15–20 min |
75 |
| 5 |
DMAP |
15–20 min |
78 |
| 6 |
DABCO |
10–15 min |
95 |
| 7 |
[H-dabco][Cl] |
15–20 min |
98 |
| 8 |
[H-dabco][OAc] |
20–25 min |
85 |
| 9 |
DABCO-CuCl |
15–20 min |
95 |
| 10 |
DABCO-CuI |
15–20 min |
96 |
Initially, the reaction was performed using various bases (Table 1) to avoid the use of piperidine as a base in the reaction, and water as medium, as a green solvent, instead of alcohol and also other solvents (DCM, toluene, 1,4-dioxane). Studies of the effects on the reaction from the excess usage of bases (alkoxides, acetates, and tert-amines) and varying the time duration were done in water as a medium at room temperature, with heating applied as needed. In the case of sodium methoxide, the formation of the product was noticed, but the reaction could not be completed even after increasing the equivalence of MeONa as well as raising the temperature (∼95 °C), and the isolated product was obtained in a yield of less than 60% (Table 1, entry 1). Similarly, sodium ethoxide resulted in almost the same yield (Table 1, entry 2); whereas sodium acetate as a base showed much less product formation even at higher temperature and with a longer reaction time (Table 1, entry 3). In search for new catalyst, previous literature reports informed us that most of the 3-acetylcoumarins were obtained using secondary/tert-amine bases other than water as a solvent. Therefore, keeping in mind the solubility of the base in water, we performed the reaction in H2O with a catalytic quantity of DBU as the base. Surprisingly, the results of the reaction were quite excellent, and the reaction proceeded smoothly with a good yield within 20 min at room temperature (Table 1, entry 4). Similarly, DMAP (Table 1, entry 5) also showed almost the same results. Further screening was performed using DABCO, with excellent product formation in a shorter time with an isolated yield of more than 95% (Table 1, entry 6). The use of DABCO showed excellent results for the synthesis of 3-acetylcoumarin derivatives, compared to DBU and DMAP.
DABCO-based catalyst
Further, the DABCO-based catalyst was explored by using a modified DABCO-ionic liquid and DABCO salts for the synthesis of the title compounds. In view of previous studies, the effects of ionic liquids in certain reactions are believed to be very interesting; hence to evaluate the profile of ionic liquids, [H-dabco][AcO] and [H-dabco][Cl] were studied in condensation reactions in the present work. These ionic liquids were synthesized according to the literature reports.33 As we can see from the results summarized in Table 1, the condensation reaction of EAA (1) with o-hydroxyaldehyde (2a) in water at room temperature with the DABCO-ionic liquid used as the catalyst was found to be excellent. The [H-dabco][Cl]-catalyzed (Table 1, entry 7) reaction showed extremely good results in terms of yield and reaction time, whereas the [H-dabco][AcO] catalyst (Table 1, entry 8) showed a lower yield and required a little more time.
However, apart from DABCO-ionic liquids, we intended to investigate whether it was possible to conduct the reaction using DABCO–CuX complex. The novel DABCO–CuX complex34 was synthesized by grinding a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalent ratio of DABCO and CuX and the using this complex for the synthesis of 3-acetylcoumarin (3a) in water as a medium using EAA (1) with o-hydroxyaldehyde (2a) at room temperature. Interestingly, the DABCO–CuX complex-catalyzed condensation reaction showed quite excellent yields (≃96%, Table 1, entries 9–10), which were nearly similar to those obtained with DABCO-ionic liquids.
:1 equivalent ratio of DABCO and CuX and the using this complex for the synthesis of 3-acetylcoumarin (3a) in water as a medium using EAA (1) with o-hydroxyaldehyde (2a) at room temperature. Interestingly, the DABCO–CuX complex-catalyzed condensation reaction showed quite excellent yields (≃96%, Table 1, entries 9–10), which were nearly similar to those obtained with DABCO-ionic liquids.
Recovering of ethanol
The solid obtained in experiments based on the model reaction was filtered thoroughly and washed with water to remove excess DABCO-based catalyst. The filtrate containing the water-soluble DABCO catalyst and by-product ethanol were separated by distillation under reduced pressure. The experiment was conducted in the gram scale in order to calculate the by-product ethanol. Salicylaldehyde 2a (5 g, 1 eq., 40.95 mmol) upon reaction with EAA 1 (5.340 g, 1 eq., 38.40 mmol) in the presence of DABCO catalyst gave 1.490 g (97%) ethanol as a by-product.
Reusability of the DABCO-based catalyst
The DABCO, DABCO-ionic liquids, and DABCO-complex catalyst screening results inspired us to extend our investigations to study the reusability of these catalysts for the reaction. As a model reaction EAA (1) reacted with salicylaldehyde (2a) in water using one equivalent of DABCO to give product 3a in a 95% yield in the first cycle (Fig. 1a). The second cycle gave almost the same percentage yields. However, upon increasing the time duration from 30–90 min for the third to fifth cycle, the percentage (%) yield formation decreased from 84% to 72%.
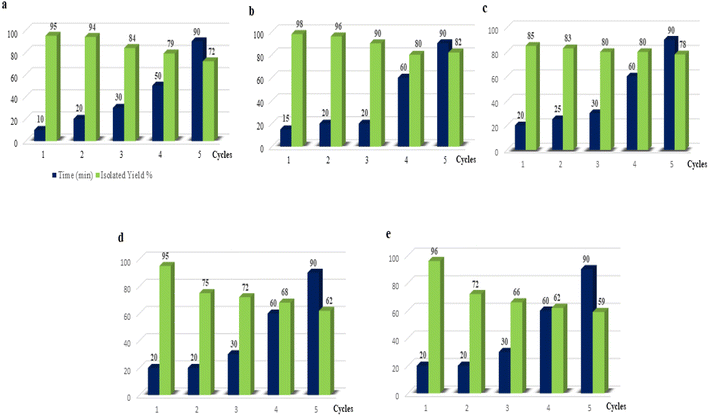 |
| | Fig. 1 Reuse of the catalysts (a) DABCO, (b) [H-dabco][Cl], (c) [H-dabco][AcO], (d) DABCO–CuCl, and (e) DABCO–CuI in the model reaction between EAA 1 and salicylaldehyde 2a in the presence of water. | |
The reusability results based on the experimental observations for the modification of DABCO into DABCO-ionic liquids [H-dabco][Cl] and [H-dabco][AcO] are summarized in Fig. 1b and c, respectively. The reuse of the [H-dabco][Cl] ionic liquid was explored for the synthesis of 3-acetyl coumarin derivatives via eco-friendly conditions by taking EAA (1) and salicylaldehyde (2a) at room temperature. Surprisingly, we noticed that the product formation of 3a was 98% and 96% I the first two cycles, respectively (Fig. 1b, cycle 1 and 2). Further catalyst reusability experiments were conducted for up to five cycles, ad it was found that the percentage of product formation decreased. For, [H-dabco][AcO] ionic liquids, smooth progress of the reaction was noticed for a certain period of time, i.e. 20–25 min, after which no progress of the reaction was observed, even with prolonged stirring and about 15–20% of the starting material remained, with the results shown in Fig. 1c. From these results, it could be observed that the ionic liquid system [H-dabco][Cl] was superior to [H-dabco][AcO] in terms of yield and time.
Further, the catalyst reusability investigations were extended by modifying DABCO to DABCO–CuX salts (X = Cl, I), in order to understand the efficiency of DABCO, and the reaction was then performed using the DABCO–CuX catalyst at room temperature in water with 1 and 2a. More interestingly, the reaction proceeded very smoothly within 20 min with a 95% conversion of the target molecule in the first cycle; while, in the second to fifth cycles, the efficiency of the catalyst was drastically reduced (Fig. 1d and e).
The catalyst reusability experimental investigation results indicated that DABCO and DABCO-ionic liquids are extremely good for clean synthesis of the title compounds compared to DABCO–CuX salts.
Catalytic efficiency
The catalytic efficiency of the DABCO-based organocatalysts was screened. A graph of the variation of the catalytic efficiency as a function of reuse times (cycles) is shown in Fig. 2. The variation in catalytic efficiency was affected by certain key properties, such as hydration effect,35a,b solvation effect,35c viscosity,35d and hydrogen bonding35e,f, leading to a decrease in the catalytic activity.
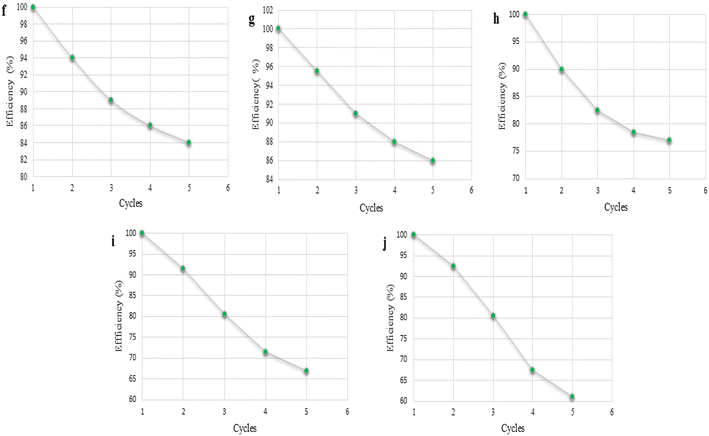 |
| | Fig. 2 Variation of the catalytic efficiency as function of the catalyst reuse times (cycles) for (f) DABCO, (g) [H-dabco][Cl], (h) [H-dabco][AcO], (i) DABCO–CuCl, and (j) DABCO–CuI in the model reaction between EAA 1 and salicylaldehyde 2a in the presence of water. | |
Scope for methodology
To test the generality of the condensation and to realize the synthesis of a small combinatorial library of 3-acetylcoumarins (3a–j) from EAA (1) and different o-hydroxyaldehydes (2a–j), we aimed to furnish the title compounds in good yields by using DABCO and DABCO-ionic liquids as a catalyst and water as a medium at room temperature (Scheme 2). Based on our experimental observations, the electron-releasing or -withdrawing group on o-hydroxyaldehydes played a major role; mainly, the electron-donating group led to excellent yield. In the case of strong electron-withdrawing groups, such as nitro substituent para-to the hydroxy group of o-hydroxyaldehydes, the reaction took longer, with a low yield (≃30% and 2–3 days) at room temperature, while under heating (65 °C-70 °C), the reaction gave a 75% yield within 2–3 h. A comparative investigation was made for the synthesis of 3-acetyl coumarins and the results are shown in Table 2. These synthesized compounds are utilized in various applications, such as material science and medicinal chemistry.
Table 2 Comparison of methods for the synthesis of 3-acetylcoumarin with present work
|
|
|
|
Reaction conditions |
Solvent |
Catalyst |
Time (min) |
Yield (%) |
Reference |
| 1 |
Stirring, r.t |
Ethanol |
Piperidine |
20 |
83.5 |
24a
|
| 2 |
Microwave irradiation, 136 °C |
Ethanol |
Piperidine |
6 |
88 |
24b
|
| 3 |
Reflux |
Ethanol |
Piperidine |
30 |
92 |
24c
|
| 4 |
Microwave irradiation |
Solvent free |
Piperidine |
— |
92.6 |
24d
|
| 5 |
Continuous-flow units, 25–130 °C |
Ethyl acetate |
Piperidine |
— |
72 |
24e
|
| 6 |
Ultrasound irradiation, 45 °C |
Solvent free |
MgFe2O4 nanoparticles |
10 |
92–96 |
25a
|
| 7 |
Microwave irradiation 120 °C or thermal, 100 °C |
Solvent free |
ZnO nano |
5–9, 36–72 |
90 |
25b
|
| 8 |
Stirring, 35–40 °C |
Ethanol |
PhI(OAc)2 |
30–60 |
82–92 |
26a
|
| 9 |
Stirring, 60–80 °C |
Dimethyl formamide |
Fe3O(BPDC)3 containing tert-butyl hydroperoxide |
180 |
65–96 |
26b
|
| 10 |
Stirring, 90 °C |
2.16% H2O |
[MMIm][MSO4] containing L-proline |
15 |
99 |
27a
|
| 11 |
Stirring, 25–30 °C |
Water |
Choline chloride |
20–45 |
90 |
27b
|
| 12 |
Stirring, 60–80 °C |
Solvent free |
L-proline |
30 |
98 |
28a
|
| 13 |
Stirring, r.t −80 °C |
Water |
L-lysine |
2880 (48 h) |
35–87 |
28b
|
| 14 |
Stirred, 80 °C |
Solvent free |
CSA |
210 (3.5 h) |
88 |
29
|
| 15 |
Reflux |
Ethanol |
HPA |
120 (2 h) |
50–98 |
30
|
| * |
Stirring, r.t. (present work) |
Water |
DABCO and DABCO-based catalysts |
10–20 |
95–98 |
— |
Experimental determination of the reaction mechanism
The versatility of the green approach encouraged us to understand the reaction mechanism, which was faster and cleaner compared to earlier reported methods. However, no such investigation was found in the literature involving DABCO organocatalyzed reactions. Hence, in order to gain an insight into the effects of the organocatalyst DABCO, we carried out a certain approach to determine the kinetic rate, and thermodynamic activation parameters in the aqueous phase. Also, a spectroscopic approach was applied for identification of the product formation quantitatively and qualitatively. The obtained experimental evidence supported the use of a stoichiometric amount of substrates, and the effects of the solvent, catalyst, temperature, and reaction time.
UV method
The absorption spectra of the reaction mixtures were measured at different concentrations of the organocatalyst DABCO, water as a solvent, and also both the reactants EAA (1) and salicylaldehyde (2a) under standard conditions. The results obtained from the absorption spectra are presented in Tables 3–5 and the reaction profile diagrams are plotted as the OD vs. time in Fig. 3–5.
Table 3 Effect of the concentration of [DABCO] and [H2O] in the synthesis of 3-acetylcoumarin from salicylaldehyde (1 eq.) with EAA (1 eq.) at 303 K
| Entry |
DABCO (eq.) |
Water (ml) |
Time (min) |
Rate |
| 1 |
1 |
10 |
300 |
0.01 |
| 2 |
1 |
5 |
120 |
0.03 |
|
3
|
1
|
1
|
10
|
0.04
|
| 4 |
0.5 |
1 |
22 |
0.03 |
| 5 |
0.1 |
1 |
28 |
0.03 |
Table 4 Effect of salicylaldehyde concentration on the reaction (EAA (1 eq.) + DABCO (1 eq.) + water (1 ml))
| Entry |
Sal (eq.) |
K
|
Log[k] |
Log[R] |
| 1 |
1 |
0.0403 |
−1.3947 |
−0.3033 |
| 2 |
0.5 |
0.0276 |
−1.5591 |
−0.6043 |
| 3 |
0.1 |
0.0076 |
−2.1191 |
−1.3036 |
Table 5 Effect of EAA concentration on the reaction (Sal (1 eq.) + DABCO (1 eq.) + water (1 ml))
| Entry |
EAA (eq.) |
k
|
Log[k] |
Log[R] |
| 1 |
1 |
0.0403 |
−1.3947 |
−0.3035 |
| 2 |
0.5 |
0.0111 |
−1.9547 |
−0.6047 |
| 3 |
0.1 |
0.0086 |
−2.0655 |
−1.3036 |
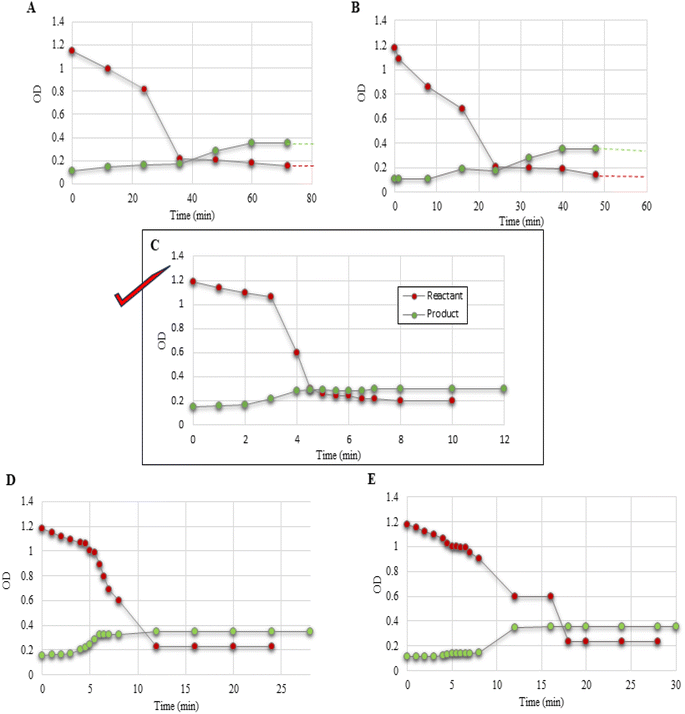 |
| | Fig. 3 Reaction profiles (A–E) for the experiments conducted and listed as entries 1–5 in Table 3. The solid red dots indicate the reactant and green dots are for the product. | |
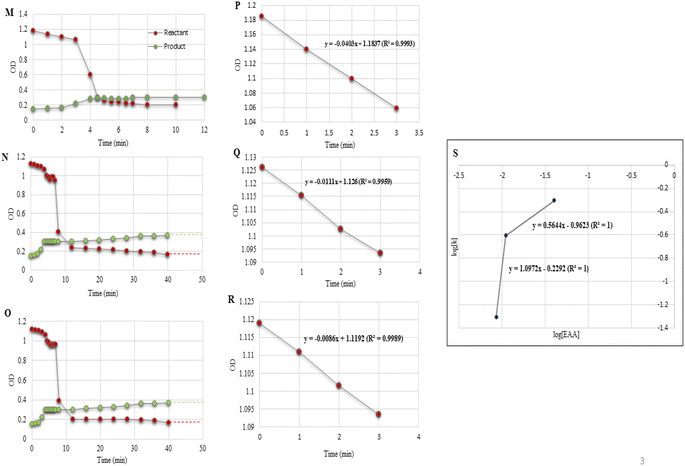 |
| | Fig. 4 (F–H) Reaction profiles for experiments conducted and listed as entries 1–3 in Table 4. (I–K) Regression plots of OD vs. time. (L) Order plot: log[k] vs. log[Sal]. Solid red dots indicate the reactant and green dots are for the product. | |
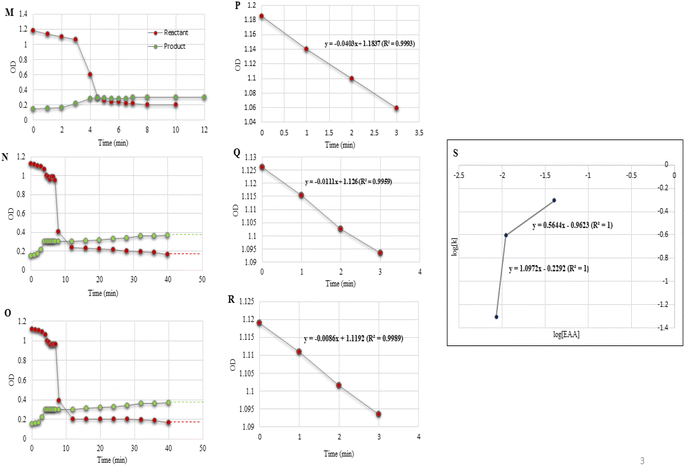 |
| | Fig. 5 (M–O) Reaction profile for experiments conducted and listed as entries 1–3 in Table 5. (P–R) Regression plots of OD vs. time. (S) Order plot: log[k] vs. log[EAA]. Solid red dots indicate the reactant and green dots are for the product. | |
Effect of water on the rate of reaction
To validate the reaction, initially the experiment was conducted using an equimolar ratio of both reactants (EAA (1), salicylaldehyde (2a)) and also DABCO catalyst as 1 eq. (Table 3, entry 1) in 10 ml of water. From the experimental results, the reaction began in the 37th min and was completed in 300 min, as monitored continuously by a UV spectrometer and TLC (Fig. 3A). Subsequently, the experiment was performed by reducing the water volume to 5 ml and 1 ml (Table 3, entries 2 and 3). The obtained results were quite excellent in the case of 1 ml water, and the reaction was completed in 10 min (Fig. 3C); while the reaction in 5 ml water needed 120 min for completion(Fig. 3B).
The aforementioned experiment illustrates that lowering the volume of water increased the reaction rate. Moreover, in raising the water volume out of our curiosity to up to 20–50 ml, it was observed that the reaction happened but took much longer for completion of the reaction (3–5 days), as monitored using TLC. Since, the solvation effect could explain the drastic drop in the reaction rate,37 the rate-determining step does not take water concentration into consideration.
Effect of DABCO on the rate of reaction
Moreover, the above investigation encouraged us to explore the reaction by varying the ratio of the organocatalyst DABCO in 1 ml of water while keeping the other reactants constant (Table 3, entries 4 and 5). The experimental results revealed that the rate of reaction was independent of the base concentration. Therefore, the base concentration term was not included in the rate-determining step. From the UV spectroscopic data, we could conclude the rate of reaction as r1eq. ≥ r0.5eq. ≥ r0.1eq. (Table 3, entries 3–5; Fig. 3C–E). Although, there was no catalyst involvement in the rate-determining step, it is essential for the reaction as it initiates as well as it controls the tautomerism of EAA, thereby starting an irreversible reaction by forming complex X with DABCO (Fig. 7, Phase Ib). The more preferred irreversible reaction step of EAA-DABCO complex formation X was confirmed using cyclic voltammetry (Fig. 12).
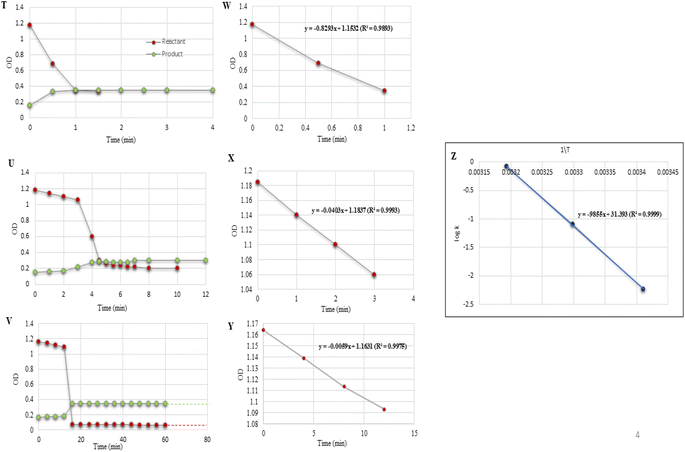 |
| | Fig. 6 (T–V) Reaction profiles for the experiments conducted and listed as entries 1–3 in Table 6. (W–Y) Regression plots of OD vs. time. (Z) Plot of log[k] vs. [1\T] in determining the activation parameters. The solid red dots indicate the reactant and the green dots are for the product. | |
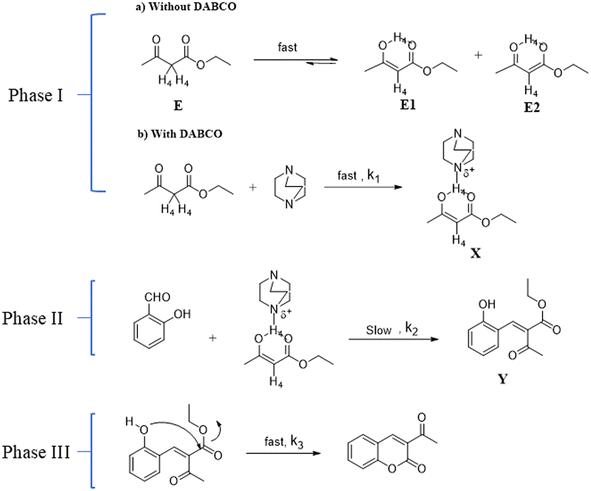 |
| | Fig. 7 Proposed reaction mechanism. | |
Effect of salicylaldehyde and EAA concentration on the rate of reaction
In the catalytic domain, both the salicylaldehyde (2a) and EAA (1) concentration effects on the rate of the reaction were tested. In the case of salicylaldehyde, the rate of reaction increased with the increase in concentration of salicylaldehyde while keeping the concentrations of the other species constant. Similarly, when the EAA concentration was increased while keeping the concentrations of the other species constant, the rate of reaction also increased. From the UV spectroscopic data, we could conclude that the rate of reaction for salicylaldehyde was r1eq. > r0.5eq. > r0.1eq. (Table 4, entries 1–3; Fig. 4F–H); and for EAA as r1eq. > r0.5eq. > r0.1eq. (Table 5, entries 1–3; Fig. 5M–O). Fig. 4I–K and Fig. 5P–R display the linear regression plot between the OD vs. time for the reaction. The correlation coefficients of the model reaction were observed as 0.9993, 0.9993, and 0.9996 for different concentration of salicylaldehyde with slopes of 0.0403, 0.0276, and 0.0076. Similarly, the correlation coefficients of the model reaction were observed as 0.9983, 0.9959, and 0.9989 for the different concentrations of EAA with slopes of 0.0403, 0.0111, and 0.0086. Fig. 4L and 5S illustrate the order of reaction from the plot of log(reactant) vs. log(k) for varying concentrations of salicylaldehyde and EAA with average slopes of 0.9008 and 0.8308 and a correlation coefficient of 1.
Rate law derivation
A kinetic investigation would typically involve the postulation of likely mechanisms and a comparison between the observed rate expression and those anticipated from theoretical approaches. It is possible to exclude from consideration any mechanisms that are incompatible with the reported kinetics.38
Based on the kinetic experimental observations, the reaction mechanism is proposed in Fig. 7. The rate law for reaction was derived by means of spectroscopic data; wherein, the formation of intermediate Y was shown to be the slowest rate-determining step (Fig. 7, Phase II). Also, by UV method, it was confirmed that only the concentration of salicylaldehyde and EAA were involved in the rate-determining step. To illustrate the development of a kinetic expression from a postulated reaction mechanism (Fig. 7), we know that the slowest step is the rate-determining step for a reaction, and the rate equation can be written as,39
Where
k2 = rate constant at Phase II.
[ED] = EAA–DABCO complex
Where [Sal]
t = concentration of salicylaldehyde at time ‘
t’.
[Sal]f = [Sal], where [Sal]f means salicylaldehyde free from all components of reactions
Where [EAA]
t = concentration of EAA at time ‘
t’.
[EAA]f = [EAA], where [EAA]f means EAA free from all components of reactions
Where k1 = rate constant at Phase I
| Rate = k2{[EAA]t − [EAA]f}[Sal] |
| = k2 {[EAA]t − [EAA]t/(1 + k1)}[Sal] |
| = k2{1 − (1/1 + k1)}[EAA]t[Sal] |
| = k2{k1/1 + k1}[EAA]t[Sal] |
| = {k1k2/(1 + k1)}[EAA][Sal] |
| Rate = k[EAA][Sal] where, k = {k1k2/(1 + k1)} |
The reaction orders were calculated from the slopes of log(kobs) vs. log(concentration) plots by varying the concentrations of salicylaldehyde (Table 4, entries 1–3; Fig. 4L) and EAA (Table 5, entries 1–3; Fig. 5S) while keeping all the other conditions constant.
Under optimized experimental conditions, the rate law can be written as, R α [salicylaldehyde]0.9008 [EAA]0.8308 order = 0.9008 + 0.8308 = 1.732.
Effect of temperature
The rate of reaction was measured at different temperatures, considering the same reaction conditions as chosen for the spectroscopic studies. Once again, the UV spectroscopic data allowed concluding the rate of reaction as r40 > r30 > r20, i.e. 0.8293 > 0.0403 > 0.0059 mol−1 L s−1 (Table 6, entries 1–3; Fig. 6(T–V)). Fig. 6(W–Y) display linear regression plots between the OD vs. time for the reactants. The correlation coefficients of the model reaction were observed as 0.9893, 0.9993, and 0.9975 for different temperatures with slopes of 0.8293, 0.0403, and 0.0059, respectively. With every 10 °C raise in temperature, the rate of the reaction increased by r40 > r30; while the rate of reaction decreased with every 10 °C drop in temperature by
Table 6 Effect of temperature on the synthesis of 3-acetyl coumarin using Sal (1 eq.) + EAA (1 eq.) + DABCO (1 eq.) + water (1 ml) in determining the activation parameters
| Entry |
Temperature (K) |
(1/T) × 1000 |
k
|
Logk |
| 1 |
313 |
3.193 |
0.8293 |
−1.859 |
| 2 |
303 |
3.298 |
0.0403 |
−3.172 |
| 3 |
293 |
3.411 |
0.0059 |
−4.007 |
A detailed thermodynamic analysis of the activation parameters for the synthesis of 3-acetyl coumarin was performed and the activation parameters obtained from the slopes of logkobsvs. 1/T plots by varying the temperature [Table 6, Fig. 6Z] for the model reaction salicylaldehyde (1 eq.), EAA (1 eq.), DABCO (1 eq.), and water (1 ml), as shown in Table 6, entries 1–3, and Fig. 6Z. Further, the activation parameters were also calculated for the slowest step of the reaction, and the activation energy was found to be Ea = 44.95 kcal mol−1. Similarly, the other activation parameters were also calculated and provided in Table 7. In the table, the positive △S# indicates that the EAA–DABCO complex was more ordered/stable than the intermediate, while the positive △H# proves the condensation reaction was endothermic and the positive △G# shows the non-spontaneity of the slowest step.
Table 7 Activation parameters with respect to the slowest step of the reaction (Ea = 44.95 kcal mol−1)
| Parameters |
Values |
| 313.16 K |
303.16 K |
293.16 K |
| ΔH# (kcal mol−1) |
44.32 |
44.34 |
44.36 |
| ΔS# (cal K−1 mol−1) |
74.39 |
73.17 |
74.45 |
| ΔG# (kcal mol−1) |
21.03 |
22.16 |
22.54 |
Spectroscopy and cyclic voltammeter evidence
The kinetic and thermodynamic experimental results prompted us to undertake investigations utilizing electrochemical techniques, such as cyclic voltammetry (CV) and spectrochemical analysis, as these tools can help instantly provide more information on the spontaneity effect of a catalyst on the rate of a reaction.
IR spectra and 1H NMR: a time-dependent study
Many methods have been adapted for clarifying the reaction mechanism, including spectroscopy for studying the reaction mechanism for homogeneous organic reactions. Spectroscopy plays a crucial role in providing evidence for organic reactions by allowing chemists to analyze the structural changes that occur during a chemical transformation. Spectroscopic techniques, such as infrared (IR) spectroscopy, can identify functional groups present in organic molecules; also, during a chemical reaction, the appearance or disappearance of certain peaks in the IR spectrum can indicate the formation or consumption of specific functional groups, providing evidence for the reaction.40 Even nuclear magnetic resonance (NMR) spectroscopy can be used to monitor the progress of a reaction in real-time. By taking NMR spectra at different time intervals during a reaction, chemists can observe changes in the chemical shifts and multiplicities of certain nuclei, which can provide insights into the formation of new products or intermediates. NMR spectroscopy provides detailed information about the connectivity and stereochemical nature of organic molecules.41 Ultimately, by making it possible to identify, characterize, and track chemical changes at the molecular level, spectroscopy is a potent tool for providing evidence for organic reactions.
There is a wide variety of intramolecular interaction that affect infrared group frequencies and band intensities. The important intra molecular factors are mass effects, electric effects (inductive and resonance effects), hydrogen bonding, symmetry, conjugation, bond angle strain, field effects, and vibrational coupling effects.42 The effects of intramolecular interactions in the infrared spectra of molecules make it possible to investigate different types of structural problems. Here, we utilized observations of the reaction changes happening by considering the peak intensities and values. We have focussed mainly on phenolic –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and aldehydic CH stretching frequencies; wherein, salicylaldehydes' aldehydic CO was observed at 1672 cm−1 (Fig. 8A), while at 4th to 8th min two CO band frequencies were found at 1736 and 1675 cm−1, which indicate the beginning of the formation of the intermediate structure Y and 3-acetyl coumarin. Hence, we rushed towards looking for aldehydic characteristic CH stretching vibration bands, which are generally observed around 2800–2650 cm−1, here as two weak bands in this region. The observed two bands at 2746 cm−1 and another at 2876 cm−1 for salicylaldehyde (Fig. 8A) disappeared during product formation. We also noticed that the hydrogen deformation vibration band of –CHO group appeared at 766 cm−1 (ref. 43) (Fig. 8A). In the IR spectra, the peak intensity diminished slowly as well as a shift of the peak towards higher wavelength was observed, from which we may explain the progress of the reaction up to the 10th min as shown in Fig. 8B–D. Moreover, the IR spectra could only explain the reaction up to a certain extent, but not precisely. Thus, we switched towards studies using time-dependent nuclear magnetic resonance (NMR) from 0 to 10 min. A technique in monitoring structural changes using time-dependent 1H NMR was proposed and then proved. All the components in the reaction, other than DABCO and H2O, were found to influence the overall kinetics, as proven in the 1H and UV studies. Three sets of standardized reaction conditions were chosen for studying the structural changes occurring in the reaction of salicylaldehyde (1 eq.) upon reaction with EAA (1 eq.) in the presence of the DABCO catalyst (1 eq.) and 1 ml of water at room temperature. Subsequently, these three sets of reactions were performed, quenched at 4, 8, and 10 min, extracted with ethyl acetate, and then the ethyl acetate was removed using a rota vapour technique, leaving behind the isolated compound, which was then analyzed by 1H NMR and compared to the reactants, i.e. salicylaldehyde and EAA.
O, and aldehydic CH stretching frequencies; wherein, salicylaldehydes' aldehydic CO was observed at 1672 cm−1 (Fig. 8A), while at 4th to 8th min two CO band frequencies were found at 1736 and 1675 cm−1, which indicate the beginning of the formation of the intermediate structure Y and 3-acetyl coumarin. Hence, we rushed towards looking for aldehydic characteristic CH stretching vibration bands, which are generally observed around 2800–2650 cm−1, here as two weak bands in this region. The observed two bands at 2746 cm−1 and another at 2876 cm−1 for salicylaldehyde (Fig. 8A) disappeared during product formation. We also noticed that the hydrogen deformation vibration band of –CHO group appeared at 766 cm−1 (ref. 43) (Fig. 8A). In the IR spectra, the peak intensity diminished slowly as well as a shift of the peak towards higher wavelength was observed, from which we may explain the progress of the reaction up to the 10th min as shown in Fig. 8B–D. Moreover, the IR spectra could only explain the reaction up to a certain extent, but not precisely. Thus, we switched towards studies using time-dependent nuclear magnetic resonance (NMR) from 0 to 10 min. A technique in monitoring structural changes using time-dependent 1H NMR was proposed and then proved. All the components in the reaction, other than DABCO and H2O, were found to influence the overall kinetics, as proven in the 1H and UV studies. Three sets of standardized reaction conditions were chosen for studying the structural changes occurring in the reaction of salicylaldehyde (1 eq.) upon reaction with EAA (1 eq.) in the presence of the DABCO catalyst (1 eq.) and 1 ml of water at room temperature. Subsequently, these three sets of reactions were performed, quenched at 4, 8, and 10 min, extracted with ethyl acetate, and then the ethyl acetate was removed using a rota vapour technique, leaving behind the isolated compound, which was then analyzed by 1H NMR and compared to the reactants, i.e. salicylaldehyde and EAA.
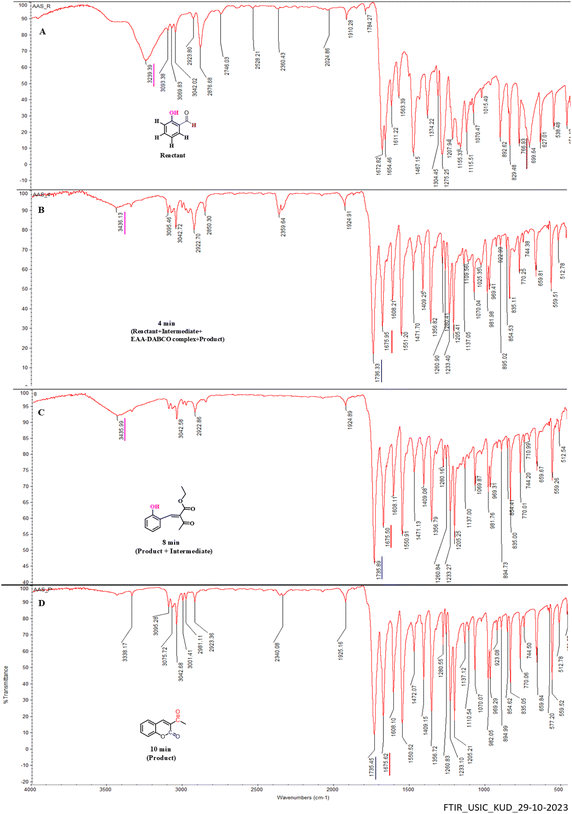 |
| | Fig. 8 Plot of % transmittance vs. wavenumber for IR spectral analysis as a time-dependent study. | |
Peaks resonating in the 1H NMR spectrum of pure EAA (Fig. 9E) indicated the presence of both EAA and the enol form of EAA species. While the choice of the perfect analytical tool is a tedious process, choosing right enables getting precise answers. As a result, we were able to tackle the issue caused in 1H NMR with the proton integration values with the use of CV. As shown in Fig. 11C3, two reductive peaks appeared in the CV for EAA caused by the formation of the two enol forms E1 and E2, respectively (see Fig. 7, Phase Ia), and these forms are impossible to distinguish using only the NMR technique. The feasibility of two H4 peaks at 4.983 (with tautomerism) and 3.462 ppm (without tautomerism) (see Fig. 7, Phase Ia) shows the reversible nature of EAA.44 For arresting this reversibility of tautomerism in EAA, a bicyclic tertiary amine base DABCO was used which holds the acidic proton of the keto-enol form, thereby making a stable six-membered ring ‘X’ (Fig. 7, Phase Ib), as proved by the disappearance of the H4 peak at 3.462 ppm and appearance of two magnetically equivalent peaks of DABCO at 3.361 and 2.691 ppm in the 1H NMR spectrum, as shown in Fig. 9F. Also, the formation of X was the fastest step, as determined through 1H NMR and CV.
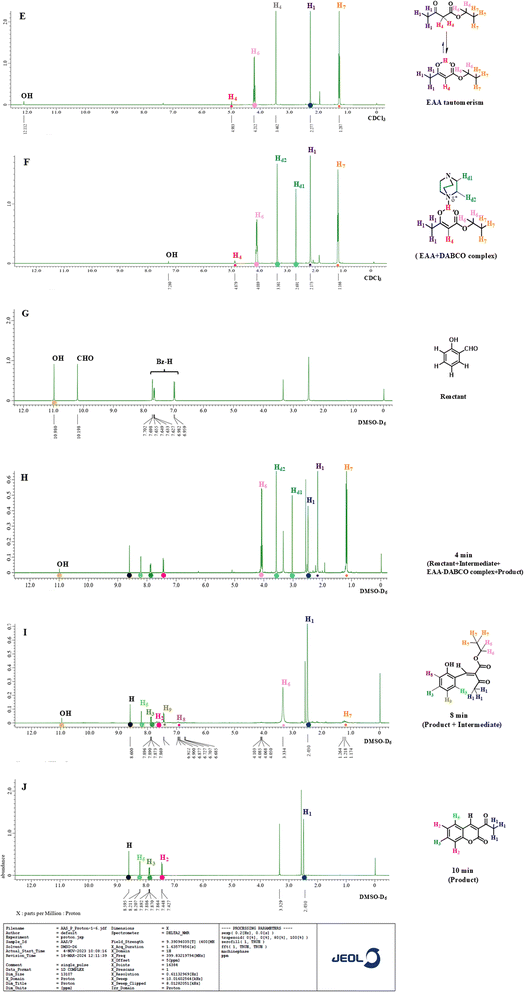 |
| | Fig. 9 Plot of abundance vs. chemical shift for 1H NMR spectral analysis as a time-dependent study. | |
The Knoevenagel condensation reaction happens between the aldehydic functional group of salicylaldehyde with the EAA-DABCO complex, resulting in the formation of intermediate ‘Y’, namely ethyl 2-(2-hydroxybenzylidene)-3-oxobutanoate (Fig. 7, Phase II). A close examination of the 1H NMR spectra allowed us to conclude that the product formation began as early as 4 min, as can be seen from Fig. 9H with the peaks for H4 and H1 resonating at 4.879 and 2.490 ppm, as well as the presence of an intermediate Y represented by H1 at 2.173, while the EAA-DABCO complex represented by Hd2, Hd1 at 3.361 and 2.691 ppm appeared at up to the 4th min. Interestingly, at the 8th min, a disappearance of H1 (2.173 ppm) was noted, wherein Hd1 and Hd2 peaks were observed, and the presence of the phenolic –OH group in intermediate Y was observed through the 1H NMR data around 11 ppm, as shown in Fig. 9I. Thus, the intermediate formation was the slowest rate-determining step in the reaction k2.45
Intramolecular ring closure of the phenolic –OH with the ethyl ester part of intermediate Y was found to be fastest step k3 in the reaction. By keenly observing the 1H NMR spectra at the 10th min (Fig. 9J), the reaction was found to be complete, showing only the product 3-acetyl coumarin peaks with the disappearance of the phenolic OH, H6, H7, H8, and H9 peaks of the intermediate at the 10th min.
Cyclic voltammeter study
Nowadays, electroanalytical methods are well recognized and applied in many chemical fields. Their typical applications include the investigation of redox processes, the study of electrochemical reactions between ions and the surface atoms of electrodes, studies to comprehend reaction intermediates and the stability of reaction products, the qualitative description of electrode reaction mechanisms, and qualitative determination of the characteristics of the charge–transfer reactions between electrolyte ions and electrons from the electrode surface.46 Since, the current in a circuit is proportional to the electrode's surface area, it is possible to measure the rate of reaction simply by measuring the current flowing through the circuit, using the formula I = dQ/dt; therefore, the rate of reduction or oxidation at the electrode surface can be determined by the current. Numerous other elements also affect this current, primarily the concentration of the redox species, the electrode's size, shape, and material, the solution resistance, the cell volume, and the quantity of electrons transported.47
Spontaneity and stability of tautomerism in EAA and its complex with DABCO catalyst
In order to equate the kinetic and thermodynamic results, cyclic voltammetry was employed, as this allows determining quick electron transfer from a molecule and allows predicting the mechanism of a reaction. In our work, we used Ag/AgCl in 1 M KCl as the reference electrode, platinum electrode as the counter electrode, and a glassy carbon electrode as the working electrode, because it was found that, at this surface (as opposed to metal electrodes such as Ag, Au, Hg, and Pt), voltammetry suffers the least interferences from adsorption processes.48 Accordingly, the reactions performed so far were chosen, and we prepared two sets of standardized solutions: EAA (1 eq.) dissolved in 10 ml phosphate buffer solution of pH 7 and another same set in the presence of DABCO catalyst (1 eq.).
As is well known, EAA having α-H4 is more acidic and undergoes tautomerism very quickly and reversibly.49 We proved this reversible nature of EAA by using cyclic voltammetry. Initially, the potential was scanned linearly over time; wherein, the potential reaches a suitably negative or positive potential, and chemical species undergo oxidation or reduction at the electrode surface, producing an electrical current. This current is monitored during the process of the potential scan in order to figure out how much of a chemical species is undergoing a redox reaction per unit time. The current increases as the potential rises through the redox-active species' half-wave potential (E1/2), making oxidation more thermodynamically favourable. Eventually, however, the oxidation process is limited by species diffusion to the electrode surface, resulting in a diffusional tail marked by a drop in current. After that, the electrode's potential sweep is reversed and scanned in the other direction until the starting potential is attained.50 For a chemically reversible electron-transfer process as denoted in Fig. 7, i.e. Phase I without the DABCO catalyst, the reduction of the electrochemically generated species resulted in a cathodic (reductive) current, leading to the voltammogram at a scan rate of 50 mV s−1 displayed in Fig. 10C1. The features of cyclic voltammograms that are generally reported in the literature are the anodic and cathodic peak heights, ipa and ipc, and the peak separation ΔEp. For an electrochemically reversible process, E1/2 is measured as halfway between the anodic and cathodic peak potentials. Here, one anodic peak around EA = +1.234 V and two mesmerising cathodic peaks around ECB = −0.6992 V and ECC = −1.350 V were observed and distinguished by CV only rather than by the NMR technique; this reveals the tautomerism of acidic α-H4 occurring on either side, in which tautomerism occurring at the keto carbonyl is more favoured (ECC) than at the ester carbonyl (ECB), as judged based on the peak area. The peak to peak separation △Ep1 = Eaa − Ecb = +1.933 V and △Ep2 = Eaa − Ecc = +2.584 V reveal the quasi-reversible nature of the reaction.51 For arresting this reversibility of tautomerism in EAA, a bicyclic tertiary amine base DABCO was used, which holds the acidic proton of the keto-enol form, thereby making a stable six-membered ring ‘X’, as proved and shown in Fig. 7, Phase Ib, leading to the voltammogram at a scan rate of 50 mV s−1 displayed in Fig. 10C2. The peak around +1.234 V remained while the two reductive peaks vanished; also the height of these peaks ≈ipa/ipc ≠1; which explains the most favoured irreversible reaction.52 Meanwhile, it was also observed that this step was the fastest step k1 (Fig. 7), as confirmed by CV (Fig. 10) and 1H NMR (Fig. 9). Curiosity prompted us to also observe the current intensity peak, whereby the intensity was increased for EAA–DABCO complex (+30.30 μA) over EAA (+16.82 μA) (see Fig. 10C1 and C2); which proves that oxidation was more thermodynamically favourable with the formation of a stable complex.
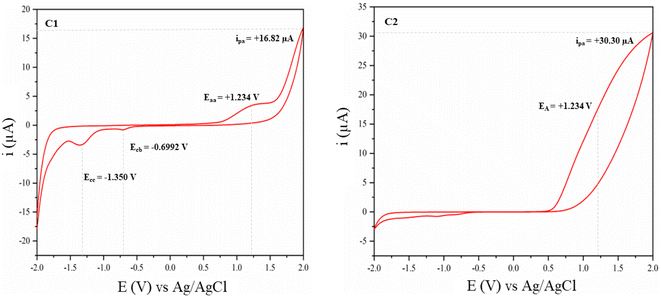 |
| | Fig. 10 Plot of the current vs. potential: C1 represents the quasi-reversible tautomerism of EAA, C2 represents the arrested form of EAA–DABCO – a favoured irreversible reaction. | |
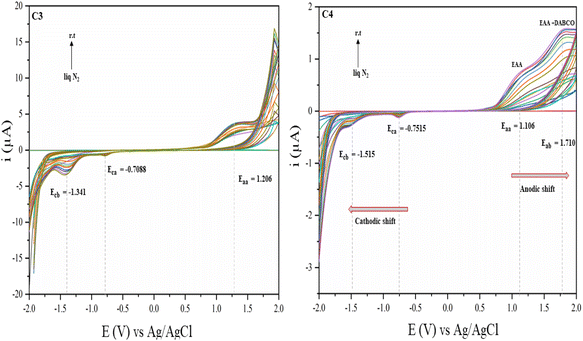 |
| | Fig. 11 Plot of the current vs. potential, where C3 represents the quasi-reversible tautomerism of EAA, and C4 represents the quasi-reversible tautomerism of EAA in the presence of a non-stoichiometric amount of DABCO. | |
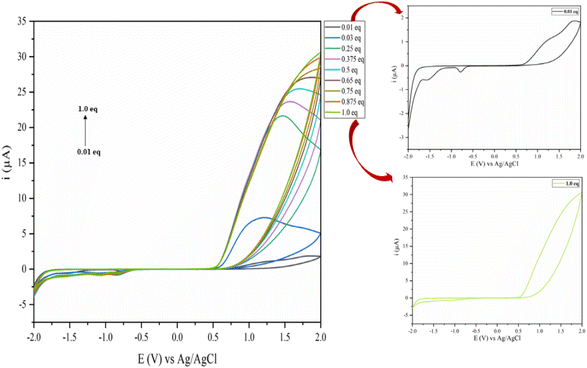 |
| | Fig. 12 Plot of the current vs. potential showing the conversation of the quasi-reversible to an irreversible process with increasing the concentration of DABCO (from 0.0125 to 1 eq.). | |
Effect of temperature over tautomerism in EAA and its complex with DABCO catalyst
Despite its significance, insufficient research has been conducted on the impact of temperature on the kinetic behaviour, leading to a lack of conclusive findings.53Fig. 11 illustrates the typical CV voltammograms at a scan rate of 50 mV s−1 for two different sets of solutions of EAA (1 eq.) and another same set in the presence of DABCO catalyst (0.0125 eq.).
The electron mobility/shift in the α-H4 acidic proton of EAA was examined (Fig. 11C3) under liquid N2 to room temperature. It was found that the tautomerism stopped in liquid N2 atmosphere and that the mesomeric effect slowing began from −10 °C with raising the temperature, which could be detected by an increase in the current. All the separations between the oxidation and reduction peak potentials (△Ep) could be observed in Fig. 11C3 and were much greater than 60 mV, indicating a quasi-reversible reaction.
In order to examine the displacement of the potential (V) of the anodic and cathodic shifts and also the intensity of the oxidative current peak during the EAA–DABCO complexation reaction, a non-stoichiometric experimental proportion of DABCO was taken. The anodic peak of pure EAA around Eaa = 1.206 V (Fig. 11C3) was shifted to Eaa = 1.106 V (Fig. 11C4), implying the formation of a complex was beginning and a new peak could thus be observed for the EAA–DABCO complex around Eab = 1.710 V. Similarly, the complexation of EAA–DABCO caused a shift in the cathodic peak of EAA around Eca = −0.7088 V (Fig. 11C3) to Eaa = −0.7515 V (Fig. 11C4) upon complexation. Second, the reductive peak abruptly changed and shifted to Ecb = −1.515 V (Fig. 11C4), revealing that the tautomerism of acidic α-H4 occurred on either side, in which the tautomerism occurring at the keto carbonyl was favoured (Ecb) over that on the ester carbonyl (Eca), as judged based on the peak area. Furthermore, the vanishing of both the anodic and cathodic peaks of EAA upon the perfect stoichiometric complexation with DABCO was studied.
Effect of DABCO concentration over tautomerism of EAA – until the favoured irreversible process
Fig. 12 illustrates the typical CV voltammograms at a scan rate of 50 mV s−1 for varying concentrations of DABCO on EAA tautomerism solution in 10 ml phosphate buffer at pH 7. Here, it can be seen that the anodic peak current (ipa) increased as the concentration of the DABCO catalyst increased. We were curious enough to check the cyclic voltammogram for each concentration, which discloses two distinct observations: first, the oxidation peak pattern changed with increasing the peak current as the concentration of DABCO increased, indicating the more thermodynamically favourable formation of a stable complex. Second, the reduction peak decreased with increasing the concentration of DABCO and straightened precisely at 1 eq. concentration, indicating the conversion of a quasi-reversible to an irreversible process. Consequently, 1 eq. DABCO concentration was the optimal quantity to hold the complete enol form of EAA–DABCO, making it irreversible by complexing.
Experimental
General information
All the reactions were monitored by TLC (Merck silica gel F-254 plates) and the isolated products were characterized by FTIR (KBr pellets; dated 29-10-2023), and 1H NMR (400 MHz; dated 04-11-2023) respectively. Chemical shifts were reported in parts per million (ppm) downfield from the TMS internal standard. Electrochemical investigations were carried out on a CHI6134E electrochemical analyzer with a standard three-electrode cell set-up (CH Instruments Ltd Co., USA).
General procedure for the synthesis of 3-acetylcoumarins (3a–h)
A mixture of EAA (1 eq.), substituted o-hydroxyaldehydes (1 eq.), and catalyst (DABCO, DABCO- ionic liquids) was stirred at room temperature until TLC monitoring analysis indicated (10–20 min) that the reaction was complete. The solid that separated was filtered and washed thoroughly with water.
Procedure for recovering of the DABCO catalyst and ethanol
Following the general procedure described above, initially, the filtrate obtained was taken and distilled under reduced pressure to recover ethanol. Moreover, the aqueous solution containing only the catalyst and free from ethanol was reused once again for the reaction. The catalyst could be reused at least 5 times.
Conclusions
From this investigation, we have shown a most useful facile green synthesis of 3-acetylcoumarins for the first time in water, offering high purity and yield under mild conditions. The condensation reaction worked well with a stoichiometric amount of catalyst in aqueous medium at room temperature. This methodology offers a good scope for the synthesis of a wide variety of 3-acetylcoumarins compared to earlier reported approaches. Also, the catalysts DABCO and DABCO-ionic liquids could be easily recovered and reused more efficiently. Meanwhile, the by-product ethanol could also be recovered and used wherever essential. We demonstrated the effect of the DABCO catalyst/reactants on the rate kinetics of the reaction and thermodynamic activation parameters. From these studies, it was revealed that the DABCO catalyst initiates the reaction very quickly and completes the reaction via holding the tautomerism, by EAA-DABCO complexation. While the interesting step of EAA-DABCO complexation and its involvement in the progress of the reaction were determined through FT-IR and time-dependent 1H NMR studies. More interestingly for the first time, the tautomeric form of EAA and its complexation with DABCO were studied in detail through CV, suggesting that the tautomerism of EAA happens fast, whereas in the EAA–DABCO complex tautomerism was arrested, leading to completion of the reaction in a shorter time. The salient features of this method are its totally eco-friendly condition, the reusability of the catalyst and by-product, and it is more economical. Moreover, this protocol can be used for the large-scale synthesis of substituted-3-acetylcoumarins; thus, the present procedure is a significant improvement over existing methods. More broadly, the exploration of this remarkable method may lead to new condensation reactions.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Author contributions
AAS conceived the ideas and carried out the experiments. SLS and LAS were involved in research investigation. All three authors contributed to the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the University Scientific Instrumentation centre (USIC) and DST-SAIF, Karnatak University, Dharwad for spectral analysis. One of the author Arpita A. Shanbhag is thankful to URS (Sr. No./KU/Fellowship/URS/2022/376 dated: 18/09/2022) and CSIR-NET- JRF New Delhi (File. No. 09/0101(15285)/2022-EMR-I dated: 27/07/2022) for the financial support. The authors express their gratitude towards Dr Atmanand Bagoji and Dr Amar Durgannavur for their valuable advice and insights.
References
- S. M. Sethna and N. M. Shah, Chem. Rev., 1945, 36, 1–62 CrossRef CAS.
- A. Vogel, Ann Phys., 1820, 64, 161–166 CrossRef.
- For general reviews on Pechmann reaction, see:
(a) T. Sugino and K. Tanaka, Chem. Lett., 2001, 110 CrossRef CAS;
(b) A. Hegedüs and Z. Hell, Catal. Lett., 2006, 112, 105–108 CrossRef;
(c) S. Sethna and P. Ragini, Org. React., 2004, 7, 1–58 Search PubMed;
(d) N. G. Khaligh, Catal. Sci. Technol., 2012, 2, 1633–1636 RSC;
(e) L. T. L. Nguyen, K. K. A. Le, H. X. Truong and N. T. S. Phan, Catal. Sci. Technol., 2012, 2, 521–528 RSC;
(f) H. V. Pechmann and J. B. Cohe, Eur. J. Inorg. Chem., 2015, 17, 2187–2191 Search PubMed;
(g) G. K. Hodgson, S. Impellizzeri and J. C. Scaiano, Chem. Sci., 2016, 7, 1314–1321 RSC;
(h) A. K. Gupta, D. De, R. Katoch, A. Garg and P. K. Bharadwaj, Inorg. Chem., 2017, 56, 4697–4705 CrossRef PubMed.
- For general reviews on Knoevenagel reaction, see:
(a) G. Brufola, F. Fringuelli, O. Piermatti and F. Pizzo, ChemInform, 1996, 27 DOI:10.1002/CHIN.199643107;
(b) L. T. L. Nguyen, K. K. A. Le, H. X. Truong and N. T. S. Phan, Catal. Sci. Technol., 2012, 2, 521–528 RSC;
(c) J. J. Clemens, J. L. Asgian, B. B. Busch, T. Coon, J. Ernst, L. Kaljevic, P. J. Krenitsky, T. D. Neubert, E. J. Schweiger, A. Termin and D. Stamos, J. Org. Chem., 2013, 78, 780–785 CrossRef CAS PubMed;
(d) P. Rammohan and S. Taradas, Int. J. Org. Chem., 2014, 04, 106–115 CrossRef;
(e) R. H. Vekariya and H. D. Patel, Synth. Commun., 2014, 44, 2756–2788 CrossRef CAS;
(f) J. V. Schijndel, L. A. Canalle, J. Smid and J. Meuldijk, Open J. Phys. Chem., 2016, 06, 101–108 CrossRef;
(g) R. C. M. A. Sobrinho, P. M. de Oliveira, C. R. M. D'Oca, D. Russowsky and M. G. M. D'Oca, RSC Adv., 2017, 7, 3214–3221 RSC;
(h) S. Crotti, N. Di Iorio, C. Artusi, A. Mazzanti, P. Righi and G. Bencivenni, Org. Lett., 2019, 21, 3013–3017 CrossRef CAS PubMed;
(i) A. Hasija, V. Prakash, M. S. Prakash, G. Krishnasamy and D. Chopra, Cryst. Struct. Theory Appl., 2020, 09, 49–62 CAS.
- For general reviews on Perkin reaction, see:
(a) J. R. Johnson, Org. React., 1942, 1, 210–265 Search PubMed;
(b) O. Yoshiro and M. Tsuchida, J. Org. Chem., 1959, 24, 78–83 CrossRef;
(c) P. M. Pawar, K. J. Jarag and G. S. Shankarling, Green Chem., 2011, 13, 2130–2134 RSC.
- For general reviews on Reformatsky reaction, see:
(a) M. Á. Fernández-Ibáñez, B. Maciá, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2008, 2571–2573 RSC;
(b) R. L. Shriner, Org. React., 2011 DOI:10.1002/0471264180.or001.01;
(c) C. Wolf and M. Moskowitz, J. Org. Chem., 2011, 76, 6372–6376 CrossRef CAS PubMed;
(d) A. Maestro, E. Martinez De
Marigorta, F. Palacios and J. Vicario, Org. Lett., 2019, 21, 9473–9477 CrossRef CAS PubMed;
(e) Qi-L. Chen, Le Mao, Yi-F. Pan, H. Cai, X.-M. Zhang, Fu-M. Zhang, Ai-J. Ma, J.-B. Peng and Y.-Q. Tu, Chem. Commun., 2023, 59, 14427–14430 RSC.
- For general reviews on Wittig reaction, see:
(a) N. S. Narasimhan and R. S. Mali, Tetrahedron, 1975, 31, 1005–1009 CrossRef CAS;
(b) T. Harayama, K. Nakatsuka, H. Nishioka, K. Murakami, Y. Ohmori, Y. Takeuchi, H. Ishii and K. Kenmotsu, Heterocycles, 1994, 38, 2729–2738 CrossRef CAS;
(c) I. Yavari, R. H. Shoar and A. Zonouzi, Tetrahedron Lett., 1998, 39, 2391–2392 CrossRef CAS;
(d) P. J. Murphy and S. E. Lee, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3049–3066 RSC;
(e) R. Robiette, J. Richardson, V. K. Aggarwal and J. N. Harvey, J. Am. Chem. Soc., 2006, 128, 2394–2409 CrossRef CAS PubMed;
(f) J. Zheng, J. Cai, J. H. Lin, Y. Guo and J. C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC;
(g) P. A. Byrne and D. G. Gilheany, Chem. Soc. Rev., 2013, 42, 6670–6696 RSC;
(h) D. Reiter, P. Frisch, T. Szilvási and S. Inoue, J. Am. Chem. Soc., 2019, 141, 16991–16996 CrossRef CAS PubMed;
(i) A. B. Cuenca and E. Fernández, Chem. Soc. Rev., 2021, 50, 72–86 RSC;
(j) A. M. Borys, E. F. Rice, G. S. Nichol and M. J. Cowley, J. Am. Chem. Soc., 2021, 143, 14065–14070 CrossRef CAS PubMed.
- V. M. Alexander, R. P. Bhat and S. D. Samant, Tetrahedron Lett., 2005, 46, 6957–6959 CrossRef CAS.
- For a reviews on coumarin synthesis by organopalladium intermediates, see:
(a) C. Jia, D. Piao, T. Kitamura and Y. Fujiwara, J. Org. Chem., 2000, 65, 7516–7522 CrossRef CAS PubMed;
(b) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992–1995 CrossRef CAS PubMed;
(c) S. Aoki, C. Amamoto, J. Oyamada and T. Kitamura, Tetrahedron, 2005, 61, 9291–9297 CrossRef CAS;
(d) B. Gabriele, R. Mancuso, G. Salerno and P. Plastina, J. Org. Chem., 2008, 73, 756–759 CrossRef CAS PubMed;
(e) T. D. A. Fernandes, B. Gontijo Vaz, M. N. Eberlin, A. J. M. Da Silva and P. R. R. Costa, J. Org. Chem., 2010, 75, 7085–7091 CrossRef CAS PubMed;
(f) K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954–10957 CrossRef CAS PubMed;
(g) S. Dharani, G. Kalaiarasi, D. Sindhuja, V. M. Lynch, R. Shankar, R. Karvembu and R. Prabhakaran, Inorg. Chem., 2019, 58, 8045–8055 CrossRef CAS PubMed.
- For a reviews on derivatives of coumarin application in laser dyes, see:
(a)
M. S. A. Abdel-Mottaleb, M. S. Antonious, M. M. Abo-Ali, L. F. M. Ismail, B. A. El-Sayed and A. M. K. Sherief, 1992, DOI: DOI:10.1007/BF02863363;
(b) M. S. A. Abdel-Mottaleb, M. S. Antonious, M. M. Abo-Aly, L. F. M. Ismaiel, B. A. El-Sayed and A. M. K. Sherief, J. Photochem. Photobiol., A, 1989, 50, 259–273 CrossRef CAS;
(c) C. Eggeling, L. Brand and C. A. M. Seidel, Bioimaging, 1997, 5, 105–115 CrossRef CAS;
(d) J. H. Richardson, L. L. Steinmetz, S. B. Deutscher, W. A. Bookless and W. L. Schmelzinger, Z. Naturforsch. A, 1978, 33, 1592–1593 CrossRef;
(e) J. R. Mannekutla, B. G. Mulimani and S. R. Inamdar, Spectrochim. Acta, Part A, 2008, 69, 419–426 CrossRef CAS PubMed;
(f) S. A. Azim, S. M. Al-Hazmy, E. M. Ebeid and S. A. El-Daly, Opt. Laser Technol., 2005, 37, 245–249 CrossRef CAS;
(g) A. Fischer, C. Cremer and E. H. K. Stelzer, Appl. Opt., 1995, 34(12), 1989–2003 CrossRef CAS PubMed.
- Z. Xu, X. Liu, J. Pan and D. R. Spring, Chem. Commun., 2012, 48, 4764–4766 RSC.
- For a reviews on derivatives of coumarin application in solution dynamic probes, see:
(a) N. M. Correa and N. E. Levinger, J. Phys. Chem. B, 2006, 110, 13050–13061 CrossRef CAS PubMed;
(b) J. A. Gutierrez, R. D. Falcone, J. J. Silber and N. M. Correa, J. Phys. Chem. A, 2010, 114, 7326–7330 CrossRef CAS PubMed.
- For a reviews on derivatives of coumarin application in dye-sensitized solar cell, see:
(a) K. Hara, K. Sayama, Y. Ohga, A. Shinpo, S. Suga and H. Arakawa, Chem. Commun., 2001, 569–570 RSC;
(b) K. Hara, Z. S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-Oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476–15482 CrossRef CAS PubMed;
(c) Z. S. Wang, Y. Cui, K. Hara, Y. Dan-Oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS;
(d) Z. S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224–7230 CrossRef CAS.
- For a reviews on derivatives of coumarin application in fluorescence brightner, see:
(a) N. S. Naik, L. A. Shastri, C. Bathula, B. Chougala, S. Shastri, M. Holiyachi and V. Sunagar, J. Fluoresc., 2017, 27, 419–425 CrossRef CAS PubMed;
(b) S. Samundeeswari, M. V. Kulkarni, J. Yenagi and J. Tonannavar, J. Fluoresc., 2017, 27, 1247–1255 CrossRef CAS PubMed.
- A. V Shanbhag, T. V Venkatesha, R. A. Prabhu and B. M. Praveen, Bull. Mater. Sci., 2011, 34(3), 571–576 CrossRef.
- For a reviews see:
(a) M. K. Li, J. Li, Y. Zhou, X. Y. Xue, X. Shi, Z. Y. Tu, Z. Hou and X. X. Luo, Res. Chem. Intermed., 2015, 41, 3289–3296 CrossRef CAS;
(b) E. Mansour, E. M. Nassar, A. F. El-Farargy and F. M. Abdelrazek, Russ. J. Bioorg. Chem., 2020, 46, 582–589 CrossRef CAS.
- V. N. Toan and N. D. Thanh, Med. Chem., 2021, 30, 1868–1885 CAS.
- For a reviews see:
(a) M. L. Low, G. Paulus, P. Dorlet, R. Guillot, R. Rosli, N. Delsuc, K. A. Crouse and C. Policar, BioMetals, 2015, 28, 553–566 CrossRef CAS PubMed;
(b) M. Mahdavi, S. Mohammadi-Izad, M. Saeedi, M. Safavi, S. E. S. Ebrahimi, A. Foroumadi and A. Shafiee, J. Iran. Chem. Soc., 2016, 13, 1139–1144 CrossRef CAS;
(c) J. B. Foo, M. L. Low, J. H. Lim, Y. Z. Lor, R. Zainol Abidin, V. Eh Dam, N. Abdul Rahman, C. Y. Beh, L. C. Chan, C. W. How, Y. S. Tor and L. Saiful Yazan, BioMetals, 2018, 31, 505–515 CrossRef CAS PubMed.
- V. Channabasappa, K. Kumara and A. Kumar Kariyappa, J. Chem. Sci., 2021, 133, 130 CrossRef CAS.
- P. Aragade, M. Palkar, P. Ronad and D. Satyanarayana, Med. Chem. Res., 2013, 22, 2279–2283 CrossRef CAS.
-
(a) N. Ingale, V. Maddi, M. Palkar, P. Ronad, S. Mamledesai, A. H. M. Vishwanathswamy and D. Satyanarayana, Med. Chem. Res., 2012, 21, 16–26 CrossRef CAS;
(b) N. Hosseini Nasab, F. Azimian, H. G. Kruger and S. J. Kim, Arabian J. Chem., 2023, 16, 104472 CrossRef CAS.
- J. K. Gupta, P. K. Sharma, R. Dudhe, A. Chaudhary, A. Singh, P. K. Verma, S. C. Mondal, R. K. Yadav and S. Kashyap, Med. Chem. Res., 2012, 21, 1625–1632 CrossRef CAS.
- A. M. Msa and M. S. Attia, Res. J. Chem. Sci., 2013, 3, 4 Search PubMed.
-
(a) A. S. Lunkad and R. L. Sawant, Int. J. Pharm. Sci. Res., 2018, 9, 2852–2858 CAS;
(b) D. Bogdal, J. Chem. Res., 1998, 468–469 RSC;
(c) K. V. Sashidhara, A. Kumar, M. Kumar, R. Sonkar, G. Bhatia and A. K. Khanna, Bioorg. Med. Chem. Lett., 2010, 20, 4248–4425 CrossRef CAS PubMed;
(d) O. O. Ajania and O. C. Nwinyi, J. Heterocycl. Chem., 2010, 47, 179 CrossRef;
(e) T. A. Hamlin and N. E. Leadbeater, J. Visualized Exp., 2015, 105 DOI:10.3791/52393.
-
(a) J. S. Ghomi and Z. Akbarzadeh, Ultrason. Sonochem., 2018, 40, 78–83 CrossRef CAS PubMed;
(b) B. V. Kumar, H. S. B. Naik, D. Girija and B. Vijayakumar, J. Chem. Sci., 2011, 123, 615–621 CrossRef CAS.
-
(a) D. Khan, S. Mukhtar, M. A. Alsharif, M. I. Alahmdi and N. Ahmed, Tetrahedron Lett., 2017, 58, 3183–3187 CrossRef CAS;
(b) T. N. Lieu, K. D. Nguyen, D. T. Le, T. Truong and N. T. S. Phan, Catal. Sci. Technol., 2016, 6, 5916–5926 RSC.
-
(a) S. B. Phadtare and G. S. Shankarling, Environ. Chem. Lett., 2012, 10, 363–368 CrossRef CAS;
(b) P. Verdía, F. Santamarta and E. Tojo, Molecules, 2011, 16, 4379–4388 CrossRef PubMed.
-
(a) K. N. Nandkishor, G. Sumit, V. Shinde, V. Sandeep and N. W. Jadhav, Chin. J. Chem., 2007, 25, 1687 Search PubMed;
(b) X. You, H. Yu, M. Wang, J. Wu and Z. Shang, Lett. Org. Chem., 2012, 9, 19–23 CrossRef CAS.
- S. S. Gholap, U. P. Deshmukh and M. S. Tambe, Iran. J. Catal., 2013, 3, 171–176 CAS.
- M. M. Heravi, S. Sadjadi, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, Catal. Commun., 2008, 9, 470–474 CrossRef CAS.
-
(a) M. Li, R. Zhang, C. Jia, Z. Liu, Y. Liu and A. Ying, Fuel, 2024, 375, 132671 CrossRef CAS;
(b) X. Lu, S. Li, L. Wang, S. Huang, Z. Liu, Y. Liu and A. Ying, Fuel, 2022, 122318 CrossRef CAS;
(c) S. Li, X. Lu, Q. Liu, L. Wang, Y. Liu, Z. Liu and A. Ying, J. Mater. Chem. A, 2022, 10, 3531–3542 RSC;
(d) A. Ying, S. Li, X. Liu, J. Wang, Y. Liu and Z. Liu, J. Catal., 2020, 391, 312–326 CrossRef CAS;
(e) N. Seyedi, F. Shirini and H. Tajik, J. Mol. Struct., 2023, 1285, 135547 CrossRef CAS;
(f) P. P. Foumani, M. Mousapour, F. Shirini, H. Tajik and S. Hassanpoor, J. Iran. Chem. Soc., 2023, 2, 359–371 Search PubMed;
(g) N. Seyedi, L. N. Nasyrmahale, F. Shirini and H. Tajik, ChemistrySelect, 2023, 8, e202204306 CrossRef CAS;
(h) N. M. Shaikh, V. Adimule, G. B. Bagihalli and R. S. Keri, Top. Catal., 2022, 1–10 Search PubMed;
(i) S. Sahrapeyma, F. Shirini and M. Mamaghani,
Iran. J. Sci.
, 2023, 47, 1553–1564 CrossRef;
(j) M. Dhama, P. Gupta and D. Sah, Res. Chem. Intermed., 2024, 50, 575–595 CrossRef CAS;
(k) J. Long, W. Dai, M. Zou, B. Li, S. Zhang, L. Yang, J. Mao, P. Mao, S. Luo and X. Luo, Microporous Mater., 2021, 318, 111027 CrossRef CAS.
- F. Scholz, ChemTexts, 2015, 1, 17 CrossRef.
- For a reviews see:
(a) Y. Q. Yu and D. Z. Xu, RSC Adv., 2015, 5, 28857–28863 RSC;
(b) N. Sahiba, A. Sethiya, P. Teli and S. Agarwal, ACS Omega, 2023, 8, 5877–5884 CrossRef CAS PubMed.
- B. Baghernejad and M. Fiuzat, Eurasian Chem. Commun., 2020, 2, 1088–1092 CAS.
-
(a) K. R. Seddon, A. Stark and M. J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS;
(b) M. J. Earle and K. R. seddon, Pure Appl. Chem., 2000, 72, 1391–1398 CrossRef CAS;
(c) P. Wasserscheid and T. Welton, Org. Process Res. Dev., 2003, 7, 223–224 Search PubMed;
(d) S. Zhang, N. Sun, X. He, X. Lu and X. Zhang, J. Phys. Chem. Ref. Data, 2006, 35, 1475–1517 CrossRef CAS;
(e) P. Nockemann, B. Thijs, S. Pittois, J. Thoen, C. Glorieux, K. V. Hecke, L. V. Meervelt, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2006, 110, 20978–20992 CrossRef CAS PubMed;
(f) T. Welton, Chem. Rev., 1999, 8, 2071–2084 CrossRef PubMed.
-
(a) H. M. Han, C. R. Lu, Y. Zhang and D. C. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, E61, o1864–o1866 CrossRef;
(b) D. Secci, S. Carradori, A. Bolasco, P. Chimenti, M. Yáñez, F. Ortuso and S. Alcaro, Eur. J. Med. Chem., 2011, 46, 4846–4852 CrossRef CAS PubMed;
(c) Z. Q. Xiong, L. Yang, S. Z. Xiao, C. Y. Yang and X. L. Nie, Z. Kristallogr., 2022, 237, 635–637 CAS;
(d) H. Valizadeh, A. Shockravi and H. Gholipur, J. Heterocycl. Chem., 2007, 44, 867 CrossRef CAS;
(e)
https://www.chembk.com/en/chem/848322-75-8
;
(f) N. N. Korgavkar and S. D. Samant, New J. Chem., 2017, 41, 12422 RSC.
-
C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, 4th edn, 2011 Search PubMed.
-
F. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part B: Reactions and Synthesis, 5th edn, 2007 Search PubMed.
-
(a) L. Jattinagoudar, S. Nandibewoor, S. Chimatadar and J. Solution, Chem, 2016, 45, 497–517 CAS;
(b) A. M. Bagoji, P. A. Magdum, S. T. Nandibewoor and J. Solution, Chem, 2016, 45, 1715–1728 CAS.
-
(a) F. Borek, J. Org. Chem., 1961, 26(4), 1292–1294 CrossRef CAS;
(b) S. Ghorai, A. Laskin and A. V. Tivanski, J. Phys. Chem. A, 2011, 115, 4373–4380 CrossRef CAS PubMed;
(c)
D. T. Bregante, P. Priyadarshini, and D. W. Flaherty, 2017, URL: https://www.sciencedirect.com/science/article/pii/S0021951717300428.
-
(a) E. J. Cone, R. H. Garner and A. A. Wallace Hayes, J. Org. Chem., 1972, 37(26), 4436–4439 CrossRef CAS;
(b) F. B. Mallory and M. B. Baker, J. Org. Chem., 1984, 49(8), 1324–1326 CrossRef;
(c) G. Bellucci, R. Bianchini, C. Chiappe, R. Ambrosetti, D. Catalano, A. J. Bennet, H. Slebocka-Tilk, G. H. M. Aarts and R. S. Brown, J. Org. Chem., 1993, 58, 3401–3406 CrossRef CAS;
(d) K. Nilsson, C. Ullenius and N. Krause, J. Am. Chem. Soc., 1996, 118, 4194–4195 CrossRef CAS;
(e) C. Puke, G. Erker, B. Wibbeling and R. Fröhlich, Eur. J. Org Chem., 1999, 8, 1831–1841 CrossRef;
(f) F. Susanne, D. S. Smith and A. Codina, Org. Process Res. Dev., 2012, 16, 61–64 CrossRef CAS;
(g) A. K. Eckhardt, Chem. Commun., 2022, 58, 8484–8487 RSC;
(h) R. Nagarjuna, A. Thakur, A. Balapure, M. S. M. Saifullah, J. Ray Dutta and R. Ganesan, Mater. Adv., 2023, 5, 593–607 RSC.
-
(a) R. C. Lord and F. A. Miller, Appl. Spectrosc., 1956, 115, 10 Search PubMed;
(b)
L. J. Bellamy, “ The Infrared Spectra of Complex Molecules.” Wiley, New York, 1958 Search PubMed.
-
(a) N. B. Colthup, J. Opt. Soc. Am., 1950, 4(6), 397 CrossRef;
(b) A. Ashdown and T. A. Kletz, J. Chem. Soc., 1948, 1454–1456 RSC.
- For a reviews on keto-enol tautomerism see:
(a) G. Allen and R. A. Dwek, J. Chem. Soc. B, 1966, 161–163 RSC;
(b) E. J. Drexler and K. W. Field, J. Chem. Educ., 1976, 53(6), 392–393 CrossRef CAS;
(c) Measuring the equilibrium constant of a keto-enol tautomerism using benchtop NMR, Application note AN52327, URL:AN52327-measuring-equilibrium-constant-keto-enol-tautomerism.pdf (thermofisher.com);;
(d) M. M. Fokendt, B. E. Weiss-Lopez, J. P. Cbauvel and N. S. True, J. Phys. Chem., 1985, 89, 3347–3352 CrossRef.
- E. V. Dalessandro, H. P. Collin, M. S. Valle and J. R. Pliego, RSC Adv., 2016, 6, 57803–57810 RSC.
-
(a) R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36(4), 706–723 CrossRef CAS;
(b)
J. Wang and N. York, Analytical Electrochemistry, 2nd edn, 2000 CrossRef.
- P. S. Joshi and D. S. Sutrave, Int. J. ChemTech Res., 2018, 11, 77–88 CrossRef CAS.
-
(a) R. D. Webster, A. M. Bond and T. Schmidt, J. Chem. Soc., Perkin Trans. 1, 1995, 2, 1365–1374 RSC;
(b) R. D. Webster, J. Chem. Soc., Perkin Trans. 1, 1999, 2, 263–269 RSC.
-
(a) M. A. Nichols and M. J. Waner, J. Chem. Educ., 2010, 87, 952–955 CrossRef CAS;
(b)
S. Farmer, D. Kennepohl, L. Morsch and L. Morsch, Keto-Enol Tautomerism, 2010, URL: 22.1: Keto-Enol Tautomerism - Chemistry LibreTexts Search PubMed.
-
(a) N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS;
(b) C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
-
(a) G. Diao and Z. Zhang, J. Electroanal. Chem., 1996, 410, 155–162 CrossRef;
(b) A. A. Al Owais, I. S. El-Hallag and E. H. El-Mossalamy, Int. J. Electrochem. Sci., 2022, 17, 220631 CrossRef CAS.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed.
-
(a) H. Liu, T. Cai, Q. Song, L. Yang, Q. Xu and C. Yan, Int. J. Electrochem. Sci., 2013, 8, 2515–2523 CrossRef CAS;
(b) W. Wang, X. Fan, J. Liu, C. Yan and C. Zeng, RSC Adv., 2014, 4, 32405–32411 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Lokesh A.
Shastri
,
Lokesh A.
Shastri














