DOI:
10.1039/D3TA07853A
(Paper)
J. Mater. Chem. A, 2024,
12, 8487-8501
Constructing a multivalent Co-confined N-doped C–Si hybrid hollow nanoreactor for synchronous pollutant mineralization and solar-driven interfacial water regeneration†
Received
19th December 2023
, Accepted 21st February 2024
First published on 26th February 2024
Abstract
In the pursuit of synchronous contaminant purification and freshwater reclamation using multifunctional nanomaterials, a broad prospect emerges. Through a tactic process involving the successive modification of ZIF67 by mesoporous silica and cobalt-incorporating polydopamine coating, followed by graphitization, the confinement of multivalent cobalt nanocrystal species within the interconnected carbon-silica hybridized porous shell has been achieved, establishing it as a multifunctional nanoreactor with abundant and well-dispersed catalytic sites. Additionally, the hybridization of the silica framework with the carbon network, facilitated by the binding of N species, has generated polar sites enhancing the capture of pollutant molecules, thereby extending the lifespan of reactive oxidative species by reducing their migration. This catalyst presents an array of metallic redox pairs, significantly enhancing electron transfer capacity and bolstering peroxymonosulfate activation. The optimized Co-SiCNO-6 catalyst exhibits the capability to degrade norfloxacin within 5 min, showcasing a reaction rate constant of about 0.917 min−1, a remarkable 5.3-fold enhancement compared to that of its ZIF67 graphitization-derived counterpart. Further studies encompassing reaction conditions, anion interference, and the catalyst's flexibility in degrading contaminants were conducted to assess its practical potential. Concurrently, exploration into the direct recovery of clean water through solar-driven interfacial water evaporation utilizing the optimized catalyst yielded an evaporation rate of 1.50 kg m−2 h−1 and an impressive 93.1% evaporation efficiency. Notably, distinctive outcomes emerged when dealing with polluted water containing norfloxacin and carbamazepine. The norfloxacin solution achieved a near 1.88 kg m−2 h−1 evaporation rate with 95% efficiency, outperforming the treatment of carbamazepine solution. The obtained Co-SiCNO-6 was further anchored onto a tailored melamine sponge by the Ca ion-triggered sodium alginate crosslinking tactic as an integrated monolith evaporator, which exhibits excellent evaporation performance (2.0 kg m−2 h−1) and rational norfloxacin degradation efficiency (30 min). This innovative hybrid hollow nanoreactor exhibits potent dual functionality in degrading contaminants and facilitating solar-driven water regeneration from polluted wastewater.
 Fu Yang | Dr Fu Yang received his PhD degree from Nanjing Tech University in industrial catalysis in 2018. He was also jointly trained at the National University of Singapore in 2016–2017. After finishing his PhD, he joined Jiangsu University of Science and Technology and was promoted to associate professor in 2020. His research focuses on the development of functional low-dimensional heterogeneous nanomaterials for the synthesis of fine chemicals, electrocatalytic synthesis of ammonia, and coupled environmental comprehensive treatment such as gas adsorption, advanced oxidation processes and photothermal water evaporation. He has published more than 100 papers with over 1500 total citations. |
1. Introduction
Swift urbanization and industrial growth have triggered a significant surge in the volume of wastewater, laden with intricate chemical compositions.1 Industrial effluents stand as the primary source of water contamination, with the organic pollutants they carry posing a severe threat to human health.2 Of particular concern are antibiotics, which, even at low concentrations, foster bacterial resistance within this environment, thereby promoting the selection of more virulent bacteria.3 Norfloxacin (NFX), oxytetracycline, and tetracycline, among others, have undergone extensive scrutiny in this regard. This pollution involving complicated contaminants has caused serious threat to human health and sustainable development.
To address this, the advanced oxidation process (AOP) has emerged, leveraging PMS activation to target organic molecules within a multiphase catalyst, establishing itself as a potent approach for treating organic wastewater.4,5 In the course of AOP treatment, a substantial quantity of reactive oxygen species (ROS) like sulphate radicals (SO4˙−), hydroxyl radicals (˙OH), superoxide ions (O2˙−), and singlet oxygen (1O2) is generated, effectively eliminating recalcitrant pollutants.6,7 The focal point of the AOP lies in the development of functional materials to drive this process. Cobalt-based catalysts have gained widespread recognition for their efficacy as heterogeneous catalysts in activating PMS within AOP investigations.8–10 While cobalt oxide (CoO) demonstrates the ability to activate permonosulfate (PMS) for contaminant removal, its catalytic performance is hampered by a limited number of active sites and the potential risk of cobalt ion leaching, limiting its efficiency.11 Carbon-based materials, owing to their abundance, non-toxicity, biocompatibility, and excellent thermal and chemical stability, have garnered significant interest.12 Recent carbonaceous materials like graphene, carbon nanotubes, and activated carbon fibers have become preferred catalyst carriers due to their substantial specific surface area, abundant surface functional groups, and impressive mechanical properties.13 Ordinarily, pristine carbon exhibits low to moderate catalytic activity. Enhancing it often involves modifying it with transition metals to introduce more favorable active sites. In homogeneous reactions, transition metal ions such as Co2+, Fe2+, Mn2+, and Ni2+ activate PMS, with the best performance seen in Co2+/PMS combinations.14 Incorporating cobalt into carbon carriers effectively mitigates cobalt ion leaching.15 In turn, cobalt nanoparticles further amplify the catalytic prowess of carbon materials in PMS activation. Thus, combining carbon-based materials with cobalt-based nanoparticles can synergistically harness the strengths of both materials, significantly boosting the oxidation effect produced by activating PMS.16 Investigating the synergies generated through ultrafine metal oxides within carbon frameworks by precisely regulating the size of carbon is a critical pursuit.17 Mesoporous silicon dioxide emerges as a promising catalyst carrier owing to its exceptional chemical stability. For instance, incorporating cobalt-iron spinel nanoparticles into porous silica carriers results in superior dispersion of metal particles, amplification of the specific surface area, and optimization of catalyst utilization.18 The surface properties of silica offer easy modification by diverse groups, and specific nanostructured silica materials demonstrate remarkable effectiveness in modulating the active state of metal species.19 However, the combination of nanocarbon mesoporous silica remains infrequent when addressing the activity optimization of metal species. In particular, the incorporation of carbon materials into mesoporous silica can cause additional photothermal properties, which are capable of extending the functionality of materials in the photothermal conversion application.
In addition to breaking down pollutants, a fascinating avenue involves regenerating freshwater directly from sewage. Typical desalination technologies, such as reverse osmosis, distillation, capacitive deionization, and battery desalination, consume large amounts of electrical or thermal energy.20–22 As an emerging desalination technology, solar-driven photoelectrochemical desalination using sustainable solar energy to remove salt ions without any external bias has gained much attention.23 Emerging interfacial photo-thermal water evaporation technologies possess the capability to confine photothermal heat at the evaporating surface, prompting evaporation of water molecules from the photothermal material to generate fresh water.24 Solar-powered desalination technologies, including solar steam generation and solar-powered membrane distillation, provide a sustainable pathway for generating clean water from wastewater.25,26 Nevertheless, the efficiency of converting natural light into water vapor remains insufficient to create a practical freshwater supply due to water's poor optical absorption and significant heat losses.27 Thus, developing a photothermal material capable of absorbing a broad spectrum of sunlight and exhibiting high photothermal conversion efficiency becomes imperative. In addition, when addressing the contaminant elimination and regeneration of freshwater by multifunctional materials, the coupled technique for synchronous pollutant mineralization and solar-driven interfacial water regeneration is rarely mentioned. During interfacial solar evaporation, sufficient light, high air–water interface temperature, and carbon-based and transition-metal photothermal materials provide the necessary conditions for the activation of PMS. Therefore, the introduction and activation of PMS in an interfacial solar evaporation system should be able to achieve both efficient solar evaporation and catalytic degradation of volatile organic compounds.28 This makes the project attractive for practical wastewater treatment when using complicated wastewater systems containing organic pollutants.
Inspired by these concepts, we modified the surface of dodecahedral ZIF67 with silica.29 Under alkaline conditions, ZIF67 dissolves and merges with SiO2, resulting in a spherical shell. Applying a dopamine coating and employing heat treatment help confine cobalt species within silica nanolayers, forming hollow photothermal nanospheres. Metal nanospecies tend to aggregate, prompting the addition of a carbon layer to ensure the uniform dispersion of metal species. However, as carbon inherently repels water, a more polar SiO2 shell acts as a support within the material.30 This, combined with the carbon layer, enhances affinity across the entire surface of the catalyst, facilitating the creation of active sites for contaminants and fostering porousness for contaminants to react within the pores. Moreover, the SiO2 shell preserves the stability of the carbon coating and ultrafine metal oxide, resulting in localized photothermal effects at the interface through interaction between the carbon coating and metal. Simultaneously, the unique structure and channel confinement effect stabilize the formed metal or metal oxide particles, rendering the material highly active. Functional metal-constrained carbon layers serve as exceptional photothermal materials for interfacial solar-thermal water evaporation, showcasing excellent photothermal conversion efficiency. This structural design ensures both efficient degradation performance and commendable photothermal properties for the catalysts. Various characterization tests were conducted to determine the physicochemical properties of the catalysts. Their degradation performance on NFX and other pollutants underwent systematic investigation. Additionally, the impact of different conditions on degradation performance was studied. The choice of a melamine sponge as a support to immobilize the catalyst is justified by its open and abundant network structure, which can effectively accommodate a greater quantity of catalysts. Additionally, the surface hydrophilicity of the melamine sponge facilitates the easy coating of alginate acid onto the skeleton, ensuring proper incorporation of the catalyst. Therefore, simulated solar water evaporation experiments for treating contaminant containing wastewater by using the obtained functional materials and integrated monolith evaporator by anchoring the nanoreactor onto the melamine sponge were conducted to gain deeper insights into the photothermal conversion performance of the catalysts, water purification, and recycling abilities.
2. Experimental section
2.1 Chemicals and reagents
tert-Butanol (99.5%, TBA), L-histidine (99%, L-his), p-benzoquinone (98%, PBQ), ethylsilicate (AR, TEOS), dopamine hydrochloride (98%, C8H12ClNO2), cobalt nitrate hexahydrate (98%, Co(NO3)2·6H2O), 2-methylimidazole (99.9%, C4H6N2), potassium peroxymonosulfate complex salt (PMS), methanol (99.9%, MeOH), p-benzoquinone (98%, p-BQ) and absolute ethanol were obtained from Energy Chemicals Co., Ltd. Cobalt acetate tetrahydrate (99.5%, Co(CH3COO)2·4H2O), monosodium phosphate (99.5%, NaH2PO4) and hexadecyl trimethyl ammonium bromide (99%, C19H42BrN) were acquired from Sinopharm Chemical Reagent Co., Ltd. Oxytetracycline (98%, OTC), tetracycline (CP, TC), carbamazepine (99%, CBZ) and norfloxacin (CP, NFX) were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Ammonium hydroxide (AR, NH3 H2O) was purchased from Chinasun Specialty Products Co., Ltd. Sodium chloride (99.7%, NaCl), sodium carbonate (AR, Na2CO3), sodium bicarbonate (99.5%, NaHCO3), sodium sulphate (99.5%, Na2SO4), sodium hydroxide (AR, NaOH), and sodium alginate (AR, (C6H7O6Na)n, SA) were obtained from Shanghai Aladdin Industrial Co., LTD. All the chemicals were used without further purification. Polyvinylidene fluoride (PVDF) filtration membranes were purchased from a filter specialist shop.
2.2. Material synthesis
2.2.1 Synthesis of ZIF67.
The zeolitic imidazolate framework ZIF-67 was synthesized according to the following method.31 Specifically, 0.668 g of cobalt nitrate and 2.242 g of 2-methylimidazole were combined in 100 mL of methanol and stirred mechanically for 30 min. This mixture was left to stand for 24 h. Afterward, the resulting purple suspension underwent centrifugation and methanol washing. The sample obtained was then dried at 70 °C in an oven and denoted as ZIF67.
2.2.2 Synthesis of the catalysts.
The acquired ZIF67 underwent successive modifications involving mesoporous silica and a strategy incorporating metal-doped polydopamine. Specifically, the process involved dissolving 1.44 g of hexadecyl trimethyl ammonium bromide (CTAB) in 45 mL of deionized water. This solution was slowly added to a mixture consisting of 0.36 g of ZIF67, 90 mL of anhydrous ethanol, and 225 mL of deionized water. Subsequently, 1.26 mL of TEOS and 3.6 mL of ammonia hydroxide were slowly added to the mixture, followed by mechanical stirring for 6 h. Afterward, 0.071 g of cobalt acetate and 0.15 g of dopamine hydrochloride were introduced and mechanically stirred for 12 h. The resulting mixture underwent washing with ethanol and water. The precipitate obtained post-centrifugation was dried at 70 °C and then treated at different temperatures (500, 600, and 700 °C) in a tube furnace at a rate of 5 °C min−1, followed by a 2 h calcination under a N2 atmosphere. The final samples were denoted as Co-SiCNO-x (where x represents the temperature: 500/600/700).
In addition to these experiments, several controlled comparative samples were prepared. In one instance, ZIF67 was directly carbonized at 600 °C in a tube furnace at a rate of 5 °C min−1 and then calcined for 2 h under a N2 atmosphere, labeled as Co-NO-6. ZIF67 coated with mesoporous silica for further treatment by the above procedure was labeled as Co-SiCNO-6. Another sample involved coating ZIF67 with cobalt-incorporating polydopamine as per a similar procedure, followed by carbonization at 600 °C in a tube furnace at a rate of 5 °C min−1 and calcination for 2 h under a N2 atmosphere, named Co-CNO-6.
2.3 Characterization
The morphology and structure of the sample were examined using field emission scanning electron microscopy (SEM, Zeiss, Germany) and high-resolution transmission electron microscopy (HR-TEM, JEOL, 2100 F, Japan). The chemical states of the surface elements were identified using X-ray photoelectron spectroscopy (XPS, ULVAC-PHI, Kanagawa, Japan). Various catalyst structures were analyzed using a Kα-ray diffractometer (XRD-6000X, Shimadzu Corporation, Tokyo, Japan) (λ = 1.54178A). Raman spectra were obtained at room temperature with a Spex 1877 D triple companion spectrometer. The catalyst surface area and pore volume were calculated using the Brunauer–Emmett–Teller (BET) method, and the pore size was determined using the Barrett–Joyner–Halenda (BJH) model. Excitation-emission-matrix spectroscopy (EEM) was performed using an F-4700 FL spectrophotometer to assess the degree of change in contaminants during degradation. Linear scanning voltammetry (LSV) and electrochemical impedance spectroscopy (EIS) were carried out on a CHI760E electrochemical workstation using a standard three-electrode method, where a platinum electrode and Ag/AgCl were used as the counter electrode and reference electrode, respectively. Photothermal testing was conducted using a solar simulator system (Solar-500T, Beijing NBeT, China) and an optical power meter (FZ400, Beijing NBeT, China). The determination of metal content in catalysts was performed using inductively coupled plasma atomic emission spectroscopy (ICP-AES).
2.4 Catalytic performance assessment
To systematically evaluate the efficacy of various catalysts in degrading contaminants, experiments were conducted to catalytically degrade NFX (initial concentration of 20 mg L−1) using PMS as an oxidizing agent. Real-time measurements of NFX concentration during the reaction were performed using a UV spectrophotometer (MAPADA P1800). The procedure involved adding a specific amount of PMS to the NFX solution (0.2 g L−1) and stirring. Subsequently, a certain quantity of catalyst was introduced to achieve a concentration of 0.2 g L−1. Samples were extracted at regular intervals from the reaction solution, and the NFX concentration was analyzed using a spectrophotometer after immediate filtration to remove the catalyst. Additionally, a control test using only the catalyst was carried out. Experimental parameters such as catalyst amount, PMS quantity, and initial NFX concentration were simultaneously optimized. To comprehend the activation mechanism of PMS by the catalyst in NFX degradation, free radical quenching experiments were conducted using various agents known for quenching free radicals. Based on parameter optimization experiments, quenchers like methanol and tert-butanol were utilized to inhibit the catalytic reaction. The concentration of NFX was analyzed using a UV spectrophotometer, and variations in reaction rates were monitored through degradation progress curves.
2.5 Photothermal water evaporation test
A photothermal test was initiated by subjecting 28.8 mg of the Co-SiCNO-6 catalyst to sonication alongside deionized water. The resultant mixture was filtered using a polyvinylidene difluoride (PVDF) filter membrane to create a photothermal test film. This film, placed alongside a beaker containing 50 mL of deionized water, was exposed to solar light irradiation. Surface temperature measurements of the sample were taken every 60 s using an infrared camera, while the mass change of the evaporated water was monitored every 100 s using an electronic balance.32 Data processing provided the photothermal conversion efficiency of the catalyst and the rate of water evaporation. Additionally, photothermal and water evaporation tests were conducted on two different aqueous solutions: a 20 mg L−1 norfloxacin solution and a 20 mg L−1 carbamazepine solution. A photothermal membrane was applied to these contaminant solutions under solar irradiation, and the mass change of the solutions was recorded. By comparing the water evaporation rate of the contaminants with that of deionized water, insights into the photothermal performance and water regeneration capacity of the catalysts in contaminant solutions were obtained.
2.6 Coupled catalytic degradation and photothermal water evaporation
To evaluate the practical relevance of the catalyst in the regeneration of fresh water and contaminant purification, an integrated floatable device for synchronous catalytic contaminant removal and water evaporation was fabricated by integration of the above obtained optimized catalyst. Typically, 30 mg of the catalyst was dispersed in an SA solution; afterwards, a tailored cylindrical sponge with 3 cm diameter was soaked in the above mixed solution and then transferred into CaCl2 solution to trigger cross-linking and anchor these introduced catalysts. This process resulted in the efficient integration of the catalyst onto the tailored circular sponge which leads to an integrated floatable evaporator for synchronous pollutant degradation and water evaporation tests. Under the established photothermal experimental conditions, the performance of the device for evaporation of water at a specific rate under sunlight was further measured. Furthermore, the device was floated in a solution containing the contaminant, and its degradation efficiency was assessed using UV spectrophotometry for the degradation of a 20 mg L−1 norfloxacin solution. The device was designed to be recyclable, signifying its considerable practical significance.
3. Results and discussion
Fig. 1a illustrates the detailed synthesis process of the targeted nanocomposite, utilizing polyhedral ZIF67 as the initial template. This was followed by coating with mesoporous silica and the successive in situ polymerization of dopamine incorporated cobalt metal cations. The resulting composite underwent further thermal treatment in an inert N2 atmosphere to yield the Co–Si/C hybrid hollow nanoreactor. Fig. 1b displays a representative SEM image of Co-SiCNO-6, showcasing an irregular nanosphere morphology characterized by a hollow configuration composed of numerous fine nanoparticles. In Fig. 1c and d, representative TEM images of Co-SiCNO-6 reveal a hollow nanospherical shape with a size distribution primarily ranging from 100 to 170 nm, with most particles clustered at around 130 nm. Additionally, the shell of the hollow nanoreactor is observed to be composed of abundant mesoporous channels without noticeable aggregated metal species, signifying high dispersity and fine size of the metal species within the mesochannel. This structure facilitates efficient diffusion and contact of molecules during the reaction process. For further characterization of the microscale structure of the nanoreactor and existing state of metal species, high-resolution transmission electron microscopy (HRTEM) was employed. As depicted in Fig. 1e and f, the lattice spacing of the observed nanocrystals measured in the selected region correspond to the (111) and (200) crystal planes of the typical CoO phase and the (111) crystal plane of metallic Co, indicating the co-existence of multiphase Co species confined in the silica-carbon composites. To determine the compositional configuration, elemental mapping analysis of several hollow nanospheres was conducted (Fig. 1g).33 The analysis revealed a uniform distribution of elements including C, Co, N, O, and Si within the hollow shell configuration. Notably, densely distributed cobalt species are evenly dispersed in the mesoporous shell without significant aggregation, confirming the homogeneous hybrid state of metal species, silica, and carbon.
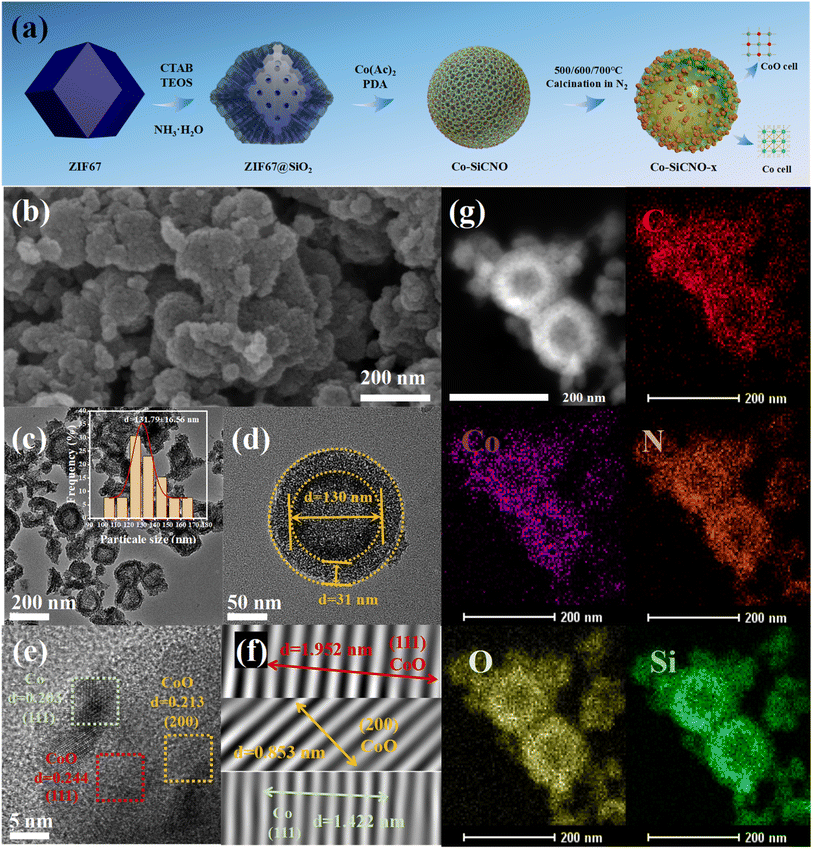 |
| | Fig. 1 (a) Schematic representation of the synthesis of Co-SiCNO-x. Representative SEM image (b), TEM image (c and d), HRTEM image (e), inverse FFT pattern of selected regions (f), HAADF-STEM image (g) and corresponding elemental mapping images of Co-SiCNO-6. | |
Fig. S1† shows the N2 adsorption/desorption isotherms and the corresponding pore size distributions of different catalysts. The adsorption/desorption isotherms of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6, and Co-NO-6 catalysts conform to type I and type IV hysteresis curves (Fig. S1a†), indicating the presence of combined micropores and mesopores in the catalysts. Table S1† summarizes microstructural properties such as specific surface area calculated by the Brunauer–Emmett–Teller (BET) method and pore size distribution. As shown in Fig. S1b, c and Table S1,† the BET specific surface areas of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6 and Co-NO-6 were 392.4 m2 g−1, 546.0 m2 g−1, 218.3 m2 g−1 and 201.4 m2 g−1, respectively. The specific surface area of Co-SiCNO-6 was obviously higher than that of Co-CNO-6 and Co-NO-6, indicating that the introduction of mesoporous silica increases the nanochannels of the material and improves the exposure and accessibility of active species for contaminants. However, the specific surface of Co-SiCNO-6 is lower than that of Co-SiNO-6 possibly because of the introduction of cobalt-containing dopamine on mesoporous silica affecting the structural properties of the material.
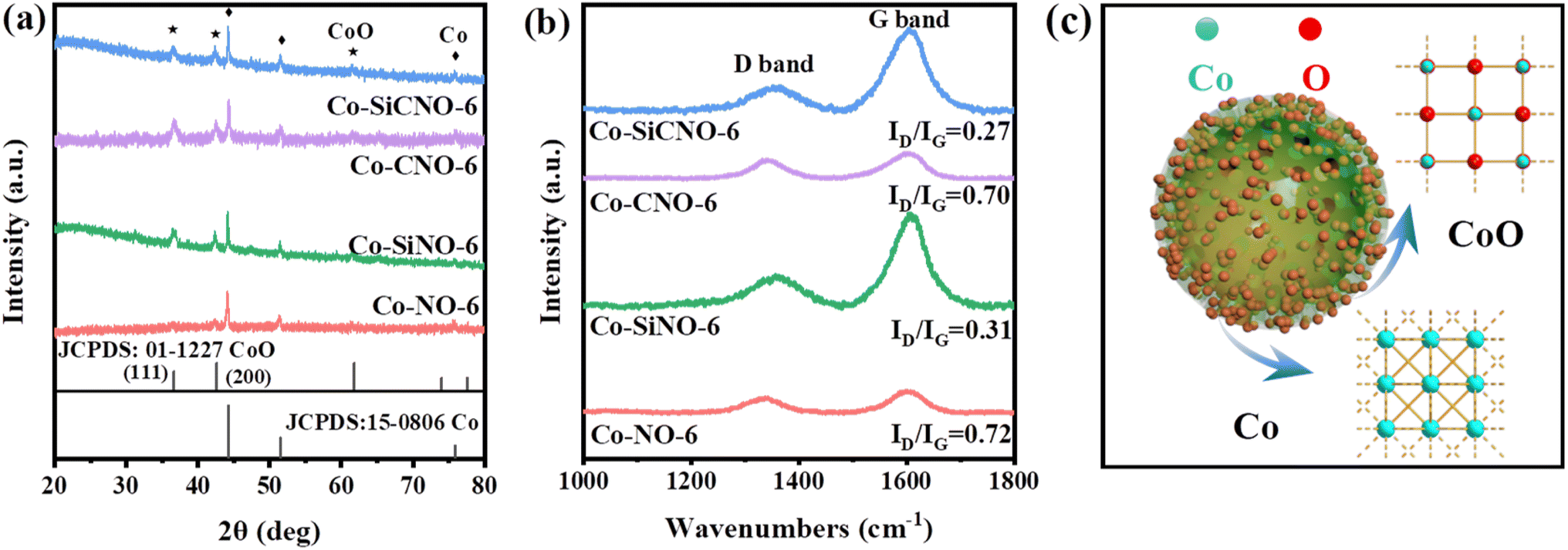
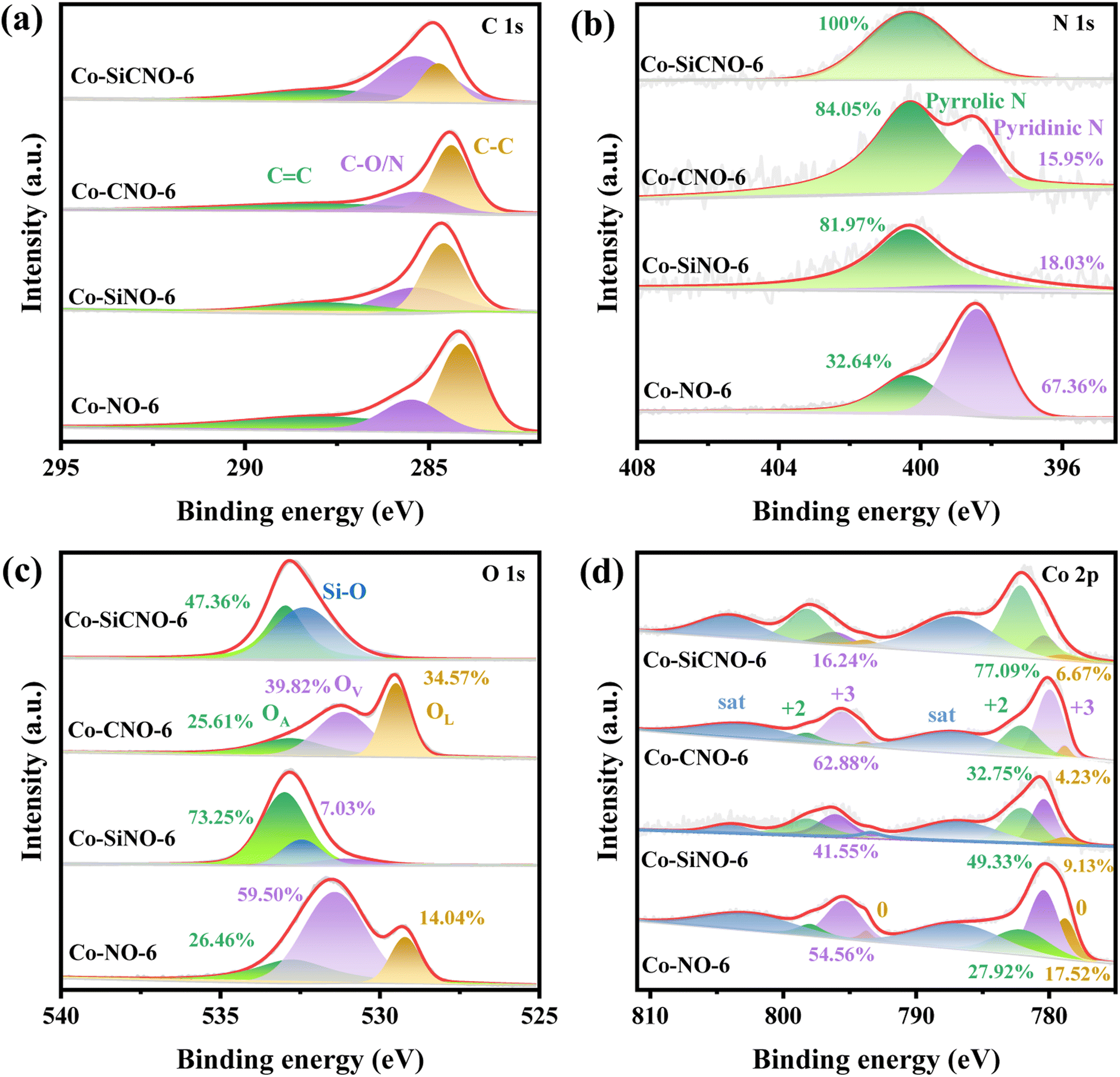
Fig. 2a depicts the wide-angle XRD patterns for the optimal sample Co-SiCNO-6, alongside comparison samples Co-CNO-6, Co-SiNO-6, and Co-NO-6. All samples exhibit robust diffraction peaks at 36.6° and 42.6°, corresponding to the (111) and (200) crystal planes of cubic CoO, aligning with the standard card (JCPDS: 01-1227). Additional peaks at 44.2° and 51.5° indicate metallic Co species (JCPDS: 15-0806), possibly resulting from the reduction of CoO to metallic Co at higher temperatures in an N2 atmosphere. Notably, Co-NO-6, derived directly from ZIF67, primarily comprises metallic Co(0). In contrast, Co-CNO-6, derived from PDA-modified ZIF67, exhibits multiple cobalt species diffractions, implying that more carbon deposition on ZIF67 reduces oxidative metal species to a metallic state. Increasing cobalt incorporation onto the PDA layer yields various cobalt species in the final nanoreactor. Fig. S2† shows XRD spectral results of comparative catalysts at 500 and 700 °C calcination temperatures. At 500 °C, Co-SiCNO-5 displays insignificant diffraction peaks due to low crystallinity and high dispersity of the introduced metal species on the carbon-SiO2 hybrid shell. Conversely, Co-SiCNO-7 exhibits diffraction peaks related to Co3O4 and Co2SiO4 at 700 °C, indicating that high temperatures lead to cobalt species reacting with silica, forming Co2SiO4. Fig. 2b illustrates the Raman spectral outcomes of comparison samples (Co-SiCNO-6, Co-CNO-6, Co-SiNO-6, and Co-NO-6). Raman peaks at around 1360 cm−1 and 1600 cm−1 correspond to the D and G bands of the carbon network, respectively. Generally, D peaks signify lattice defects in carbon atoms, while G peaks represent in-plane stretching vibrations of sp2 hybridized C atoms. The relative intensity ratios of the D and G bands (ID/IG) elucidate atomic crystal defects or the degree of graphitization in carbon materials.34 The lower ID/IG in Co-SiCNO-6 and Co-SiNO-6 compared to the other catalysts likely stem from increased graphitized carbon layers due to SiO2 introduction, enhancing carbon graphitization. To gain deeper insights into the surface elemental composition and states, Fig. S3† presents comprehensive XPS survey results for comparison samples: Co-SiCNO-6, Co-CNO-6, Co-SiNO-6, and Co-NO-6, outlining the predominant elemental compositions of C, O, N, and Co. In Fig. 3a, the C 1s core level spectra across different samples reveal distinct peaks corresponding to three types of carbon assignments: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (287.8 eV), C–O/N (285.4 eV), and C–C (284.6 eV), indicating the presence of heteroatoms in the carbon framework. Fig. 3b shows N 1s core level peaks at 398.4 eV and 400.3 eV, representing pyridine N and pyrrole N, respectively, confirming the successful nitrogen species doping within the carbon network. Interestingly, the SiO2-doped catalyst predominantly exhibits pyrrole N species, while the direct calcination of ZIF67 results in an increased presence of pyridine N, suggesting that PDA modification facilitates the formation of pyrrole N species, potentially contributing to more adsorptive sites for polar contaminant molecules. Moving to Fig. 3c, the O 1s spectra exhibit three peaks corresponding to lattice oxygen OL (529.5 eV), oxygen vacancy OV (531.1 eV), and adsorbed oxygen OA (532.8 eV). Specifically, Co-SiCNO-6 and Co-SiNO-6 show Si–O and adsorbed oxygen, while Co-CNO-6 and Co-NO-6 exhibit CoOx-related lattice oxygen and oxygen vacancy species, indicating the increased presence of Co species within the mesoporous silica shell due to silica introduction. Fig. 3d displays Co 2p core level XPS spectra for different catalysts, revealing Co(II) and Co(III) in the Co 2p1/2 and Co 2p3/2 core energy levels, consistent with typical crystalline species. Notably, characteristic peaks of zero-valent cobalt appear, alongside two satellite peaks of Co 2p, indicating the presence of multivalent cobalt oxide species. Co-SiCNO-6 notably shows a significantly higher Co(II) content (77.09%) compared to Co-CNO-6 (32.75%), Co-SiNO-6 (49.33%), and Co-NO-6 (27.92%). This higher Co(II) content makes Co-SiCNO-6 advantageous for PMS activation, as Co(II) often serves as the primary active center, potentially initiating more ROS for efficient NFX degradation.35 In Fig. 3c, three distinct peaks in the O 1s spectra correspond to lattice oxygen OL (529.5 eV), oxygen vacancy OV (531.1 eV), and adsorbed oxygen OA (532.8 eV).36 Notably, the O 1s spectra of Co-SiCNO-6 and Co-SiNO-6 reveal adsorbed oxygen and Si–O assignments at 533.0 eV and 532.4 eV, respectively. Conversely, Co-CNO-6 and Co-NO-6 showcase lattice oxygen and oxygen vacancy species related to CoOx, suggesting that the introduction of silica leads to an increased presence of Co species within the internal structure of the mesoporous silica shell. Moving to Fig. 3d, the XPS spectra of the Co 2p core level for different catalysts indicate the presence of Co(II) and Co(III) in the Co 2p1/2 and Co 2p3/2 core energy levels, consistent with the elemental composition of typical crystalline species. Specifically, the deconvoluted binding energy at 780.4 eV and 795.5 eV corresponds to the Co 2p3/2 and Co 2p1/2 of Co(II), respectively, while the energy level peaks at 782.1 eV and 798.2 eV align with the Co 2p3/2 and Co 2p1/2 of Co(III). Notably, two distinct peaks of zero-valent cobalt emerge at 778.8 eV and 793.8 eV, along with two satellite peaks of Co 2p, indicating the presence of multivalent cobalt oxide species. Significantly, the calculated relative Co(II) content of Co-SiCNO-6 (77.09%) surpasses that of other catalysts-Co-CNO-6 (32.75%), Co-SiNO-6 (49.33%), and Co-NO-6 (27.92%). This higher Co(II) content makes Co-SiCNO-6 uniquely advantageous for PMS activation, as Co(II) often serves as the primary active center, potentially initiating more ROS for efficient degradation of NFX. In addition, the cobalt contents of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6 and Co-NO-6 were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) to be 5.69%, 9.73%, 67.45% and 21.74%, respectively (Table S2†). Table S2† also reveals that the cobalt leaching of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6 and Co-NO-6 reaches 5.5%, 5.64%, 17.97% and 14.65%, respectively. This result indicates that the introduction of a silica framework in the carbon network could efficiently reduce the leaching of Co during the reaction, which further highlights the advantage of silica for stabilizing the metal species confined in carbon.
C (287.8 eV), C–O/N (285.4 eV), and C–C (284.6 eV), indicating the presence of heteroatoms in the carbon framework. Fig. 3b shows N 1s core level peaks at 398.4 eV and 400.3 eV, representing pyridine N and pyrrole N, respectively, confirming the successful nitrogen species doping within the carbon network. Interestingly, the SiO2-doped catalyst predominantly exhibits pyrrole N species, while the direct calcination of ZIF67 results in an increased presence of pyridine N, suggesting that PDA modification facilitates the formation of pyrrole N species, potentially contributing to more adsorptive sites for polar contaminant molecules. Moving to Fig. 3c, the O 1s spectra exhibit three peaks corresponding to lattice oxygen OL (529.5 eV), oxygen vacancy OV (531.1 eV), and adsorbed oxygen OA (532.8 eV). Specifically, Co-SiCNO-6 and Co-SiNO-6 show Si–O and adsorbed oxygen, while Co-CNO-6 and Co-NO-6 exhibit CoOx-related lattice oxygen and oxygen vacancy species, indicating the increased presence of Co species within the mesoporous silica shell due to silica introduction. Fig. 3d displays Co 2p core level XPS spectra for different catalysts, revealing Co(II) and Co(III) in the Co 2p1/2 and Co 2p3/2 core energy levels, consistent with typical crystalline species. Notably, characteristic peaks of zero-valent cobalt appear, alongside two satellite peaks of Co 2p, indicating the presence of multivalent cobalt oxide species. Co-SiCNO-6 notably shows a significantly higher Co(II) content (77.09%) compared to Co-CNO-6 (32.75%), Co-SiNO-6 (49.33%), and Co-NO-6 (27.92%). This higher Co(II) content makes Co-SiCNO-6 advantageous for PMS activation, as Co(II) often serves as the primary active center, potentially initiating more ROS for efficient NFX degradation.35 In Fig. 3c, three distinct peaks in the O 1s spectra correspond to lattice oxygen OL (529.5 eV), oxygen vacancy OV (531.1 eV), and adsorbed oxygen OA (532.8 eV).36 Notably, the O 1s spectra of Co-SiCNO-6 and Co-SiNO-6 reveal adsorbed oxygen and Si–O assignments at 533.0 eV and 532.4 eV, respectively. Conversely, Co-CNO-6 and Co-NO-6 showcase lattice oxygen and oxygen vacancy species related to CoOx, suggesting that the introduction of silica leads to an increased presence of Co species within the internal structure of the mesoporous silica shell. Moving to Fig. 3d, the XPS spectra of the Co 2p core level for different catalysts indicate the presence of Co(II) and Co(III) in the Co 2p1/2 and Co 2p3/2 core energy levels, consistent with the elemental composition of typical crystalline species. Specifically, the deconvoluted binding energy at 780.4 eV and 795.5 eV corresponds to the Co 2p3/2 and Co 2p1/2 of Co(II), respectively, while the energy level peaks at 782.1 eV and 798.2 eV align with the Co 2p3/2 and Co 2p1/2 of Co(III). Notably, two distinct peaks of zero-valent cobalt emerge at 778.8 eV and 793.8 eV, along with two satellite peaks of Co 2p, indicating the presence of multivalent cobalt oxide species. Significantly, the calculated relative Co(II) content of Co-SiCNO-6 (77.09%) surpasses that of other catalysts-Co-CNO-6 (32.75%), Co-SiNO-6 (49.33%), and Co-NO-6 (27.92%). This higher Co(II) content makes Co-SiCNO-6 uniquely advantageous for PMS activation, as Co(II) often serves as the primary active center, potentially initiating more ROS for efficient degradation of NFX. In addition, the cobalt contents of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6 and Co-NO-6 were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) to be 5.69%, 9.73%, 67.45% and 21.74%, respectively (Table S2†). Table S2† also reveals that the cobalt leaching of Co-SiCNO-6, Co-SiNO-6, Co-CNO-6 and Co-NO-6 reaches 5.5%, 5.64%, 17.97% and 14.65%, respectively. This result indicates that the introduction of a silica framework in the carbon network could efficiently reduce the leaching of Co during the reaction, which further highlights the advantage of silica for stabilizing the metal species confined in carbon.
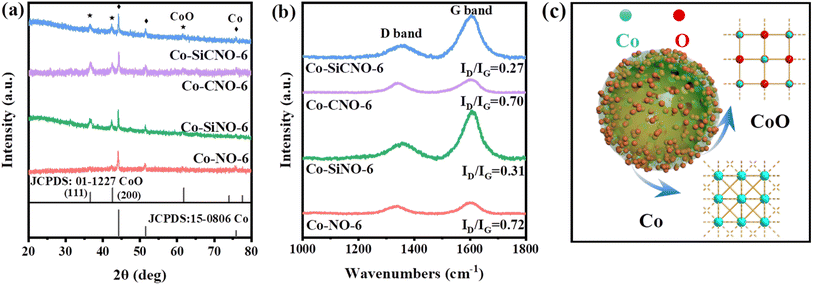 |
| | Fig. 2 (a) Wide-angle XRD patterns and (b) Raman spectra of Co-SiCNO-6, Co-CNO-6, Co-SiNO-6 and Co-NO-6; (c) crystal cell structure of multiple Co species in the catalyst. | |
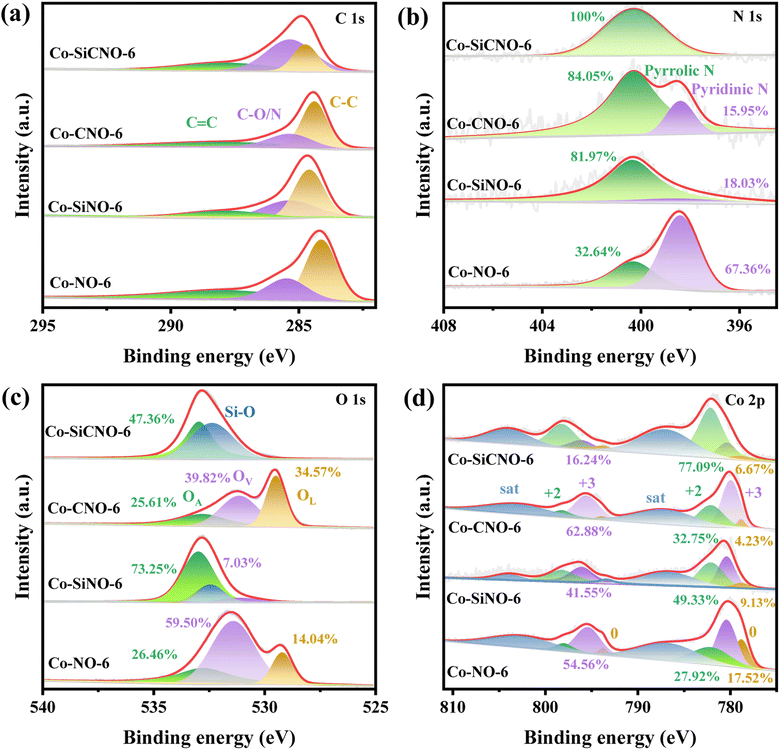 |
| | Fig. 3 XPS spectra of (a) C 1s, (b) N 1s, (c) O 1s and (d) Co 2p core levels of Co-SiCNO-6, Co-CNO-6, Co-SiNO-6, and Co-NO-6. | |
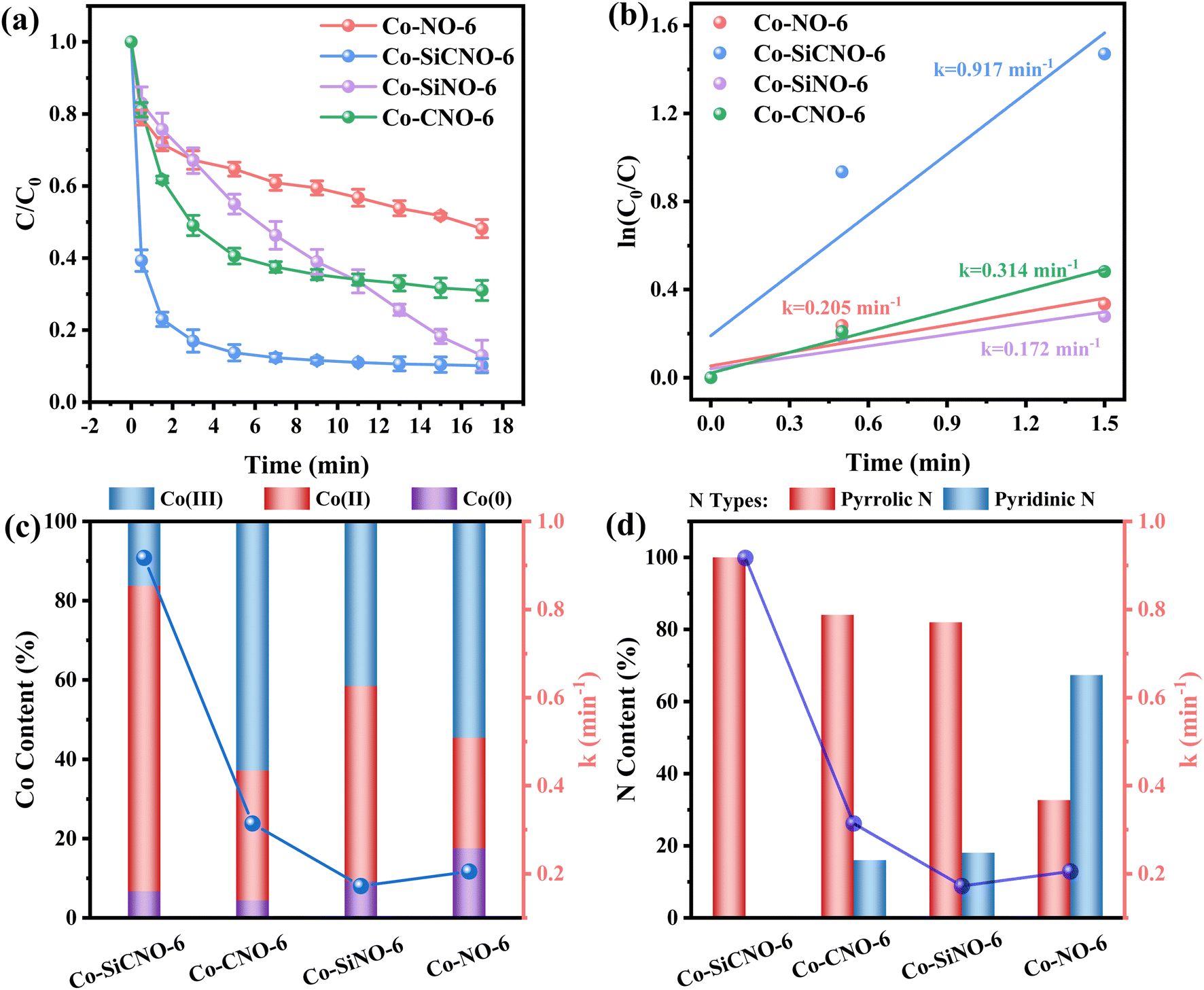
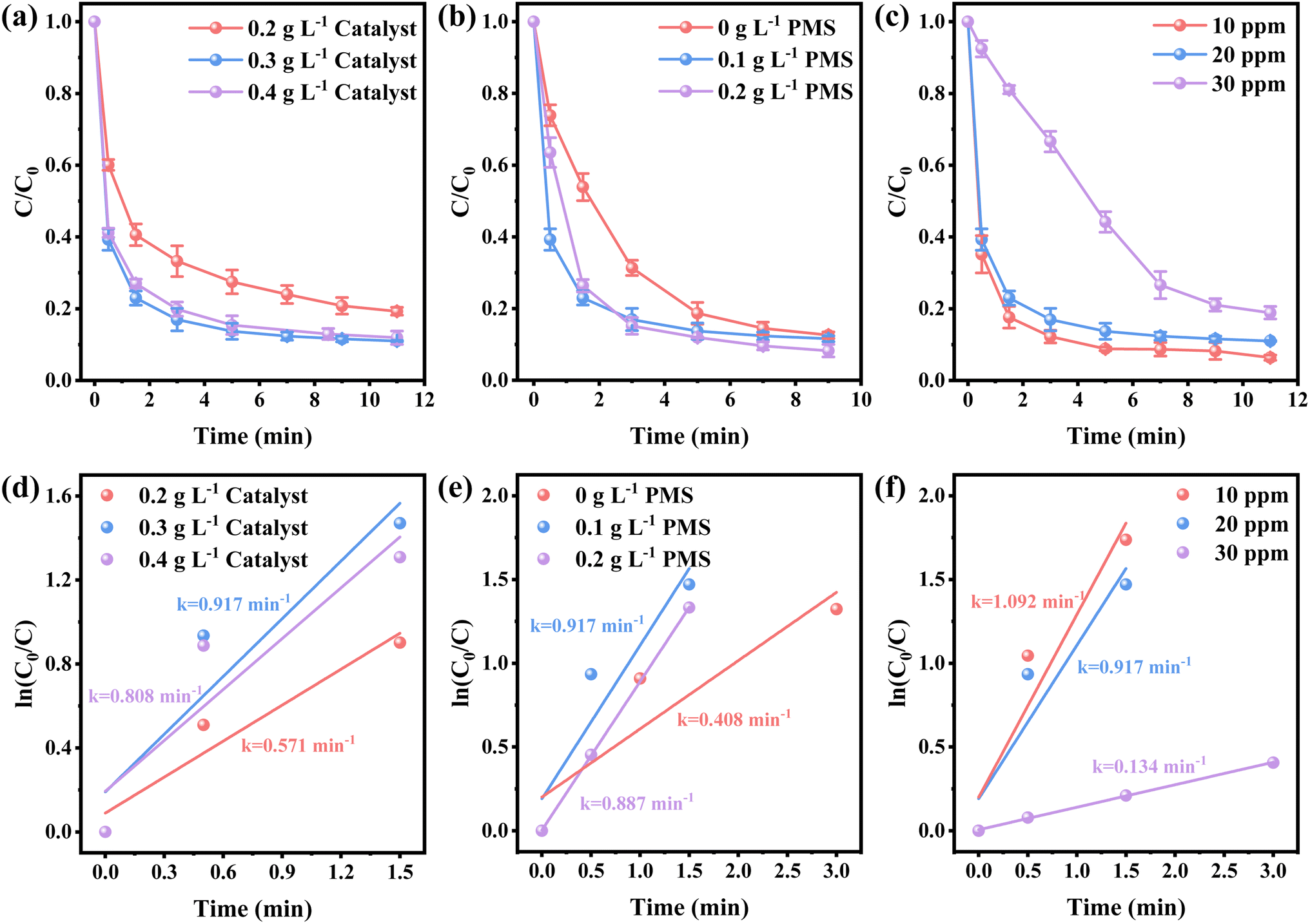
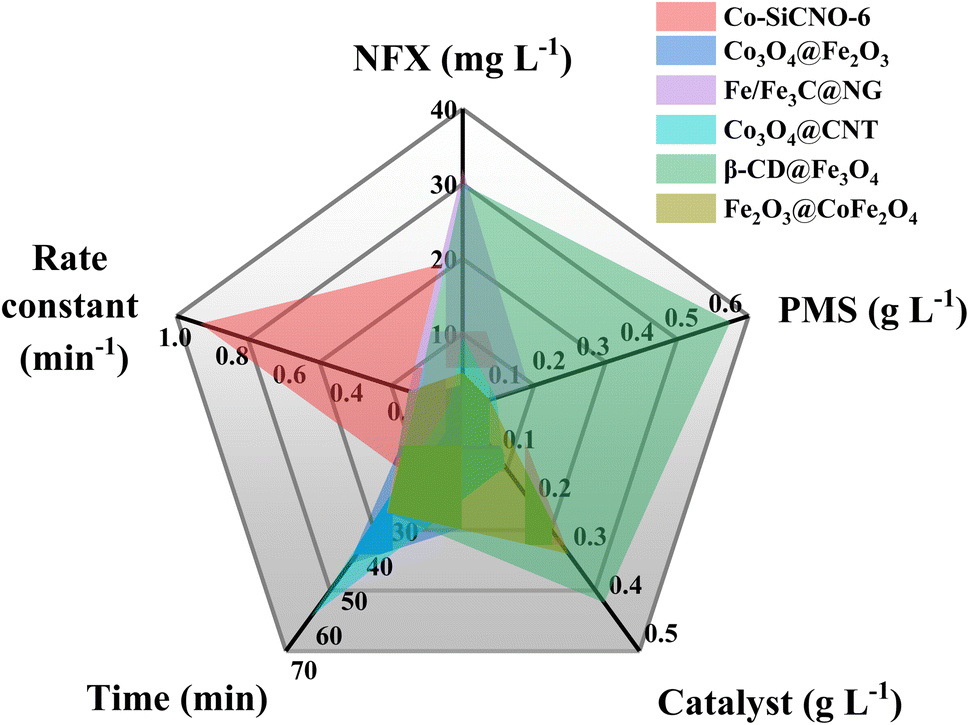
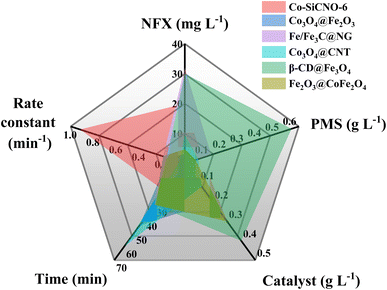
The efficiency of various samples in catalyzing PMS to degrade NFX was assessed. In Fig. S4,† the degradation curves of NFX over time for catalysts derived at different calcination temperatures are displayed. Notably, Co-SiCNO-6 exhibits superior catalytic degradation performance, showing the highest first-order kinetic constant (Kabs = 0.426 min−1), establishing it as the most effective catalyst for NFX degradation. Fig. 4a highlights the disparity in NFX degradation among comparison samples obtained at the optimal calcination temperature. Even when compared to the samples lacking modification with metal-incorporating PDA, Co-SiCNO-6 demonstrates significantly enhanced reaction activity. Furthermore, based on the first-order kinetic fitting analysis (Fig. 4b), the sequence of reaction kinetic constants for the four catalysts stands as follows: Co-SiCNO-6 (0.917 min−1) > Co-CNO-6 (0.314 min−1), Co-NO-6 (0.205 min−1) > Co-SiNO-6 (0.172 min−1). This underscores that the metal-incorporating carbon layer on the SiO2 shell over ZIF67 effectively confines and stabilizes active metal species, thereby enhancing the efficiency of PMS activation. To elucidate the activity origin of the optimized catalyst, Fig. 4c illustrates the correlation between different cobalt valences and reaction rate constants. Remarkably, the optimal catalyst Co-SiCNO-6, possessing the highest Co(II) fraction, exhibits the highest reaction rate, emphasizing the pivotal role of Co(II) in catalytic PMS activation. The correlation is clear: a higher valence metal fraction, especially higher Co(II) content, leads to a faster reaction rate. Interestingly, while the primary reaction kinetics of Co-SiNO-6 are slower than those of Co-CNO-6 and Co-NO-6, the degradation efficiency and degree of degradation of Co-SiNO-6 surpass those of Co-CNO-6 and Co-NO-6 at seventeen minutes. This discrepancy might stem from the absence of cobalt-incorporating polydopamine coating in Co-SiNO-6, where the introduced SiO2 reduces the exposure and reactivity of active cobalt species to some extent. Fig. 4d further illustrates the relationship between nitrogen species types and content versus the reaction rate. Notably, the primary kinetics of the four catalysts increase as the pyrrolic N content increases. The optimal catalyst containing solely pyrrolic N species exhibits the fastest reaction rate, indicating a positive correlation between pyrrolic N and primary reaction kinetics. More surface pyrrolic N species are recognized as polar sites that enrich polar contaminants around the reaction sites, shortening the migration distance of generated ROS towards contaminants and thereby enhancing their activity lifespan. A sequence of experiments was conducted to establish the optimal parameters for the catalytic elimination of NFX utilizing Co-SiCNO-6. These experiments were methodically designed to optimize catalyst dosage, PMS dosage, and NFX concentration. The outcomes, as depicted in Fig. 5a and d, elucidate the influence of varying catalyst dosages exclusively on the catalytic degradation of NFX while maintaining controlled variables.37 Notably, an increase in catalyst dosage from 0.2 g L−1 to 0.3 g L−1 resulted in an 88% degradation of NFX within a nine-minute timeframe. However, a further increase to 0.4 g L−1 did not enhance the reaction rate, indicating that excessive catalyst amounts render reactive sites unavailable for efficient NFX elimination. Furthermore, optimal PMS quantities were determined by solely varying PMS additions while keeping other variables constant, as illustrated in Fig. 5b and e.38 The catalyst without PMS exhibited the slowest NFX removal, attributed predominantly to adsorption effects rather than inherent catalytic function. Conversely, both 0.1 g L−1 and 0.2 g L−1 PMS contributed effectively to NFX degradation, to similar extents, although the initial-stage reaction kinetic constant for 0.1 g L−1 PMS was higher. This suggests that insufficient PMS underutilizes active sites, while excessive amounts might inhibit the reaction due to competitive processes. Interestingly, even in the absence of PMS, the catalyst eliminated 80% of NFX within 9 min, potentially due to the presence of abundant pyrrole nitrogen species enhancing ROS generation. This augmentation facilitated the attack on pollutants around the catalyst surface, thereby improving catalytic efficiency by reducing ROS diffusion distances. Fig. 5c and f indicate a gradual reduction in the catalytic activity of Co-SiCNO-6 as NFX concentration increased from 10 mg L−1 to 30 mg L−1. Maintaining a lower NFX concentration appeared more favorable for the catalyst, with a pronounced reduction in the reaction rate observed at 30 mg L−1. Moreover, qualitative analysis conducted as shown in Fig. S5†via 3D fluorescence assay confirmed the decreased NFX content during the degradation reaction, corroborating results from UV-absorption spectroscopy assays. Subsequent experiments at various temperatures established the low reaction activation energy (Ea) of Co-SiCNO-6 to be 1.420 kJ mol−1 (Fig. S6†), showing its distinctive advantage in NFX degradation. Table S3† shows the total organic carbon (TOC) content before and after the degradation of NFX by different catalysts, reflecting the degree of mineralization of organic pollutants. Specifically, Co-SiCNO-6 shows the highest TOC removal of 50.96%, which was higher than that of Co-SiNO-6 (33.36%), Co-CNO-6 (37.48%) and Co-NO-6 (31.84%). This basically agrees with the degradation activity trend of the above several comparative catalysts. In Fig. 6 and Table S4,† a comparative assessment of Co-SiCNO-6 and previously reported catalysts underscores its exceptional performance, demonstrating superiority in NFX degradation with respect to concentration, reaction rate, time, PMS, and catalyst dosage. Co-SiCNO-6 emerges as a catalyst showcasing superior comprehensive degradation performance for NFX across all evaluated parameters.39–44
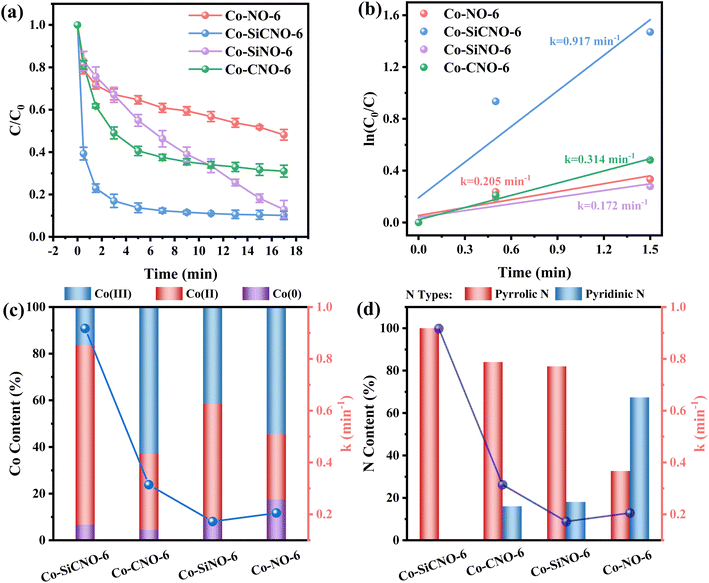 |
| | Fig. 4 Reaction time dependent NFX degradation course curves (a) and ln(C0/C) versus reaction time of NFX degradation (b) over various catalysts; reaction conditions: [NFX] = 20 mg L−1, [PMS] = 0.1 g L−1, [catalyst] = 0.3 g L−1; the relationship between the reaction rate constant and valence-determined element composition state of Co 2p (c) and N 1s (d) of various catalysts. | |
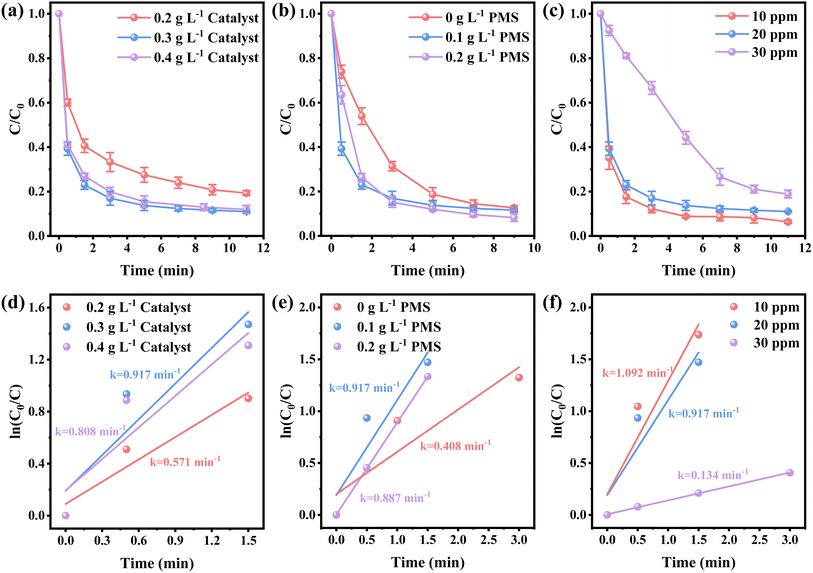 |
| | Fig. 5 The catalyst dosage effect on the degradation process (a) and ln(C0/C) versus reaction time (d) of Co-SiCNO-6; reaction conditions: [NFX] = 20 mg L−1, [PMS] = 0.1 g L−1. The PMS usage effect on the degradation process (b) versus ln(C0/C) and reaction time (e) of Co-SiCNO-6 catalyst; reaction conditions: [NFX] = 20 mg L−1, [catalyst] = 0.3 g L−1. The NFX concentration effect on degradation process curves (c) and ln(C0/C) versus reaction time (f) on the Co-SiCNO-6 catalyst; reaction conditions: [PMS] = 0.1 g L−1, [catalyst] = 0.3 g L−1. | |
 |
| | Fig. 6 Comprehensive performance comparison of NFX degradation based on optimal Co-SiCNO-6 and several reported comparative catalysts. | |
To explore the impact of anions on the degradation of NFX catalyzed by the catalyst, control experiments were conducted by introducing various inorganic salts like NaOH and Na2CO3 under optimized conditions (Fig. S7†). Observations reveal that Cl− exerts a noticeable inhibitory effect on the NFX degradation process. This inhibition could be attributed to the oxidation of Cl− by SO4−˙ and ˙OH, generating less reactive Cl˙ and HOCl−˙ (eqn (1)–(3)).45 Conversely, SO42− shows minimal influence on the degradation of NFX by Co-SiCNO-6, while OH− significantly diminishes the efficiency of the catalyst in degrading NFX, indicating that an excessively alkaline environment severely impedes PMS activation. Phosphates are known for their role as complex ions, engaging in chelation reactions by binding to –OH and Me–O groups on the surface. Additionally, they function as quenchers for ˙OH (eqn (4)).46 The introduction of H2PO4− somewhat curtails the activity of the reacting species, resulting in decreased catalytic performance. Moreover, the presence of HCO3− and CO32− inhibits NFX degradation due to their competition with reactive SO4−˙ or ˙OH, ultimately diminishing the population of reactive radicals (eqn (5)–(8)).47
| | | HOCl−˙ + H+ → Cl˙ + H2O | (2) |
| | | Cl− + SO4−˙ → SO42− + Cl˙ | (3) |
| | | ˙OH + H2PO4− → OH− + H2PO4−˙ | (4) |
| | | ˙OH + HCO3− → CO3−˙ + OH− | (5) |
| | | ˙OH + CO32− → CO3−˙ + OH− | (6) |
| | | SO4−˙ + HCO3− → CO3−˙ + HSO4− | (7) |
| | | SO4−˙ + CO32− → CO3−˙ + SO42− | (8) |
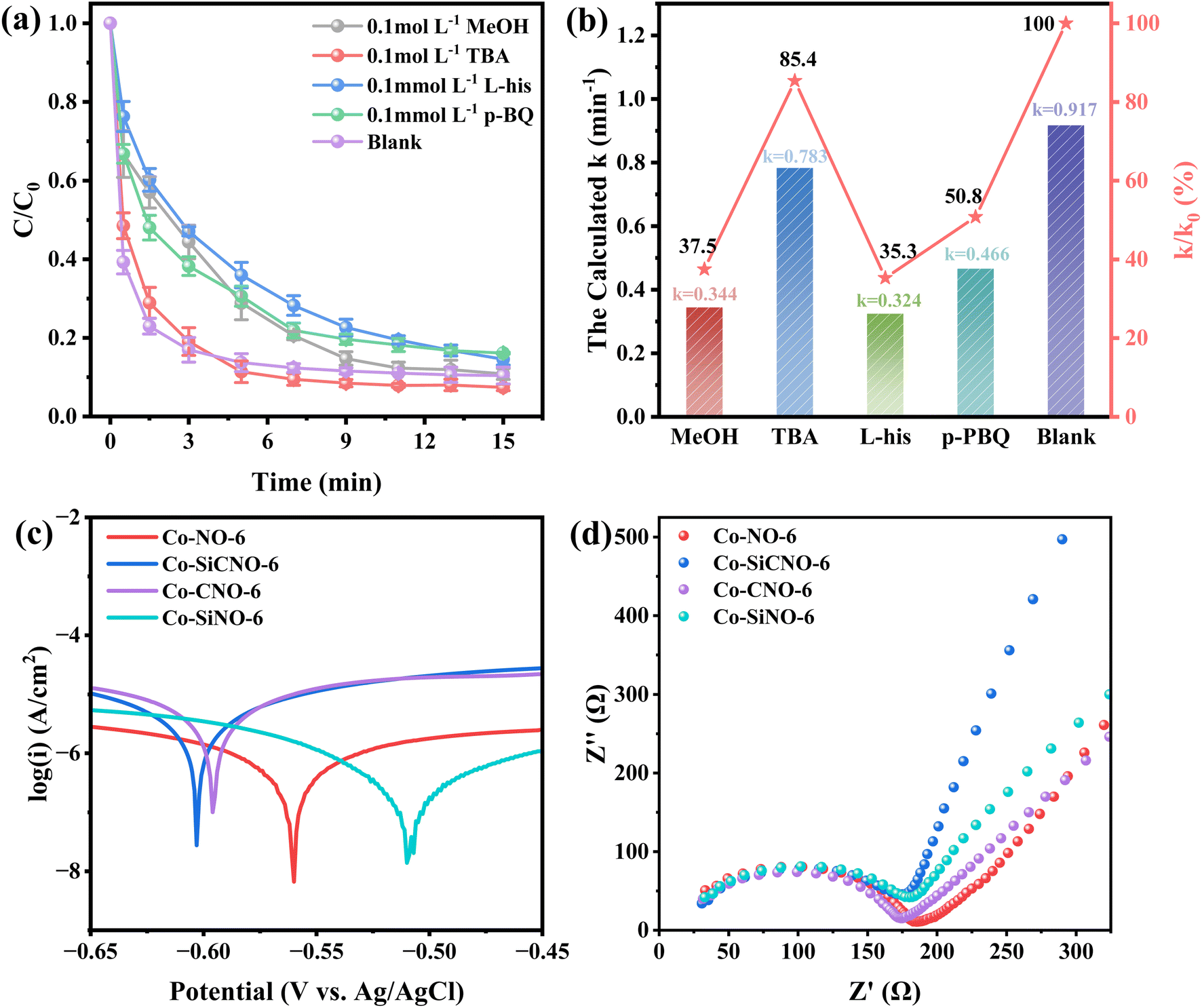
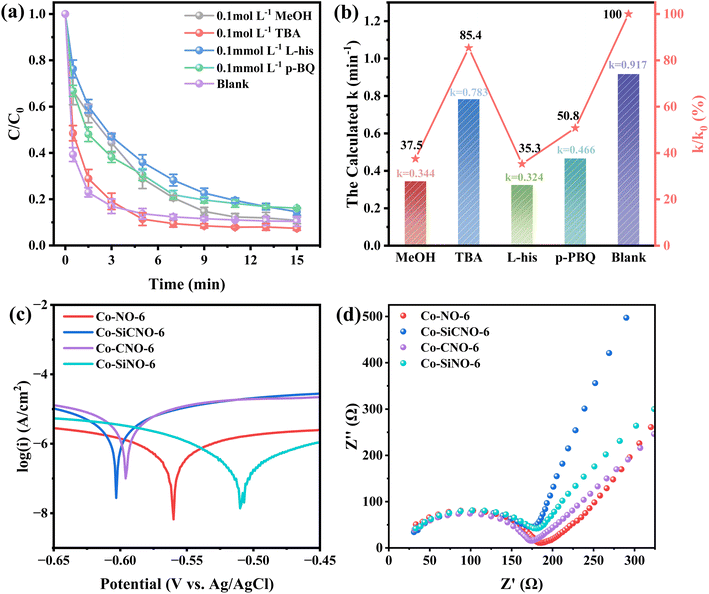
To gain deeper insights into the roles and reaction mechanisms of the primary active substances in NFX degradation through catalyst-activated PMS, free radical quenching experiments were performed. Methanol (MeOH), tert-butanol (TBA), L-histidine (L-his), and p-benzoquinone (p-BQ) were employed to quench specific free radicals. Broadly, MeOH effectively reacts with ˙OH (hydroxyl radical) and ˙SO4− (sulfate radical), while TBA exclusively interacts with ˙OH but not with ˙SO4−. Conversely, p-BQ and L-his exhibit evident scavenging effects on ˙O2− (superoxide radical) and 1O2 (singlet oxygen), respectively. The results presented in Fig. 7a and b indicate a reduction in the reaction rate constants of the optimal catalyst to 0.344 min−1, 0.783 min−1, 0.324 min−1, and 0.466 min−1 upon the addition of MeOH, TBA, L-his, and p-BQ to NFX, respectively. Remarkably, TBA displays the weakest effect after consuming ˙OH, while MeOH and L-his exhibit the most prominent impact, suggesting that ˙SO4− and 1O2 predominantly drive NFX removal. In a bid to assess the electron transfer capabilities of various catalyst composites, linear sweep voltammetry (LSV) (Fig. 7c) and electrochemical impedance spectroscopy (EIS) (Fig. 7d) data were collected. Fig. 7c illustrates heightened electron transport in Co-SiCNO-6, resulting in an accelerated oxidation reaction and exceptional electron transport capacity. Additionally, Fig. 7d portrays a steeper slope for Co-SiCNO-6 in the low-frequency region, indicating lower charge transfer resistance and swifter interfacial charge transfer, potentially contributing to the activity advantage of Co-SiCNO-6 from this perspective. Furthermore, to investigate the role of Co-SiCNO-6 in authentic water sources, NFX degradation experiments were conducted in tap water and Lake Haiyun water from Zhenjiang. These were compared with degradation experiments in deionized water (Fig. S8a†). Degradation rates in tap water and lake water were slightly lower than those in deionized water. This decline can be attributed to Cl− in tap water, which diminishes active radicals, consequently reducing the reaction rate. In lake water, various anions impact the catalyst, while other substances compete for active sites, further diminishing the reaction rate. However, Co-SiCNO-6 exhibited high catalytic performance for different water qualities, stably degrading over 80% of NFX within 19 min. The recyclability of Co-SiCNO-6 was assessed (Fig. S8b†), demonstrating the catalyst's capability to degrade almost 80% of NFX even after three cycles, affirming its stability and reproducibility. Moreover, to explore the versatility of Co-SiCNO-6 in degrading other pollutants, control degradation experiments (Fig. S8c†) were conducted with this catalyst for p-nitrophenol (PNP), bisphenol A (BPA), carbamazepine (CBZ), tetracycline (TC), and oxytetracycline (OTC). These experiments, conducted using optimal reaction parameters for NFX, displayed compelling results: PNP was completely degraded in eleven minutes, and CBZ, TC, and NFX were degraded to nearly 90% within the same timeframe, while BPA and OTC were degraded by 70%. This underscores the versatility of Co-SiCNO-6 in the degradation of various common organic pollutants.
 |
| | Fig. 7 (a) Quenching experimental results of NFX degradation of Co-SiCNO-6 using various quenching agents; reaction conditions: [NFX] = 20 mg L−1, [PMS] = 0.1 g L−1, [catalyst] = 0.3 g L−1; (b) reaction rate constant and relationship of catalytic systems of Co-SiCNO-6 involving addition of various quenching agents; linear sweep voltammetry (LSV) (c), and electrochemical impedance spectroscopy (EIS) (d) of the comparative catalysts. | |
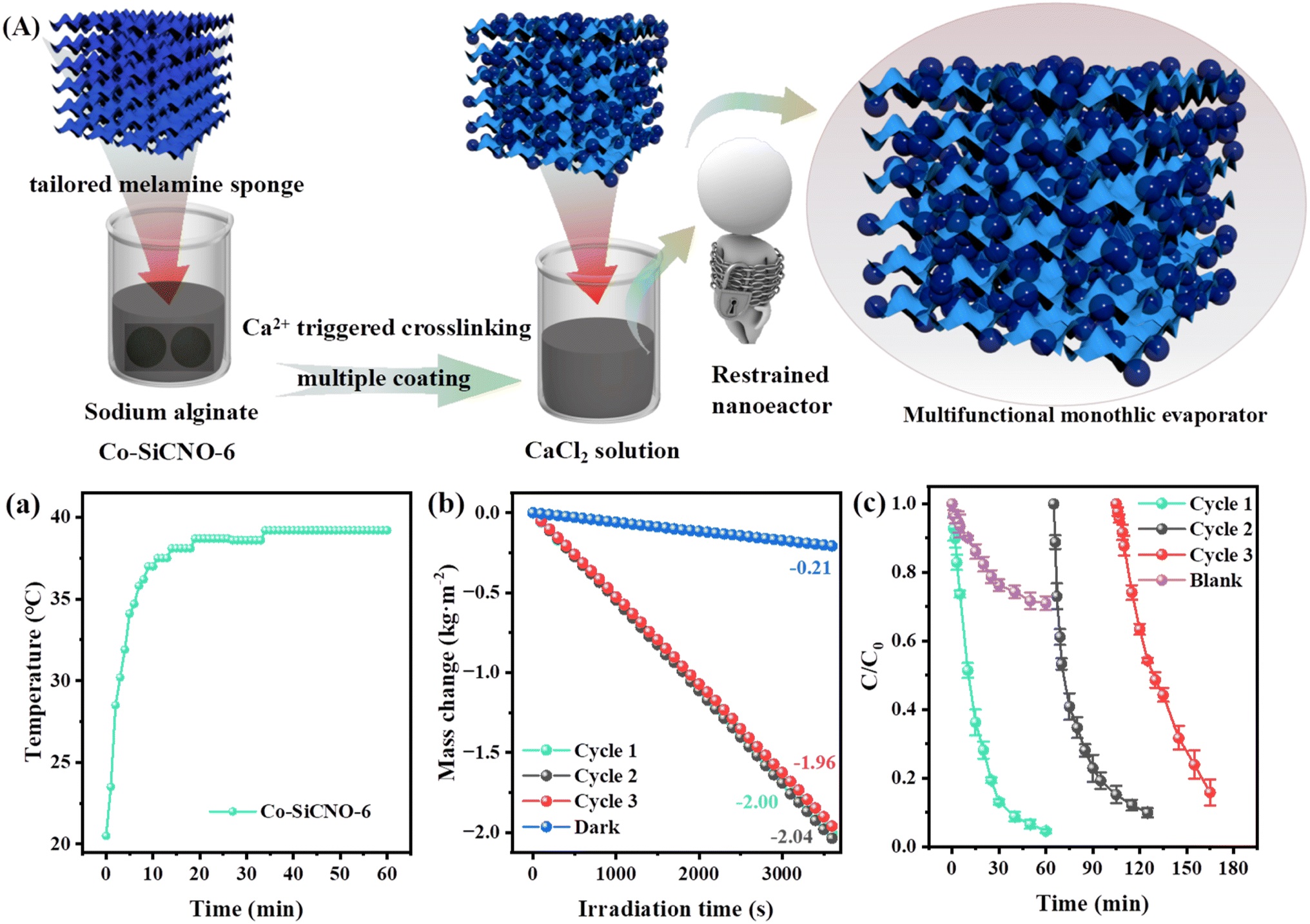
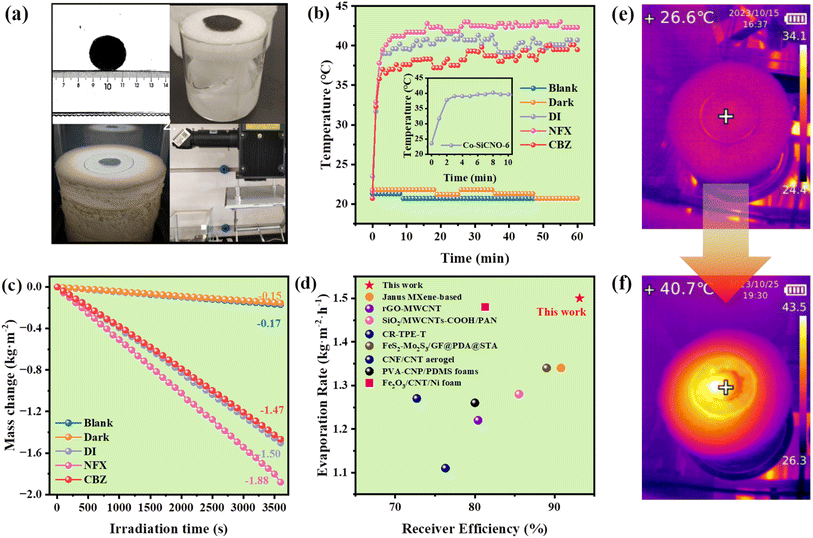
Besides effectively mineralizing contaminants, the potential for reclaiming clean water from wastewater is alluring. The presence of carbon and plasmonic metallic Co amplifies the photothermal attributes, a facet of particular interest. Specifically, the photothermal conversion of specialized materials is gaining traction in solar-driven interfacial water evaporation, harnessing sunlight to generate heat on photothermal materials. The amalgamation of carbon layers and metal species within the nanoreactor notably aids in surface heating. Additionally, the nanoreactor's mesoporous channels and hollow configuration expedite water vapor diffusion towards air, enhancing water evaporation.48Fig. 8a shows digital photos of the photothermal membrane prepared for water evaporation. Evaluating the Co-SiCNO-6 nanoreactor's photothermal properties involved measuring the integrated catalyst membrane's surface temperature using an infrared thermal imager. Infrared photothermal images depicted in Fig. S9a† reveal an increase in the nanoreactor's surface temperature from 26.6 °C to 71.2 °C within 30 min of irradiation. Fig. S9b† illustrates photothermal images of the wet sample, where Co-SiCNO-6 reaches 39.6 °C after 10 min, remaining at around 40 °C for the next 50 min. Moreover, the photothermal membrane's surface temperature significantly increases under sunlight exposure when soaked in NFX and CBZ solutions, ultimately reaching 42.3 °C and 39.5 °C, respectively. These results underscore Co-SiCNO-6's commendable photothermal conversion performance, likely stemming from Co metal's incorporation into the carbon layer, fostering exceptional photothermal conversion efficiency. In addition, the UV-vis absorption spectrum of Co-SiCNO-6 further validates the excellent sunlight absorption of the constructed materials compared to the natural light spectrum (Fig. S10†). Fig. 8b shows the temperature variation during the water evaporation process for different pollutants and deionized water. Its inset highlights Co-SiCNO-6's temperature variation in deionized water under light irradiation within 10 minutes, swiftly increasing from room temperature to 39.6 °C and stabilizing under the catalyst's influence. As depicted in Fig. 8c, the blank group data indicate water evaporation occurring directly under natural conditions, registering an evaporation rate of 0.17 kg m−2 h−1. In the absence of light, Co-SiCNO-6 exhibits a lower water evaporation rate of 0.15 kg m−2 h−1 compared to the blank group due to the nanoreactor's membrane aggregation impeding water molecule diffusion to some extent.49 However, under light conditions, the Co-SiCNO-6 membranes achieve a water evaporation rate of 1.50 kg m−2 h−1, boasting a photothermal evaporation efficiency of 93.14%. In a comparison of evaporation rates or efficiency with those in prior studies (Fig. 8d and Table S5†), Co-SiCNO-6 emerges as the top performer in photothermal capabilities during the evaporation process.50–57Fig. 8e and f show the infrared-thermal images of the membrane initially and after irradiation for 60 min under one sun irradiation. In addition, the water evaporation rate of Co-SiCNO-6 reached 1.88 kg m−2 h−1 for NFX and 1.47 kg m−2 h−1 for CBZ when NFX and CBZ solutions were used as the evaporating substances, respectively. This variance may be attributed to the influence of contaminant-bearing interfacial water's thermal conductivity on the photothermal materials due to the addition of macromolecular contaminants. The cyclic evaporation experiments further demonstrate the excellent efficiency of the Co-SiCNO-6 photothermal material (Fig. S11†).
 |
| | Fig. 8 (a) Digital combined photos of the prepared evaporation membrane of Co-SiCNO-6 and the sunlight solar simulator. (b) Temperatures variation of different systems including blank control, a wet Co-SiCNO-6 membrane in the dark or under light conditions soaked in water or, NFX/CBZ solution over a prolonged time. (c) Time-dependent mass variation of water during the photothermal interfacial water evaporation course. (d) The comparison of the evaporation rate versus evaporation efficiency of Co-SiCNO-6 and reported materials under one sun simulated irradiation. Infrared thermal images of (e) initial and (f) 60 min post-irradiation 2D membranes. Operation conditions: temperature: 20 ± 5 °C, relative humidity: 60 ± 10%. | |
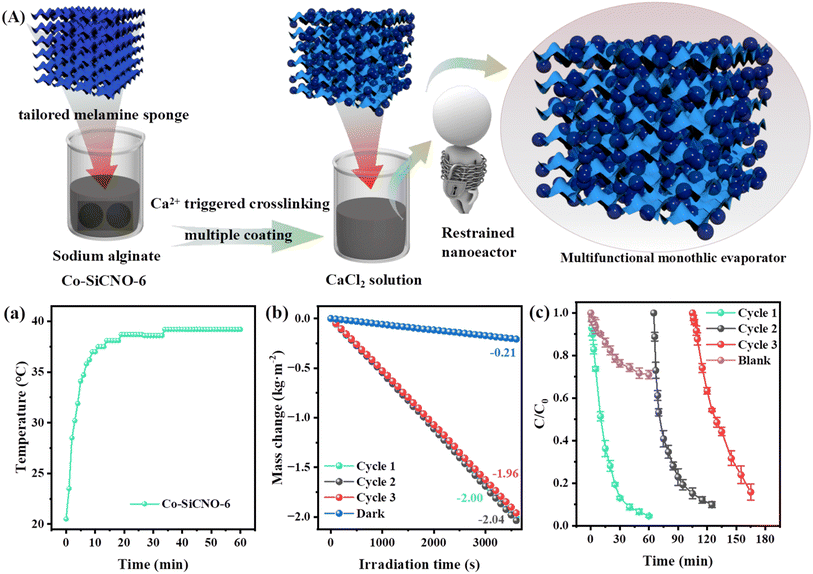
Due to the limitation of a separated 2D membrane for treating complex wastewater containing contaminants, in particular, the residual organic pollutants cannot be removed during the evaporation process. For practical applications utilizing photothermal materials, 30 mg of Co-SiCNO-6 catalyst was anchored onto a circular melamine sponge of 3 cm diameter to evaluate its performance in degrading pollutants and regenerating water. The combination of plasmonic metals and carbon materials enables efficient photothermal conversion in the composite. We anchored the photothermal catalyst onto the sponge evaporator through Ca ion-triggered crosslinking of SA. This integrated sponge evaporator offers a well-developed network, facilitating easy water diffusion and evaporation. Interfacial solar-assisted evaporation, achieved by constructing the evaporator from photothermal materials, is considered a feasible and promising method. The preparation is depicted in Fig. 9A; specifically, the obtained samples were dispersed in the SA solution, and the tailored sponge was soaked in the above mixture to adsorb them and further crosslink by Ca ion interaction. As a result, the integrated monolith evaporator composed of the functional materials and sponge can be placed in polluted wastewater as a floatable device, which can achieve solar-driven water evaporation and synchronous contaminant degradation on adding PMS as an oxidant. Impressively, this integrated device not only degrades pollutants but also achieves water evaporation at a high rate exceeding 2.0 kg m−2 h−1. Fig. 9a illustrates the photothermal performance of the integrated photothermal evaporator. The surface temperature increases from 20.2 °C to 30.2 °C within 3 min under one-sun irradiation, reaching 37 °C within 10 min, with a gradual increase and eventual stabilization. Additionally, under these conditions, Fig. 9b shows that the device exhibits water evaporation rates of 2.00 kg m−2 h−1, 2.04 kg m−2 h−1 and 1.96 kg m−2 h−1 in one hour during the three cycles, indicating the good stability and reusability of the integrated evaporator. The energy loss of the integrated sponge evaporator and separated 2D membrane involved in water evaporation was assessed (Table S6†). The calculated result reveals that the integrated sponge evaporator affords a high radiation loss and convection loss compared to the separated 2D membrane; in contrast, the integrated sponge evaporator contributes to an obviously lower conduction loss than that of the separated 2D membrane. Taken together, the total energy loss of the integrated sponge evaporator is higher than that of the separated 2D membrane. The catalytic degradation measurement, shown in Fig. 9c, confirms that the integrated monolith evaporator can degrade NFX by about 95% in one hour under sunlight and still degrades NFX by nearly 90% after three cycles, validating the reusability of the integrated evaporator in multiple contaminant degradation. The control group reveals that the NFX removal by virtue of the device without the addition of Co-SiCNO-6 and crosslinked with sodium alginate only reaches nearly 30%, indicating that Co-SiCNO-6 plays a crucial role in this real NFX degradation. Given the catalyst's integration with the sponge and its excellent recyclability, it aligns with practical requirements for water evaporation and contaminant removal.
 |
| | Fig. 9 (A) Fabrication of the integrated evaporator for contaminant purification and water evaporation, (a) temperature variation versus time of Co-SiCNO-6 anchored on sponges under light conditions in the presence of NFX. (b) Variation of water mass with time during three cycles of evaporation of NFX at the photothermal interface. (c) The cycling experiment results of the Co-SiCNO-6 integrated monolith evaporator for the activation of PMS for NFX degradation and blank control group using only a catalyst-free sponge. Reaction conditions: [NFX] = 20 mg L−1, [PMS] = 0.1 g L−1. Operation conditions: temperature: 20 ± 5 °C, relative humidity: 60 ± 10%. | |
4. Conclusions
In essence, employing ZIF67 as a template, followed by modifying mesoporous silica and applying a metal-incorporating PDA coating, yields a hollow nanoreactor with dispersed multivalent Co species and a carbon-silica hybrid mesoporous shell. This configuration distributes and confines metal species within the mesoporous shell, enriched with abundant N species modifications, offering ample catalytic and polar adsorptive sites. Moreover, the silica network contributes to the stability of the carbon framework and active metal species, ensuring structural integrity and reducing the leaching of metal during catalytic reactions. The notable presence of Co(II) species in the nanoreactor accelerates PMS activation, while the incorporation of pyrrole nitrogen into the carbon framework efficiently concentrates contaminants around the active sites, reducing ROS migration and enhancing utilization efficiency. Through interactions involving silica, a carbon thin layer, and cobalt species, this design achieves not only the removal of organic pollutants like norfloxacin but also the evaporation of interfacial water. The integrated photothermal monolith evaporator, employing Co-SiCNO-6, demonstrates impressive water purification and evaporation rates of 2.0 kg m−2 h−1 and a photothermal conversion efficiency of 93.14%, respectively. This catalyst holds practical significance, creating a unified system that adeptly combines water pollutant degradation and clean water evaporation. This research introduces a novel approach using photothermal materials for both pollutant degradation and water evaporation, offering unique insights for environmental remediation.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by the China National Natural Science Foundation (21908085), the China Postdoctoral Science Foundation (2023M731422), and the Ministry of Education, Singapore, under its Academic Research Fund Tier 1 (RG10/22). We appreciate the characterization support from the Instrumental Analysis Center, Jiangsu University of Science and Technology.
References
- Y. Huang, Y. You, M. Wu, M. Han, J. Zhang, W. Gao, D. Xie, H. Chen, H. Ou, N. Song, C. Song, W. Zhuang, J. Li, Z. Lei, B. Jin, Z. Zhou and M. Li, Environ. Res., 2023, 229, 115980 CrossRef CAS PubMed.
- O. B. Akpor, D. A. Otohinoyi, D. T. Olaolu and B. I. Aderiye, Inter. J. Enviro. Res. Earth Sci., 2014, 3(3), 050–059 Search PubMed.
- F. Hernández, J. V. Sancho, M. Ibáñez and C. Guerrero, TrAC, Trends Anal. Chem., 2007, 26(6), 466–485 CrossRef.
- M. P. Rayaroth, M. Marchel and G. Boczkaj, Sci. Total Environ., 2023, 857, 159043 CrossRef CAS PubMed.
- D. Ma, H. Yi, C. Lai, X. Liu, X. Huo, Z. An, L. Li, Y. Fu, B. Li, M. Zhang, L. Qin, S. Liu and L. Yang, Chemosphere, 2021, 275, 130104 CrossRef CAS PubMed.
- Y. Deng and R. Zhao, Curr. Pollut. Rep., 2015, 1, 167–176 CrossRef CAS.
- F. Ghanbari and M. Moradi, Chem. Eng. J., 2017, 310, 41–62 CrossRef CAS.
- P. Hu and M. Long, Appl. Catal., B, 2016, 181, 103–117 CrossRef CAS.
- S. Gupta, R. Fernandes, R. Patel, M. Spreitzer and N. Patel, Appl. Catal., A, 2023, 119254 CrossRef CAS.
- J. Hou, X. He, S. Zhang, J. Yu, M. Feng and X. Li, Sci. Total Environ., 2021, 770, 145311 CrossRef CAS PubMed.
- J. Dan, Q. Wang, P. Rao, L. Dong, M. Zhang, X. Zhang, Z. He, N. Gao and J. Deng, Chem. Eng. J., 2022, 429, 132189 CrossRef CAS.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30(17), 1909265 CrossRef CAS.
- P. Hu, M. Long, X. Bai, C. Wang, C. Cai, J. Fu, B. Zhou and Y. Zhou, J. Hazard. Mater., 2017, 332, 195–204 CrossRef CAS PubMed.
- Z. Tian, Q. Chen, S. Ren, H. Zhang, W. Tian, H. Sun and S. Wang, Chem. Eng. J., 2023, 143856 CrossRef CAS.
- Y. Bao, M. Tian, S. K. Lua, T. T. Lim, R. Wang and X. Hu, Chemosphere, 2020, 245, 125407 CrossRef CAS PubMed.
- J. Yang, L. Bai, J. Zhao, Y. Liu, H. Wang, H. Zhang, G. Li and H. Liang, Sep. Purif. Technol., 2023, 316, 123820 CrossRef CAS.
- S. Zhang, H. Gao, X. Xu, R. Cao, H. Yang, X. Xu and J. Li, Chem. Eng. J., 2020, 381, 122670 CrossRef CAS.
- F. Meng, Y. Wang and Q. Cao, Chemosphere, 2023, 337, 139441 CrossRef CAS PubMed.
- H. Cai, X. Li, D. Ma, Q. Feng, D. Wang, Z. Liu, X. Wei, K. Chen, H. Lin, S. Qin and F. Lu, Sci. Total Environ., 2021, 764, 144200 CrossRef CAS PubMed.
- X. Xu, M. Eguchi, Y. Asakura, L. Pan and Y. Yamauchi, Energy Environ. Sci., 2023, 16(5), 1815–1820 RSC.
- X. Liu, X. Xu, X. Xuan, W. Xia, G. Feng, S. Zhang, Z.-G. Wu, B. Zhong, X. Guo, K. Xie and Y. Yamauchi, J. Am. Chem. Soc., 2023, 145(16), 9242–9253 CrossRef CAS PubMed.
- Z. Xing, X. Xuan, H. Hu, M. Li, H. Gao, A. Alowasheeir, D. Jiang, L. Zhu, Z. Li, Y. Kang, J. Zhang, X. Yi, Y. Yamauchi and X. Xu, Chem. Commun., 2023, 59(30), 4515–4518 RSC.
- Y. Lu, H. Zhang, Y. Wang, X. Zhu, W. Xiao, H. Xu, G. Li, Y. Li, D. Fan, H. Zeng, Z. Chen and X. Yang, Adv. Funct. Mater., 2023, 33(21), 2215061 CrossRef CAS.
- T. Ding, Y. Zhou, W. L. Ong and G. W. Ho, Mater. Today, 2021, 42, 178–191 CrossRef CAS.
- D. Wei, C. Wang, J. Zhang, H. Zhao, Y. Asakura, M. Eguchi, X. Xu and Y. Yamauchi, Adv. Mater., 2023, 35(47), 2212100 CrossRef CAS PubMed.
- Y. Peng, Y. Wang, W. Li and J. Jin, J. Mater. Chem. A, 2021, 9(17), 10678–10684 RSC.
- L. Zhu, M. Gao, C. K. N. Peh and G. W. Ho, Nano Energy, 2019, 57, 507–518 CrossRef CAS.
- L. Cui, C. Ma, P. Wang, H. Che, H. Xu and Y. Ao, Appl. Catal., B, 2023, 122988 CrossRef CAS.
- T. Meng, Z. Li, Z. Wan, J. Zhang, L. Wang, K. Shi, X. Bu, S. M. Alshehri, Y. Bando, Y. Yamauchi, D. Li and X. Xu, Chem. Eng. J., 2023, 452, 139193 CrossRef CAS.
- W. Yu, F. Shu, Y. Huang, F. Yang, Q. Meng, Z. Zou, J. Wang, Z. Zeng, G. Zou and S. Deng, J. Mater. Chem. A, 2020, 8(39), 20677–20686 RSC.
- G. Zhong, D. Liu and J. Zhang, J. Mater. Chem. A, 2018, 6(5), 1887–1899 RSC.
- T. Gao, Y. Wang, X. Wu, P. Wu, X. Yang, Q. Li, Z. Zhang, D. Zhang, G. Owens and H. Xu, Sci. Bull., 2022, 67(15), 1572–1580 CrossRef CAS PubMed.
- H. Yang, J. Ouyang and A. Tang, J. Phys. Chem. B, 2007, 111(28), 8006–8013 CrossRef CAS PubMed.
- X. Sun, H. Qi, S. Mao and Z. Sun, Chem. Eng. J., 2021, 423, 130169 CrossRef CAS.
- H. Li, Z. Zhao, J. Qian and B. Pan, Environ. Sci. Technol., 2021, 55(9), 6397–6406 CrossRef CAS PubMed.
- Q. Wang, Z. Xu, Y. Cao, Y. Chen, X. Du, Y. Yang and Z. Wang, Chem. Eng. J., 2022, 427, 131953 CrossRef CAS.
- S. Yang, W. Zhang, M. Liu, H. Zhao, H. Lu, H. Li, Z. Guo, A. Yuan, J. Yang, J. Pan and F. Yang, J. Environ. Chem. Eng., 2023, 11(1), 109190 CrossRef CAS.
- H.-Y. Zhu, M.-T. Liu, G. Wang, R.-R. Du, H.-Y. Zhao, H. Lu, S.-Q. Yang, S. Tang, Z.-J. Guo, J. Yang, C.-Z. Zhu and F. Yang, Rare Met., 2023, 1–16 Search PubMed.
- L. Chen, X. Zuo, S. Yang, T. Cai and D. Ding, Chem. Eng. J., 2019, 359, 373–384 CrossRef CAS.
- Q. Peng, Y. Ding, L. Zhu, G. Zhang and H. Tang, Sep. Purif. Technol., 2018, 202, 307–317 CrossRef CAS.
- B. Liu, W. Song, H. Wu, Z. Liu, Y. Teng, Y. Sun, Y. Xu and H. Zheng, Chem. Eng. J., 2020, 398, 125498 CrossRef CAS.
- Y. Qin, A. Ahmed, S. Iqbal and M. Usman, Mol. Catal., 2023, 547, 113368 CrossRef CAS.
- M. Kohantorabi, M. Hosseinifard and A. Kazemzadeh, New J. Chem., 2020, 44(10), 4185–4198 RSC.
- Y. Luo, X. Huang, Y. Li, Y. Fu, Z. Wang, J. Lu and J. Zhang, Sep. Purif. Technol., 2020, 252, 117476 CrossRef CAS.
- Z. Yang, Y. Li, X. Zhang, X. Cui, S. He, H. Liang and A. Ding, Chem. Eng. J., 2020, 384, 123319 CrossRef CAS.
- F. Qi, W. Chu and B. Xu, Chem. Eng. J., 2014, 235, 10–18 CrossRef CAS.
- C. Tan, N. Gao, D. Fu, J. Deng and L. Deng, Sep. Purif. Technol., 2017, 175, 47–57 CrossRef CAS.
- R. Du, H. Zhu, H. Zhao, H. Lu, C. Dong, M. Liu, F. Yang, J. Yang, J. Wang and J. Pan, Environ. Res., 2023, 222, 115365 CrossRef CAS PubMed.
- R. Du, H. Zhu, H. Zhao, H. Lu, C. Dong, M. Liu, F. Yang, J. Yang, J. Wang and J. Pan, J. Alloys Compd., 2023, 940, 168816 CrossRef CAS.
- J. Su, Y. Xie, P. Zhang, R. Yang, B. Wang, H. Zhao, Y. Xu, X. Lin, J. Shi and C. Wang, Desalination, 2023, 566, 116905 CrossRef CAS.
- Y. Wang, C. Wang, X. Song, S. K. Megarajan and H. Jiang, J. Mater. Chem. A, 2018, 6(3), 963–971 RSC.
- Q. Qi, W. Wang, Y. Wang and D. Yu, Sep. Purif. Technol., 2020, 239, 116595 CrossRef CAS.
- G. Chen, J. Sun, Q. Peng, Q. Sun, G. Wang, Y. Cai, X. Gu, Z. Shuai and B. Z. Tang, Adv. Mater., 2020, 32(29), 1908537 CrossRef CAS PubMed.
- H. Sang, C. Tang, K. Ma and X. Li, J. Water Process Eng., 2023, 56, 104403 CrossRef.
- F. Jiang, H. Liu, Y. Li, Y. Kuang, X. Xu, C. Chen, H. Huang, C. Jia, X. Zhao and E. Hitz, ACS Appl. Mater. Interfaces, 2018, 10(1), 1104–1112 CrossRef CAS PubMed.
- S. Wang, S. M. Almenabawy, N. P. Kherani, S. N. Leung and P. G. O'Brien, ACS Appl. Energy Mater., 2020, 3(4), 3378–3386 CrossRef CAS.
- S. Han, J. Yang, X. Li, W. Li, X. Zhang, N. Koratkar and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2020, 12(11), 13229–13238 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Edison Huixiang
Ang
a,
Edison Huixiang
Ang