
Surface oxygen vacancy engineering in weak Bi–O bonded ferroelectric bismuth sodium titanate for boosting the photocatalytic CO2 reduction reaction†
Xiao
Liu
 *a,
Jicong
Wang
a,
Fangyuan
Zhu
b,
Yanrui
Li
a,
Wenchao
Tian
c,
Weijia
Wang
d,
Ruiyun
Guo
a,
Laijun
Liu
e and
Jing
Shi
*c
*a,
Jicong
Wang
a,
Fangyuan
Zhu
b,
Yanrui
Li
a,
Wenchao
Tian
c,
Weijia
Wang
d,
Ruiyun
Guo
a,
Laijun
Liu
e and
Jing
Shi
*c
aSchool of Materials Science and Engineering, Xi'an University of Science and Technology, Xi'an 710054, China. E-mail: liuxiao@xust.edu.cn
bShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China
cState Key Laboratory of Electromechanical Integrated Manufacturing of High-Performance Electronic Equipments, School of Mechano-Electronic Engineering, Xidian University, Xi'an 710071, China. E-mail: jshi@xidian.edu.cn
dState Key Laboratory of Solidification Processing, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, China
eKey Laboratory of New Processing Technology for Nonferrous Metal & Materials (MOE), College of Materials Science and Engineering, Guilin University of Technology, Guilin 541006, China
First published on 13th March 2024
Abstract
Photocatalytic conversion of CO2 into renewable fuels is an exciting and challenging approach to mitigate carbon emissions and the energy crisis. The efficiency is hindered by the insufficient carrier transport capacity and high electron–hole pair complexation of the catalysts. The inherent spontaneous polarization in ferroelectric materials can affect the generation, transportation, and isolation of intermediates within catalytic reactions in modulating the pace and effectiveness of photocatalytic processes. Herein, ferroelectric bismuth sodium titanate (BNT) decorated with oxygen vacancies (OVs) via in situ reduction is first explored to promote the photocatalytic CO2 reduction reaction (CO2RR). The chemical states and coordination environment of the cations and OVs are elaborated by using the X-ray absorption fine structure to probe weakened Bi–O bonds and enhanced lattice distortion. Besides built-in electric fields originating from intrinsic spontaneous polarization, carrier dynamics and reactive sites are optimized with extended visible light absorption thanks to the special morphology and OVs. Remarkably, oxygen defected catalysts show 18 times higher CO reduction yield than pristine BNT, outperforming state-of-the-art bulk ferroelectrics for photocatalytic CO2RR. Interestingly, the product selectivity can be tailored after a poling treatment. This work provides a systematic approach for designing efficient ferroelectric photocatalysts, and meanwhile, expands the ferroelectric photocatalyst family for advancing CO2 conversion.
1. Introduction
The reduction of CO2 into chemicals and carbon-based fuels is a sustainable strategy to combat the energy crisis and global warming through the use of renewable energy.1–3 The unlimited solar energy as input energy for CO2 conversion (i.e. photocatalytic CO2 reduction) has already attracted intensive research interest.4–6 Through a semiconductor-based photocatalytic process, CO2 can be converted by light immobilization into valuable green solar fuels including CO, CH4, CH3OH, and HCOOH. A typical semiconductor photocatalytic process consists of three key steps:7–9 charge generation, charge separation, and surface reaction. However, the photocatalytic performance of a number of photocatalysts (e.g. TiO2,10–12 BiWO3,13–15 and BiOCl16–18) is still limited by poor visible light absorption, carrier recombination, and high energy barrier to trap and activate thermodynamically stable CO2 molecules. Therefore, many strategies have been developed to overcome these limitations including doping,19–22 heterojunction structures,23–26 and surface modification techniques,27–29 which have been shown to achieve effective photocatalytic CO2 reduction.Ferroelectric materials have compelling potential for applications in photocatalysis due to their unique electrical properties and polarization effects. These materials have an intrinsic polarized electric field that can promote the separation of photogenerated electron–hole pairs, thereby increasing the efficiency of photocatalytic reactions.30–32 In addition, the lattice defects on the surface, and multifunctional properties, of ferroelectric materials will help to increase the catalytically active sites and improve the photocatalytic effect. For instance, ultrasonication and annealing heat treatment of ferroelectric Bi0.5Na0.5TiO3 nanoparticles have been reported to decompose organic pigments in water by Ashok et al.33 The oxygen vacancies, catalyst size, and internal electric field play significant roles in enhancing the electron–hole separation, thus boosting the catalytic reaction. The strong polar electric field generated by controlling the thickness of SrTiO3 at the interface of n-TiO2 and p-Ag2O heterojunctions can accelerate the separation of photoexcited carriers, prolong the carrier lifetime, and fundamentally improve the performance of photoelectric devices.34 The ferroelectric polarization in a high-quality ferroelectric heterojunction built by epitaxial growth of Ti4O5 surface reconstruction on the BaTiO3 surface can alter the contact potential between the interfaces and speed up the electron transport, resulting in an almost tenfold increased CO reduction yield of 11.8 μmol g−1 h−1.35 It is worth mentioning that only a few ferroelectric materials with or without piezoelectric energy input demonstrate promotion in the CO2RR after considering their energy band structure, and the catalysis mechanism is proposed although it is still in the early stage of research. By making full use of the polarization specialty, developing or exploring new photocatalyst-based ferroelectric materials is surely exciting for advancing the CO2RR.
As a typical A-site composite perovskite, lead-free bismuth sodium titanate (Bi0.5Na0.5TiO3)-based materials (BNTs) have been extensively studied in piezoelectric, ferroelectric, and dielectric applications. Also, the catalysis performances in the water splitting or pollutant degradation were reported in the previous reports.33,36–38 Interestingly, the disordered structure of the ferroelectric BNTs can achieve significant oxygen ion conductivity and low oxygen vacancy jump barriers (activation energy from ∼1.8 eV to ∼0.4 eV) when subjected to primary non-stoichiometric doping or process modulation. This is mainly due to the weak Bi–O bonding resulting from the high polarization of Bi3+ with 6s2 lone pair electrons.39,40 The intrinsic spontaneous polarization properties of the crystal provide an internal electric field that drives the displacement of electrons and holes in opposite directions, providing a natural driving force for the separation and transport of photogenerated carrier pairs.41–44 The polarization charge distribution on both sides simultaneously causes the energy band to bend downwards and upwards, increasing the reaction potential, and the photogenerated charge is effectively separated promoting carrier lifetime extension. Moreover, the material exhibits a high polarization strength, accompanied by a large leakage current. In traditional BNT studies, oxygen vacancies or leakage currents are a disadvantage and a source of increased losses during electric field-induced domain reversal and thus need to be avoided.45,46 However, from a new perspective, its higher ionic conductivity in catalysis can facilitate the charged defect migration steadily or the in situ surface stress modulation and species control. Therefore, polarity-enhanced BNTs together with their oxygen vacancy hopping capacity that refrains from metastable vacancy consumption arouse interest and have potential as catalysts for photocatalytic or piezoelectric and other combined catalytic research areas.
Introducing oxygen vacancies (OVs) into photocatalysts emerges as a versatile approach to enhance carbon dioxide adsorption and activation, and improve photocatalytic conversion via non-homogeneous catalysis. For instance, optimizing the concentration of OVs on the surface of BiOIO3 nanostrips establishes a local electric field for charge migration to catalytic sites that significantly enhances the photocatalytic CO2 reduction performance.47 Additionally, Bi2O3 nanosheets, rich in oxygen vacancies, not only provide more local electrons but also reduce CO2 adsorption energies within the Bi2O3 atomic layer. This property makes them efficient catalysts for CO2 photofixation into valuable long-chain chemicals.48 Furthermore, the controlled introduction of suitable OV concentrations on the surface of BiOBr/Bi2WO6 heterojunction nanoflowers, achieved by regulating the duration of low-temperature annealing, positively influences the effective carrier separation and CO2 adsorption. This underscores the pivotal role of bismuth-based semiconductor materials in solar-driven CO2 conversion.13
The spontaneous polarization and oxygen vacancy behaviors could bring advantages in the photocatalytic CO2RR process, respectively. Moreover, considering its alterable oxygen vacancy hopping barriers and conduction-mechanism characteristics due to the highly polarized Bi3+, in this work, bismuth-based ferroelectric bismuth sodium titanate is first explored to investigate its photocatalytic CO2 reduction capacity as related work on BNTs is still absent. Several strategies have been developed to increase OVs in catalysts, such as heat treatment, chemical reduction, light irradiation, doping, and so on.49–54 Among them, chemical reduction is a simple method to adjust the OV content by altering the concentration of the reducing agent. Thus, mild reductants were employed to prepare BNT catalysts with appropriate OV concentrations. Nanomaterials with different morphologies and oxygen vacancy states are obtained via controlling the hydrothermal synthesis process and additive species. The photocatalytic performance is significantly improved, yielding CO and CH4 as the primary reduction products with a noteworthy preference for selectivity. The enhanced role of oxygen vacancy engineering in charge carrier kinetics and performance promotion in the CO2RR is elaborated by investigating the structure evolution and chemical environment of both the bulk and surface of ferroelectric BNTs.
2. Experimental section
2.1 Synthesis of Bi0.5Na0.5TiO3
The BNTs were synthesized by using Bi(NO3)3·5H2O (99%, Sinopharm) as the bismuth source and C16H36O4Ti (98%, Sinopharm) as the titanate source based on the following process: first, 10 mmol Bi(NO3)3·5H2O was dissolved in 10 mL deionized water and dispersed by ultrasonication for 30 min. Then, 0.02 mol of C16H36O4Ti was added to the solution and stirred for 1 h to obtain a dispersed faint yellow suspension. A certain amount of NaOH (99%, Innochem) as the mineralizer was dissolved in 50 mL deionized water to achieve a NaOH concentration of 10 mol L−1. The mineralizer solution was added to the precursor solution under stirring conditions, stirring was continued for 2 h to obtain a uniform solution, and the suspension was transferred to a 100 mL Teflon-lined autoclave and heated at 180 °C for 24 h. Following this reaction, the products were washed several times with deionized water, until the pH reached around 7, and dried at 60 °C for 12 h. The manipulation of various morphologies can be accomplished by altering the concentration of the mineralizing agent, sodium hydroxide. The samples are labelled BNT, sphere, and cube, corresponding to concentrations of 10 mol L−1, 14 mol L−1, and 16 mol L−1, respectively. The powders synthesized through a solid-phase reaction are named bulk, with Na2CO3 (99%, Sinopharm), Bi2O3 (99.9%, Aladdin), and TiO2 (99.9%, Aladdin) as raw powders.2.2 Synthesis of Bi0.5Na0.5TiO3−δ
Using glyoxal (OHCCHO, 40%, Daimao Reagent) as a mild reducing agent, 300 mg of BNT powder was added to 30 mL of aqueous solution containing a certain amount of glyoxal, and ultrasonically stirred for 1 h to disperse and mix evenly. The final mixture was hydrothermally treated at 120 °C for 6 h in a 50 mL Teflon-lined autoclave with a filling degree of 60%. The solution was washed with deionized water and centrifuged and the light-yellow powder was dried in a vacuum drying oven at 60 °C for 24 h. The different concentrations of oxygen vacancies in BNT can be achieved by controlling the amount of glyoxal added. The optimized initial Bi0.5Na0.5TiO3 and oxygen vacancy decorated Bi0.5Na0.5TiO3−δ samples are named BNT and BNT–VO, respectively.2.3 Electrochemical and photoelectrochemical testing
Testing was conducted with an electrochemical system (CHI-760E, Chenhua, China) using a three-electrode system, with the prepared catalyst sample as the working electrode, a platinum plate as the counter electrode, Ag/AgCl (saturated KCl solution) as the reference electrode and 0.1 M Na2SO4 (99%, Kermel) as the electrolyte. Measurements were conducted at room temperature at open-circuit potential. EIS tests were performed at frequencies ranging from 106 Hz to 1 Hz. Mott–Schottky curves were measured in the potential window from −1 to 1 V vs. Ag/AgCl. Photocurrent responses were obtained in the same three-electrode electrochemical cell using a 300 W xenon lamp as the light source. The working electrode was prepared as follows: 10 mg of the photocatalyst was dispersed in 100 μL N,N-dimethylformamide (HCON(CH3)2, DMF, Innochem) solution for 2 h to obtain a uniform emulsion. Using a pipette gun, a 50 μL drop of the suspension was applied to a 1 × 1 cm conductive ITO glass and dried at 473 K for 2 h to remove the organic solvent.2.4 Photocatalytic CO2 reduction
Photocatalytic CO2 reduction tests were carried out in a custom-built 50 mL atmospheric pressure quartz photocatalytic reactor with a sealed rubber ring, a 300 W xenon lamp as the light source, an Agilent gas chromatograph with TCD and FID detectors for gas product analysis and ultra-high purity argon (99.999%) as the carrier gas. 30 mg of catalyst was evenly dispersed on a quartz fiber filter plate and the reactor was sealed. The reactor was flushed with high-purity CO2 (99.999%) for 1 h before exposure to light, and during exposure the gas product was extracted with a 1 mL gas syringe at 1 h intervals and injected into a gas chromatograph for gas analysis. Blank experimental groups were drilled under the following conditions: a photocatalyst in high purity argon and light, a catalyst in carbon dioxide without light, a carbon dioxide atmosphere and light but without a catalyst. No significant gaseous products were detected, and these blank experiments confirmed the absence of carbon impurity effects in the experimental system.2.5 Characterization
The crystalline structure of the catalysts obtained was measured by X-ray diffraction (XRD, D8, Bruker, Germany) using Cu-Kα radiation. The surface morphology and grain size of the samples were observed using scanning electron microscopy (SEM, Gemini 300, Zeiss, Germany) and transmission electron microscopy (TEM, H-8100IV, Hitachi, Japan). X-ray photo electron spectroscopy (XPS) was carried out using a spectrometer (Thermo Scientific K-Alpha, United States), and the spectra were fitted by Advantage software. Direct tracking of unpaired single electrons in materials was carried out by electron paramagnetic resonance spectroscopy (EPR, EMXplus-6/1, Bruker, Germany). The transmittance and absorption spectra of the samplewereobtained using a UV-vis near infrared spectrophotometer (UV-3600iplus, Shimadzu, Japan). Photoluminescence (PL, FLS1000, Edinburgh, UK) measurements were carried out by excitation at 375 nm. The amplitude, phase images, and localized electrical responses (phase-voltage hysteresis loops and amplitude-voltage butterfly curves) of the samples were acquired using piezoelectric microscopy (PFM, MFP-3D, Asylum Research, CA, USA). The piezoelectric properties were investigated using dual AC resonance tracking AFM mode with a Pt/Ir coated Si tip. The powdered samples were dispersed on the surface of a Ag paste coated Si sheet using alcohol and dried at 50 °C in an oven. X-ray absorption fine structure (XAFS) characterization: Bi LIII-edge, Ti L-edge, and O K-edge XAFS measurements were performed at beamline 14W1 at the Shanghai synchrotron radiation facility, China. The X-ray was monochromatized by using a double-crystal Si(111) monochromator. The storage ring was operated at 3.5 GeV with a current of 300 mA. The acquired XAFS data were processed according to standard procedures using the Demeter.553. Results and discussion
3.1 Spatial separation of photogenerated charges in OV-modified ferroelectric catalysts
Ferroelectric polarization in the non-centrosymmetric structure of BNTs can impact electron behavior and significantly expedite its transportation. The integrated electric field can enhance the separation efficacy of photogenerated carriers, thus promoting photocatalytic reactions. Besides, the introduction of oxygen vacancies in ferroelectric BNTs is proposed to create vacancy-defective energy levels that can inhibit the compounding of carriers during the migration process, ultimately achieving the goal of reducing the charge hopping potential barrier. The moderate OVs on the surface of the material will cause excess local electrons and ligand unsaturation, offering active sites for the adsorption of small CO2 molecules, thus promoting molecule activation through the injection of light-generated electrons.56–59 Herein, the OV concentration and morphology are carefully optimized after extensive experiments according to the catalysis performance. Promising oxygen vacancy decorated BNT-VO is achieved by thermal reduction with an appropriate amount of solvent glyoxal and synthesis route, taking into account the defect association of the lattice. The schematic illustration and perovskite structure are presented in Fig. 1a and b. In rhombohedral BNTs with a−a−a− octahedral tilts, the cations are displaced parallel to each other to give the spontaneous polarization direction of the c axis that is parallel to [111]pc. Due to the hybridization of the bismuth 6s lone pair electrons and the oxygen 2p orbitals, weak Bi–O covalency is proposed with off-centering of Bi ions and a reduction in the coordination number that leads to a variety of Bi–O bond lengths for the non-cubic polyhedron.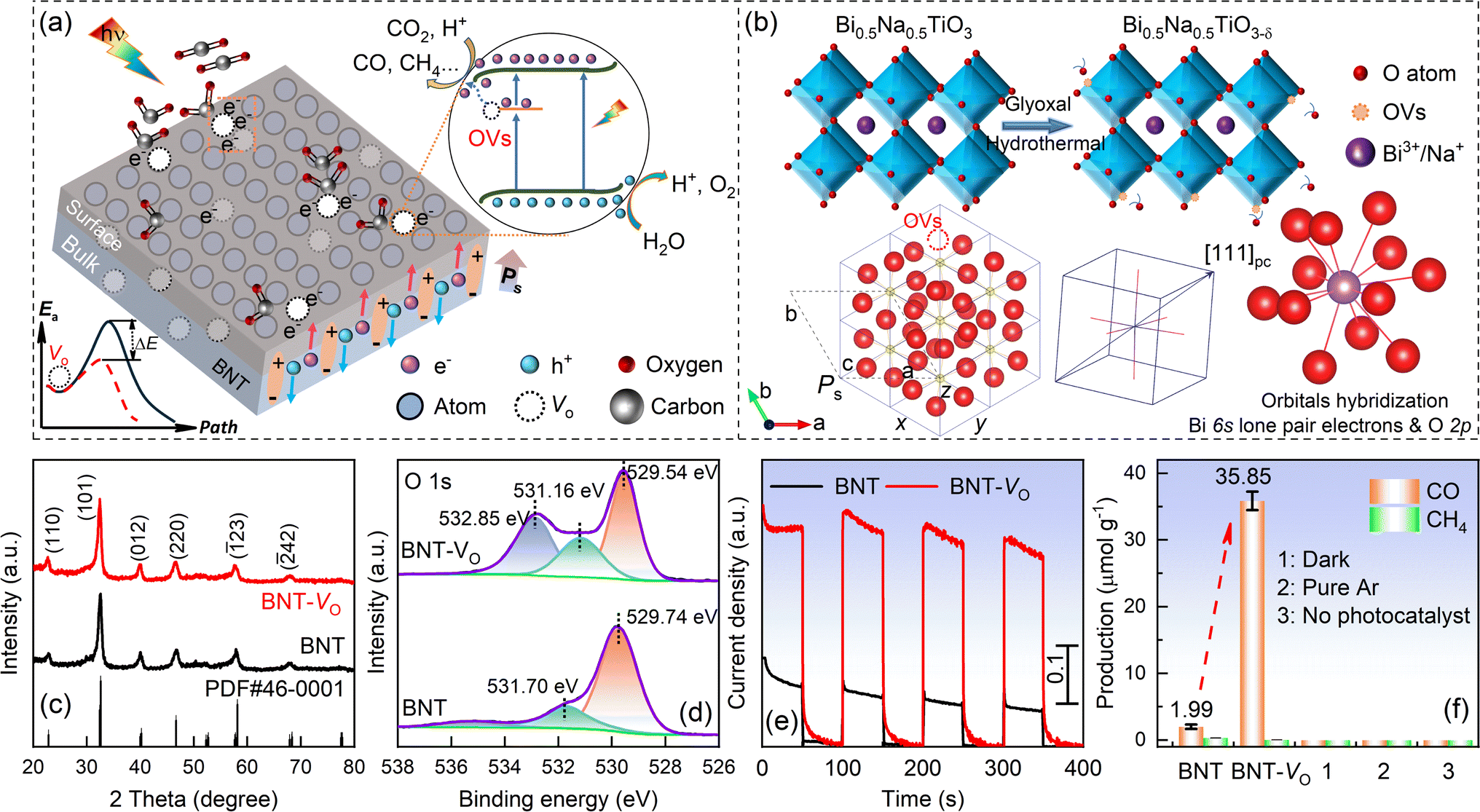 | ||
| Fig. 1 (a) Schematic illustration of the BNT photocatalytic CO2RR based on OV modification. (b) Schematic diagram of OV formation on BNT, the crystalline rhombohedral structure taken along the [001] zone axis, the spontaneous polarization of [111]pc in the pseudocubic structure, and the nonequal distance of Bi/Na–O bonds in 12 coordination. (c) XRD patterns, (d) O 1s XPS spectra, and (e) photocurrent response curve of BNT and BNT-VO. (f) CO and CH4 reduction over BNT and BNT-VO under simulated sunlight irradiation for 1 h. Error bars represent the standard deviation. | ||
The diffraction peaks of both BNT and BNT-VO in the XRD pattern match well with the rhombohedral-phase polar non-centrosymmetric space group R3c (PDF#46-0001) in Fig. 1c. All the hydrothermal-synthesized samples exhibit a characteristic ABO3-type perovskite structure without discerned impurities. In the perovskites, Na+ and Bi3+ ions co-occupy the A-site, surrounded by oxygen ions in an octahedral coordination. Meanwhile, Ti4+ ions occupy the B-site and are hexahedrally coordinated with oxygen ions, forming a [TiO6] coordination configuration (Fig. 1b). This arrangement leads to the displacement of positive and negative charge centers in the octahedron, generating spontaneous polarization. The intrinsic spontaneous polarization induces anisotropic charges to migrate in opposite directions, driven by the built-in electric field, thereby achieving spatial separation of carriers in the bulk phase (Fig. 1a). The modification of OVs on the material's surface does not alter the main crystalline phase structure, as evidenced by the unchanged positions of the diffraction peaks (Fig. 1c). The half-peak widths also remain relatively unchanged, indicating no significant secondary growth of the catalyst grains at lower hydrothermal temperatures and an invisible change in grain size. The wider half-peak widths of the diffraction peaks suggest the nanosizing effect of the grains.
The surface elements and chemical states of both BNT and BNT-VO are studied by XPS (Fig. S1a†). The XPS survey spectra of both samples present the peaks corresponding to Bi 4f, Ti 2p, O 1s, and Na 1s. For O 1s, it displays a main asymmetric peak that can be fitted by using the Gaussian curves (Fig. 1d). The peak at ∼529.74 eV can be ascribed to lattice oxygen (OL), while the peak located at a higher binding energy of ∼531.16 eV is associated with oxygen binding in the oxygen-deficient region, as reported in the previous studies.1,33 Interestingly, a brand new peak at a binding energy of 532.85 eV, corresponding to water adsorption (–OH), emerges due to the enrichment of OVs on the catalyst surface. Together with the enhanced intensity at 531.16 eV (OVs), this confirms the higher concentrations of oxygen vacancies in BNT-VO. A semi-quantitative determination of the surface OV content employs a percentage of the inverse convolution peak area of the OVs.49,53,60,61 By fitting the data, it was shown that the convolution peak area of the OVs accounts for 34% in BNT-VO, and 18% in BNT. Furthermore, the reduction-induced removal of more coordinated oxygen atoms results in a local imbalance charge around the oxygen defects. Consequently, more than two electrons are needed for the compensation, leading to a transition to a lower chemical valence state of the coordination metal atom.
The photogenerated carrier separation efficiency significantly influences the photocatalytic performance of a photocatalyst (Fig. 1e). Transient photocurrent response curves are commonly used to characterize the photo-exciton separation of catalysts in photoreactions. The photocurrents generated for all samples show excellent reproducibility. BNT-VO exhibits the highest photocurrent density and the strongest photocurrent response compared to others. The enhancement can be attributed to the abundance of oxygen vacancies on its catalyst surface, which effectively facilitates the separation of the photogenerated electron–hole pairs in BNT-VO. The photocatalytic CO2 reduction capacity is evaluated in a gas–solid configuration using a quartz mass-sealed photoreactor with simulated sunlight as the light source (Fig. 1f). The initial BNT exhibits CO and CH4 production rates of 1.99 μmol g−1 and 0.32 μmol g−1 for an hour, respectively. Remarkably, BNT-VO demonstrates superior CO2 reduction performance with CO and CH4 yields of 35.85 μmol g−1 and 0.04 μmol g−1, respectively, which is superior to that of the other recently reported ferroelectric catalysts by using the light source only, where most are <22 μmol g−1.30,35,62–68 The CO reduction rate with small repeatability deviation is increased by 18 times associated with an ultrahigh selectivity of 99.88%, proving the key characteristics of OVs in the CO2RR of the ferroelectrics. To verify the catalytic reaction initiated by the BNT-VO catalyst and the photoreduction of CO2 as the source of CO production, blank experimental groups are investigated under dark conditions, without a catalyst, and in an argon atmosphere, respectively. No specific gas product is detected under these conditions, thereby confirming the catalytic role of the BNT-VO catalyst initialized by spontaneous polarization and excluding the mechanically piezoelectric effect in facilitating CO2 photoreduction in the absence of applied stress or vibration during the catalytic process.
3.2 Microscopic morphology, exposing facets and ferroelectricity
The SEM and TEM images reveal the mixed microscopic morphology, consisting of nanospherical agglomerates, ranging from 200 nm to 400 nm in diameter with stepped surfaces, and thin nanobelts with long aspect ratios (Fig. S2†). The EDS mappings show that all areas consist of Bi, Na, Ti, and O, and each element is uniformly distributed. In Fig. 2, the surface of the nanosphere is formed by cubic initial nanoparticles, each with an average diameter of 10 to 20 nm, and the edges of the nanoparticles are visible in the HR-TEM image. For the thin nanobelts, the plate-like structure is gradually transformed as indicated by the blue lines. The stripe spacings of 0.195 nm and 0.274 nm in HR-TEM image from different morphologies of BNT-VO can be matched well with the (220) and (101) crystal planes in the R3c phase of BNTs in agreement with the XRD patterns. Selected area electron diffraction (SAED) indicates the polycrystalline form of nanospheres, while the single-crystal in nanobelts is constituted by a thin layer of a superposition structure.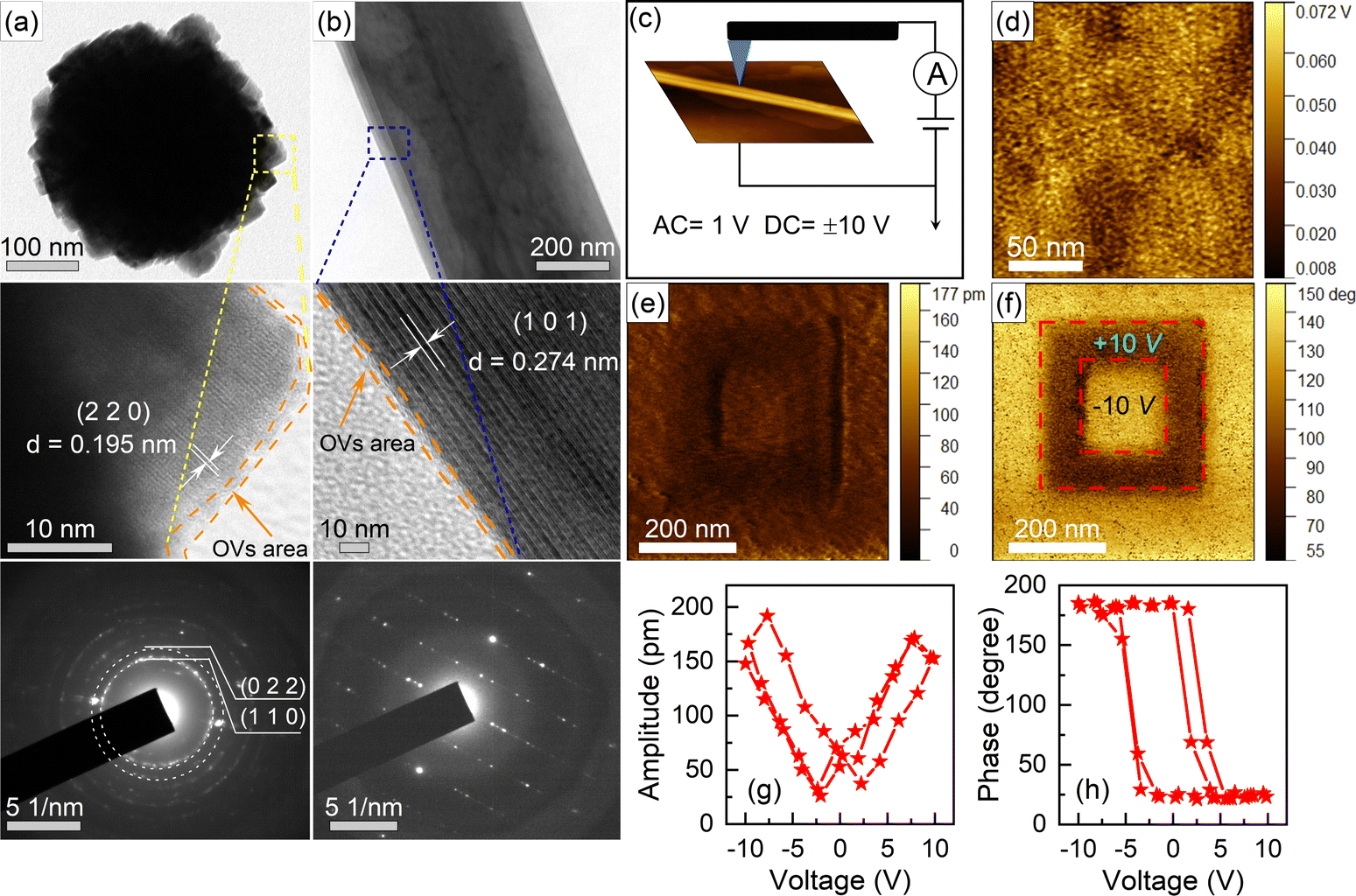 | ||
| Fig. 2 (a and b) Micromorphology, local HR-TEM images and the corresponding electron diffraction patterns of nanospheres and nanobelts, respectively, for BNT-VO. (c) Schematic for PFM measurement of a nanobelt on a Ag/Si substrate. (d) KPFM potential images. (e) PFM amplitude and (f) phase scans of BNT with a box-in-box pattern obtained by the applications of reverse dc biases. (g) Amplitude butterfly and (h) phase hysteresis loops versus voltage. | ||
The unique morphology of the nanosphere surface can provide more catalytically active sites. Additionally, the long aspect ratio nanobelts not only provide a larger specific surface area but also promote rapid charge migration along the shorter vertical direction, facilitating the separation of photogenerated electron–hole pairs driven by the spontaneous polarization of the built-in electric field. It is worth mentioning that both the nanospheres and nanobelts in BNT-VO exhibit damaged edges, evidenced by the disordered and disorganized lattice fringes, which will be related to the richer oxygen vacancies on the surface of BNT-VO.69 In addition, more morphologies (Fig. S3†) of the nanostructure are obtained by modulating the mineralizer concentration to evaluate the catalytic performances of BNTs, and the morphology formation mechanism is illustrated in Fig. S4.† All samples have a mesoporous structure, and the specific surface area is greatly improved in BNT and BNT-VO as further evidenced by BET (Fig. S5†). The introduction of OVs does not significantly alter its specific surface area. Comparison of the UV-vis spectral curves reveals that the absorption capacity of BNTs for visible light remains hardly unchanged despite the differences in morphology (Fig. S5c†).
To verify their ferroelectric properties, the piezoelectric response of BNT is probed by using dual AC resonance tracking AFM mode in the respective areas (Fig. 2c). The surface potential maps show different potentials on the surface of BNT, suggesting the formation of a polarized electric field between the dark and light regions (Fig. 2d). The intrinsic electric field generated by the spontaneous polarization causes a redistribution of charges that move in opposite directions. This process favors the separation of photogenerated electron–hole pairs. The box-in-box pattern obtained using positive and negative voltage tip bias shows clear reversal of phase and amplitude contrast, verifying the switchable polarization under the application of an electric field, and the remnant polarization when the electric field is removed (Fig. 2e and f). The uniform phase contrast indicates complete polarization switching. Typical amplitude-voltage butterfly and phase hysteresis loops with 160° phase change against rotation further confirm the ferroelectric switching behaviors (Fig. 2g and h).
3.3 Elemental coordination environment and valence state
X-ray adsorption fine spectroscopy is employed to gain insights into the changes in the coordination environment and the chemical valence states of metallic elements caused by the presence of OVs in BNT. The Bi-LIII edge of BNT-VO has a slight shift towards lower energy, suggesting a change in the coordination of Bi due to the absence of the oxygen atom (Fig. 3a).70,71 The edge shift is a typical indication of a chemical valence change from the highly oxidized state Bi3+ to the reduced state Bi(3−x)+. Due to the unsaturated coordination nature of the metal atoms neighboring the OVs, the presence of OVs can be confirmed by coordination information, such as the coordination number and the strength of the M–O bond.72,73 The local environment of Bi is further understood at the atomic level by Fourier transformation (Fig. 3b). The main peak of BNT, corresponding to the Bi–O bond, is located at around 1.7 Å. The formation of OVs reduces the peak intensity by losing O atoms coordinated to Bi atoms. The difference in the oscillation function, the shift of the main peak, and the apparent decrease in intensity collectively suggest a disordered local lattice arrangement with coordination-unsaturated Bi atoms. Additionally, the main Bi–O bond length at ∼1.7 Å is shortened by 0.02 Å whereas the Bi–O bond length at ∼2.65 Å is increased by 0.11 Å as a result of the distorted contractive lattice. The EXAFS wavelet transforms (WT) also confirm the states of Bi–O radial distance and species (Fig. 3d and e). The inverse change in both the peak shift and intensity ratio proves the anabatic variety in the non-equidistant Bi–O bond, revealing the enhanced structural distortion.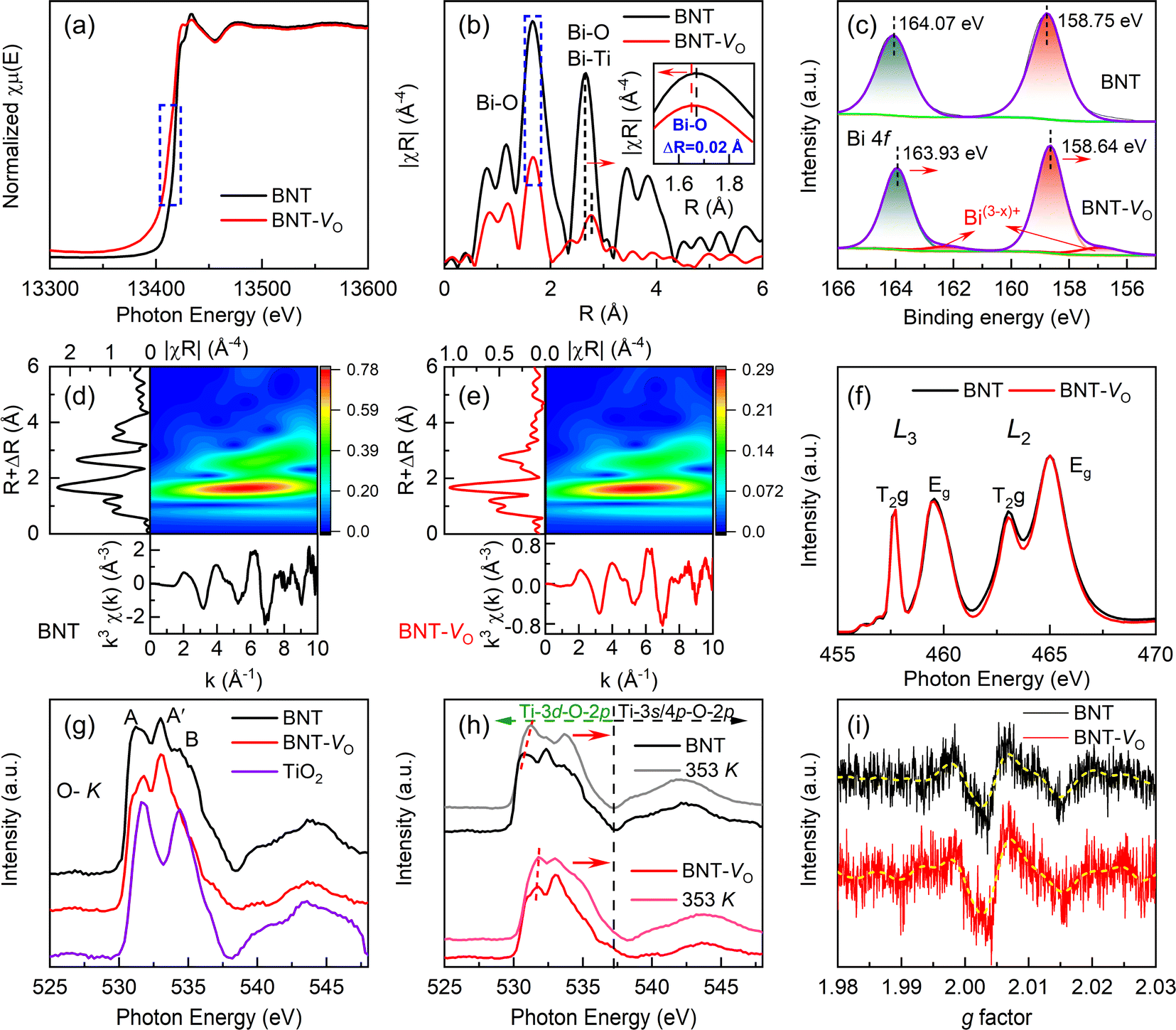 | ||
| Fig. 3 Characterization of the surface composition and chemical state. (a) Normalized Bi-LIII-edge XAFS spectra. (b) Fourier transformed profiles for Bi coordination environments. ΔR represents the difference in the Bi–O bond distance between BNT and BNT-VO. (c) Bi 4f XPS spectra. Wavelet transforms for the k3-weighted EXAFS signals of (d) BNT and (e) BNT-VO. (f) The spectra of Ti-L2,3 edges from XANES spectra. (g and h) High-resolution X-ray absorption spectra of O–K in BNT, BNT-VO and TiO2 at room temperature and 353 K. (i) ESR spectra of BNT-VO and BNT at room temperature. | ||
The chemical state of Bi is further determined from the Bi 4f XPS spectra (Fig. 3c). The two main spectral peaks at binding energies of 164.07 eV and 158.75 eV correspond to Bi 4f5/2 and Bi 4f7/2, respectively, which can be assigned to Bi3+.74,75 The peaks shift towards lower binding energies when compared to those of BNT implying the weaker Bi–O bond. Moreover, as the produced OVs could be accompanied by the quantitative reduction in the coordination number of Bi ions in average weakened Bi–O bonds, the smaller pair peaks, indicated by arrows, with a calculated proportion of 0.4% at lower binding energies of 162.4 eV and 157.3 eV are consistently attributed to Bi(3−x)+.76,77 The L2,3-edges of the samples in Fig. 3f show mainly dipole transitions from Ti 2p to the unoccupied Ti 3d state.78 The splitting at the Ti-L2,3 edge serves as an indicator of the strength of Ti–O hybridization and the interactions within the ligand-field atomic environment. The intensity of T2g/Eg ratios at the L3 edge remains stable while exhibiting small variation at the L2 edge. The reduced ratio of the splitting peaks indicates the subtle change in Ti–O bond strength and Ti4+ reduction regardless of the OVs that will lead to the distortion of the [TiO6] octahedron and loss of its symmetry. This agrees with the slightly low binding energy shift of Ti 2p1/2 and Ti 2p3/2 peaks for Ti4+ at 463.94 eV and 458.10 eV, respectively, without discernible Ti3+ peaks in XPS (Fig. S1b†).
The O–K XAS originates from a dipole transition process between O 1s and 2p states as shown in Fig. 3g. The two subbands T2g and Eg, labeled peaks A and B, correspond to the crystal field splitting at the Ti L2,3 edge according to a pair of cleavage peaks seen in the TiO2 curve, where O 2p hybridizes with Ti 3d orbitals in the conduction band to form a hybridized band. The feature A′ emerges from a heterozygous state jump between O 2p and A-site elements.79 Due to the diffuse nature, the ratio of T2g/Eg in O–K XAS is hard to be determined to reveal the difference in dipole transitions related to oxygen defects, but the obvious enhancement in height around A′ confirms the significant role of A–O bonds in BNT-VO. Meanwhile, the absence of oxygen reduces the valence state of the transition metal, leading to a decrease in the effective nuclear charge (proton number) of the metal and an increase in the energy level of the occupied state. While the O 1s energy level remains almost unchanged, slightly higher energy is required to excite the O 1s electron to the unoccupied state. As a result, the characteristic peaks produce a shift towards higher energy in BNT-VO. At elevated temperatures of 353 K, the weakness of bond strength is revealed due to the vibration of lattice oxygen and expanded cells (Fig. 3h). BNT-VO containing more oxygen defects exhibits a lower peak shift than BNT, illustrating the thermal stability. This agrees with the easier oxygen loss with increasing temperatures in the stoichiometric rhombohedral BNTs as compared with nominal oxygen-deficient compositions.46 Additionally, compression or expansion of the Ti–O bond length, resulting from the oxygen vacancies, causes pronounced [TiO6] octahedral distortion. Consequently, cleavage peaks shift more prominently in the high-energy direction of the phase.
The oxygen defects will lead to an increase in free electrons within the BNT as demonstrated by the electron spin resonance spectrum (Fig. 3i). The ESR signal peak of BNT-VO shows a slight increase, which is attributed to the more uncoordinated lone electrons on the surface of BNT-VO, mainly unbound or weakly bound under the defect. The increase in electron spin resonance is further linked to the low valence metal-induced spin enhancement, originating from the electric field polarization around the oxygen defect.53,77,80 Combined with the considerable OVs reflected in XPS, this suggests that the intrinsic OVs are more easily distributed on the surface than bulk. The conduction mechanism is not significantly changed despite its particular sensitivity to oxygen deficiency in non-stoichiometric BNTs.
3.4 Charge transfer capability and the material energy band structure
The transport capacity of the intrinsic charge in the catalyst can be illustrated by the Nyquist curve,81,82 where the small semicircle shown in Fig. 4a represents charge transfer resistivity. The resistance is 28.83 Ω for BNT-VO, an approximately two times decrease as compared to 60.37 Ω for BNT through a circuit fitting using the Z-view. The lower charge transfer impedance in oxygen vacancy-modified BNT-VO is further evidenced by the reduced CO2 reduction overpotential with a lower initial voltage and higher circuit density (Fig. 4b). To clarify the location of the energy band boundaries of BNT and BNT-VO, the Fermi energy levels (EF) are obtained through the electrochemical Mott–Schottky curves to identify their conduction band minima (Fig. 4c). The positive slope of the curve indicates that both samples exhibit an n-type dominant conduction mechanism. Its intersection with the x-axis is investigated to obtain the flat-band potentials of −0.96 V and −0.87 V for BNT and BNT-VO, respectively (Fig. S6†). In the electrolytes, EF is very close to the material flat-band potential. Thus, the EF values of BNT and BNT-VO are −0.76 V and −0.67 V vs. NHE (pH = 7) based on the Nernst equation ENHE/V = EAg/AgCl/V + 0.197 V. Typically, the potential of EF is 0 to 0.1 V lower than the minimum potential of the conduction band (ECB) in n-type semiconductors.49 Therefore, the conduction band minima for BNT and BNT-VO are −0.86 V and −0.77 V, respectively, and both the conduction band potentials fully meet the reduction potential requirements of CO2/CO and CO2/CH4 (Fig. 4e).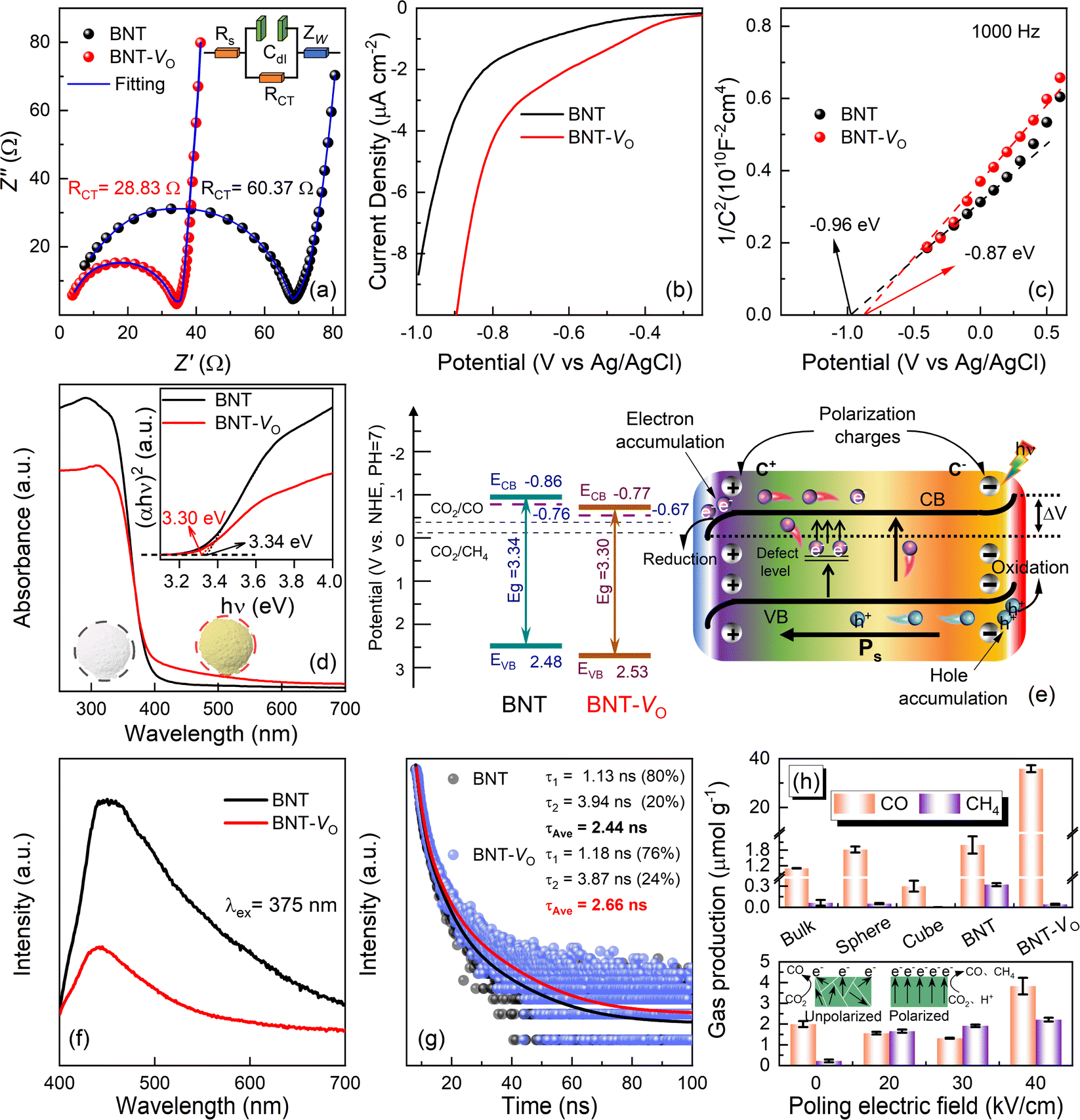 | ||
| Fig. 4 Charge transfer capability and optimization of the oxygen vacancy energy band structure. (a) Electrochemical impedance curves. (b) Linear sweep voltammetry plots. (c) Mott–Schottky curves. (d) UV-vis spectra and the corresponding band gap widths. (e) Schematic diagram of the band structure of the defective energy level and the catalytic mechanism of a redox reaction under band bending and a polarization field. (f) PL spectra. (g) Time-resolved decay photoluminescence. (h) Yield of CO and CH4 over pristine bulk, sphere, cube, BNT and BNT-VO for 1 h, and BNT with different poling electric fields. | ||
The UV-vis diffuse reflectance spectra indicate that the initial absorption limit is at about 450 nm for BNT, suggesting that the main absorbed photons are from the UV band, which restricts the energy conversion of the catalyst (Fig. 4d). In contrast, the absorption range of BNT-VO extends to visible wavelengths, proving the enhanced absorption capacity in the visible region, making it more favorable for the photocatalytic reduction reaction to improve the CO2 reduction capacity. The inset shows that the BNT and BNT-VO are macroscopically white and yellowish, respectively, due to the different OV concentrations in the materials since there are no additional bond species such as Bi metal atoms according to the Bi-LIII-edge EXAFS spectra. The band gaps of BNT and BNT-VO are determined to be 3.34 eV and 3.30 eV, respectively, from the transformed Kubelka–Munk function (inset of Fig. 4d), leading to the valence bands of 2.48 eV and 2.54 eV (V vs. NHE), respectively. The OVs do not significantly narrow the band gap but still enhance strong absorption with an extended visible light response range, due to the formation of adventitious energy levels by electrons localized in the oxygen vacancies.83 The oxygen vacancies form defective energy levels in the BNT band gap so that the excited electrons in the conduction band are trapped and the direct complexation of charge carriers is suppressed,84 thereby promoting the photocatalytic performance.
3.5 Charge separation and CO2 photocatalytic properties
When utilizing ferroelectric BNT as a photocatalyst, the intrinsic electric field originates from spontaneous polarization (Ps) and results in the formation of positively charged regions, referred to as C+ domains, and negatively charged regions referred to as C− domains. In the C+ domains, the surface energy levels will bend downwards, which facilitates the transfer of electrons and allows them to accumulate in specific regions to contribute to the reduction reaction. Conversely, in the C− domains, the surface energy levels will bend upwards which enhances the transfer of holes.85 The presence of an internal electric field will determine the degree of bending of the energy band structure at the interface of the material. By appropriately controlling the internal electric field, the photo-excited e− and h+ can be effectively localized at different interfaces. This helps to achieve efficient charge separation and allows electrons and holes to participate in the catalytic reaction separately, giving rise to the improved efficiency of the photocatalyst.PL spectra can qualitatively show the separation of excitation charges. In particular, BNT-VO exhibits a weak PL peak with the fluorescence emission peak intensity being 2.6 times smaller than that of BNT (Fig. 4f). This suggests the lower complex efficiency of photogenerated electron–hole pairs. Additionally, time-resolved decay photoluminescence is investigated under λ = 375 nm excitation to clarify the specific carrier dynamics. The average lifetime of photogenerated charges, directly inferred from the fluorescence decay data (Fig. 4g), in BNT-VO is 2.66 ns, which is slightly longer than that of BNT (2.44 ns). The lower fluorescence decay rate of BNT-VO compared to the former indicates that photogenerated electrons survive longer on its surface. These results further confirm that a suitable OV concentration can be effective for trapping photogenerated electrons, mitigating charge recombination of carriers, and facilitating separation and migration within the ferroelectric materials, thereby leading to beneficial photocatalytic properties.
Nanostructured BNTs having different morphologies of bulk, sphere, cube, etc. all exhibit limited catalytic capacity. Due to the richer oxygen vacancy modification on the surface, the photocatalysis capability is greatly promoted in BNTs with microstructures constituted by a mixture of nanospheres and nanobelts, and the electrons generated by the photoreaction mainly contribute to the CO2 to CO conversion pathway (close to 100% CO selectivity). The product selectivity is consistent with those in reported photocatalyst studies modified by oxygen vacancies.86,87 The catalyst remains active with acceptable degradation on extending the reaction time. It is worth mentioning that the CO yield does not increase noticeably over time, on the one hand, due to the adsorption of CO2 and the desorption of reduction products. Most importantly, it is affected by the surface oxygen vacancy state of the catalyst which is, actually, still a challenge in most oxygen vacancy-modified oxide catalysts that can meet the obstacles of defect reoxidation and the formation of electron–hole complex sites. In addition, BNT with different OVs by controlling the amount of glyoxal reflects less enhanced photocatalytic performance when the OV content is low as a result of fewer defects in the absence or on introduction of defect energy levels. It is not conducive to obtaining more photon energy due to the weak absorption of visible light. In the high OV case, the photocatalytic performance does not improve significantly because too many defect sites in the catalysts will increase the probability of the formation of electron–hole complex sites, leading to the extinction of photogenerated electrons and holes.
Furthermore, the poled BNT powders are examined, and all the CH4 yields are improved, producing 1.60, 1.95, and 2.28 μmol g−1 after poling at electric fields of 20, 30, and 40 kV cm−1, respectively (Fig. 4h). This enhancement is attributed to the increase in the well-lined built-in electric field that facilitates the process of multi-electron transfer. When a DC voltage is applied, the domains reorient and progressively align in the same direction, thereby creating a robust electric field, which can remain because of the irreversible phase transition in rhombohedral BNTs. As demonstrated in the inset, the random orientation of domains when unpolarized can result in the offset annihilation of electrons and holes. After polarization, the orientation of the polarized electric field is the same, which facilitates the accumulation of electrons caused by the reverse migration of electrons and holes. The conversion of CO2 to CH4 requires the participation of more free electrons than the conversion of CO. Therefore, the accumulation of electrons after polarization is more conducive to this reaction. Additionally, the OVs will also hinder the reverse reorientation of ferroelectric domains, establishing a more potent and enduring ferroelectric electric field.30 Consequently, as the poling electric field increases, the orientation of the ferroelectric domains results in a heightened potential difference and an increased degree of energy band bending.37,85 This effect provides a more substantial driving force for charge separation, consequently enhancing the multi-electron photocatalytic CO2 reduction reaction to CH4.
4. Conclusions
In conclusion, the photocatalytic CO2 reduction reaction is first explored in ferroelectric BNTs. The self-assembly of mixed nanobelt and nanosphere morphology, oxygen vacancies, and intrinsic ferroelectric polarization associated with weak Bi–O bonds co-play the key roles in promoting photocatalysis: (a) the energy band structure is optimized by the defect energy level, reducing the barrier for carrier-leap and broadening absorption within the visible light range. (b) The coordination unsaturation point of the cations provides a local enrichment of electrons, while the ferroelectric spontaneous polarization encourages the segregation and migration of photogenerated electron–hole pairs in the opposite direction. (c) Manipulation of polarization in the ferroelectrics produces a directional orientation of ferroelectric domains and bending of energy bands, resulting in a favorable impact on multi-electron mediated reactions. Particularly, oxygen vacancies change the local chemical environment, and the bonding capacity leads to the elongation or contraction of coordination bonds, which in turn causes lattice distortion and enhances the intrinsic spontaneous strength to some extent. Oxygen vacancy-defected BNT shows great enhancement in CO yield and product selectivity as compared to other ferroelectric photocatalysts. Well-modulated by the surface and polarization states in perovskite oxides, it opens up a new route for the design of efficient lead-free ferroelectric photocatalysts for the CO2RR in a synergistic manner.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation (51702249 and 51602252), the Natural Science Basic Research program of Shaanxi (2023-JC-YB-397 and 2024JC-YBMS-265), and the Guangxi Key Laboratory of Optical and Electronic Materials and Devices Foundation (22KF-3).Notes and references
- J. Zhou, J. Li, L. Kan, L. Zhang, Q. Huang, Y. Yan, Y. Chen, J. Liu, S. L. Li and Y. Q. Lan, Nat. Commun., 2022, 13, 4681 CrossRef CAS PubMed.
- Y. Zhang, X. Zhi, J. R. Harmer, H. Xu, K. Davey, J. Ran and S. Z. Qiao, Angew. Chem., Int. Ed., 2022, 61, e202212355 CrossRef CAS PubMed.
- L. Hurtado, A. Mohan, U. Ulmer, R. Natividad, A. A. Tountas, W. Sun, L. Wang, B. Kim, M. M. Sain and G. A. Ozin, Chem. Eng. J., 2022, 435, 134864 CrossRef CAS.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- Y. Wang, X. Li, Y. Chen, Y. Li, Z. Liu, C. Fang, T. Wu, H. Niu, Y. Li, W. Sun, W. Tang, W. Xia, K. Song, H. Liu and W. Zhou, Adv. Mater., 2023, 35, e2305257 CrossRef PubMed.
- X. Guo, F. Yang, X. Sun, C. Han, Y. Bai, G. Liu, W. Liu and R. Wang, J. Mater. Chem. A, 2022, 10, 3146–3158 RSC.
- F. Xu, B. Zhu, B. Cheng, J. Yu and J. Xu, Adv. Opt. Mater., 2018, 6, 1800911 CrossRef.
- Y. Shen, C. Ren, L. Zheng, X. Xu, R. Long, W. Zhang, Y. Yang, Y. Zhang, Y. Yao, H. Chi, J. Wang, Q. Shen, Y. Xiong, Z. Zou and Y. Zhou, Nat. Commun., 2023, 14, 1117 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Zhu, H. Liu, J. Xu and J. Yu, Adv. Funct. Mater., 2019, 29, 1904256 CrossRef CAS.
- Z. Jiang, X. Xu, Y. Ma, H. S. Cho, D. Ding, C. Wang, J. Wu, P. Oleynikov, M. Jia, J. Cheng, Y. Zhou, O. Terasaki, T. Peng, L. Zan and H. Deng, Nature, 2020, 586, 549–554 CrossRef CAS PubMed.
- D. Wu, Q. Liang, H. Si, X. Yan, H. Huang, Z. Li and Z. Kang, J. Mater. Chem. A, 2022, 10, 24519–24528 RSC.
- J. Wu, K. Li, S. Yang, C. Song and X. Guo, Chem. Eng. J., 2023, 452, 139493 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- J. Hu, D. Chen, Z. Mo, N. Li, Q. Xu, H. Li, J. He, H. Xu and J. Lu, Angew. Chem., Int. Ed., 2019, 58, 2073–2077 CrossRef CAS PubMed.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- L. Chen, Z. Mao, Y. Wang, Y. Kang, Y. Wang, L. Mei and X. Ji, Sci. Adv., 2022, 8, eabo7372 CrossRef CAS PubMed.
- S. Cheng, Z. Sun, K. H. Lim, T. Zhang, E. Hondo, T. Du, L. Liu, M. Judd, N. Cox, Z. Yin, G. K. Li and S. Kawi, ACS Appl. Nano Mater., 2023, 6, 3608–3617 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- Q. Wang, C. Yang, G. Zhang, L. Hu and P. Wang, Chem. Eng. J., 2017, 319, 39–47 CrossRef CAS.
- C.-C. Lin, T.-R. Liu, S.-R. Lin, K. M. Boopathi, C.-H. Chiang, W.-Y. Tzeng, W.-H. C. Chien, H.-S. Hsu, C.-W. Luo, H.-Y. Tsai, H.-A. Chen, P.-C. Kuo, J. Shiue, J.-W. Chiou, W.-F. Pong, C.-C. Chen and C.-W. Chen, J. Am. Chem. Soc., 2022, 144, 15718–15726 CrossRef CAS PubMed.
- P. Reñones, F. Fresno, F. E. Oropeza, G. Gorni and V. A. de la Peña O'Shea, J. Mater. Chem. A, 2022, 10, 6054–6064 RSC.
- M. Wang, S. Zeng, A. R. Woldu and L. Hu, Nano Energy, 2022, 104, 107925 CrossRef CAS.
- W. Shi, J.-C. Wang, X. Guo, X. Qiao, F. Liu, R. Li, W. Zhang, Y. Hou and H. Han, Nano Res., 2022, 15, 5962–5969 CrossRef CAS.
- H. Yin, F. Dong, D. Wang and J. Li, ACS Catal., 2022, 12, 14096–14105 CrossRef CAS.
- M. Laghaei, M. Ghasemian, W. Lei, L. Kong and Q. Chao, J. Mater. Chem. A, 2023, 11, 11925–11963 RSC.
- J. Di, C. Chen, C. Zhu, R. Long, H. Chen, X. Cao, J. Xiong, Y. Weng, L. Song and S. Li, Adv. Energy Mater., 2021, 11, 2102389 CrossRef CAS.
- H. Zhang, Y. Wang, S. Zuo, W. Zhou, J. Zhang and X. W. D. Lou, J. Am. Chem. Soc., 2021, 143, 2173–2177 CrossRef CAS PubMed.
- Y. Zhang, X. Wang, P. Dong, Z. Huang, X. Nie and X. Zhang, RSC Adv., 2018, 8, 15991–15998 RSC.
- H. Yu, F. Chen, X. Li, H. Huang, Q. Zhang, S. Su, K. Wang, E. Mao, B. Mei, G. Mul, T. Ma and Y. Zhang, Nat. Commun., 2021, 12, 4594 CrossRef CAS PubMed.
- J. Ma, S. Jing, Y. Wang, X. Liu, L. Y. Gan, C. Wang, J. Y. Dai, X. Han and X. Zhou, Adv. Energy Mater., 2022, 12, 2200253 CrossRef CAS.
- K. Das, R. Das, M. Riyaz, A. Parui, D. Bagchi, A. K. Singh, A. K. Singh, C. P. Vinod and S. C. Peter, Adv. Mater., 2023, 35, 2205994 CrossRef CAS PubMed.
- A. Ranjan, K. Y. Hsiao, C. Y. Lin, Y. H. Tseng and M. Y. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 35635–35644 CrossRef CAS PubMed.
- Y. Liu, Q. Li, Z. Lian, J. Fan, Y. Tao, G. Li and H. Li, Chem. Eng. J., 2022, 437, 135132 CrossRef CAS.
- R. Su, Z. Sun, C. He, S. Wei, L. Chen, D. Zhang, Z. Wang, X. An and F. Li, Inorg. Chem. Front., 2023, 10, 3947–3954 RSC.
- H. Xiao, J. He, X. Lu, F. Wang and Y. Guo, Chemosphere, 2022, 306, 135543 CrossRef CAS PubMed.
- H. Xiao, W. Dong, Q. Zhao, F. Wang and Y. Guo, J. Hazard. Mater., 2021, 416, 125808 CrossRef CAS PubMed.
- L. Wang and W. Wang, Int. J. Hydrogen Energy, 2012, 37, 3041–3047 CrossRef CAS.
- X. Liu, H. Fan, J. Shi and Q. Li, Sci. Rep., 2015, 5, 12699 CrossRef CAS PubMed.
- X. Liu, R. Rao, J. Shi, J. He, Y. Zhao, J. Liu and H. Du, J. Alloys Compd., 2021, 875, 159999 CrossRef CAS.
- T. Lv, J. Li, N. Arif, L. Qi, J. Lu, Z. Ye and Y.-J. Zeng, Matter, 2022, 5, 1–37 CrossRef.
- Z. Liang, C.-F. Yan, S. Rtimi and J. Bandara, Appl. Catal., B, 2019, 241, 256–269 CrossRef CAS.
- C.-P. Hu, S. Tu, N. Tian, T. Ma, Y. Zhang and H. Huang, Angew. Chem., Int. Ed., 2021, 60, 16309–16328 CrossRef CAS PubMed.
- L. Ju, J. Shang, X. Tang and L. Kou, J. Am. Chem. Soc., 2019, 142, 1492–1500 CrossRef PubMed.
- M. Li, M. J. Pietrowski, R. A. De Souza, H. Zhang, I. M. Reaney, S. N. Cook, J. A. Kilner and D. C. Sinclair, Nat. Mater., 2014, 13, 31–35 CrossRef CAS PubMed.
- J. Shi, X. Liu, F. Zhu, W. Tian, Y. Xia, T. Li, R. Rao, T. Zhang and L. Liu, J. Materiomics, 2022, 8, 719–729 CrossRef.
- F. Chen, Z. Ma, L. Ye, T. Ma, T. Zhang, Y. Zhang and H. Huang, Adv. Mater., 2020, 32, 1908350 CrossRef CAS PubMed.
- S. Chen, H. Wang, Z. Kang, S. Jin, X. Zhang, X. Zheng, Z. Qi, J. Zhu, B. Pan and Y. Xie, Nat. Commun., 2019, 10, 788 CrossRef CAS PubMed.
- D.-M. Liu, J.-T. Zhang, C.-C. Jin, B.-B. Chen, J. Hu, R. Zhu and F. Wang, Nano Energy, 2022, 95, 106975 CrossRef CAS.
- X. Zu, Y. Zhao, X. Li, R. Chen, W. Shao, Z. Wang, J. Hu, J. Zhu, Y. Pan, Y. Sun and Y. Xie, Angew. Chem., Int. Ed., 2021, 60, 13840–13846 CrossRef CAS PubMed.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- W. Jiang, H. Loh, B. Q. L. Low, H. Zhu, J. Low, J. Z. X. Heng, K. Y. Tang, Z. Li, X. J. Loh, E. Ye and Y. Xiong, Appl. Catal., B, 2023, 321, 122079 CrossRef CAS.
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef PubMed.
- C. Mao, H. Cheng, H. Tian, H. Li, W.-J. Xiao, H. Xu, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 228, 87–96 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- R. Schaub, E. Wahlström, A. Rønnau, E. Lægsgaard, I. Stensgaard and F. Besenbacher, Science, 2003, 299, 377–379 CrossRef CAS PubMed.
- M. Setvín, U. Aschauer, P. Scheiber, Y.-F. Li, W. Hou, M. Schmid, A. Selloni and U. Diebold, Science, 2013, 341, 988–991 CrossRef PubMed.
- M. Setvin, J. Hulva, G. S. Parkinson, M. Schmid and U. Diebold, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, E2556–E2562 CrossRef CAS PubMed.
- H. Yu, J. Li, Y. Zhang, S. Yang, K. Han, F. Dong, T. Ma and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 3880–3884 CrossRef CAS PubMed.
- X. Duan, Y. Jiang, B. Liu, Z. Duan, Y. Zhang, Z. Yuan and H. Tai, Sens. Actuators, B, 2024, 402, 135136 CrossRef CAS.
- Y. Guo, B. Liu, Z. Duan, Z. Yuan, Y. Jiang and H. Tai, Mater. Chem. Phys., 2023, 302, 127768 CrossRef CAS.
- J. He, X. Wang, S. Lan, H. Tao, X. Luo, Y. Zhou and M. Zhu, Appl. Catal., B, 2022, 317, 121747 CrossRef CAS.
- G. Yang, S. Wang, Y. Wu, H. Zhou, W. Zhao, S. Zhong, L. Liu and S. Bai, ACS Appl. Mater. Interfaces, 2023, 11, 14228–14239 Search PubMed.
- Q. Xu, J. Jiang, X. Sheng, Q. Jing, X. Wang, L. Duan and H. Guo, Inorg. Chem. Front., 2023, 10, 2939–2950 RSC.
- L. Liu, H. Huang, Z. Chen, H. Yu, K. Wang, J. Huang, H. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 18303–18308 CrossRef CAS PubMed.
- S. Tu, Y. Zhang, A. H. Reshak, S. Auluck, L. Ye, X. Han, T. Ma and H. Huang, Nano Energy, 2019, 56, 840–850 CrossRef CAS.
- H. Yu, H. Huang, A. H. Reshak, S. Auluck, L. Liu, T. Ma and Y. Zhang, Appl. Catal., B, 2021, 284, 119709 CrossRef CAS.
- S. Li, F. Chen, S. Chu, Z. Zhang, J. Huang, S. Wang, Y. Feng, C. Wang and H. Huang, Small, 2022, 19, 2203559 CrossRef PubMed.
- H. Liu, P. Chen, X. Yuan, Y. Zhang, H. Huang and F. Dong, Chin. J. Catal., 2019, 40, 620–630 CrossRef CAS.
- J. Duan, T. Liu, Y. Zhao, R. Yang, Y. Zhao, W. Wang, Y. Liu, H. Li, Y. Li and T. Zhai, Nat. Commun., 2022, 13, 2039 CrossRef CAS PubMed.
- S. Peng, X. Han, L. Li, S. Chou, D. Ji, H. Huang, Y. Du, J. Liu and S. Ramakrishna, Adv. Energy Mater., 2018, 8, 1800612 CrossRef.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- Y. Huang, Y. Yu, Y. Yu and B. Zhang, Sol. RRL, 2020, 4, 2000037 CrossRef CAS.
- R. Huang, J. Wu, E. Lin, Z. Kang, N. Qin and D. Bao, Nanoscale Adv., 2021, 3, 3159–3166 RSC.
- G. Ren, S. Liu, Z. Li, H. Bai, X. Hu and X. Meng, Sol. RRL, 2022, 6, 2200154 CrossRef CAS.
- L. Ye, L. Zan, L. Tian, T. Peng and J. Zhang, Chem. Commun., 2011, 47, 6951–6953 RSC.
- Y. Shi, Z. Yang, L. Shi, H. Li, X. Liu, X. Zhang, J. Cheng, C. Liang, S. Cao and F. Guo, Environ. Sci. Technol., 2022, 56, 14478–14486 CrossRef CAS PubMed.
- E. Eberg, A. T. J. van Helvoort, R. Takahashi, M. Gass, B. Mendis, A. Bleloch, R. Holmestad and T. Tybell, J. Phys. Chem. A, 2011, 109, 322–327 Search PubMed.
- A. Chassé, S. Borek, K. M. Schindler, M. Trautmann, M. Huth, F. Steudel, L. Makhova, J. Gräfe and R. Denecke, Phys. Rev. B, 2011, 84, 195135 CrossRef.
- Y. Shi, H. Shou, H. Li, G. Zhan, X. Liu, Z. Yang, C. Mao, J. Cheng, X. Zhang and Y. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202302286 CrossRef CAS PubMed.
- D. Yu, Z. Liu, J. Zhang, S. Li, Z. Zhao, L. Zhu, W. Liu, Y. Lin, H. Liu and Z. Zhang, Nano Energy, 2019, 58, 695–705 CrossRef CAS.
- X. H. Jiang, L. S. Zhang, H. Y. Liu, D. S. Wu, F. Y. Wu, L. Tian, L. L. Liu, J. P. Zou, S. L. Luo and B. B. Chen, Angew. Chem., Int. Ed., 2020, 59, 23112–23116 CrossRef CAS PubMed.
- H. Wang, D. Yong, S. Chen, S. Jiang, X. Zhang, W. Shao, Q. Zhang, W. Yan, B. Pan and Y. Xie, J. Am. Chem. Soc., 2018, 140, 1760–1766 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- M. A. Khan, M. A. Nadeem and H. Idriss, Surf. Sci. Rep., 2016, 71, 1–31 CrossRef CAS.
- Z. Yang, H. Zhang, J. Zhao, H. Shi, Y. Liu, H. Yang and P. Yang, ChemSusChem, 2022, 15, e202200260 CrossRef CAS PubMed.
- J. Xiang, T. Zhang, R. Cao, M. Lin, B. Yang, Y. Wen, Z. Zhuang and Y. Yu, Sol. RRL, 2021, 5, 2100703 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full and core level Ti 2p XPS spectra of the catalysts, SEM, BET and UV-vis of different morphologies of BNTs, schematic of the formation mechanisms for BNT nanoparticles and nanobelts, Mott–Schottky plots of the catalysts at frequencies of 500 Hz, 1000 Hz and 1500 Hz of BNTs. See DOI: https://doi.org/10.1039/d4ta01030b |
| This journal is © The Royal Society of Chemistry 2024 |