DOI:
10.1039/D3TA07815A
(Paper)
J. Mater. Chem. A, 2024,
12, 9672-9680
Oxygen vacancy redistribution and ferroelectric polarization relaxation on epitaxial perovskite films during an electrocatalytic process†
Received
17th December 2023
, Accepted 10th March 2024
First published on 12th March 2024
Abstract
Ferroelectrics hold great promise in catalysis because of their ability to modulate intermediate adsorption strength and facilitate electron transfer on polarized surfaces. However, the effect of ferroelectric polarization on catalytic performance remains unclear. Here, an epitaxial BaTiO3 film was fabricated by pulsed laser deposition, which was utilized as a proof-of-concept material to reveal the polarization-dependent oxygen evolution reaction (OER). We show that negative ferroelectric polarization significantly improves the OER performance, due to enhanced adsorption of OH− and accelerated charge transfer by the redistribution of oxygen vacancies, and the additive effect of the built-in electric field. Interestingly, we find a periodic oscillation in the chronoamperometry test, which is probably attributed to the adsorption of reactants, gradual accumulation and sudden release of products, and the dynamical evolution of ferroelectric polarization. This work not only provides valuable insights into the origin of the controllable OER activity of ferroelectrics, but also sheds light on the relaxation of ferroelectric polarization in catalytic processes.
Introduction
Ferroelectric oxide films with spontaneous polarization have attracted increasing interest because of their potential applications in clean-energy and environmental protection. For example, ferroelectrics have been applied in photocatalysis because spontaneous polarization could promote the efficient separation of photogenerated carriers.1–3 Ferroelectrics have also been utilized in piezo-catalysis because a piezoelectric field can harvest vibration energy for triggering a redox reaction.4–6 Additionally, ferroelectric materials with pyroelectric properties could generate localized heat through thermoplastic nanostructures for pyro-catalysis.7,8
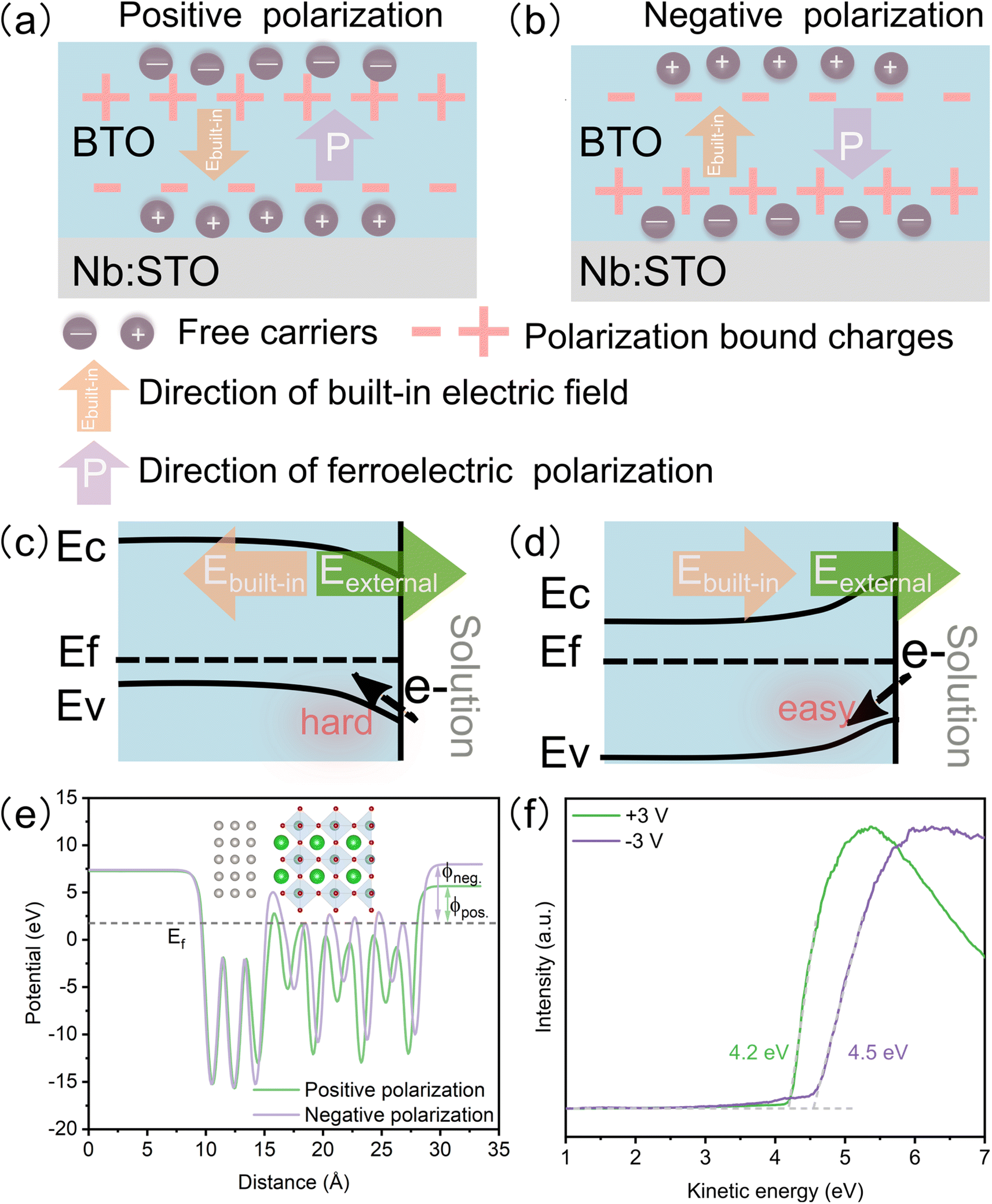
For ferroelectric materials, ferroelectric polarization can be reversed through an external field, inducing polarization bound charges at the surface. The polarization bound charges could create a depolarized field, namely a built-in electric field, in the opposite direction of ferroelectric polarization. To stabilize the ferroelectric polarization, free carriers (holes and electrons) as compensation charges are redistributed to reduce the depolarized field.9 Regarding ferroelectric catalysis, there are two pivotal factors, which are usually overlooked in previous research: (1) oxygen vacancies would form spontaneously as compensated charges to stabilize the polarization in ferroelectric oxide films,10,11 acting as active sites for electrocatalysis; (2) the built-in electric field that can be added or subtracted from the external electric field. Previous theoretical studies reported that ferroelectric polarization reconstructed the surface potential and affected the molecular adsorption energy greatly.12–14 However, nanoscale ferroelectrics were mostly used in ferroelectric catalysis, posing a great challenge to study the effect of ferroelectric polarization on catalysis in experiments and usually making the reaction mechanism unclear.5,6,15,16 For example, single-phase Sr0.5Ba0.5Nb2O6 showed that large polarization could induce strong adsorption and band-tilting, which accelerated the electron transfer in the oxygen evolution reaction (OER).16 Additionally, it has been reported that oxygen vacancies on the negatively polarized surface of PbTiO3 nanosheets were the active sites for water oxidation, and there was a strong correlation among oxygen vacancies, ferroelectric polarization, and kinds of products.3 Ren et al. also reported that oxygen vacancies could be well confined at the negatively polarized surface of PbTiO3 nanoplates as the active sites for enhanced CO oxidation kinetics.17 In comparison with nanoparticles or nanosheets, the epitaxial ferroelectric oxide film with a single phase has a well-defined and uniform surface structure, which provides an ideal platform for modulating and understanding the ferroelectric polarization, as well as unraveling the effect of the chemical environment on the ferroelectric polarization. Pedram Abbasi et al. reported that the upward polarization in the epitaxial ferroelectric film reduced the work function and lowered the hydrogen evolution reaction barrier.18 However, the OER as a complicated four-electron process has not been investigated based on the epitaxial ferroelectric film. Especially, the redistribution of surface oxygen vacancies under the ferroelectric polarization and the mechanism for the enhanced OER performance in epitaxial ferroelectric oxide films have not been clarified.
Meanwhile, the ferroelectricity of films is strongly affected by the electrochemical environment.19–21 For instance, the ferroelectric polarization can be reversibly and reproducibly switched by varying the partial oxygen pressure above its free surface.22 The surface adsorbates could induce screening charge, change the structure distortion of the outer atomic layer, and improve the stability of ferroelectricity.14,23,24 Therefore, the chemical environment must be taken into account when studying the ferroelectric polarization of oxide films without a top electrode in the catalytic process.
Herein, we fabricated an epaxial single-crystal BaTiO3 (BTO) ferroelectric film by pulsed laser deposition (PLD). The effects of oxygen vacancy redistribution induced by the ferroelectric polarization and the built-in electric field on the enhanced OER performance were investigated in detail. Interestingly, we found a periodic oscillation of current density during a chronoamperometry (CA) test and proposed that the evolution of ferroelectric polarization is responsible for the observed phenomena during the OER process.
Results and discussion
BTO epitaxial film growth
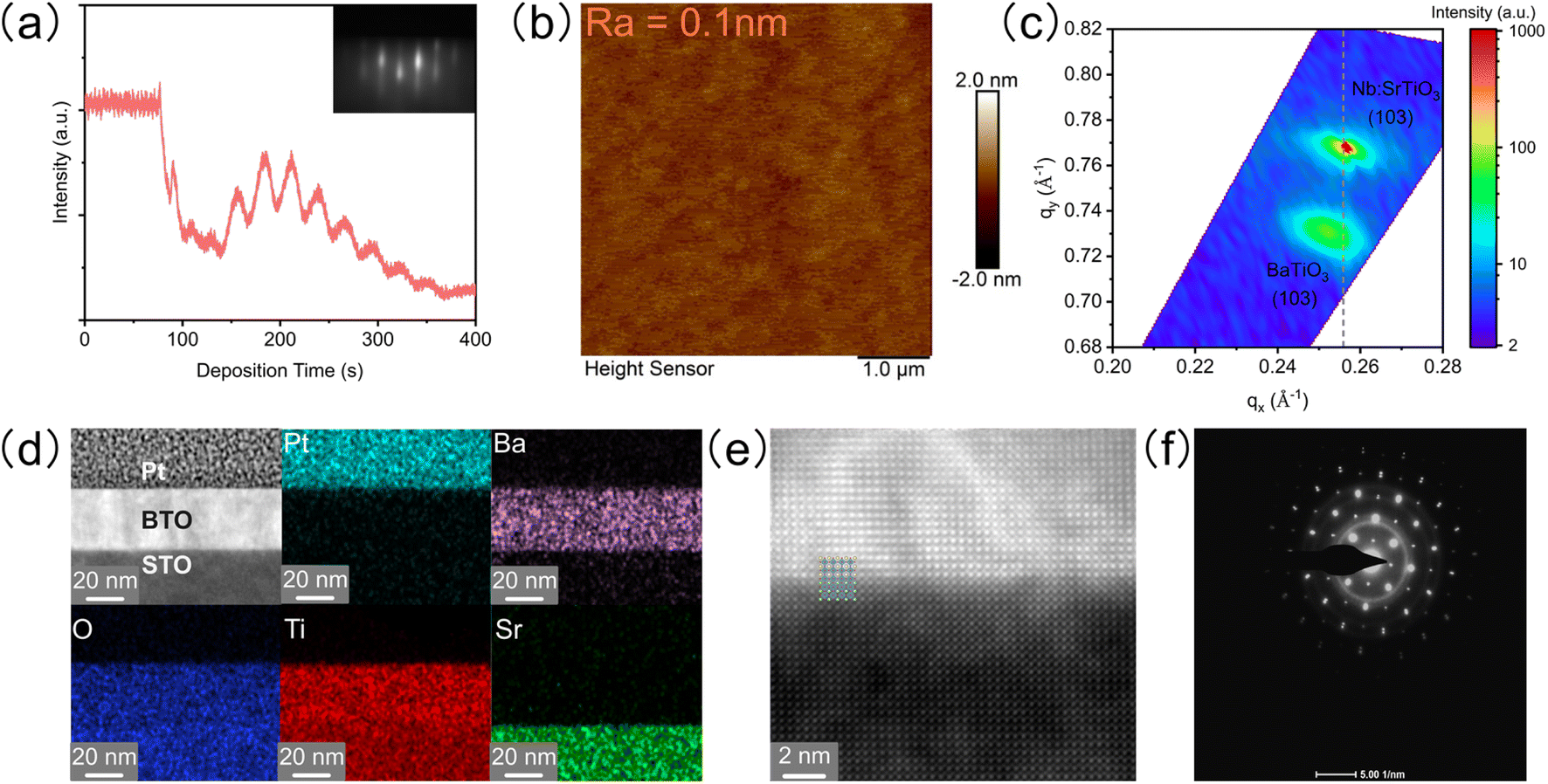
The epitaxial single crystalline BTO film with a smooth surface was grown on a Nb-doped SrTiO3 (STO) substrate by PLD and systematically characterized. Firstly, the distinct oscillation and pattern of reflection high-energy diffraction (RHEED) show layer-by-layer growth (Fig. 1a). Secondly, the atomic force microscope (AFM) image shows that the root mean square roughness is only 0.1 nm, indicating an atomically smooth surface (Fig. 1b). Thirdly, reciprocal space mapping (RSM) around the (103) peak of the film and substrate presents that the BTO film has the same in-plane lattice parameters as the substrate under slight compression (Fig. 1c). Finally, a high-resolution transmission electron microscope (HRTEM) confirms the atomic distribution and arrangement on the cross section of the epitaxial BTO film (Fig. 1d and e): (1) the energy dispersive spectra (EDS) show the sharp interface of elements between BTO and STO (Fig. 1d); (2) the atomic resolution high-angle annular dark field (HAADF) demonstrates a high-quality epitaxial BTO film with perfect periodic arrangement of ions, which matches with the STO substrate well (Fig. 1e); (3) the selected area electron diffraction (SAED) pattern shows sharp, narrow, and round spots (Fig. 1f), indicating the single crystalline form. The thickness of the BTO film is about 30 nm (Fig. S1†). Additionally, both 2θ–ω X-ray diffraction (XRD) and HAADF confirm that the BTO epitaxial film is in the tetragonal phase with a = 3.91 Å and c = 4.11 Å (Fig. S2a–c†).
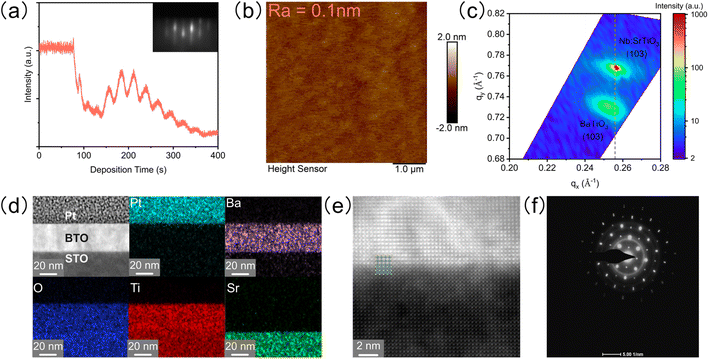 |
| | Fig. 1 Characterization of the epitaxial BTO film. (a) Oscillation of RHEED intensity during the film growth. Inset in (a) shows the RHEED diffraction patterns after the film growth, showing the layer-by-layer growth model. (b) AFM image. (c) RSM along (103) diffraction. (d) Cross-sectional HAADF-STEM and EDS mappings of Pt/BTO/STO. (e) Atomic resolution HAADF image. (f) SAED pattern. | |
Ferroelectric properties
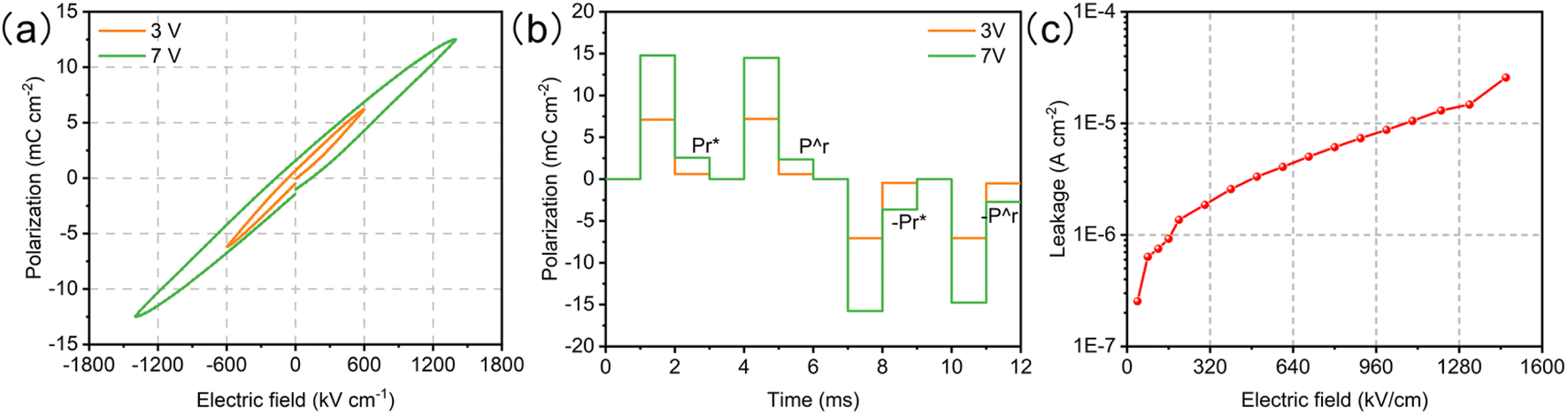
The ferroelectricity of the BTO film was characterized by the hysteresis loop (P–E loop), positive-up negative-down (PUND), and leakage with a Precision Multiferroic Tester. The P–E loop clearly indicates the ferroelectric polarization of the BTO film (Fig. 2a). To accurately obtain the remanent polarization (Pr), the PUND tasks were carried out with the applied voltages of 3 and 7 V (Fig. 2b). The Pr values are 0.02 and 0.21 μC cm−2 at 3 and 7 V, respectively. Although the Pr values are not large, the difference between the two polarization fields is strong enough to explore the effect of ferroelectric polarization on the OER. The leakage current (the surface polarization charge transfer under an external field) only increases from 4.1 × 10−6 A cm−2 at 3 V to 2.0 × 10−5 A cm−2 at 7 V (Fig. 2c). Thus, the high-quality epaxial BTO film has strong and stable ferroelectricity, which can serve as an ideal platform for revealing the mechanism of the catalytic activity in the OER process.
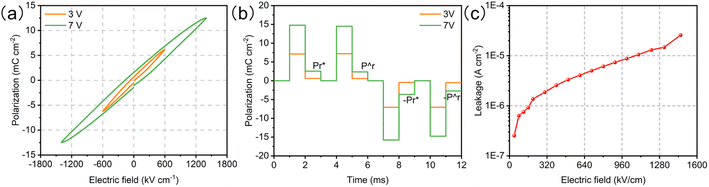 |
| | Fig. 2 Ferroelectricity measurements of the BTO film. (a) P–E loops measured at 1 kHz. (b) PUND tasks under 3 and 7 V. (c) Leakage current as a function of voltage. | |
Redistribution of oxygen vacancies
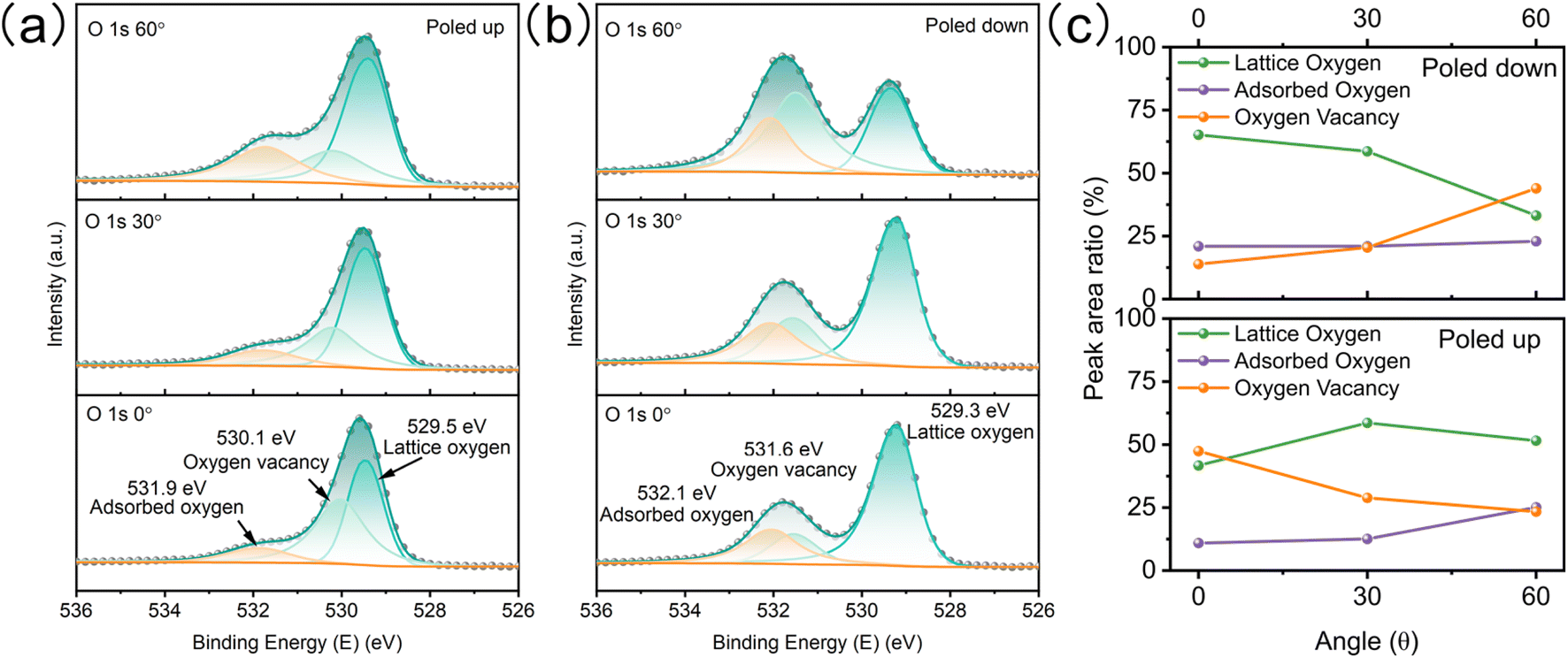
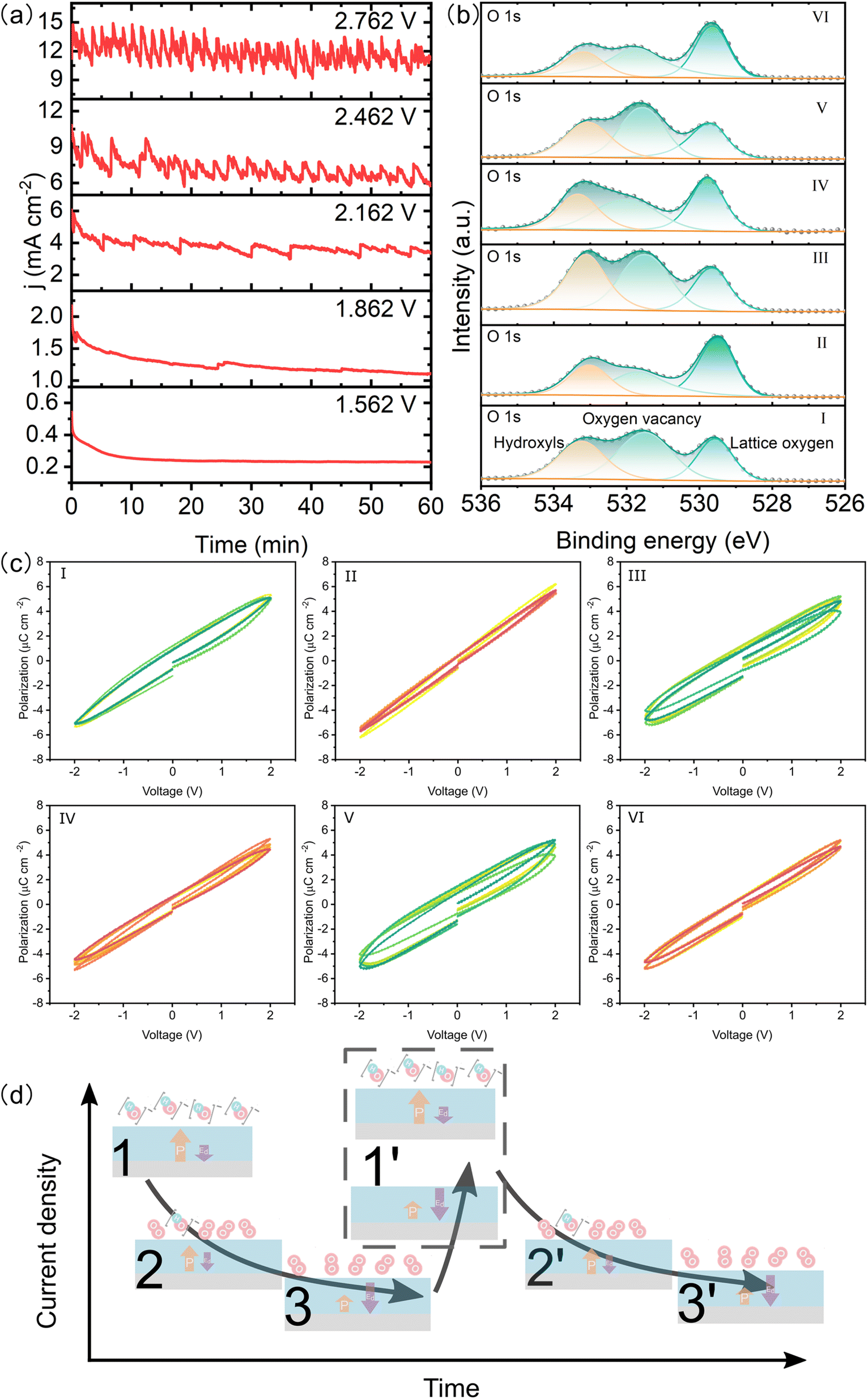
To study the redistribution of surface oxygen vacancies controlled by ferroelectric polarization, the BTO film was poled up or poled down by an electrochemical method.18 The direction and intensity of the ferroelectric polariton were adjusted by applying a steady-state voltage ( ±3 V) in a nonaqueous electrolyte without a metallic electrode contact (Fig. S3†). Angle-resolved XPS (AR-XPS) can assess the composition and chemical state as a function of depth and be used to reveal the surface chemical state (Fig. S4†). AR-XPS of the BTO film was carried out after being poled up (positive ferroelectric polarization) and poled down (negative ferroelectric polarization), respectively. The high-resolution spectra of O 1s are well-fitted with three components: lattice oxygen, oxygen vacancy, and adsorbed oxygen (Fig. 3a).25,26 Significantly, the binding energy of oxygen vacancies after being poled down shifts to a higher binding energy compared to that after being poled up (Fig. 3b), because the positive valence oxygen vacancies accumulate at the surface after being poled down, resulting in a stronger screening of the core hole after photoionization.27,28 The peak area ratios for the three components (Fig. 3c) indicate an increase in oxygen vacancies on the surface of the poled-down BTO film as the sample tilt increases. In contrary, the oxygen vacancies decrease when the BTO film is poled up. The evolution of lattice oxygen and oxygen vacancies with increasing sample tilt is opposite. The adsorbed oxygens on the BTO film slightly increased at high tilt angles after being poled up and poled down. Additionally, the high-resolution XPS spectra of Ti 2p demonstrate less variation after different polarization (Fig. S5†) compared to Ba. Ba exists in the ionic state, while Ti exists in the covalent state in the BTO film.18,29,30 Therefore, the Ba 3d core levels are greatly sensitive to the concentration of oxygen vacancies. The high-resolution XPS spectra of Ba 3d have two different core level features because of the spin–orbit coupling and are fitted to Ba I (778.2 eV and 793.5 eV) and Ba II (779.7 eV and 794.9 eV) (Fig. S5a and b†). Ba I and Ba II are attributed to BaO and BaO2, respectively.31–33 As the tilt angle increases on the poled-down BTO film, the concentration of oxygen vacancies also increases, resulting in an increase in the intensity of Ba II, as expected. AR-XPS analysis further demonstrates that the negative ferroelectric polarization can drive oxygen vacancies to migrate and accumulate at the BTO film surface.
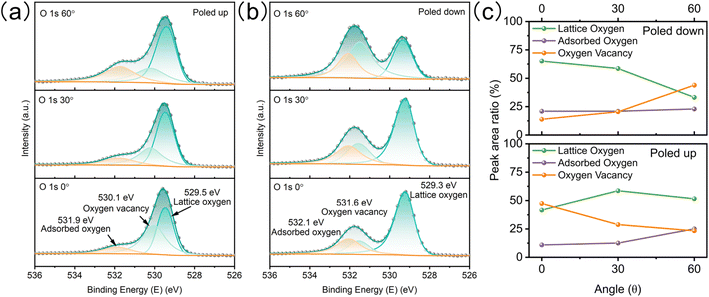 |
| | Fig. 3 Electronic structure characterization of the BTO film with different ferroelectric polarizations via AR-XPS. High-resolution XPS spectra of O 1s from 0° to 60° after being poled up (a) and poled down (b). (c) Evolution of three oxygen components with the sample tilt angle. | |
Enhanced OER performance via negative ferroelectric polarization
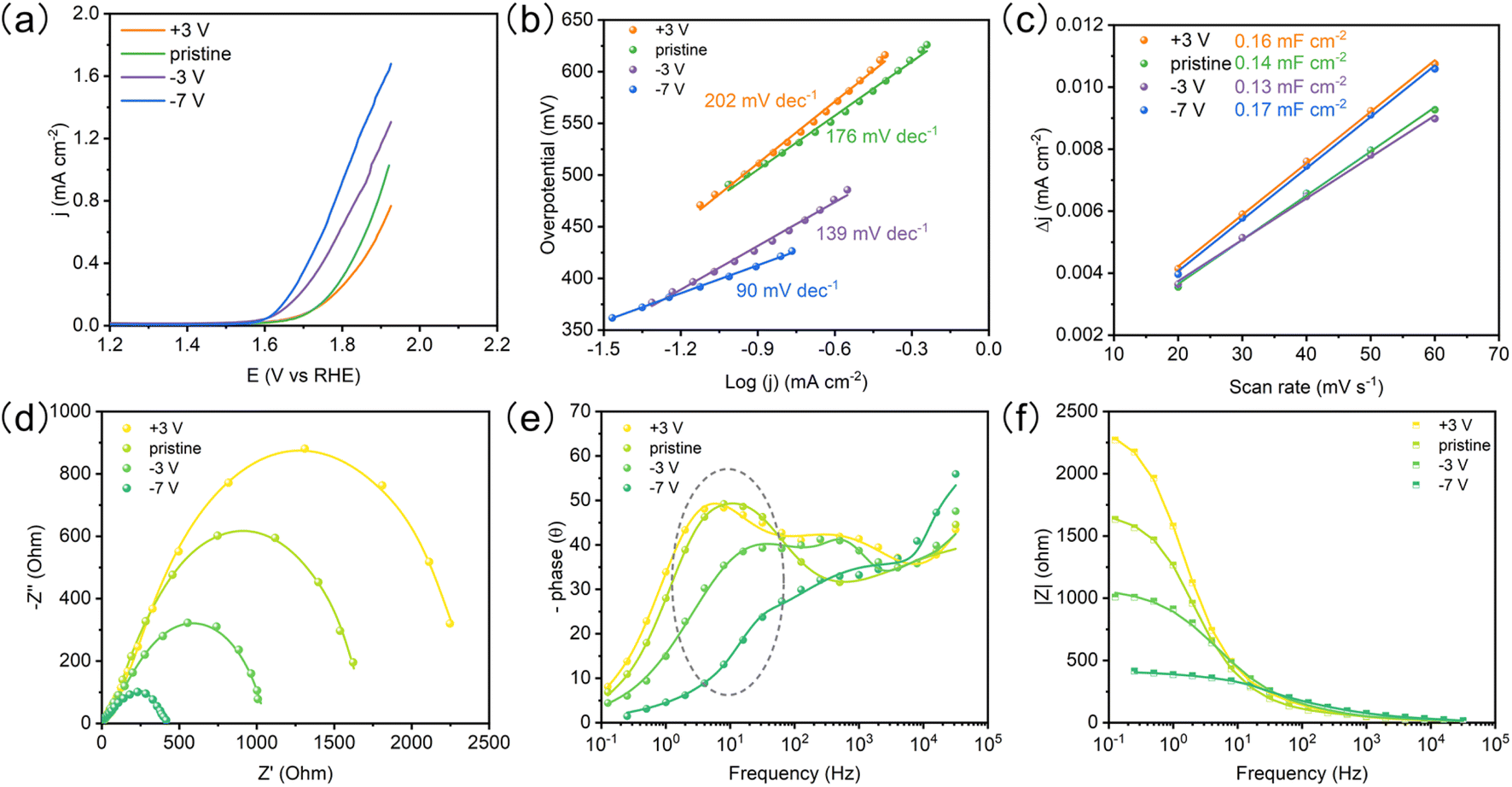
The OER performances of the BTO film with different ferroelectric polarizations were evaluated by linear sweep voltammetry (LSV) in 1 M KOH electrolyte (Fig. 4a). The poled-down BTO film shows higher OER activity than pristine and poled-up samples at the same potential. We see that the OER activity can be further improved by increasing polarization bias (−7 V). The BTO film shows a lower Tafel slope with the increasing poled-down bias voltage (Fig. 4b and Table S2†), indicating that the negative ferroelectric polarization promotes electron transfer. The ECSA was estimated from the electrochemical double-layer capacitance (Cdl) of the catalytic surface,34 which can be determined by measuring the non-faradaic capacitive current (Fig. S6†). The electrochemically active surface area (ECSA) is not affected by the ferroelectric polarization (Fig. 4c). To probe the OER kinetics of the BTO film under different ferroelectric polarizations, electrochemical impedance spectroscopy (EIS) was performed. Combining the corresponding Bode phase and resistance, the Nyquist plots were fitted with an equivalent circuit (Fig. 4d–f and S7†).35 The charge transfer resistance (Rct) and pseudo-capacitance (CPEct) could be utilized to describe the adsorption reaction of reactants (OH−) and interfacial charge transfer.35 The poled-down polarization remarkably reduces the semicircle of Nyquist plots, indicating faster charge transfer, while poled-up polarization works in the opposite way (Fig. 4d and Table S1†), indicating that the negative ferroelectric polarization accelerates the OH− adsorption and charge transfer. Meanwhile, the CPEct presents the same trend as Rct (Table S1†). The large CPEct on the poled-down BTO film is favorable to the catalytic driving force during the OER.35,36 Significantly, the peaks of the Bode phase have a positive shift and lower phase angle for the poled-down BTO film at the middle frequency (1 Hz–102 Hz), indicating that the oxygen vacancies as the active sites accelerate the OH− adsorption17,35,37–39 (Fig. 4f). Therefore, the redistributed oxygen vacancies induced by the negative ferroelectric polarization accumulate on the surface, in favor of improving the OH− adsorption and accelerating charge transfer for enhanced OER performance.
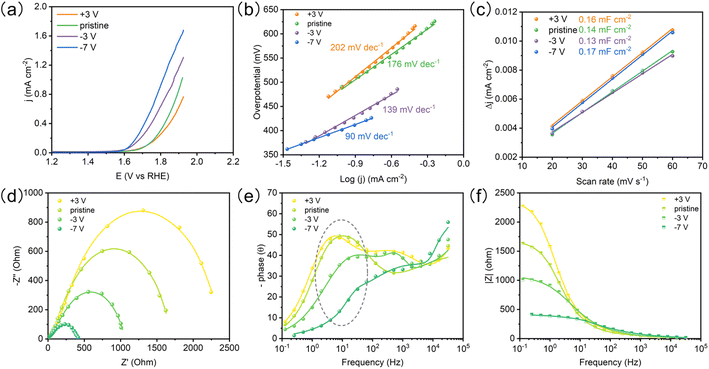 |
| | Fig. 4 The OER performance of the BTO film. (a) LSVs of BTO films after the ferroelectric polarization under different bias voltages. (b) Tafel slopes. (c) Double layer capacitances. (d) Nyquist plots for BTO films with different ferroelectric polarizations, (e) Bode phase, and (f) resistance. | |
Higher surface potential and built-in electric field
In ferroelectric oxide films, the polarization bond charge is formed under ferroelectric polarization, leading to a built-in electric field opposite to the ferroelectric polarization direction. To stabilize the ferroelectric polarization field, the free carriers are redistributed at the surface (Fig. 5a and b).9 The built-in electric field results in band-tilting and varying surface potentials. Negative ferroelectric polarization causes band tilting and generates a higher surface potential than positive ferroelectric polarization (Fig. 5c and d). For the OER process, the higher surface potential is beneficial to the electron transfer.38 Additionally, because the oxidation of the reactant happens at the anode, the positive reaction potential (external electric field) and the built-in electric field of negative ferroelectric polarization are consistent, leading to a superimposed reaction potential, which increases the reaction potential and enhances the charge transfer greatly. The DFT calculation (Fig. 5e) and work function measured by ultraviolet photoemission spectroscopy (UPS) (Fig. 5f) confirm that the negative polarized film has a higher work function than the positive polarized one. Meanwhile, the DFT calculation shows that the BTO slab with negative ferroelectric polarization exhibits a lower energy barrier in the rate-limiting step of the OER (oxidation of *O to *OOH) than that with positive ferroelectric polarization (Fig. S8a†). The lattice oxygen mechanism (LOM) may be responsible for the dominant OER mechanism because LOM shows the lowest over potential (Fig. S8b†). The BTO film also shows many corrosion pits after a long-term OER test (Fig. S9†), which may result from the dissolution of lattice oxygen.
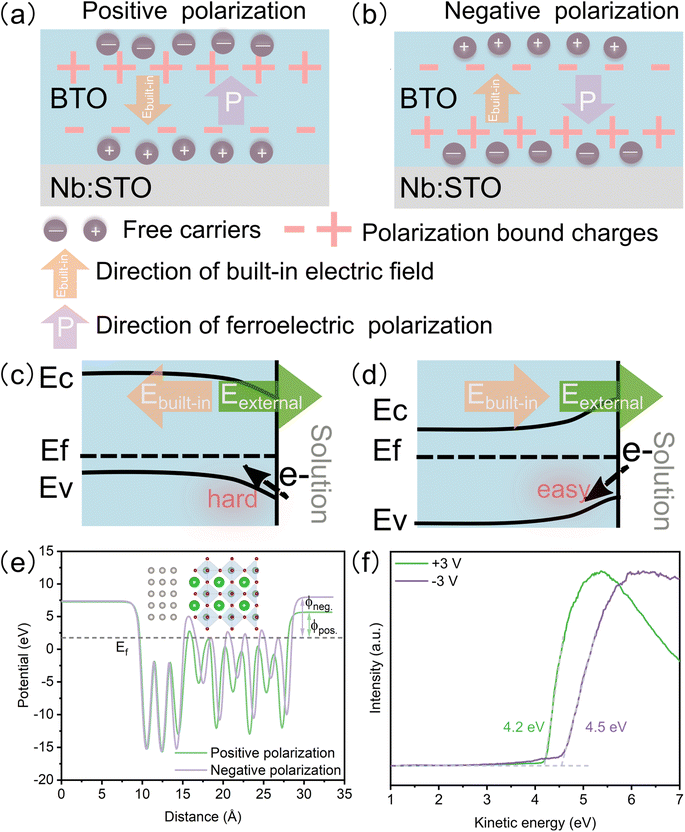 |
| | Fig. 5 Evolution of surface potential with different ferroelectric polarizations. Schematic diagram of carrier and charge redistribution: (a) positive and (b) negative ferroelectric polarizations. Schematic diagram of the band structure: (c) negative and (d) positive ferroelectric polarizations. (e) Electrostatic potential energy profiles under positive and negative ferroelectric polarizations. Inset picture shows the negative ferroelectric polarization structure. (f) Work functions of the BTO film after +3 v and −3 V poling. | |
Evolution of ferroelectric polarization in the OER process
As the applied potential increases, the LSV curve of the BTO film shows an oscillating increment (Fig. S10†), indicating the rapid accumulation of reactants on the catalyst surface. The CA responses are oscillated periodically under constant voltage and the period decreases with increasing potential (Fig. 6a), which is different from the typical stable CA response. We took six points on the CA response for measuring the XPS and P–E loop, where the I, III, and V points represent the highest current density and the others are the lowest one (Fig. S11†). The XPS spectra clearly show higher concentrations of surface adsorbed hydroxyls and oxygen vacancies at the highest current density compared to the lowest current density (Fig. 6b & S12†). Interestingly, we find that the BTO films exhibit higher remanent polarization at I, III, and V points as compared to the other points (Fig. 6c and S13†), indicating the dynamic fluctuation of ferroelectric polarization in the OER process. It has been reported that the relaxation of ferroelectric polarization is greatly influenced by the depolarized field of the ferroelectric film.23,39,40 This field can be reduced by the surface oxygen vacancies and adsorbed polar molecules, such as hydroxyls, which provide charge compensation.23 Additionally, the adsorptions of *OH and *O can induce an upward polarization by causing the displacement of atoms at the top-layer,23 which increases the ferroelectric polarization. The time dependent CVs also suggest that the positive ferroelectric polarization is more stable (Fig. S14†). However, the depolarization field gradually increases as the hydroxyls convert to O2, leading to reduced ferroelectric polarization. The evolution of surface adsorbed hydroxyls dynamically alters the chemical environment of the ferroelectric film, resulting in a dynamic relaxation of ferroelectric polarization during the OER performance. The evolution of ferroelectric polarization influences the adsorption of intermediates in return. Therefore, we propose that a large number of reactants are absorbed onto the BTO film to participate in the OER, which results in sharply increasing current density. Subsequently, the gradually decreasing current density results from the gradual accumulation of products (adsorbed O2), which is accompanied by decreasing ferroelectric polarization. Finally, the strength of ferroelectric polarization weakens until the products are suddenly released. The active sites on the surface are exposed, and reactants are adsorbed on the surface again (Fig. 6d). The periods of reactant adsorption and product accumulation/release are reduced as the reaction potential increases (Fig. 6a). Therefore, the novel CA response results from the adsorption of reactants and the accumulation/release of oxygens as well as their effect on ferroelectric polarization.
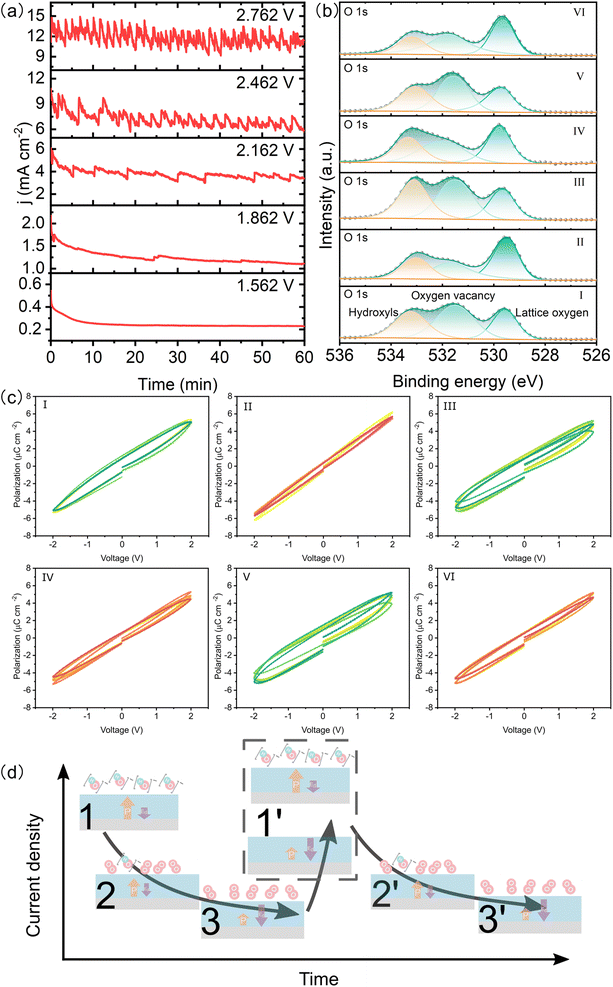 |
| | Fig. 6 Ferroelectric relaxation in the catalysis process. (a) CA response of the BTO film with increasing potentials (vs. RHE). High-resolution XPS spectra of O 1s (b) and P–E loops (c) of the BTO film sampled according to termination points of the CA test. (d) Schematic scheme for the evolution of reactant adsorption and product accumulation on the BTO surface. The evolution of ferroelectric polarization and the built-in electric field also is shown. The yellow arrow is ferroelectric polarization. The purple arrow is the built-in electric field. | |
Conclusion
In summary, we synthesized an epitaxial single-crystal BTO film with robust ferroelectric polarization using PLD. The redistribution of surface oxygen vacancies was induced by the ferroelectric polarization. When the ferroelectric polarization is negative, the oxygen vacancies accumulate on the surface and act as the active sites to accelerate the OH− adsorption and charge transfer. Additionally, the negative ferroelectric polarization generates a higher surface potential and a built-in electric field in the same direction as the external electric field. The ferroelectric polarization modulates the adsorption of intermediates and is influenced by the adsorption/desorption of reactants/products in return. Interesting, we found a novel phenomenon about potential-dependent periodic oscillation during CA test. Interestingly, a periodic oscillation occurs in the CA test under constant voltage. These results offer valuable insight into how oxygen vacancies, higher surface potential, and built-in electric field affect the improved performance of OER under negative ferroelectric polarization. They also shed light on the behavior of ferroelectric materials during the catalytic process and provide new opportunities to design ferroelectric catalysts that are highly active and stable.
Methods
Epitaxial film synthesis and characterization
The BTO epitaxial film was grown on a 0.7% Nb-doped STO (001) substrate via a PLD system with a KrF laser (LPX 300, Coherent). The film was fabricated at 800 °C under 1 mTorr O2 oxygen pressure. The laser repetition rate was 3 Hz and the laser power was set to 300 mJ. The growth was monitored in situ with a RHEED system (kSA400) operated at 20 kV (1.4 A). After the deposition, the sample was held under 760 torr O2 for 1 h, and then the annealing rate was 10 °C min−1 to room temperature. RSM and 2θ–ω XRD measurements were performed in a parallel-beam geometry using a Rigaku SmartLab 9 kW with Cu Kα radiation (λ = 0.154 nm). The cross-section of the BTO film was prepared using a focused ion beam scanning electron microscope (FIB-SEM, Cross Beam, Carl Zeiss). HRTEM and EDS were carried out on a Talos F200S system. The AR-XPS measurement was taken to obtain the high-resolution spectra using an electron analyzer with a monochromatic X-ray (Al, K-α, hν = 1486.7 eV) under ultrahigh vacuum (1.0 × 10−9 mbar). All the spectra were calibrated with C 1s, which is assumed to have a binding energy of 284.8 eV.
Ferroelectric measurement
To ensure that the external electric field was applied to the BTO films, insulation paste was applied to the back and side of the substrate. The BTO film was poled up or down in a non-aqueous electrolyte (0.1 M LiClO4 in polycarbonate solvent) for about 1 min. The counter electrode is Pt. After polarization switching, the sample was washed repeatedly with acetone and deionized water. The P–E loops were measured using a Precision Multiferroic Tester (Radiant Technologies) at a frequency of 1 kHz. For the PUND task, the pulse delay and width were set to 1000 and 1 ms, respectively. The first pulse is for repolarization with 1 ms. The first read pulse (Pr*) is a positive switching pulse. The second read pulse (P^r) is measured, which is an unswitching pulse, after a 1000 ms delay time. Likewise, the third and fourth are negative switching and unswitching measured with the formula:
The remanent polarization (Pr) was calculated from the PUND task. The leakage current as a function of increasing voltage was determined with 1000 ms soak time and 1000 ms measure time.
Electrochemical measurement
The OER performance was determined using a standard three-electrode cell with an electrochemical workstation (ModuLab XM) in 1 M KOH electrolyte. Standard Hg/HgO (1 M KOH) and a graphite rod were the reference and counter electrodes, respectively. The BTO epitaxial film was the working electrode. LSV with 5 mV s−1 was performed versus RHE. The RHE potential was converted via the Nernst equation (E vs. RHE = E vs. Hg/HgO + 0.0591 × pH + 0.098 V). EIS was performed at 1.76 V (vs. RHE) in the frequency range of 1−1–105 Hz with an amplitude of 10 mV. The scan-rate dependent CVs were tested in a non-faradaic potential range (1.27 V–1.32 V vs. RHE) at different scan rates of 20, 30, 40, 50, and 60 mV s−1. The Cdl was estimated by plotting Δj at 1.295 vs. RHE. The ECSA was calculated using the following equation:where Cs is the specific capacitance of the planar surface.
DFT calculation
The slab model with a 15 Å vacuum was built based on a 2 × 2 × 3 BTO supercell. Referring our epitaxial film to previous studies, the Ti–O surface along (001) with the lowest surface energy was selected as the chemistry-active surface.18,41 To simulate the surface of the ferroelectric, three layers of Pt were put at the bottom of BTO as an electron reservoir,13,14 and the bottom four atomic layers of BTO were fixed during the structural optimization. Meanwhile, dipole correction was added in the z-axis direction to avoid the spurious electric field across the vacuum layer. The first-principles calculation was performed via the Vienna Ab initio Simulation Package.42,43 The generalized gradient approximation was used with the Perdew–Burke–Ernzerhof exchange-correlation functional.44–46 The plane-wave cutoff energy was set to 550 eV, and the Monkhorst–pack K-point grid was set to 4 × 4 × 1 for calculation.47–49 The Hubbard U is critical for 3d orbital localized electrons.50 Thus, an on-site U = 4 eV was adopted for the Ti ion.19 The threshold for energy and force convergence was limited to 10−6 eV and 10−2 eV, respectively.
Conflicts of interest
The authors declare no commitment to financial interests.
Acknowledgements
This work was supported by the Science and Technology Development Fund (FDCT) from Macau SAR (0071/2019/AMJ, 0050/2023/RIB2, 0023/2023/AFJ, 006/2022/ALC, and 0111/2022/A2), Multi-Year Research Grants (MYRG-CRG202200016-ICMS, MYRG-GRG2023-00010-IAPME and MYRG2022-00026-IAPME) from Research & Development Office at University of Macau, and Shenzhen-Hong Kong-Macao Science and Technology Research Programme (Type C) (SGDX20210823103803017) from Shenzhen. The DFT calculations were performed in the High-Performance Computing Cluster (HPCC) of Information and Communication Technology Office (ICTO) at the University of Macau and Bianshui Riverside Supercomputing Center.
References
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425 CrossRef CAS PubMed
 .
.
- Y. Liu, M. Zhang, Z. Wang, J. He, J. Zhang, S. Ye, X. Wang, D. Li, H. Yin, Q. Zhu, H. Jing, Y. Weng, F. Pan, R. Chen, C. Li and F. Fan, Nat. Commun., 2022, 13, 4245 CrossRef CAS PubMed .
- G. Wan, L. Yin, X. Chen, X. Xu, J. Huang, C. Zhen, H. Zhu, B. Huang, W. Hu, Z. Ren, H. Tian, L. Wang, G. Liu and H. M. Cheng, J. Am. Chem. Soc., 2022, 144, 20342 CrossRef CAS PubMed .
- M. Wang, B. Wang, F. Huang and Z. Lin, Angew. Chem., Int. Ed. Engl., 2019, 58, 7526 CrossRef CAS PubMed .
- R. Su, Z. Wang, L. Zhu, Y. Pan, D. Zhang, H. Wen, Z. D. Luo, L. Li, F. T. Li, M. Wu, L. He, P. Sharma and J. Seidel, Angew. Chem., Int. Ed. Engl., 2021, 60, 16019 CrossRef CAS PubMed .
- H. You, Z. Wu, L. Zhang, Y. Ying, Y. Liu, L. Fei, X. Chen, Y. Jia, Y. Wang, F. Wang, S. Ju, J. Qiao, C. H. Lam and H. Huang, Angew. Chem., Int. Ed. Engl., 2019, 58, 11779 CrossRef CAS PubMed .
- H. You, S. Li, Y. Fan, X. Guo, Z. Lin, R. Ding, X. Cheng, H. Zhang, T. W. B. Lo, J. Hao, Y. Zhu, H. Y. Tam, D. Lei, C. H. Lam and H. Huang, Nat. Commun., 2022, 13, 6144 CrossRef CAS PubMed .
- Y. Zhang, P. T. T. Phuong, E. Roake, H. Khanbareh, Y. Wang, S. Dunn and C. Bowen, Joule, 2020, 4, 301 CrossRef CAS .
- Y. Watanabe, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 789 CrossRef CAS .
- Y. Yao and H. Fu, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 064112 CrossRef .
- A. V. Kimmel, P. M. Weaver, M. G. Cain and P. V. Sushko, Phys. Rev. Lett., 2012, 109, 117601 CrossRef PubMed .
- K. Garrity, A. Kakekhani, A. Kolpak and S. Ismail-Beigi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 045401 CrossRef .
- A. Kakekhani and S. Ismail-Beigi, ACS Catal., 2015, 5, 4537 CrossRef CAS .
- J. H. Lee and A. Selloni, Phys. Rev. Lett., 2014, 112, 196102 CrossRef PubMed .
- R. Su, H. A. Hsain, M. Wu, D. Zhang, X. Hu, Z. Wang, X. Wang, F. T. Li, X. Chen, L. Zhu, Y. Yang, Y. Yang, X. Lou and S. J. Pennycook, Angew. Chem., Int. Ed. Engl., 2019, 58, 15076 CrossRef CAS PubMed .
- X. N. Li, Y. M. Du, L. B. Ge, C. Y. Hao, Y. Bai, Z. P. Fu, Y. L. Lu and Z. X. Cheng, Adv. Funct. Mater., 2023, 33, 2210194 CrossRef CAS .
- Z. Ren, L. Ruan, L. Yin, K. Akkiraju, L. Giordano, Z. Liu, S. Li, Z. Ye, S. Li, H. Yang, Y. Wang, H. Tian, G. Liu, Y. Shao-Horn and G. Han, Adv. Mater., 2022, 34, e2202072 CrossRef PubMed .
- P. Abbasi, M. R. Barone, M. de la Paz Cruz-Jauregui, D. Valdespino-Padilla, H. Paik, T. Kim, L. Kornblum, D. G. Schlom, T. A. Pascal and D. P. Fenning, Nano Lett., 2022, 22, 4276 CrossRef CAS PubMed .
- S. M. Yang, A. N. Morozovska, R. Kumar, E. A. Eliseev, Y. Cao, L. Mazet, N. Balke, S. Jesse, R. K. Vasudevan, C. Dubourdieu and S. V. Kalinin, Nat. Phys., 2017, 13, 812 Search PubMed .
- D. Hernandez-Martin, F. Gallego, J. Tornos, V. Rouco, J. I. Beltran, C. Munuera, D. Sanchez-Manzano, M. Cabero, F. Cuellar, D. Arias, G. Sanchez-Santolino, F. J. Mompean, M. Garcia-Hernandez, A. Rivera-Calzada, S. J. Pennycook, M. Varela, M. C. Munoz, Z. Sefrioui, C. Leon and J. Santamaria, Phys. Rev. Lett., 2020, 125, 266802 CrossRef CAS PubMed .
- K. P. Kelley, A. N. Morozovska, E. A. Eliseev, Y. Liu, S. S. Fields, S. T. Jaszewski, T. Mimura, S. Calderon, E. C. Dickey, J. F. Ihlefeld and S. V. Kalinin, Nat. Mater., 2023, 22, 1144 CrossRef CAS PubMed .
- R. V. Wang, D. D. Fong, F. Jiang, M. J. Highland, P. H. Fuoss, C. Thompson, A. M. Kolpak, J. A. Eastman, S. K. Streiffer, A. M. Rappe and G. B. Stephenson, Phys. Rev. Lett., 2009, 102, 047601 CrossRef CAS PubMed .
- D. D. Fong, A. M. Kolpak, J. A. Eastman, S. K. Streiffer, P. H. Fuoss, G. B. Stephenson, C. Thompson, D. M. Kim, K. J. Choi, C. B. Eom, I. Grinberg and A. M. Rappe, Phys. Rev. Lett., 2006, 96, 127601 CrossRef CAS PubMed .
- Y. Tian, L. Wei, Q. Zhang, H. Huang, Y. Zhang, H. Zhou, F. Ma, L. Gu, S. Meng, L. Q. Chen, C. W. Nan and J. Zhang, Nat. Commun., 2018, 9, 3809 CrossRef PubMed .
- Y. Wang, R. Yang, Y. Ding, B. Zhang, H. Li, B. Bai, M. Li, Y. Cui, J. Xiao and Z. S. Wu, Nat. Commun., 2023, 14, 1412 CrossRef CAS PubMed .
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J. M. Chen, S. Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2022, 13, 7754 CrossRef CAS PubMed .
- J. Zhou, J. Palisaitis, J. Halim, M. Dahlqvist, Q. Tao, I. Persson, L. Hultman, P. O. A. Persson and J. Rosen, Science, 2021, 373, 801 CrossRef CAS PubMed .
- G. Greczynski, R. T. Haasch, N. Hellgren, E. Lewin and L. Hultman, Nat. Rev. Methods Primers, 2023, 3, 40 CrossRef CAS .
- C. Sousa and F. Illas, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 13974 CrossRef CAS PubMed .
- A. E. Bocquet, T. Mizokawa, K. Morikawa, A. Fujimori, S. R. Barman, K. Maiti, D. D. Sarma, Y. Tokura and M. Onoda, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 1161 CrossRef CAS PubMed .
- I. Spasojevic, G. Sauthier, J. M. Caicedo, A. Verdaguer and N. Domingo, Appl. Surf. Sci., 2021, 565, 150288 CrossRef CAS .
- T. C. Droubay, L. Kong, S. A. Chambers and W. P. Hess, Surf. Sci., 2015, 632, 201 CrossRef CAS .
- S. Chakrabarti, S. Ginnaram, S. Jana, Z. Y. Wu, K. Singh, A. Roy, P. Kumar, S. Maikap, J. T. Qiu, H. M. Cheng, L. N. Tsai, Y. L. Chang, R. Mahapatra and J. R. Yang, Sci. Rep., 2017, 7, 4735 CrossRef PubMed .
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed .
- Z. Xiao, Y. C. Huang, C. L. Dong, C. Xie, Z. Liu, S. Du, W. Chen, D. Yan, L. Tao, Z. Shu, G. Zhang, H. Duan, Y. Wang, Y. Zou, R. Chen and S. Wang, J. Am. Chem. Soc., 2020, 142, 12087 CrossRef CAS PubMed .
- H. Y. Wang, S. F. Hung, H. Y. Chen, T. S. Chan, H. M. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 36 CrossRef CAS PubMed .
- X. Zhang, H. Tian, X. Wang, G. Xue, Z. Tian, J. Zhang, S. Yuan, T. Yu and Z. Zou, Mater. Lett., 2013, 100, 51 CrossRef CAS .
- S. Zhang, C. Tan, R. Yan, X. Zou, F. L. Hu, Y. Mi, C. Yan and S. Zhao, Angew. Chem., Int. Ed. Engl., 2023, 62, e202302795 CrossRef CAS PubMed .
- D. J. Kim, J. Y. Jo, Y. S. Kim, Y. J. Chang, J. S. Lee, J. G. Yoon, T. K. Song and T. W. Noh, Phys. Rev. Lett., 2005, 95, 237602 CrossRef CAS PubMed .
- J. H. Lee, R. H. Shin and W. Jo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 094112 CrossRef .
- N. Vonruti and U. Aschauer, J. Chem. Phys., 2020, 152, 024701 CrossRef CAS PubMed .
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef .
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed .
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS .
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed .
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS .
- P. E. Blochl, Phys.
Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed .
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef .
- V. V. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Lulu
Qiao
a,
Xingshuai
Lv
a,
Lianyi
Shao
a,
Jinxian
Feng
a,
Lulu
Qiao
a,
Xingshuai
Lv
a,
Lianyi
Shao
a,
Jinxian
Feng