
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced hydrogen production via assisted biomass gasification using lithium manganate as a bifunctional material†
Carlos
Hernández-Fontes
 ,
Nan
Wang
,
Nayeli
Gómez-Garduño
and
Heriberto
Pfeiffer
*
,
Nan
Wang
,
Nayeli
Gómez-Garduño
and
Heriberto
Pfeiffer
*
Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito exterior s/n, Cd. Universitaria, Del. Coyoacán, CP. 04510, Ciudad de México, Mexico. E-mail: pfeiffer@materiales.unam.mx
First published on 22nd April 2024
Abstract
The rising energy demand, among other economic and technological factors, has resulted in an increase in greenhouse gas emissions. Therefore, it is crucial to develop technologies to produce clean energy, such as hydrogen (H2) generation from biomass sources. In this context, the use of alkaline ceramics has been reported to show promising results for pyrolysis and gasification processes. Thus, the present study aimed to investigate hydrogen production based on the bifunctional activity of lithium manganate (Li2MnO3) using glucose and cellulose molecules as biomass models. Furthermore, the effect of the heating rate and biomass![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ceramic molar ratio was evaluated. The results for glucose showed that the addition of Li2MnO3 during its pyrolysis highly enhanced and shifted H2 production to lower temperatures through an assisted gasification process, reducing Mn4+ ions to Mn3+ and Mn2+. Besides, solid products evidenced carbon capture, which mainly contributed to improving H2/COx ratios. Thereafter, during cellulose evaluation, under optimal glucose:Li2MnO3 experimental pyrolytic conditions, the results corroborated the bifunctional application of the ceramic. Thus, further studies on the biomass assisted-gasification process using modified Li–Mn-based ceramics have significance to enhance the H2 production and purity, while reducing the emission of carbon oxides.
:ceramic molar ratio was evaluated. The results for glucose showed that the addition of Li2MnO3 during its pyrolysis highly enhanced and shifted H2 production to lower temperatures through an assisted gasification process, reducing Mn4+ ions to Mn3+ and Mn2+. Besides, solid products evidenced carbon capture, which mainly contributed to improving H2/COx ratios. Thereafter, during cellulose evaluation, under optimal glucose:Li2MnO3 experimental pyrolytic conditions, the results corroborated the bifunctional application of the ceramic. Thus, further studies on the biomass assisted-gasification process using modified Li–Mn-based ceramics have significance to enhance the H2 production and purity, while reducing the emission of carbon oxides.
1. Introduction
Globally, the energy demand has been increasing as a result of the rapidly increasing population and the development of technology.1–3 Moreover, the dependence on fossil fuels to fulfill this energy demand has caused a continuous increase in CO2 emissions, which have reached 423.7 ppm, causing several and severe health and environmental problems.4–6 Therefore, governments have started focusing on several strategies to mitigate greenhouse gas (GHG) emissions.7,8 In this context, the transition to renewable energy sources appears to be one of the most promising solutions.9 However, clean energies and alternative fuels are associated with several economic, technological, social and environmental challenges, such as limited accessibility, dependence on weather conditions, low government funding and the use of engine technologies.10,11From this perspective, hydrogen has been attracting attention as a potential clean energy resource owing to its zero emissions and high energy density as well as its diverse production sources,12,13 such as methane reforming, water electrolysis, bioproduction, photoelectrochemical water splitting and biomass conversion.14–18 In fact, biomass conversion through pyrolysis and gasification processes has been widely recognized as a key process to meet the world energy demand considering its low cost, neutral carbon print increment and local accessibility.19–22
Biomass feedstock is defined as any organic solid material susceptible to combustion or transformation into end products (modern biomass), such as syngas mixtures (H2 + CO), bio-oils and/or bio-char.23–26 Conversely, pyrolysis is a thermochemical process occurring in the absence of oxygen, which can be performed at around 250–850 °C, producing bio-oils; bio-char; and different gaseous products such as hydrogen (H2) and carbon oxides (COx), in addition to other light organic gases.27,28 Alternatively, depending on biomass composition, low amounts of sulfur oxides and/or nitrogen oxides can be produced.29,30 Moreover, gasification is a partial thermal oxidation process, where an oxygen source must be supplied (air, steam, supercritical water and/or CO2).28,31,32 Generally, this process is carried out at higher temperatures than pyrolysis, where the main product is a syngas mixture (H2 + CO), together with CO2, water and gaseous hydrocarbons.33,34 However, some low quantities of secondary compounds such as tar and char (mixture of carbon or ash) may be produced.33
Although H2 production via the thermochemical conversion of biomass is a neutral CO2 emission process, as it is produced as syngas, limiting its application. For instance, to feed fuel cells and generate electric energy, given that they are prone to poisoning by CO, the H2 supply must be subjected to costly purification techniques, namely cryogenic separation, pressure swing adsorption (PSA), and membrane selective separation, to reduce the content of CO impurities to <10 ppm.35,36 Moreover, it has been reported that the addition of different metal oxides or supported metal-particles as catalysts in addition to CO2 sorbents during the pyrolysis and gasification processes enhance the production and increases the purity of H2.19,37–41
In this context, alkaline ceramics have demonstrated high capture capabilities for CO2 and/or bifunctional properties for consecutive processes, such as CO oxidation and subsequent chemisorption. In fact, Li4SiO4 and Na2ZrO3 have been reported as bifunctional catalyst-sorbent materials during the pyrolysis process, enhancing the H2 production and chemically trapping CO2 as carbonates.42,43 However, the catalytic properties of Zr or Si are poor or non-existent. Therefore, the study of different bifunctional ceramics with better catalytic properties for the biomass pyrolysis process is crucial.44 In this case, only lithium manganate (Li2MnO3) has demonstrated selective CO catalytic (T < 500 °C) and chemisorption (T ≥ 500 °C, reaction (1), maintaining some catalytic activity) capabilities under inert or non-oxidative conditions.45,46 Moreover, this material was tested using a synthetic syngas mixture for H2 purification, showing a very low ceramic interaction with H2.47
| Li2MnO3(s) + CO(g) → Li2CO3(s) + MnO(s) | (1) |
Based on this, this work aimed to analyze the catalytic and sorption effects of Li2MnO3 added during biomass pyrolysis using glucose and cellulose as model biomass molecules.28,48,49 This study was performed using a thermobalance and a catalytic flow reactor system, with complementary techniques.
2. Experimental
2.1. Synthesis and characterization
Li2MnO3 was synthesized via a solid-state method using manganese(II) oxide (MnO, Meyer) and lithium oxide (Li2O, Aldrich) as reactants, following a previously reported methodology.45,47,50 Briefly, both powders were mechanically mixed with 5 wt% excess lithium.51,52 The resultant mixture was pelletized (40 MPa) and heat-treated at 900 °C for 12 h in air. Afterwards, the product was pulverized and fully characterized by X-ray diffraction (XRD) and N2 adsorption–desorption analysis. The XRD analysis was performed at room temperature (RT) using a D5000 diffractometer (Siemens) coupled to a copper anode X-ray tube (Cu Kα-radiation). Prior to the N2 adsorption–desorption measurement, the sample was degassed under vacuum at RT for 12 h. Then, a Minisorp II instrument (BEL Japan) was used to perform the analysis at 77 K. Besides, the specific surface area (SBET) was determined using the Brunauer–Emmett–Teller (BET) model.2.2. Catalytic and carbon oxide sorption measurements
The effect of Li2MnO3 on the biomass pyrolysis process was studied. The glucose (C6H12O6, Aldrich) molecule was selected as the initial model. Therefore, different homogeneous Li2MnO3:glucose mixtures were prepared (Table 1). Each mixture, labeled as LMO-G, was mechanically mixed in an agate mortar for 10 min.
| Measurement type | Sample label | Li2MnO3 (wt%) | Glucose (wt%) | Mass ratio (ceramic/glucose) | Molar ratio (ceramic/glucose) |
|---|---|---|---|---|---|
| Thermogravimetry (thermobalance) | LMO-G 5-95 | 5 | 95 | 0.0526 | 0.08 |
| LMO-G 25-75 | 25 | 75 | 0.3333 | 0.34 | |
| LMO-G 50-50 | 50 | 50 | 1.0000 | 0.61 | |
| LMO-G 75-25 | 75 | 25 | 3.0000 | 0.82 | |
| Gas evolutions (catalytic reactor) | LMO-G 5-95 | 5 | 95 | 0.0526 | 0.08 |
| LMO-G 10-90 | 10 | 90 | 0.1111 | 0.15 | |
| LMO-G 15-85 | 15 | 85 | 0.1765 | 0.21 | |
| LMO-G 20-80 | 20 | 80 | 0.2500 | 0.28 | |
| LMO-G 25-75 | 25 | 75 | 0.3333 | 0.34 |
To identify the influence of Li2MnO3 on the glucose decomposition behavior, a broad range of Li2MnO3 contents (from 5 to 75 wt%) was analyzed by thermogravimetry, including glucose alone for comparison. These experiments were performed using a Q550 thermobalance (TA Instruments) in an inert atmosphere (flowing 60 mL min−1 of N2, Praxair grade 4.8) from 30 °C to 950 °C, at 5 °C min−1. Then, the LMO-G 25-75 sample was selected to evaluate the influence of the heating rate (HR) on the gaseous products, employing a fixed-bed catalytic reactor (Bel-Rea, from Bel Japan) attached to a cooler (water trap), an FTIR gas-cell spectrometer (ALPHA-Platinum, from Bruker) and a gas chromatography system (GC-2014 with a Carboxen-1000 column, from Shimadzu). The sample (200 mg) was placed on a quartz wool support and dynamically heated from 30 °C to 850 °C under an N2 flow of 60 mL min−1 at various heating rates (5 °C min−1, 10 °C min−1, 20 °C min−1 and 30 °C min−1). Additionally, the optimized conditions (60 mL min−1 of flowing N2 with a heating rate of 30 °C min−1) were selected to evaluate the glucose sample for comparison. Complementarily, the condensable volatile products from the glucose and LMO-G 25-75 samples were recovered using a cooler system and analyzed through GC-mass spectrometry (GC-MS) (GCMS-QP 2010 SE, from Shimadzu). These samples were diluted in 1 mL of methanol, and then an aliquot of 1 μL was injected into the instrument in split mode using helium (He 4.5 grade, Praxair) as the carrier gas and an Rtx®-200 column (Resteck). The mass spectra of the samples were compared with the NIST11 database.
Thereafter, the effect of the Li2MnO3 amount in the ceramic-biomass mixtures on the gaseous products during the pyrolysis of biomass was evaluated. For this purpose, the Li2MnO3 content was reduced in the ceramic-glucose mixtures from 25 to 5 wt% (see Table 1).
These experiments were performed in the catalytic reactor system using N2 (60 mL min−1) and the best heating rate conditions (30 °C min−1). Complementarily, to elucidate the effect of the presence of lithium in the ceramic composition, a sample containing only manganese(IV) oxide (MnO2, Meyer) and glucose was evaluated under the best Li2MnO3:glucose conditions (25:75, heating rate of 30 °C min−1 in N2 flow of 60 mL min−1).
Based on all the results, the best heating conditions (30 °C min−1 in N2 flow) and the best ceramic:biomass ratio (0.3333) were selected to study the whole pyrolysis process involving Li2MnO3 and glucose, involving two-step procedure, as follows: (i) the sample was dynamically heated from RT to the target temperature of the study (between 500 °C and 700 °C at 50 °C intervals), and (ii) the sample was isothermally treated at the target temperature for 2 h. Moreover, all the isothermal solid products as well as the as-prepared mixture (LMO-G 25-75) were characterized by XRD and attenuated total reflectance infrared spectroscopy (ATR-FTIR, ALPHA-Platinum, from Bruker).
After determining the best conditions for glucose pyrolysis (30 °C min−1 and 0.3333 mass ratio of ceramic in N2 flow), they were used to evaluate the pyrolysis of cellulose. A mixture of Li2MnO3 and cellulose ((C6H10O5)n, Aldrich) was mechanically mixed in an agate mortar for 10 min (labeled as LMO-C). Then, LMO-C was thermogravimetrically analyzed and tested in a catalytic reactor, as described above. It must be mentioned that all the experiments described above were repeated at least three times to evaluate their reproducibility.
2.3. GC data processing
The GC data was processed to calculate the flow (mL min−1) of each compound based on its concentration (gas species flow/total flow) according to eqn (2), where Fx is the flow of each gas, Cx is the concentration of gas and FT is the total flow of each aliquot. The concentration of each compound (Cx) was determined by calibration curves, which were experimentally determined using different mixtures of N2 (Infra, grade 4.8), CO (Praxair, 5% N2 balanced), CO2 (Praxair, grade 4.8), H2 (Praxair, grade 4.5) and CH4 (Infra, grade 4.8), respectively. Alternatively, the total flow (FT) was determined from the concentration and flow of the inert gas (N2) in the analyzed aliquot. Then, the collected flow data was normalized to the biomass (glucose or cellulose) content (eqn (3)), where FN,x, Fx and m are the normalized flow of each gas, the gas flow in mL min−1 and the biomass mass in g, respectively.| Fx = Cx(FT) | (2) |
| FN,x = Fx/m | (3) |
Moreover, according to the collected data, the total volume of gaseous products normalized per g of biomass (V) was calculated through the integration of the flow normalized data as a function of time (FN,x(t), eqn (4)), from initial (t0) and final time (tf).
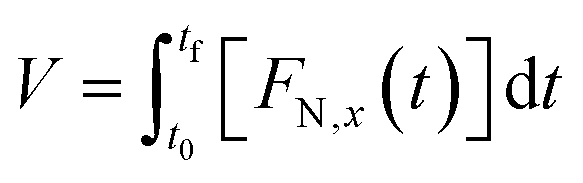 | (4) |
2.4. Kinetic parameter calculation
The Flynn–Wall–Ozawa integration method was implemented to calculate the kinetic parameters of the glucose and cellulose pyrolysis processes in presence or absence of Li2MnO3 at a mass ratio of 0.3333. These samples were thermogravimetrically analyzed by modifying the HR from 10 °C min−1 to 30 °C min−1 at 10 °C min−1 intervals using an N2 flow (60 mL min−1). All collected data were fitted to eqn (5) and (6), where Ea, A, R, α, β, T, a and b are the Arrhenius activation energy, pre-exponential factor, gas constant, conversion value of decomposition, heating rate, and temperature, where Ea/T and T/Ea are numerical integration constants. Ea and pre-exponential factor were determined as the average of the obtained α values from 0.08 to 0.13.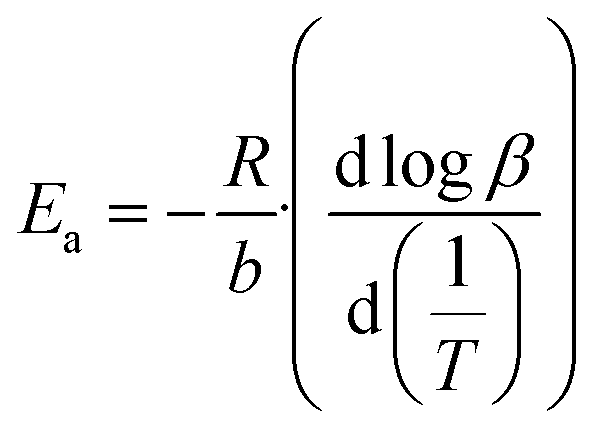 | (5) |
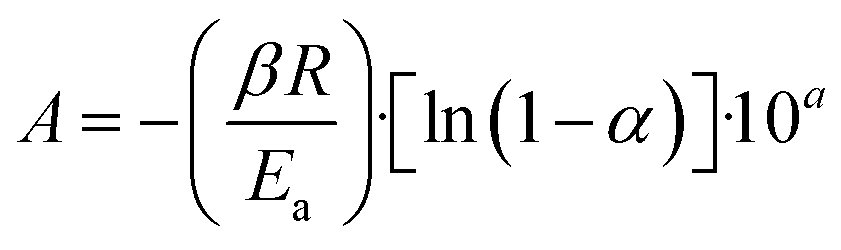 | (6) |
3. Results and discussion
Li2MnO3 was synthesized following the previously reported methodology.45,47,50 Herein, the data characterization of the material is provided in the ESI (Fig. S1†), where its XRD pattern matched the 01-081-1953 file from the PDF database, corresponding to the monoclinic Li2MnO3 crystal structure without any other secondary phase. In addition, the N2 adsorption–desorption isotherm depicted a type II isotherm according to the IUPAC classification, without the presence of any hysteresis loop.53,54 Besides, it was determined to possess a specific surface area (SBET) of less than 1 m2 g−1. These results are in agreement with previous solid-state synthesis reports on this material.45,46,50After the characterization of Li2MnO3, the effect of its addition to the glucose pyrolysis process was evaluated through dynamic thermogravimetric analyses (Fig. 1), where different mixtures of Li2MnO3 and glucose were tested (see Table 1), including a pristine glucose sample. Fig. 1A shows noticeable differences in the expected weight loss, depending on the Li2MnO3:glucose ratio. Indeed, in the case of pristine glucose, most of the mass was lost (∼77.5%) in a single and continuous step between 170 °C and 550 °C. Afterwards, it slowly showed 4 wt% weight loss in the remaining temperature range (550 °C to 950 °C), where the final solid product was only a mixture of carbon (C) and ash traces, i.e., so-called char.55 Conversely, when Li2MnO3 was added, not only the temperature in which the process started was reduced, depending on the Li2MnO3 amount, but also the process was split into three steps. For instance, after the first weight loss, it was stabilized at 480 °C for LMO-G 5-95, and this weight stabilization temperature diminished as a function of Li2MnO3 up to 430 °C for the LMO-G 75-25 sample. Besides, after the weight stabilization, a second decomposition process was observed from 580 °C to 625 °C for the LMO-G 5-95 and LMO-G 75-25 samples, respectively. Then, the third weight loss seemed to be stabilized in the samples with 25 and 50 wt% of Li2MnO3, while the samples with a lower ceramic content did not reach a plateau zone. Evidently, the final mass loss produced by each sample depended on the content of glucose, given that Li2MnO3 has been reported to be a thermal stable material between RT and 650 °C in an N2 flow.45 In fact, during the last decomposition process, superficial oxygen release from the Li2MnO3 crystal structure may be involved, given that it was lower than 1.8 wt% considering only Li2MnO3.45
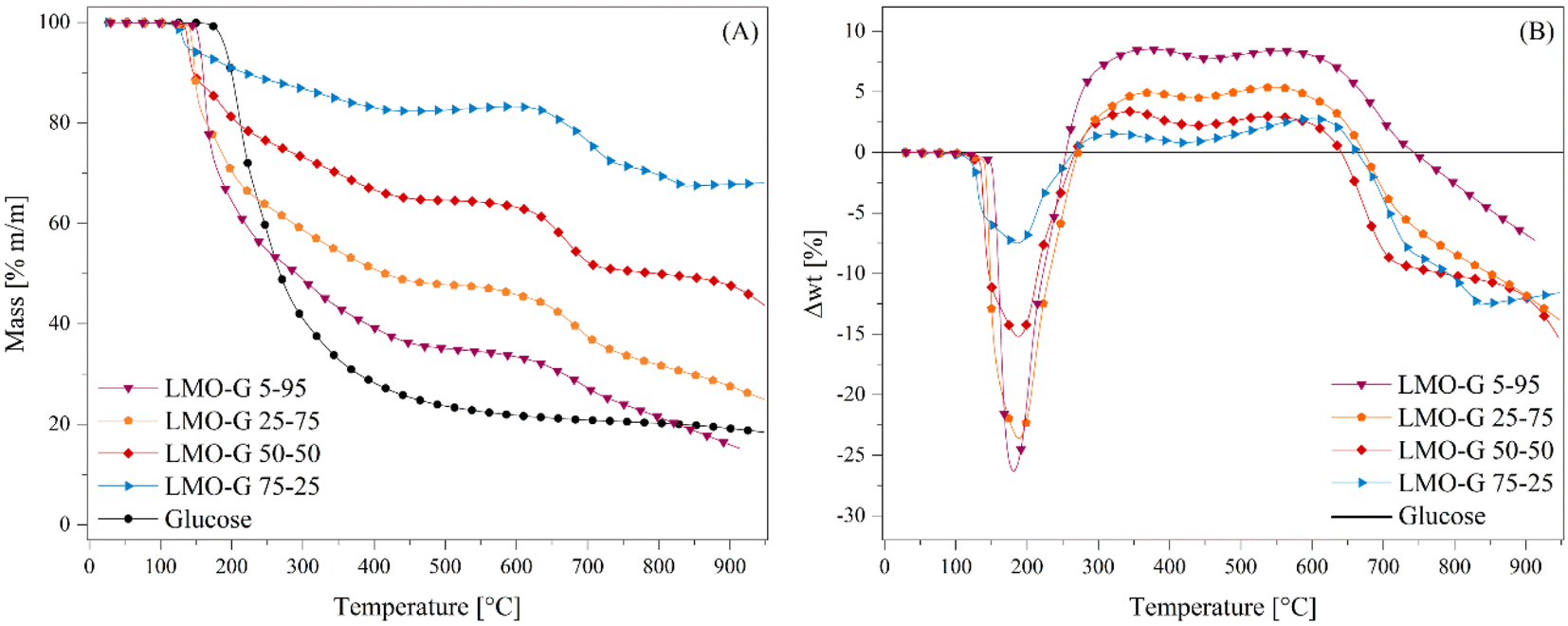 | ||
| Fig. 1 (A) Thermogravimetric analysis of mixtures of different Li2MnO3 and glucose ratios at 5 °C min−1 in N2 flow. (B) Normalized weight loss percentages to glucose content compared with glucose pyrolysis alone (black line at zero position). | ||
To understand the processes involved during the pyrolysis of Li2MnO3:glucose, the expected weight loss of glucose for each sample was determined (Fig. S2†). Moreover, all the thermogravimetric data were treated according to eqn (7) to determine the differences in the weight loss behavior of each sample in comparison to the pristine glucose sample (Δwt%), where wts%(T) and wtg%(T) are the weight percentages of each LMO-G sample and glucose, respectively, as a function of temperature. Moreover, mg/m and mc/m are the mass ratio of glucose and ceramic in each LMO-G sample, respectively.
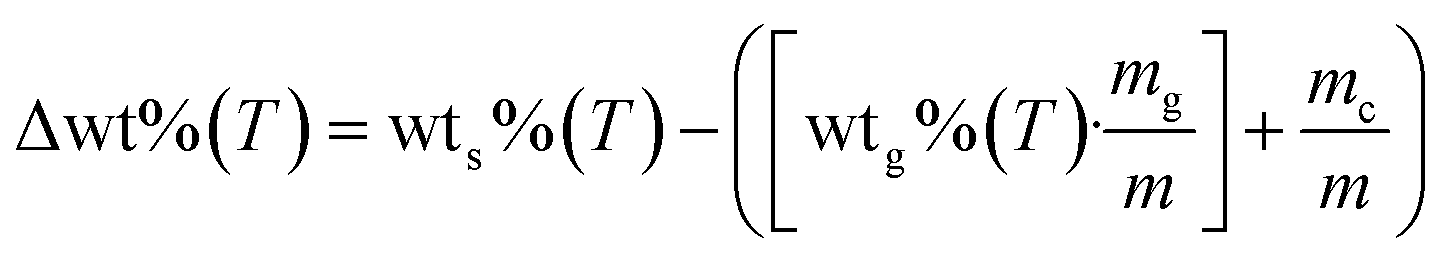 | (7) |
The zero line in Fig. 1B represents the glucose behavior during the pyrolysis process (as described by eqn (7)). Therefore, the negative values between 100 °C and 270 °C can be ascribed to the catalytic effect during the pyrolysis process due to the addition of Li2MnO3, which was narrower in this temperature range and more pronounced with a decrease in the ceramic content. Conversely, the positive values (from 250 °C to 740 °C) must be related to the retained mass that was not released as a gas during the pyrolysis process. It seems that CO and/or CO2 were being chemically sorbed as lithium carbonate (Li2CO3).45,47 It should be noted that the retained amount was higher in the sample with the lowest ceramic content (8.6 wt% for LMO-G 5-95 sample). Moreover, as the Li2MnO3 content increased, the retained gas amounts decreased. It must be considered that the carbon oxide (COx) concentration diminished simply as a consequence of the lower glucose content. Therefore, the solid–gas interphase must be modified, decreasing the CO capture, and finally resulting in lower positive mass percentages.46 Furthermore, two crests are depicted in this temperature range. The first one (∼250 °C and 440 °C) can be ascribed to the surface COx sorption-desorption equilibrium, while the latter (∼440 °C and 740 °C) should involve COx chemical capture controlled by the Li+ and O2− diffusion mechanisms,56 together with the occurrence of some thermal stress.57
Nevertheless, these positive values can be also attributed to a stabilizing process where other compounds derived from glucose could be produced, such as bio-oils.58,59 In this approach, it was observed that between 250 °C and 440 °C the glucose pyrolysis was not finished, and then as the positive values did not continuously increase, some bio-oils were also undergoing several decomposition processes, increasing the production of gas.60 In contrast, from 440 °C to 600 °C, most of the pyrolysis decomposition occurred, implying that bio-oil may be thermally stable in that temperature range. The last observed negative values can be associated with a further decomposition process, i.e., Li2CO3 decomposition involving the loss of oxygen from the crystal structure (reaction (8)) and/or the so-called Boudouard reaction (reaction (9)),61,62 or partial carbon oxidation taking oxygen from the crystal structure (reaction (10)). In this last step, it is also plausible that Li2MnO3 (or a derived solid) can be reacting with formed bio-oils in a partial oxidation reaction, where the oxygen source comes from the ceramic, decreasing the final produced char. This is consistent with the negative value observed in comparison to glucose pyrolysis.
| Li2CO3(s) → Li2O(s) + CO2(g) | (8) |
| C(s) + CO2(g) → CO(g) | (9) |
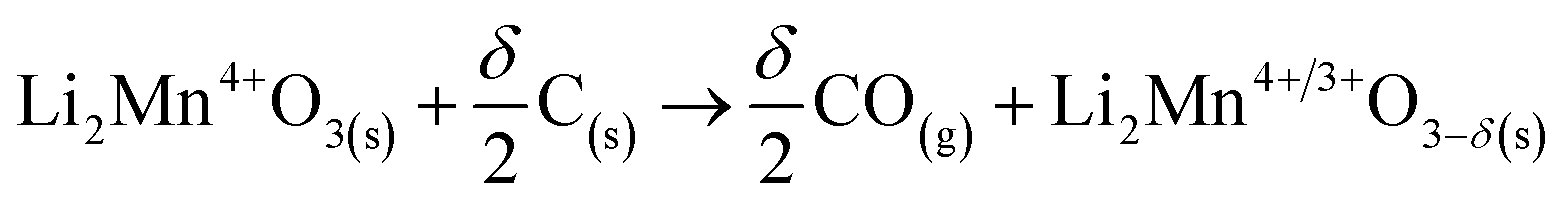 | (10) |
All these TG analyses suggest the end formation of gaseous compounds during the decomposition of glucose. Therefore, the gas evolution was evaluated using the LMO 25-75 sample to determine out the best H2 production conditions. Moreover, HR was modified from 5 °C min−1 to 30 °C min−1 to enhance the production of gases.63 Glucose pyrolysis produced H2, CO, CO2 and CH4, with all different trends depending on the heating rate (Fig. 2). In the case of H2 (Fig. 2A), it was barely produced from 200 °C to 400 °C. Afterwards, its production significantly increased from 400 °C to 850 °C, regardless the heating rate. However, as the heating rate increased, the flow of H2 per g of glucose increased as well, from 3.2 to 14.8 mL min−1 gglucose−1 when using the heating rates of 5 °C min−1 and 30 °C min−1, respectively. Moreover, two peaks were observed during H2 formation, similar to the two crests observed in the thermogravimetric analyses (see Fig. 1B). In fact, these two peaks were more evident as a function of the heating rate, and they were observed at higher temperatures than those using lower heating rates, perhaps due to a non-equilibrium condition.
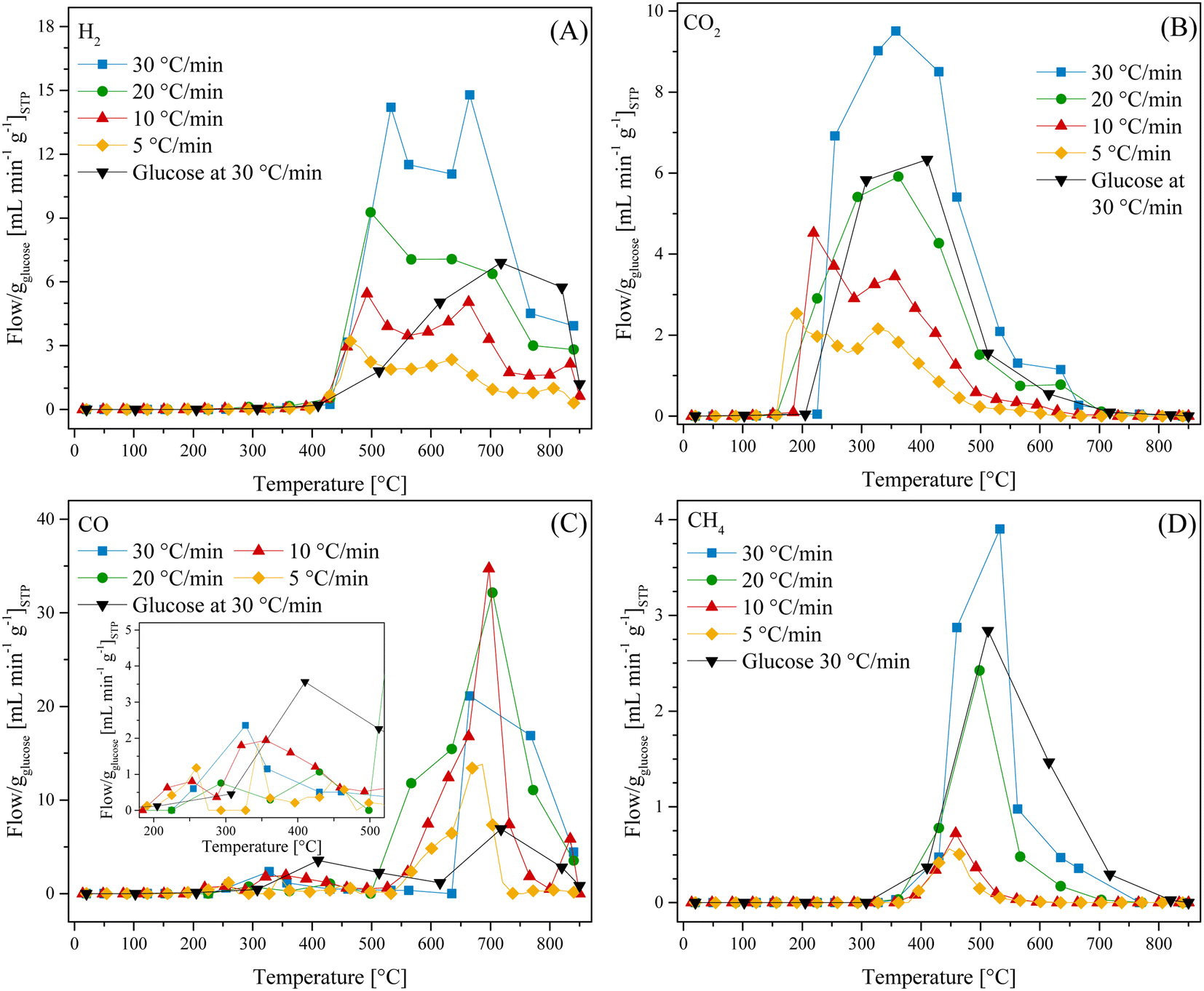 | ||
| Fig. 2 Thermal evolution of different gases (H2 (A), CO2 (B), CO (C) and CH4 (D)) for LMO-G 25-75 and glucose samples from 30 °C to 850 °C using different heating rates (5, 10, 20 and 30 °C min−1), all normalized per gram of glucose. | ||
Similarly, the HR enhanced the CO2 production (Fig. 2B), shifting its maximum to higher temperatures from 2.5 to 9.5 mL min−1 gglucose−1 at 190 °C and 357 °C, respectively. Moreover, the temperature range for CO2 production (150–600 °C) was also shifted by around 80 °C to higher temperatures when the HR was 5 °C min−1 and 30 °C min−1, respectively. It should be noted that CO2 was mainly produced at lower temperatures than H2.
In contrast, CO formation (Fig. 2C) presented interesting behavior. CO was produced starting at 190 °C up to the end of every dynamic analysis. However, it must be pointed out that this production depicted two crests, where the first was between 190 °C and 500 °C with a maximum production of 3.3 mL min−1 of CO per g of glucose at 330 °C for the 30 °C min−1 case. It was observed that CO production was always lower than CO2 production in this temperature range. Moreover, it must be noted that in the case of the first crest, the starting CO production temperature was lower than that where Li2MnO3 can chemisorb it (420–705 °C).45 Then, at higher temperatures (525–850 °C), a second crest was observed, reaching a maximum production at around 700 °C, depending on the ceramic:glucose ratio. For instance, the maximum CO production was depicted using an HR of 10 °C min−1 at 697 °C (34.7 mL min−1 gglucose−1). Besides, as the HR increased from 5 °C min−1 to 20 °C min−1, the CO production significantly increased and this crest seemed to become broader. However, this trend was not observed when using an HR of 30 °C min−1. Furthermore, the second CO production crest matched with H2 production. It must be emphasized that in this temperature range, Li2MnO3 selectively reacts with CO.45,47 In addition, the produced carbon or any possible bio-oil must be mainly oxidized to CO, given that its production is higher than that for CO2, reducing manganese species. In fact, H. Zhang et al.64 reported that MnO2 can be reduced to MnO by reacting with volatiles (tar) from the pyrolysis of biomass, producing H2, CO and some light carbons. At last, CH4 production (Fig. 2D) was observed between 390 °C and 705 °C. In this case, the maximum methane production also increased and right-shifted by 100 °C as a consequence of the increase in the HR, which was less than 1 or 4 mL min−1 gglucose−1 for HR of 5 and 30 °C min−1, respectively.
Based on all these results, the best heating conditions seemed to be 30 °C min−1, given that this condition resulted in the highest H2 production. Thus, for comparison, the glucose sample was tested, as shown in Fig. 2, depicting that the addition of Li2MnO3 not only highly enhanced the H2 production but also decreased its starting production (Fig. 2A). For instance, in the absence of the ceramic, the best H2 production was 6.9 mL min−1 gglucose−1 at 717 °C. Moreover, the addition of Li2MnO3 seemed to modify the production of carbon oxides. In the case of CO (Fig. 2C), in the presence of Li2MnO3 the temperature range of CO production shifted by 80 °C to lower temperatures. Furthermore, its addition modified the CO production in two different stages. In the first stage, between 410 °C and 615 °C, the CO production was lower in the presence of Li2MnO3, matching the temperature range of CO capture in Li2MnO3.45–47 Despite this phenomenon, in the second stage (T > 615 °C), the CO production increased twice in the presence of Li2MnO3. This must be explained due to the release of oxygen from the crystal lattice, which oxidizes the glucose pyrolysis byproducts, diminishing the available oxygen. Moreover, the Boudouard reaction should occur due to CO2 production from carbonate decomposition and as an intermediate from the oxidation-capture of CO.45,47 Additionally, in the case of CO2 (Fig. 2B), the addition of Li2MnO3 increased the overall production and left-shifted the maximum CO2 production in comparison to the pristine glucose. This is also a consequence of CO oxidation produced by Li2MnO3. Then, in the case of CH4 (Fig. 2D), the glucose sample depicted a broader temperature range production, suggesting that addition of Li2MnO3 inhibits CH4 formation.
Complementarily, all the gaseous products were analyzed through an FTIR gas-cell (Fig. S3†). For instance, CO vibration bands were observed at 2120 and 2170 cm−1, whereas CO2 vibration bands were depicted at 670, 2350, 2360, 3600, 3630, 3700 and 3740 cm−1. Moreover, the formation of water was observed (bands at 1500, 1700, 3400 and between 3670 and 3850 cm−1), regardless the implemented heating rate. Furthermore, some vibration bands associated with C–H bonds were identified between 1400 and 1800 cm−1 as well as at 2950 cm−1, all of which were different from the methane vibration bands (1300 and 3010 cm−1). Hence, some bio-oils or organic compounds must be produced during the pyrolysis, which are described as a mixture of aldehydes, ketones, phenols, aromatic compounds, heterocyclic compounds, etc.19,65 Indeed, in the present case, the recovered bio-oils from the LMO-G 25-75 and glucose dynamic experiments at 30 °C min−1 were identified as mixtures of these compounds. These identifications were performed by chromatography coupled to mass spectrometry (Fig. S4†).
To further understand the effect of the HR on the production of gases, the H2/CO ratio was calculated as a function of temperature (Fig. 3A). Fig. 3A shows that in the presence of Li2MnO3, the H2/CO ratio increased in two different stages, between 430 °C and 580 °C and after 720 °C. In the first stage, the H2/CO ratio was not larger than 11.3 for the HR of 5 °C min−1, 10 °C min−1 and 20 °C min−1. However, at 30 °C min−1, the H2/CO ratio was 40.0 at 532 °C. Afterwards, for all HR, the H2/CO ratio diminished significantly (around 0.10 at 700 °C), reaching a second increment stage at 800 °C (between 2 and 3.3). These results show that using a HR of 30 °C min−1 highly increased the H2/CO ratio. The increase in HR induced higher CO capture. Indeed, a decrease in COx partial pressure may compromise its capture by modifying the solid–gas interphase.66–69 Furthermore, by comparing the H2/CO ratio with the that obtained in the glucose case, it depicted a considerably decrement and right shift, which was 4.3 at 615 °C. Consequently, the addition of Li2MnO3 during the pyrolysis process did not only increase the production of H2, but also enhanced its purity.
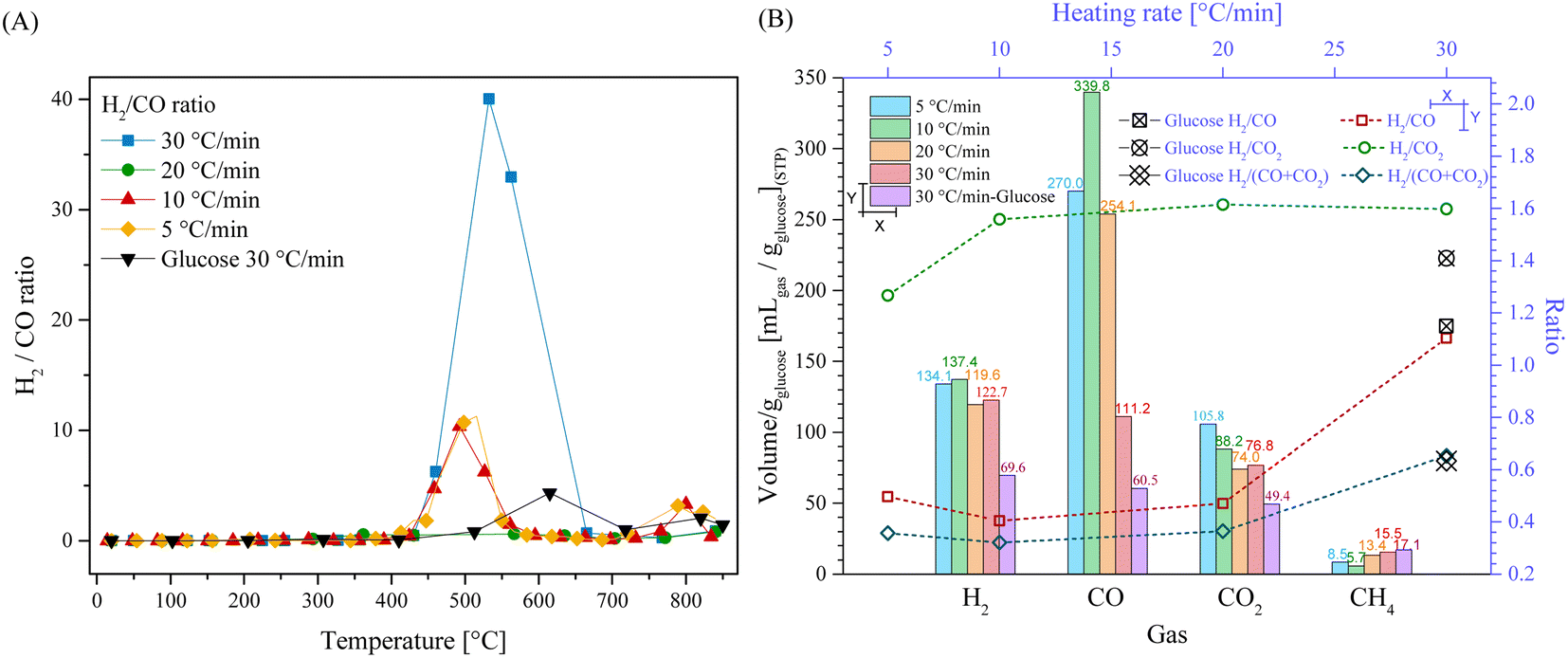 | ||
| Fig. 3 H2/CO ratio evolution from 30 °C to 850 °C for LMO-G 25-75 and glucose samples at different heating rates (A). Total volume of the gaseous products (B) from the corresponding pyrolytic processes, normalized per gram of glucose (bottom and left axes) and the respective H2:COx ratios as a function of the heating rate (top and right axes). | ||
Additionally, considering the whole process, in Fig. 3B the amounts of all produced gases were plotted. In the case of H2, it was shown that the total amount of H2 was not significantly affected (variations lower than 11%) by the heating rate, which was 134.1 mL gglucose−1 at 5 °C min−1 and 122.7 mL g−1glucose at 30 °C min−1. It must be mentioned that the dynamic experiments depicted higher H2 production flows at high HR (see Fig. 2A), although the production over time was similar. This phenomenon must be related to the pyrolysis kinetics, where with an increase in the heating rate, glucose was pyrolyzed faster, preventing equilibrium being reached over the respective gaseous products. This trend suggests that at certain temperatures, different products were obtained in higher amounts, although final equilibrium over the H2 production must be limited by thermodynamics or by the specific path of pyrolysis decomposition when Li2MnO3 was added to the process.
On the contrary, the thermodynamic equilibrium during CO and CO2 production was highly affected by the HR after the addition of Li2MnO3. For instance, the CO production was 339.9 and 270.0 mL gglucose−1 at 10 °C min−1 and 5 °C min−1. Conversely, the CO production diminished at higher heating rates, which was 254.1 and 111.2 mL gglucose−1 at 20 °C min−1 and 30 °C min−1, respectively. These results suggest that at low heating rates (5 and 10 °C min−1), the interaction between Li2MnO3 and biomass induced the higher release of oxygen from the crystal lattice structure. These implications between the two lowest HR also induced a reduction in the oxygen available for the formation of CO2, as evidenced by the CO2 reduction from 105.8 to 88.2 mL gglucose−1 at 5 °C min−1 and 10 °C min−1, respectively. Simultaneously, at HRs of 20 and 30 °C min−1, the CO amounts considerably diminished as well as the CO2 amounts, which were ∼28.7% lower. In fact, at 30 °C min−1, the total CO and CO2 amounts were 111.2 and 76.8 mL gglucose−1, respectively. Besides, CH4 formation increased as a function of HR from 8.5 to 15.5 mL gglucose−1 at 5 °C min−1 and 30 °C min−1, respectively, except for the HR of 10 °C min−1, which is associated with the large selectivity for glucose pyrolysis to CO. Based on these results, the increase in HR must kinetically enhance the release of oxygen and induce the formation of CH4. Moreover, as a reason for the non-equilibrium conditions, at higher HRs, the material must have achieved high temperatures faster than the release of CO from the pyrolysis process, favoring CO capture.
Moreover, all the H2/COx ratios were evaluated at different heating rates (Fig. 3B). The results depicted that the best ratios were observed using an HR of 30 °C min−1. Conversely, other HR conditions resulted in similar H2/CO and H2/(CO + CO2) ratios of around 0.5 and 0.36, respectively. Moreover, the H2/CO2 ratios were similar for all the HR (around 1.59) except for that of 5 °C min−1 (1.26).
Then, by comparing the gas amounts from LMO-G 25-75 with that from pristine glucose, it was evident that the H2, CO and CO2 amounts were greater in the presence of Li2MnO3. In fact, the addition of the ceramic increased the H2 and CO amounts by 1.8 times, while increasing the CO2 amount by 1.6 times. Meanwhile, the addition of Li2MnO3 decreased the formation of CH4. Complementarily, the H2/CO and H2/COx ratios for the glucose sample were not very different in comparison to that obtained for the LMO-G 25-75 sample. In the case of H2/CO2, it increased due to the addition of Li2MnO3, mainly due to the increase in H2 production. All these results indicate that Li2MnO3 modified the mechanism of glucose pyrolysis, enhancing H2 production and inducing the formation of carbon oxides, but significantly increased the H2/CO ratio within a specific temperature range (430–580 °C), while diminishing the production of other hydrogenated compounds (H2O, CH4 and tar).
Based on all the previous experiments, the heating rate of 30 °C min−1 was selected to further study the effect of Li2MnO3 on the pyrolysis process. Thus, different LMO-G mixtures were prepared with an increasing amount of glucose (Table 1). H2, CO, CO2 and CH4 flows, normalized per g of glucose, are presented in Fig. 4. As can be observed, the Li2MnO3 content increased the production of H2 (Fig. 4A) from 8.3 to 14.2 mL min−1 gglucose−1 at 532 °C for 5 and 25 wt% of Li2MnO3, respectively. This trend must be related to the increase in the surface contact between glucose and Li2MnO3, enhancing the selectivity of glucose decomposition towards H2.
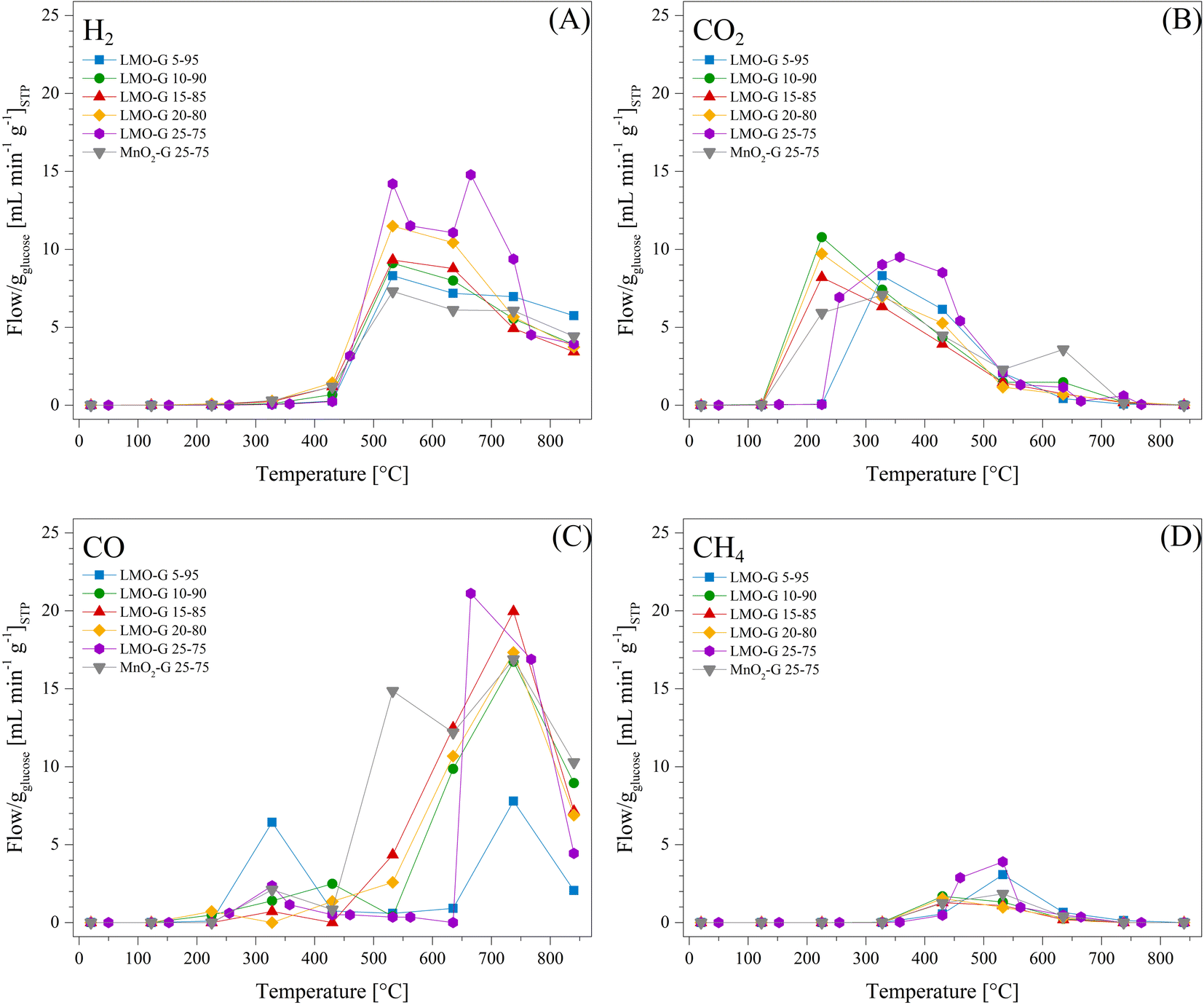 | ||
| Fig. 4 Thermal evolution of the produced gases ((A) H2, (B) CO2, (C) CO and (D) CH4) for LMO-G and MnO2-G samples, from 30 °C to 850 °C in N2, at a heating rate of 30 °C min−1. All samples normalized per gram of glucose. | ||
On the other hand, the production of carbon oxides depicted slight differences depending on the Li2MnO3 content. In the case of CO2, it was produced from 122 °C to 840 °C, reaching the maximum at around 225 °C (Fig. 4B). It must be noted that the CO2 production shifted to higher temperatures for the 5 and 25 wt% Li2MnO3 samples. In fact, the 5 wt% sample was shifted to 327 °C due to its low Li2MnO3 content. This temperature is closer to the peak CO2 production temperature in the case of the glucose sample (410 °C). So, the 25 wt% sample shifted to 357 °C given that part of the CO2 produced is due to the oxidation of CO. These data show that low amounts of ceramic tended to behave similarly to the gas evolution of glucose decomposition, as expected.
In addition, during the production of CO (Fig. 4C), again it depicted two different behaviors, depending on the ceramic content. In the first CO production trend (between 225 °C and 430 °C), as the lower the ceramic content, the higher the CO production. In contrast, in the second CO production thermal range, the CO production increased inversely to the percentage of Li2MnO3. These phenomena must be intertwined as a function of CO oxidation-capture and oxygen availability during pyrolysis, given that both processes were favored with Li2MnO3. However, as the oxygen availability increased, the capability of Li2MnO3 to react with some byproducts increased. Alternatively, it is also plausible that CO production during the second thermal process was enhanced due to the higher production of lithium carbonate, which reacts with char.
In the case of CH4, the samples with 10, 15 and 20 wt% of Li2MnO3 depicted shifts to lower temperatures and diminished production. Moreover, they all produced lower CH4 amounts than glucose. In a previous study, it was evidenced that CH4 formation is not favored by Li2MnO3 in the presence of H2 and CO.47 Hence, this result implies that CH4 formation from glucose pyrolysis is mainly related to the formation of carbon oxides. In fact, the formation of methane is favored once the available oxygen diminished. Moreover, given that the formation of CH4 increased in the samples with higher H2 production, a methanation process (reaction (11)) must be involved.70,71
| 2H2(g) + C(s) → CH4(g) | (11) |
Complementarily, it was necessary to elucidate the effect of lithium, determining if the described behaviors were only related to the catalytic effect of manganese or also the properties of lithium. It must be mentioned that lithium oxide (Li2O) was not complementarily tested due to its high corrosivity and toxicity levels, which would discourage its use. Hence, MnO2 was evaluated under the same conditions where the highest H2 production was obtained (25 wt% ceramic content and HR of 30 °C min−1). This sample was labeled as MnO2-G 25-75. Fig. 4 shows that using MnO2 maintained the H2 production thermal range, but decreased its production and concentration in comparison to the Li2MnO3-containing samples. Conversely, CO production depicted a triple peak behavior, where the first was slightly lower than that observed for the LMO-G 25-75 sample. The second peak showed a significantly higher CO production at 532 °C in comparison to all the Li2MnO3-containing samples. Then, the third CO production peak matched the temperature of the second peak for the Li2MnO3-containing samples, where the CO production was lower in the case of MnO2. It must be mentioned that in comparison to LMO-G 25-75, the CO2 production by the MnO2-G sample was higher at T ≤ 225 °C, but it became lower between 252 °C and 532 °C, then increasing again at higher temperatures.
In the case of MnO2-G, the behaviors of CO and CO2 seem to be intertwined, given that the CO2 production was higher at low temperatures (T ≤ 225 °C), which is probably due to the increase in the availability of surface oxygen. Conversely, as the temperature increased (from 252 °C to 532 °C), the availability of surface oxygen may decrease, diminishing the oxidation of CO. Moreover, given that MnO2 did not exhibit COx capture capabilities, the CO production was higher than that using Li2MnO3. Then, at higher temperatures (T > 532 °C), a carbon source (char or tar) and/or CO were oxidized to CO2 through the core oxygen lattice of MnO2.
In the case of CH4, the implementation of MnO2 diminished the methane production, but its formation temperature range fitted with the highest H2 and CO formations. It must be noted that in the presence of any Mn4+ source, namely MnO2 or Li2MnO3, the methane formation was lower in comparison to the glucose sample. Consequently, the reaction pathway for the formation of carbon oxides induced H2 production, both modifying the formation of CH4. Indeed, COx production led to the low formation of methane and carbon. However, the increase in H2 production and high temperatures in the presence of carbon induced a weak methanation reaction.70,71 Based on all these results, it was indirectly determined that the presence of lithium in the crystal structure of Li2MnO3 did promote higher H2 production, obviously resulting in COx capture, and thus better H2/COx selectivity.
For comparison, Fig. 5 shows the overall production of gases normalized to g of glucose for all the LMO-G samples. The total amount of H2 significantly increased with an increase in the content of Li2MnO3 content from 69.2 to 122.7 mL gglucose−1. Moreover, the sample with the lowest content of Li2MnO3 (LMO-G 5-95) produced a similar H2 amount to that of the glucose sample (around 69.6 mL gglucose−1), as was expected. Alternatively, MnO2-G produced only 79.4 mL gglucose−1, which is significantly lower than that by Li2MnO3 with the same mass content, indicating that the Li-based ceramic did present improvements in H2 production.
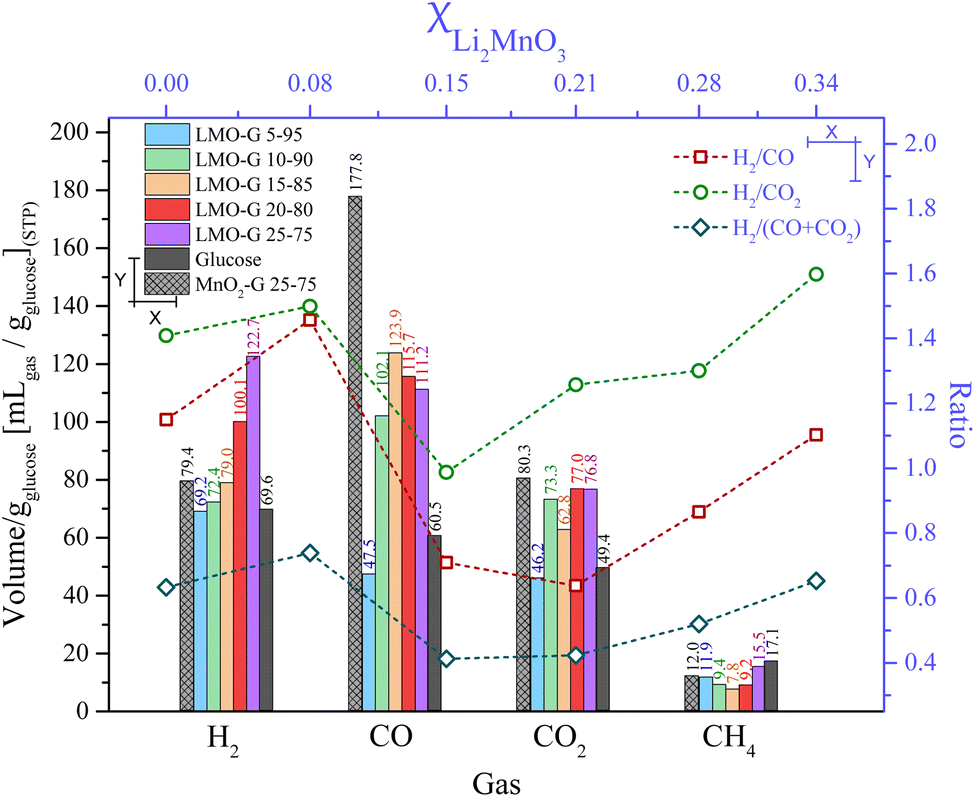 | ||
| Fig. 5 Total volume of the gaseous products from pyrolytic processes of the LMO-G, MnO2-G and glucose samples, all normalized per gram of glucose (bottom and left axes), as well as H2 and carbon oxide ratios obtained as a function of molar ratio (top and right axes). | ||
As can be observed, the LMO-G and MnO2-G samples showed higher CO and CO2 production in comparison to the pristine glucose. Furthermore, when the production of carbon oxides by MnO2-G and LMO-G (both at 25 wt%) was compared, it can be observed that the CO and CO2 production by the LMO-G sample was lower by 37.4% and 4.4%, respectively. All these results show that the Li2MnO3 content, even if the H2 production was not dramatically enhanced, diminished the overall production of carbon oxides during the pyrolysis of glucose.
Lastly, in the case of CH4, it diminished from 11.9 to 7.8 mL gglucose−1 for the LMO-G 5-95 and LMO-G 15-85 samples. Afterwards, it increased up to 15.5 mL gglucose−1 for the LMO-G 25-75 sample. Besides, the MnO2-G sample produced lower CH4 amounts (12.0 mL gglucose−1) than the LMO-G 25-75 sample. It must be noted that regardless of the manganese source and weight content, the CH4 amount was always lower than that obtained from the glucose sample. Hence, the hydrogen available from glucose to produce CH4 is reduced, modifying the reaction pathways in which glucose decomposes.
In addition, Fig. 5 shows the overall H2/COx ratio as a function of the molar fraction of Li2MnO3, depicting that the highest H2/CO ratio (1.45) was observed for the sample with the lowest proportion of ceramic (LMO-G 5-95, XLi2MnO3 = 0.08). At high Li2MnO3 contents, the H2/CO ratios were not as high, reaching the minimum at 0.21 molar content (15 wt% of ceramic) and increasing again with the ceramic content. This trend must be related to the capability of Li2MnO3 to release some oxygen at high temperatures (T > 700 °C),45 inducing the partial oxidation of the carbon species to CO during the pyrolysis of glucose. This statement is supported by the previous dynamic experiments, where CO was mainly produced over 650 °C (see Fig. 4C). Alternatively, the H2/CO2 ratios depicted an interesting behavior as a function of the molar fraction of ceramic. At a ceramic molar fraction of 0.08, the H2/CO2 ratio was slightly higher in comparison to the glucose sample (1.50 and 1.40, respectively). At the molar fraction of 0.15, the H2/CO2 ratio decreased to 0.99. Then, higher Li2MnO3 contents tended to increase the H2/CO2 ratio up to 1.60. Finally, the H2/(CO + CO2) ratio displayed a similar trend as that of H2/CO2. As it was previously discussed, the formation of carbon oxides must be intertwined with the ceramic content, favoring the CO oxidation-capture process.
Based on all the previous sections, the LMO-G 25-75 sample was selected to perform the dynamic-isothermal experiments. Fig. 6 shows the dynamic-isothermal profiles of the gaseous products in the range of 500 °C to 700 °C. Herein, most of the gaseous products were mainly obtained during the heating process, such as H2, CO2 and CH4. H2 production (Fig. 6A) in all cases started at the sixth minute (T ≥ 240 °C), reaching the maximum at the respective target temperature. It must be noted that H2 was mainly produced during the initial 31 min, and after that, its production decreased to around 0.5 mL min−1 g−1, regardless of the isothermal target. Moreover, given that all these measurements were carried out at different fixed isothermal temperatures, the best H2 production was obtained in the isothermal experiment performed at 500 °C, in good agreement with the dynamic experiments.
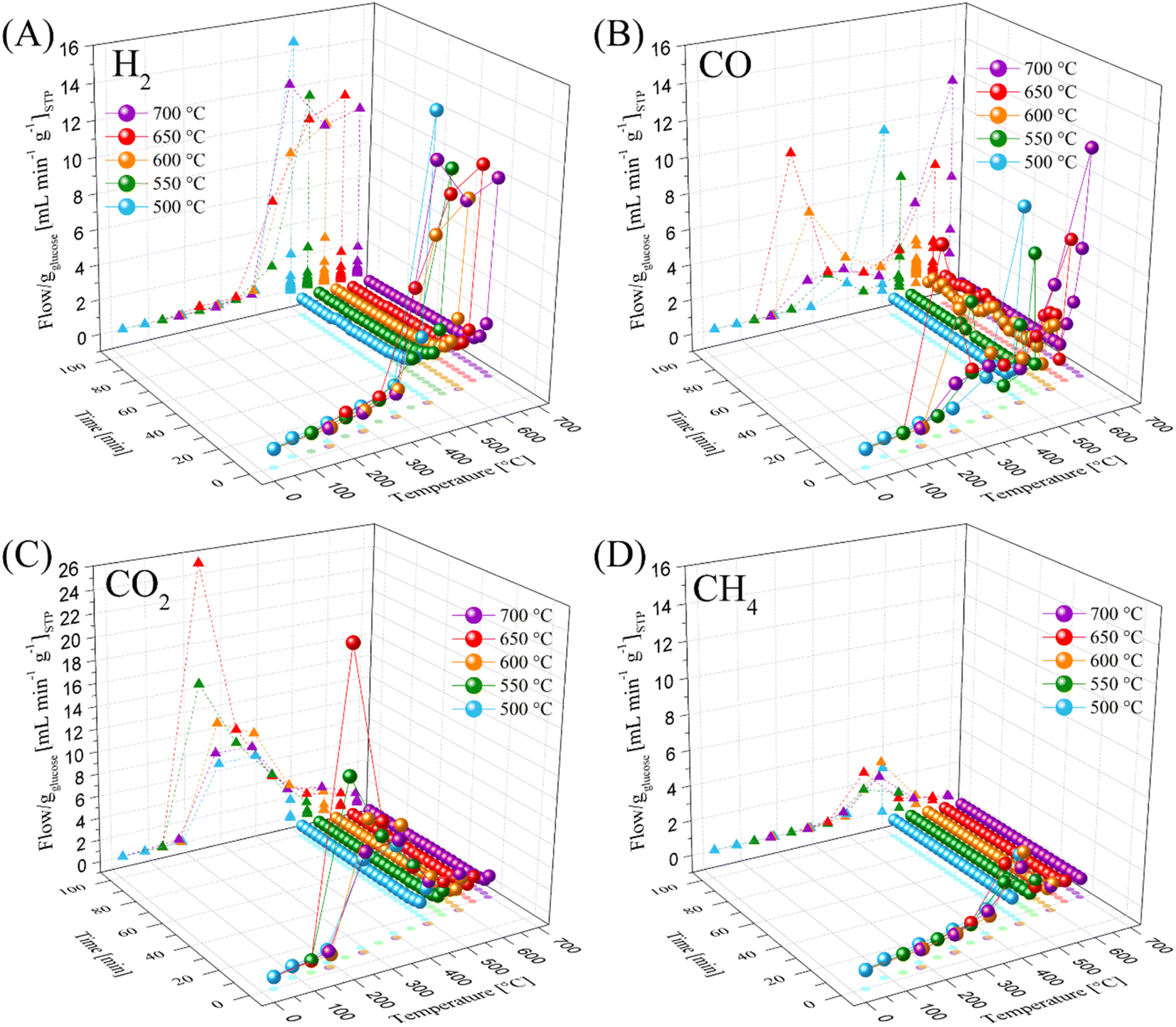 | ||
| Fig. 6 Thermal and time gas evolution of H2 (A), CO (B), CO2 (C) and CH4 (D) for LMO-G 25-75 sample, during different dynamic-isothermal processes between 500 °C and 700 °C, normalized per gram of glucose. Spheres and solid lines represent gas flow as a function of temperature and time, respectively. For easier visualization, two projections were carried out, flow as a function of temperature (triangles and dotted lines) and temperature as a function of time (shadowed circles). | ||
In the case of CO (Fig. 6B), three production peaks were observed at 240 °C, 500 °C and 700 °C. Moreover, the lowest CO productions were observed at around 400 °C and 600 °C. It must be noted that CO production was presented within the same temperature range for the oxidization and chemical capture of CO in the Li2MnO3.45 Alternatively, the CO production significantly diminished after ∼20 min of the isothermal step. It must be noted that at the highest temperatures, CO production was observed after H2 production was completed, showing that Li2MnO3 reacted with one of the products of the glucose pyrolysis, for instance char, inducing the partial reduction of Mn4+.
Complementarily, the main CO2 production (Fig. 6C) was observed after 3 min (T ≥ 130 °C), reaching the maximum at 240 °C. Then, the CO2 production significantly decreased as a function of temperature and time, becoming negligible after 24 min. It must be mentioned that below 450 °C, CO2 production was larger than that of CO. Perhaps, the thermal decomposition of glucose shifted to the formation of water and carbon dioxide at moderate temperatures (T ≤ 450 °C) due to the large oxygen content. Additionally, CH4 production (Fig. 6D) was observed after the initial 10 min (T ≥ 340 °C), reaching the maximum at around 500 °C (15.6 min). In contrast to the H2 results, CH4 formation was negligible once the isothermal target temperatures were reached, indicating a weak methanation process.70,71
Fig. 7 depicts the overall gases production and H2/COx ratios depending on the isothermal target temperatures before starting the isothermal steps (Fig. 7A) and after the whole process (Fig. 7B). In fact, the overall production of all the gases increased as a function of temperature (Fig. 7A). Specifically, in the experimental sections reaching 500 °C and 700 °C, the H2 and CO production amounts depicted the highest increase. In the case of H2, these values increased from 19.7 to 90.3 mL gglucose−1, whereas in the case of CO, from 17.7 to 54.2 mL gglucose−1. It must be noted that although the CO2 production increased as a function of temperature, the increment was not higher than 13% in the tested range, which was 81.7 to 91.9 mL gglucose−1 between 500 °C and 700 °C. Again, as was explained, most of the CO2 production occurred below 430 °C. In contrast, the CH4 production merely increased from 7.4 to 11.1 mL gglucose−1 at 500 °C and 700 °C, respectively. Thus, CH4 formation did not significantly change as a function of the initial step, i.e., the dynamic process.
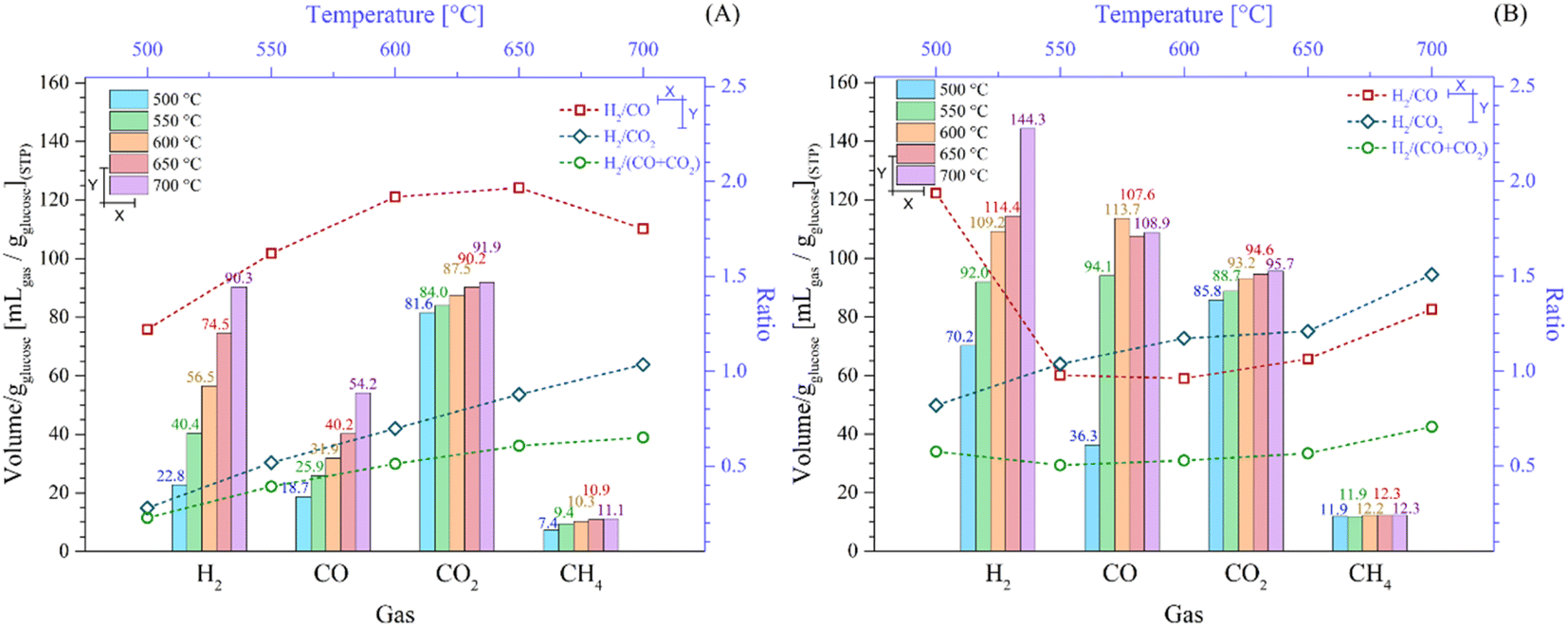 | ||
| Fig. 7 Total volume of the gaseous products from dynamic-isothermal process (bottom and left axes) of the LMO-G 25-75 sample between 500 °C and 700 °C after the heating dynamic steps (A) and after the whole process (B), all normalized per gram of glucose. Corresponding H2 and carbon oxide ratios are presented as a function of the target temperature (top and right axes). | ||
Additionally, Fig. 7A shows the H2/COx ratios once the target temperature was reached. All the H2/COx ratios increased as a function of the final dynamic temperature, except for the H2/CO ratio at the highest final dynamic temperature. In the case of H2/CO, it increased from 1.22 at 500 °C to 1.97 at 650 °C, diminishing to 1.75 at 700 °C. This sudden change in the CO increment was related to the oxygen release from Li2MnO3, which is produced at high temperature, as already described. On the contrary, the H2/CO2 ratio increment trend (from 0.28 at 500 °C to 1.04 at 700 °C) must be related to the fact that CO2 is mostly produced at low temperatures (T < 430 °C). In contrast, when both carbon oxides were considered (H2/COx ratio), it equally increased with temperature from 0.23 at 500 °C to 0.65 at 700 °C, given that it was simply the addition of the two previous cases. Based on these results, it would be of interest to avoid collecting gases below 500 °C to diminish the CO2 and CO contents by 88.9% and 32.7%, respectively, although it would diminish the H2 amount by 21.8% during the dynamic heating process reaching 700 °C.
Fig. 7B shows the production of gases once all the processes were completed, including isothermal sections. It was observed that the amount of H2 significantly increased in comparison to the dynamic heating steps, for example 90.3 and 144.3 mL gglucose−1 in the experiment performed at 700 °C before and after the isothermal step, respectively. In fact, it corresponds to an increment of 59.9%. These results indicate that some of the pyrolytic glucose byproducts decomposed during the isothermal steps and/or because of the pyrolysis process continuing at the target temperature. Likewise, the isothermal steps enhanced the CO amounts, depicting relevant behavior. For instance, in the experiments performed isothermally at 500 °C, 550 °C and 600 °C, the CO production was 36.3, 94.1 and 113.7 mL gglucose−1, respectively. Afterwards, it diminished to 107.6 and 108.9 mL gglucose−1 for the isothermals steps performed at the two highest temperatures (650 °C and 700 °C), respectively. All these CO production amounts are larger than that observed during the dynamic heating steps. Thus, during the isothermal steps, Li2MnO3 promoted the reactions with the pyrolytic byproducts through the release of oxygen. Conversely, the CO2 production only increased by around 6.5% after the isothermal processes in comparison to that produced during the dynamic heating steps. Thus, the oxygen released from the material mostly produced CO. Finally, the CH4 production was almost the same (difference lower than 3.3%), regardless of the isotherm temperature. Therefore, methane was produced in the same amount from the pyrolysis of glucose as long as the process was carried out at T ≥ 500 °C due to the weak methanation reaction.
Complementarily, all the H2/COx ratios, including the dynamic and isothermal steps, are shown in Fig. 7B. The H2/CO2 ratio was the only one that increased with temperature from 0.82 at 500 °C to 1.51 at 700 °C. This phenomenon must be related to the poor CO2 production during the isothermal step in comparison to the enhanced H2 production. Alternatively, the H2/CO ratio diminished from 1.94 to 0.96 at 500 °C and 600 °C, respectively. Then, the H2/CO ratio increased again to 1.33 at 700 °C. In the experiments performed isothermally at 650 °C and 700 °C, the ceramic was capable of capturing CO faster and more efficiently than in the lower isothermal experiments, although the production of CO increased as well during the isothermal tests. In the case of H2/COx, its value was around 0.55 in the experiments between 500 °C and 650 °C, slightly increasing to 0.71 at 700 °C. This trend was clearly marked by the larger CO production in comparison to the CO2 amounts, which was greater than the H2 concentration.
Complementarily, to identify the solid evolution as a function of temperature, the dynamic-isothermal solid products were recovered and characterized by XRD and ATR-FTIR (Fig. 8). According to the XRD results (Fig. 8A), it can be observed that the glucose signals were not detectable in either of the solid products. Conversely, Li2MnO3 evolved as a function of the isothermal temperature. At 500 °C, Li2MnO3 seemed to be the main crystalline phase, although other crystal phases were detected, i.e., t-LiMnO2, Li1+xMn2−xO4, o-LiMnO2 and MnO, evidencing the partial reduction of Mn4+ ions (Li2MnO3) to Mn3+ (LiMnO2) and Mn2+ (MnO). Furthermore, the presence of t-LiMnO2 and spinel Li1+xMn2−xO4 is consistent with the superficial activation of Li2MnO3.45 Nevertheless, at T > 500 °C, Li2MnO3 was not detected, only LiMnO2, MnO and Li2CO3, confirming the chemical capture of COx. Furthermore, at T ≥ 650 °C, the formation of Li2O and MnO and the presence of SiO2 (quartz wool used as support in the implemented reactor) were identified. Complementarily, the FTIR spectra of the same solid products are presented in Fig. 8B. The vibration bands observed for this sample were assigned as follows: 3800–3000 cm−1 to OH stretching, 2960–2860 cm−1 to CH stretching, 1880–1590 cm−1 to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, 1525–1345 cm−1 to COC (ester) and COH (alcohol) deformations, 1165–965 cm−1 to CO and CC stretching, and 860–590 cm−1 to CH out-plane vibrations.72–75 Moreover, the presence of carbonates was identified for all the dynamic-isothermal products (vibration bands at 1500–1400 and 800 cm−1),76,77 confirming the formation of carbonates, for instance Li2CO3. In fact, Li2CO3 was detected in all the samples, even in those where it was not clearly identified by XRD. Furthermore, all the samples depicted a vibration band in the range of 2392–2297 cm−1, which was assigned to environmental CO2. All these results corroborate the fact that Li2MnO3 can trap COx while it chemically evolves. Only at 500 °C Li2MnO3 acted as a catalyst, while at the other temperatures, it catalyzed and reacted with COx. The Li2MnO3 to LiMnO2 evolution must contribute to the limited CO capture, given that the latter has slower kinetics.45 Moreover, the partial reduction of Mn4+ to Mn3+ and Mn2+ was identified, with the corresponding release of oxygen, evidencing that the enhancement in H2 production is due to a partial gasification reaction rather than a catalytic effect during the pyrolysis process.
O stretching, 1525–1345 cm−1 to COC (ester) and COH (alcohol) deformations, 1165–965 cm−1 to CO and CC stretching, and 860–590 cm−1 to CH out-plane vibrations.72–75 Moreover, the presence of carbonates was identified for all the dynamic-isothermal products (vibration bands at 1500–1400 and 800 cm−1),76,77 confirming the formation of carbonates, for instance Li2CO3. In fact, Li2CO3 was detected in all the samples, even in those where it was not clearly identified by XRD. Furthermore, all the samples depicted a vibration band in the range of 2392–2297 cm−1, which was assigned to environmental CO2. All these results corroborate the fact that Li2MnO3 can trap COx while it chemically evolves. Only at 500 °C Li2MnO3 acted as a catalyst, while at the other temperatures, it catalyzed and reacted with COx. The Li2MnO3 to LiMnO2 evolution must contribute to the limited CO capture, given that the latter has slower kinetics.45 Moreover, the partial reduction of Mn4+ to Mn3+ and Mn2+ was identified, with the corresponding release of oxygen, evidencing that the enhancement in H2 production is due to a partial gasification reaction rather than a catalytic effect during the pyrolysis process.
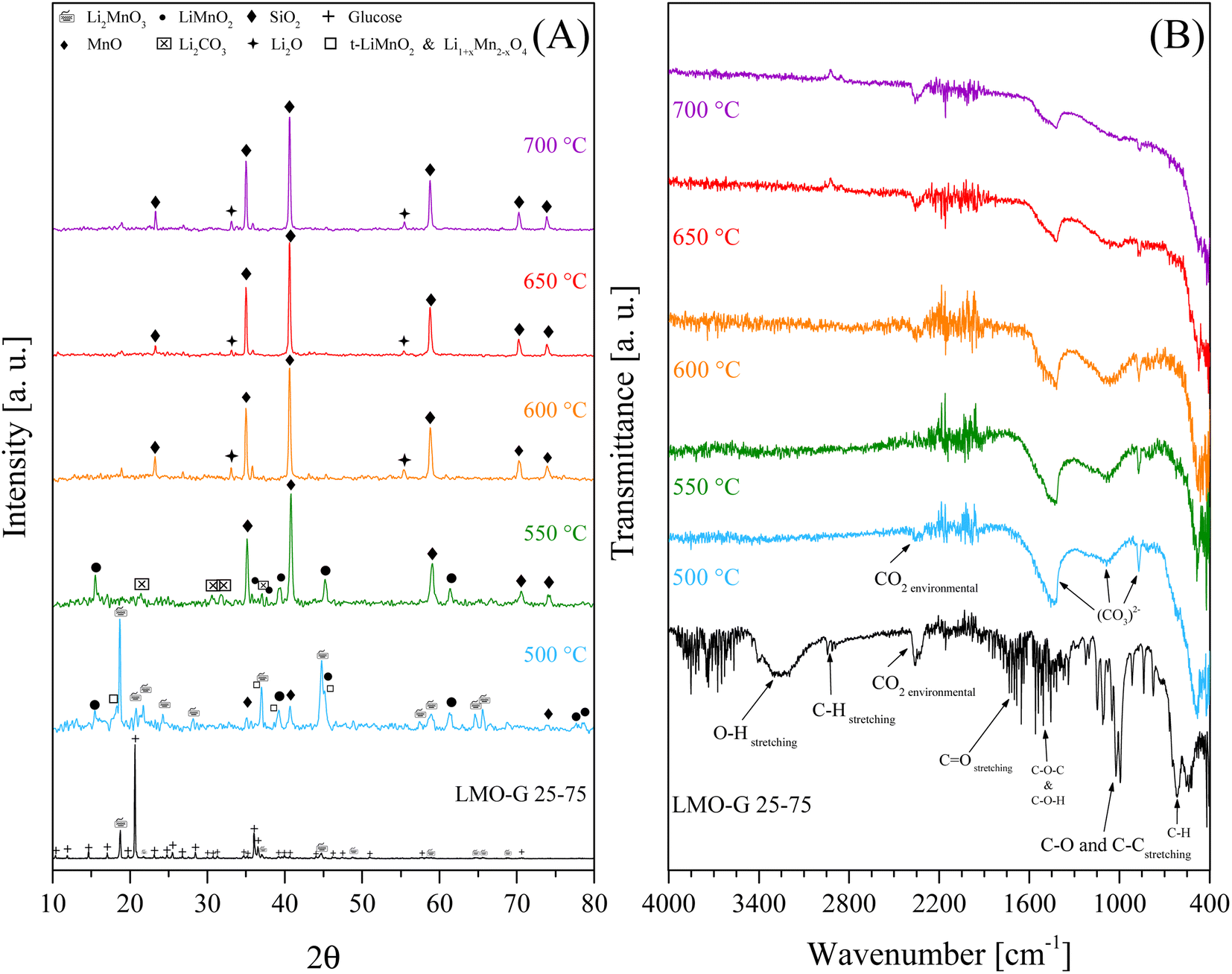 | ||
| Fig. 8 XRD patterns of the solid products of dynamic-isothermal processes using LMO-G 25-75 (A) and respective ATR-FTIR spectra (B), where LMO-G sample is included for comparison. | ||
The following reactions for the pyrolysis of glucose are proposed in the absence and presence of Li2MnO3 (reactions (12)–(15)) based on the thermogravimetric, chromatographic, XRD and infrared analyses.
| C6H12O6(s) → 0.56H2(g) + 0.49CO(g) + 0.40CO2(g) + 0.14CH4(g) + dH2O(l) + C2.0H10.32−2dO4.71−d(l) + 2.97C(s) | (12) |
In the pyrolysis of glucose, in the absence of ceramic (reaction (12)), the assigned coefficients of all the gas compounds were determined by GC (see Experimental section). The carbon (C) coefficient was determined by the final weight observed during the dynamic TG (see Fig. 1A), given that only carbon is expected to be formed due to the high temperature reached (850 °C). It must be mentioned that this proposal was made considering the qualitative identification of water (observed in the cooler and by FTIR). In addition, the bio-oils determined by GC-mass spectrometry (Fig. S4†) were not quantified. Therefore, it was proposed that an unknown organic compound, C2.00H10.32−2dO4.71−d, representing this bio-oil mixture, be used to adjust this equation by introducing the coefficient factor “d” for water and this unknown organic compound.
Likewise, for the proposed reaction of glucose pyrolysis in the presence of Li2MnO3 (reactions (13)–(15)), some considerations must be noted. In this case (LMO-G 25-75 sample), the proposed reaction was considered at different temperatures due to the differences in the gas phase composition and solid products. Specifically, the first reaction for the dynamic-isothermal process performed at 500 °C (reaction (13)), the second for the dynamic-isothermal process performed at 700 °C (reaction (14)) and the last for the whole dynamic process up to 850 °C (reaction (15)).
| C6H12O6(s) + 0.51Li2MnO3(s) → 0.56H2(g) + 0.29CO(g) + 0.69CO2(g) + 0.10CH4(g) + dH2O(l) + C4.92−h−w−δwH10.48−2dO4.84−d+w−(0.51)δ(l) + w(1 − δ)Li2CO3(s) + (0.51 − w)Li2MnO3−δ(s) + 2δwLiMnO2(s) + w(1 − 2δ)MnO(s) + h C(s) | (13) |
| C6H12O6(s) + 0.51Li2MnO3(s) → 1.16H2(g) + 0.88CO(g) + 0.77CO2(g) + 0.10CH4(g) + dH2O(l) + C4.25−h−wH9.28−2dO4.09−d−2w(l) + hC(s) + (0.51 − w)Li2O(s) + wLi2CO3(s) + 0.51MnO(s) | (14) |
| C6H12O6(s) + 0.51Li2MnO3(s) → 0.99H2(g) + 0.89CO(g) + 0.62CO2(g) + 0.12CH4(g) + dH2O(l) + C4.37−hH9.54−2dO4.38−d(l) + hC(s) + 0.51Li2O(s) + 0.51MnO(s) | (15) |
In all these cases, the gas products (H2, CO, CO2, and CH4) were determined by GC, while the solid products were identified by XRD or ATR-FTIR. It must be explained in detail that the coefficient factor “w” is related to the lithium content in the solid products. Thus, it must not be higher than 0.51 mol. In addition, to adjust the reactions, an unknown organic compound, CxHyOz, was proposed, which represents the bio-oils. Here, the coefficient factors “h”, “w” and “d” are related to the C, H and O relationship in the different gaseous and solid compounds to fit the reaction, respectively. In contrast to the case of glucose, the carbon coefficients were not fitted to the thermogravimetric analyses due to the oxidation of this species by the oxygen from the Li2MnO3 framework or through a reaction with lithium carbonate. Then, at 500 °C, given that Li2MnO3 was identified as well as solid products (see Fig. 8A), it was proposed that this activated crystal phase id Li2MnO3−δ. Conversely, at 700 °C and 850 °C, considering that all the manganese cations (Mn4+) must be reduced at the end of the pyrolysis process into Mn2+, MnO must exhibit the same molar proportion to the LMO-G 25-75 sample (0.51 mol of manganese).
Based on all the previous results, this study was expanded. Li2MnO3 was tested for the pyrolysis of cellulose using a mass ratio of ceramic of 0.3333 (or 25 wt%, sample labelled as LMO-C). Fig. 9 shows the dynamic thermogravimetric analyses of the LMO-C and cellulose samples at 30 °C min−1 in an N2 flow (60 mL min−1). In the case of cellulose, two different weight loss (steps) were observed, where the first one depicted from 70 °C to 140 °C (around 1.0 wt%) is attributed to water evaporation. Then, most of the mass was lost (∼83.2 wt%) in a single and continuous step, similar to the glucose behaviour but at a higher temperatures (between 230 °C and 415 °C). At T > 415 °C, the cellulose sample slowly lost 6.4 wt%, being the final solid product char. In contrast, in the presence of Li2MnO3, the process was apparently modified from two steps into three well-defined steps. Indeed, the initial weight loss was merely the dehydration process described above. Then, the second thermal step was slightly shifted (∼15 °C) to lower temperatures. This weight loss slowly diminished until stabilized from 560 °C to 650 °C. Thus, after that weight stabilization, a third decomposition process was observed from 650 °C to 820 °C. All these behaviors are similar to that observed as a consequence of the addition of Li2MnO3 to the pyrolysis of glucose, suggesting that similar processes occurred during the pyrolysis of glucose and cellulose, namely the catalytic function (second decomposition step), CO capture (second stabilized zone) and oxygen release from the crystal structure (third decomposition step).
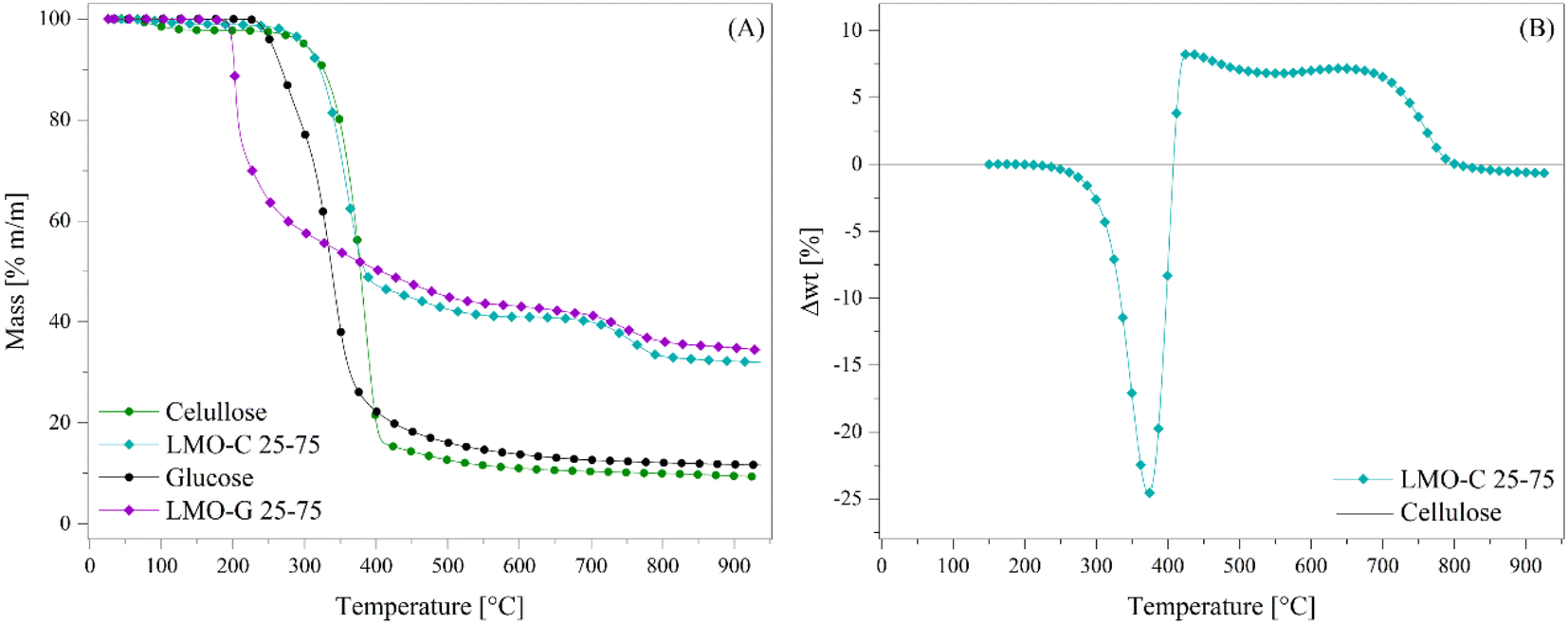 | ||
| Fig. 9 Thermogravimetric analyses of the cellulose mixture with or without Li2MnO3 at 30 °C min−1 in N2 flow (A). Normalized weight loss percentages to cellulose content (B) compared with cellulose pyrolysis alone (black line at zero position). Glucose and LMO-G 25-75 results are added in the panel (A) for comparison. | ||
Therefore, data was treated according to eqn (7) to determine the differences in the weight loss behavior of the LMO-C sample in comparison to the expected weight loss of the pristine cellulose (the latter represented as the zero line in Fig. 9B). The obtained trend is similar to that observed for the LMO-G samples, and thus the phenomena involved during the pyrolysis of cellulose must be closely related. In this case, the negative values between 215 °C and 408 °C are entirely associated with the catalytic effect during the pyrolysis process due to the addition of Li2MnO3. Alternatively, the positive values (from 408 °C to 800 °C) depicted the same two crests, which are related to CO and/or CO2 chemical capture with the formation of Li2CO3 and/or to the formation of some thermally stable bio-oils within this temperature range. Besides, the last negative values (between 803 °C and 950 °C) are related to the oxygen release from the crystal structure.
Additionally, the LMO-C and cellulose samples were catalytically tested. Fig. 10 shows the evolution of the gases normalized to the cellulose content. H2 production was carried out from 225 °C to 900 °C, which was always higher in the presence of Li2MnO3 mostly between 225 °C and 532 °C, where the maximum of 10.9 mL min−1 was observed. In fact, this amount is significantly lower and shifted to lower temperatures than that observed for the case of LMO-G. CO2 production presented an interesting behaviour, which was significantly lower (23.9 mL min−1) in the presence of ceramic in comparison to pristine cellulose (43.0 mL min−1). It must be noted that CO2 production was observed in a higher but narrower temperature range than that of glucose. From T > 440 °C, the CO2 production diminished, but always higher in the presence of Li2MnO3. Besides, at 635 °C, a second local maximum was observed, which may be associated with the CO oxidation-capture process.
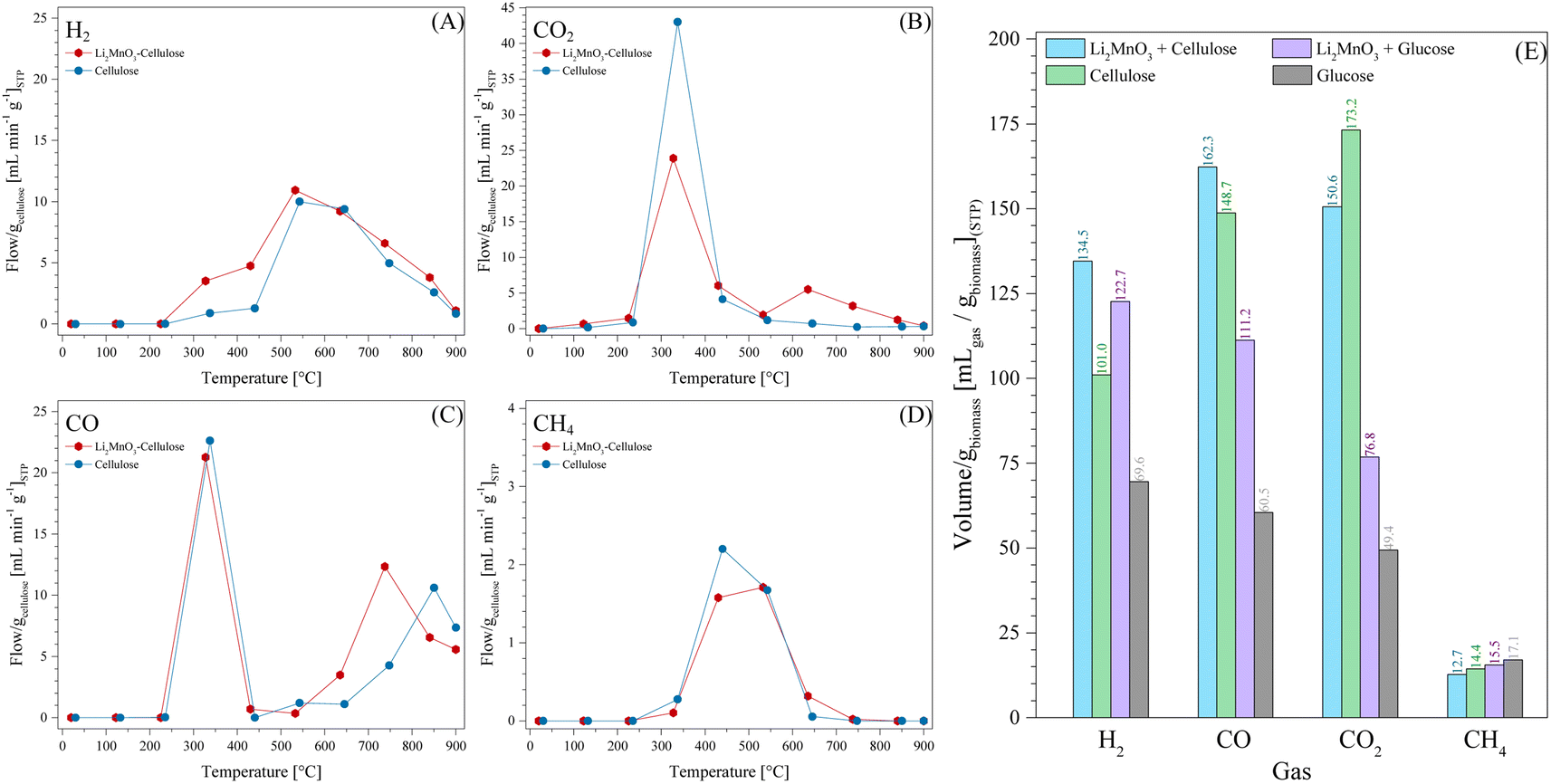 | ||
| Fig. 10 Thermal evolution of the produced gases (H2 (A), CO2 (B), CO (C) and CH4 (D)) for LMO-C and cellulose samples, from 30 °C to 900 °C in N2, using a heating rate of 30 °C min−1, normalized per gram of cellulose. Total volume of the gaseous products (E) normalized per gram of biomass. Glucose and LMO-G 25-75 results are added for comparison. | ||
CO production depicted a similar behaviour in the presence or absence of Li2MnO3 from room temperature to T < 532 °C. Particularly, at 532 °C, the CO production was lower for LMO-C, and then it increased up to 800 °C. This trend is in good agreement with the previous discussion presented for the case of LMO-G. Thereby, the temperature increment not only activated the CO capture process, but also the release of oxygen from the ceramic structure, increasing the production of CO and CO2. Here, as the CO production increased from 635 °C to 800 °C, the CO2 production diminished, indicating that a fraction of CO2 was captured. However, it must be emphasized that the CO production at T < 400 °C was larger in the cellulose sample, whereas at T > 600 °C, it was larger in the case of glucose, which will be explained below. In the case of CH4, it was produced from 372 °C to 737 °C, being in general slightly lower in the presence of the ceramic. In comparison to the case of glucose, the CH4 production temperature range was broader for cellulose pyrolysis and it did not fit with high H2 and CO production. Thus, CH4 formation may be mainly because of the cellulose decomposition mechanism.
It must be mentioned that the differences in the gas evolution observed for the cellulose case must be closely related to the presence of O-glycosidic bonds, modifying the H2 production to lower temperatures, while increasing the formation of carbon oxides at T < 400 °C. Moreover, the lower content of hydroxyl groups reduced the probability of water production. Therefore, the oxygen atoms in the cellulose polymer are prone to produce the most thermodynamically stable compound (CO2).
Considering the whole process, the overall amounts of produced gases were plotted, while normalized per g of biomass (Fig. 10E). As can be observed, the pyrolysis of cellulose produced high contents of H2, CO and CO2, while that of CH4 decreased. Thus, the O-glycosidic bond highly modified the decomposition mechanism. Moreover, the addition of Li2MnO3 to both types of biomasses induced the production of larger amounts of H2 and CO, which had a lower effect on cellulose, although its H2 and CO amounts were higher. Conversely, the CO2 amount diminished for the LMO-C sample in comparison to pristine cellulose, although it was again higher than that for glucose. Similarly, the addition of Li2MnO3 diminished the production of CH4 in both biomass samples. Hence, the addition of Li2MnO3 enhanced the H2 production by an assisted gasification process, inducing the oxidation of biomass to CO and CO2, being partially captured as Li2CO3. Moreover, the O-glycosidic bonds modify the mechanism of carbon oxide formation. Based on all these results, the cellulose decomposition reactions in the absence or presence of Li2MnO3 were proposed (reactions (16) and (17), respectively), following the same methodology as the glucose-containing samples, considering the formation of water and bio-oils, with the latter represented as CxHyOz, where “x”, “y” and “z” vary depending on the gaseous and solid products. Moreover, the coefficient factors “d” and “h” were added to fit the reaction.
| (C6H10O5)n(s) → n(0.73H2(g) + 1.08CO(g) + 1.25CO2(g) + 0.10CH4(g) + dH2O(l) + C2.17H8.14−2dO1.42−d(l) + 1.40C(s)) | (16) |
| (C6H10O5)n(s) + 0.46Li2MnO3(s) → n(0.98H2(g) + 1.18CO(g) + 1.09CO2(g) + 0.09CH4(g) + dH2O(l) + C3.64−hH7.68−2dO2.1−d(l) + hC(s) + 0.46Li2O(s) + 0.46MnO(s)) | (17) |
Considering all this information, the kinetic parameters for the pyrolysis of glucose and cellulose were determined using the Flynn–Wall–Ozawa integration method,78 in the presence or absence of Li2MnO3 (Fig. S5† shows the TG of each experiment). As expected, the decomposition activation energy is higher for the cellulose-containing samples (Table 2), which is attributed to the polymer stability generated through the O-glycosidic bonds. In fact, the difference between the glucose and cellulose activation energies (32.9 kJ mol−1) matches the Gibbs energy difference reported by Y. Nishimura et al.79 (33.18 kJ mol−1). Moreover, the addition of the ceramic reduced the pyrolysis activation energy for both biomasses, confirming all the catalytic and carbon oxide sorption properties described above. Complementarily, the H2/COx ratios were also calculated, which were also enhanced for both types of biomass by the addition of Li2MnO3. In addition, it was observed that the H2/COx ratios for the cellulose-containing samples were lower in comparison to the glucose-containing samples simply because of the greater formation of carbon oxides.
| Sample | E a (kJ mol−1) | lnA |
H2/CO ratio | H2/CO2 ratio | H2/(CO + CO2) ratio | H2 yield (%) | H2 selectivitya (%) | H2 selectivityb (%) |
|---|---|---|---|---|---|---|---|---|
| a H2 yield was calculated as: H2 yield = (mol of produced H2/max mol of H2 from reactions (18) and (19)) × 100. b H2 selectivity was calculated as: H2 selectivity = (mol of produced hydrogen/mol of all gaseous carbon species) × (carbon for hydrogen ratio) × 100. Carbon for the hydrogen ratio in H2 selectivity was calculated as follows: mol of ideal gaseous carbon species/mol of ideal hydrogen, being 0.5 for a (reactions (18) and (19)), and 1.0 for b (reactions (20) and (21)) calculations. c Calculated values corresponding to generated data from dynamic-isothermal process at 700 °C. d Calculated values corresponding to generated data from dynamic-isothermal process at 500 °C. | ||||||||
| Glucose | 64.7 ± 1.4 | 12.3 ± 0.3 | 1.15 | 1.41 | 0.63 | 9.32 | 27.40 | 54.80 |
| LMO-G | 61.0 ± 1.2 | 13.5 ± 0.3 | 1.10 | 1.60 | 0.65 | 16.45 | 30.14 | 60.27 |
| 1.33c | 1.51c | 0.71c | 19.35c | 33.28c | 66.55c | |||
| 1.94d | 0.82d | 0.58d | 9.42d | 26.21d | 52.43d | |||
| Cellulose | 97.6 ± 0.5 | 17.2 ± 0.1 | 0.68 | 0.58 | 0.31 | 14.62 | 15.01 | 30.03 |
| LMO-C | 94.4 ± 2.9 | 17.1 ± 0.6 | 0.83 | 0.89 | 0.43 | 19.48 | 20.66 | 41.31 |
The H2 yield was calculated considering the ideal H2 production from glucose and cellulose, reactions (18) and (19), where only the formation of carbon and CO2 was proposed, or reactions (20) and (21), involving exclusively the formation of CO, respectively. As can be seen, cellulose exhibited ∼1.6 times higher H2 yields than glucose (Table 2), which is attributed to its lower hydroxyl group content prone to be released as water. Furthermore, the addition of Li2MnO3 increased the H2 yields by 1.8 and 1.3 times in comparison to the glucose and cellulose samples, respectively. This be ascribed to its catalytic effect on the formation of carbon oxides, diminishing the available oxygen in the sample to produce water, in addition to the unique properties of the ceramic, resulting in a preferential reaction with carbon species than with hydrogen species.
| C6H12O6(s) → 6H2(g) + 3CO2(g) + 3C(s) | (18) |
| (C6H10O5)n(s) → n(5H2(g) + 2.5CO2(g) + 3.5C(s)) | (19) |
| C6H12O6(s) → 6H2(g) + 6CO(g) | (20) |
| (C6H10O5)n(s) → n(5H2(g) + 5CO(g) + C(s)) | (21) |
Finally, the H2 selectivity was estimated as a function of the type of biomass (Table 2). Similar to the H2/COx ratios, the cellulose-containing samples depicted lower H2 selectivity than the glucose-containing samples, merely because of the larger amounts of produced gaseous. As mentioned, the O-glycosidic bonds in cellulose modified the reaction pathway. However, the addition of Li2MnO3 increased the H2 selectivity for both biomass pyrolysis processes due to the catalytic and sorption properties, as already explained. It should be mentioned that Table 2 includes the dynamic-isothermal data for glucose pyrolysis for comparison. Accordingly, it can be observed that these processes showed a lower H2/CO ratio with an increase in temperature. Moreover, the obtained H2 yield and selectivity at 500 °C was almost the same that the obtained for the pyrolysis of glucose, but the latter shifted to 350 °C. Obviously, as the temperature increased, the production of H2 and CO increased, diminishing the H2/CO ratio, while increasing the H2/CO2 ratio, H2 yield and H2 selectivity.
4. Conclusion
In summary, Li2MnO3 was studied as a bifunctional material (catalytic and sorption), testing its activity in the biomass pyrolysis process, using glucose and cellulose. The effects of the heating rate and molar ratio were studied as variables, evidencing that the best H2 production performance was depicted at 30 °C min−1 and an Li2MnO3/glucose molar ratio of 0.34. Besides, it was demonstrated that the addition of Li2MnO3 to glucose during the pyrolysis process enhanced the H2 production and formation of carbon oxides through a gasification process, in which the Li2MnO3 was partially reduced to other crystal structures. Furthermore, the partial chemical capture of the produced CO as Li2CO3 was corroborated by XRD and FTIR. However, CO chemical capture was not as high as in previous reports, which is mainly due to the low CO partial pressure. In addition, at T ≥ 635 °C, the Boudouard reaction occurred as well as carbon gasification due to the decomposition of Li2CO3 and oxygen release from Li2MnO3, reducing the purity of H2. Finally, when cellulose was tested, the data showed similar results to that obtained in the case of glucose. The O-glycosidic bonds present in cellulose modified the mechanism for the formation of carbon oxides, enhancing the production of H2 but reducing its purity. Moreover, the kinetic analysis showed that the addition of Li2MnO3 reduced the activation energy of all these processes, while increasing the H2 yield and selectivity. Based on this whole analysis, it seems that Li2MnO3 is a promising material to be further studied for H2 production from biomass sources through assisted gasification processes.Conflicts of interest
There are not conflicts to declare.Acknowledgements
This work was financially supported by the PAPIIT-UNAM project IN-205823. Authors thank to A. Tejeda for her technical assistance. Moreover, CHF, NW and NGG thank to CONAHCYT for their Ph D scholarships.References
- J. Gaona-Cumbicos, K. Naula-Duchi, P. Álvarez-Lloret, W. Mejía-Galarza, B. Bernal-Pesántez and L. Jara-Cobos, Catalysts, 2023, 13, 1323 CrossRef CAS
.
- A. G. Olabi and M. A. Abdelkareem, Renewable Sustainable Energy Rev., 2022, 158, 112111 CrossRef CAS
- M. Cai, L. Xu, J. Guo, X. Yang, X. He and P. Hu, J. Mater. Chem. A, 2024, 12, 592–612 RSC
-
X. Lan, P. Tans and K. W. Thoning, Trends in Globally-Averaged CO2 Determined from NOAA Global Monitoring Laboratory Measurements, 2023 Search PubMed
- Y. Zhou, Z. Liu, C. Luo, Z. Han, D. Lai, F. Wu, X. Li and L. Zhang, Carbon Capture Sci. Technol., 2024, 10, 100168 CrossRef CAS
- R. Fuller, P. J. Landrigan, K. Balakrishnan, G. Bathan, S. Bose-O'Reilly, M. Brauer, J. Caravanos, T. Chiles, A. Cohen, L. Corra, M. Cropper, G. Ferraro, J. Hanna, D. Hanrahan, H. Hu, D. Hunter, G. Janata, R. Kupka, B. Lanphear, M. Lichtveld, K. Martin, A. Mustapha, E. Sanchez-Triana, K. Sandilya, L. Schaefli, J. Shaw, J. Seddon, W. Suk, M. M. Téllez-Rojo and C. Yan, Lancet Planet. Heal., 2022, 6, e535–e547 CrossRef PubMed
- N. Djellouli, L. Abdelli, M. Elheddad, R. Ahmed and H. Mahmood, Renewable Energy, 2022, 183, 676–686 CrossRef CAS
- L. Yuping, M. Ramzan, L. Xincheng, M. Murshed, A. A. Awosusi, S. I. BAH and T. S. Adebayo, Energy Rep., 2021, 7, 4747–4760 CrossRef
- J. Goldemberg and S. Teixeira Coelho, Energy Policy, 2004, 32, 711–714 CrossRef
- J. D. D. Holladay, J. Hu, D. L. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS
- A. Q. Al-Shetwi, Sci. Total Environ., 2022, 822, 153645 CrossRef CAS PubMed
- W. Liu, H. Jiang, X. Zhang, Y. Zhao, S. Sun and J. Qiao, J. Mater. Chem. A, 2019, 7, 27236–27240 RSC
- J. Fermoso, F. Rubiera and D. Chen, Energy Environ. Sci., 2012, 5, 6358 RSC
- W. Cui, H. Luo and G. Liu, Waste Manage., 2023, 171, 173–183 CrossRef CAS PubMed
- R. Yukesh Kannah, S. Kavitha, Preethi, O. Parthiba Karthikeyan, G. Kumar, N. V. Dai-Viet and J. Rajesh Banu, Bioresour. Technol., 2021, 319, 124175 CrossRef CAS PubMed
- J. Turner, G. Sverdrup, M. K. Mann, P.-C. C. Maness, B. Kroposki, M. Ghirardi, R. J. Evans and D. Blake, Int. J. Energy Res., 2008, 32, 379–407 CrossRef CAS
- Y. Yan, V. Manovic, E. J. Anthony and P. T. Clough, Energy Convers. Manage., 2020, 226, 113530 CrossRef CAS
- G. Glenk, P. Holler and S. Reichelstein, Energy Environ. Sci., 2023, 16, 6058–6070 RSC
- Z. Sun, X. Wu, C. K. Russell, M. D. Dyar, E. C. Sklute, S. Toan, M. Fan, L. Duan and W. Xiang, J. Mater. Chem. A, 2019, 7, 1216–1226 RSC
- M. Z. Hossain, M. R. Karim, S. Sutradhar, M. B. I. Chowdhury and P. A. Charpentier, Int. J. Hydrogen Energy, 2023, 48, 39791–39804 CrossRef CAS
- H. Haykiri-Acma, S. Yaman and S. Kucukbayrak, Energy Convers. Manage., 2006, 47, 1004–1013 CrossRef CAS
- V. Kirubakaran, V. Sivaramakrishnan, R. Nalini, T. Sekar, M. Premalatha and P. Subramanian, Renewable Sustainable Energy Rev., 2009, 13, 179–186 CrossRef
- E. Madadian, M. Lefsrud, C. A. P. Lee and Y. Roy, J. Green Eng., 2014, 4, 101–116 CrossRef
- R. Zhou, Y. Zhao, R. Zhou, T. Zhang, P. Cullen, Y. Zheng, L. Dai and K. (Ken) Ostrikov, Carbon Energy, 2023, 5, e260 CrossRef
- J. Ren, Y. L. Liu, X. Y. Zhao and J. P. Cao, J. Energy Inst., 2020, 93, 1083–1098 CrossRef CAS
- A. Demirbas and G. Arin, Energy Sources, 2002, 24, 471–482 CrossRef CAS
- S. Meng, W. Li, Z. Li and H. Song, Fuel, 2023, 353, 129169 CrossRef CAS
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 RSC
- T. M. Roberge, S. O. Blavo, C. Holt, P. H. Matter and J. N. Kuhn, Top. Catal., 2013, 56, 1892–1898 CrossRef CAS
- L. Frigge, G. Elserafi, J. Ströhle and B. Epple, Energy and Fuels, 2016, 30, 7713–7720 CrossRef CAS
- D. Hendry, C. Venkitasamy, N. Wilkinson and W. Jacoby, Bioresour. Technol., 2011, 102, 3480–3487 CrossRef CAS PubMed
- Z. Fang, T. Minowa, C. Fang, R. L. Smith, H. Inomata and J. A. Kozinski, Int. J. Hydrogen Energy, 2008, 33, 981–990 CrossRef CAS
- A. Molino, S. Chianese and D. Musmarra, J. Energy Chem., 2016, 25, 10–25 CrossRef
-
V. S. Sikarwar and M. Zhao, in Encyclopedia of Sustainable Technologies, Elsevier, 2017, pp. 205–216 Search PubMed
- A. Yokozeki and M. B. Shiflett, Appl. Energy, 2007, 84, 351–361 CrossRef CAS
- J. Zou and W. S. W. W. Ho, J. Chem. Eng. Jpn., 2007, 40, 1011–1020 CrossRef CAS
- C. Cao, Y. Zhang, W. Cao, H. Jin, L. Guo and Z. Huo, Catal. Lett., 2017, 147, 828–836 CrossRef CAS
- S. Chimpae, S. Wongsakulphasatch, S. Vivanpatarakij, T. Glinrun, F. Wiwatwongwana, W. Maneeprakorn and S. Assabumrungrat, Processes, 2019, 7, 349 CrossRef CAS
- M. Z. Memon, G. Ji, J. Li and M. Zhao, Ind. Eng. Chem. Res., 2017, 56, 3223–3230 CrossRef CAS
- Y. Wu, H. Wang, H. Li, X. Han, M. Zhang, Y. Sun, X. Fan, R. Tu, Y. Zeng, C. C. Xu and X. Xu, Renewable Energy, 2022, 196, 462–481 CrossRef CAS
- D. González-Varela, C. Hernández-Fontes, N. Wang and H. Pfeiffer, Carbon Capture Sci. Technol., 2023, 7, 100101 CrossRef
- M. Zhao, M. Z. Memon, G. Ji, X. Yang, A. K. Vuppaladadiyam, Y. Song, A. Raheem, J. Li, W. Wang and H. Zhou, Renewable Energy, 2020, 148, 168–175 CrossRef CAS
- A. K. Vuppaladadiyam, M. Z. Memon, G. Ji, A. Raheem, T. Z. Jia, V. Dupont and M. Zhao, ACS Sustain. Chem. Eng., 2019, 7, 238–248 CrossRef CAS
- N. K. Gupta, C. Hernández-Fontes and S. N. Achary, Mater. Today Chem., 2023, 27, 101329 CrossRef CAS
- C. Hernández-Fontes and H. Pfeiffer, Chem. Eng. J., 2022, 428, 131998 CrossRef
- C. Hernández-Fontes, F. Plascencia Hernández and H. Pfeiffer, J. Phys. Chem. C, 2023, 127, 6670–6679 CrossRef
- C. Hernández-Fontes, D. G. Araiza, G. Díaz and H. Pfeiffer, React. Chem. Eng., 2023, 8, 229–243 RSC
- A. Messori, G. Martelli, A. Piazzi, F. Basile, J. De Maron, A. Fasolini and R. Mazzoni, Chempluschem, 2023, 88, e202300357 CrossRef CAS PubMed
- N. Di Fidio, S. Fulignati, I. De Bari, C. Antonetti and A. M. Raspolli Galletti, Bioresour. Technol., 2020, 313, 123650 CrossRef CAS PubMed
- C. Hernández-Fontes and H. Pfeiffer, React. Chem. Eng., 2022, 7, 1573–1588 RSC
- T. Sata, Ceram. Int., 1998, 24, 53–59 CrossRef CAS
- R. H. Lamoreaux and D. L. Hildenbrand, J. Phys. Chem. Ref. Data, 1984, 13, 151–173 CrossRef CAS
-
F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids, Academic Press, San Diego, 1999, vol. 11 Search PubMed
-
S. Lowell, J. E. Shields, M. A. Thomas and M. Thommes, Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density, Springer Netherlands, Dordrecht, 2004, vol. 16 Search PubMed
- T. R. Carlson, J. Jae, Y. C. Lin, G. A. Tompsett and G. W. Huber, J. Catal., 2010, 270, 110–124 CrossRef CAS
- K. Oh-Ishi, Y. Matsukura, T. Okumura, Y. Matsunaga and R. Kobayashi, J. Solid State Chem., 2014, 211, 162–169 CrossRef CAS
- J. Sun, C. Liang, X. Tong, Y. Guo, W. Li, C. Zhao, J. Zhang and P. Lu, Fuel, 2019, 239, 1046–1054 CrossRef CAS
- A. Stegarescu, I. Lung, C. Leostean, I. Kacso, O. Opris, M. D. Lazăr, L. Copolovici, S. Gutoiu, M. Stan, A. Popa, O. Pană, A. S. Porav and M.-L. Soran, Waste Biomass Valorization, 2020, 11, 5003–5013 CrossRef CAS
- N. Rinaldi, A. A. Dwiatmoko and A. Kristiani, IOP Conf. Ser.: Mater. Sci. Eng., 2021, 1011, 012033 CAS
- M. Koike, C. Ishikawa, D. Li, L. Wang, Y. Nakagawa and K. Tomishige, Fuel, 2013, 103, 122–129 CrossRef CAS
- D. A. Fox and A. H. White, Ind. Eng. Chem., 1931, 23, 259–266 CrossRef CAS
- J. Y. Hwang, J. H. Yu and K. Kang, Curr. Appl. Phys., 2015, 15, 1580–1586 CrossRef
- A. Wang, D. Austin and H. Song, Fuel, 2019, 246, 443–453 CrossRef CAS
- H. Zhang, G. Zhu, H. Yan, T. Li and Y. Zhao, Metall. Mater. Trans. B, 2013, 44, 889–896 CrossRef CAS
- V. Seshadri and P. R. Westmoreland, J. Phys. Chem. A, 2012, 116, 11997–12013 CrossRef CAS PubMed
- Q. Zhang, D. Peng, S. Zhang, Q. Ye, Y. Wu and Y. Ni, AIChE J., 2017, 63, 2153–2164 CrossRef CAS
- H. A. Lara-García, P. Sanchez-Camacho, Y. Duan, J. Ortiz-Landeros and H. Pfeiffer, J. Phys. Chem. C, 2017, 121, 3455–3462 CrossRef
- H. A. Lara-García and H. Pfeiffer, Chem. Eng. J., 2017, 313, 1288–1294 CrossRef
- E. Vera, S. García, M. M. Maroto-Valer and H. Pfeiffer, Adsorption, 2020, 26, 781–792 CrossRef CAS
- C. Liu, J. Luo, H. Dong, Z. Zhao, C. Xu, S. Abuelgasim, A. Abdalazeez, W. Wang, D. Chen and Q. Tang, Sep. Purif. Technol., 2022, 300, 121912 CrossRef CAS
- B. Lecker, L. Illi, A. Lemmer and H. Oechsner, Bioresour. Technol., 2017, 245, 1220–1228 CrossRef CAS PubMed
- O. Anjos, M. G. Campos, P. C. Ruiz and P. Antunes, Food Chem., 2015, 169, 218–223 CrossRef CAS PubMed
- K. Aydıncak, T. Yumak, A. Sınağ and B. Esen, Ind. Eng. Chem. Res., 2012, 51, 9145–9152 CrossRef
- S. Sedaghat, E. Arshadi, A. Nafar and R. Dabbagh, Rev. Roum. Chim., 2019, 64, 409–413 CrossRef
- M. Ibrahim, M. Alaam, H. El-Haes, A. F. Jalbout and A. De Leon, Eclét. Quím., 2006, 31, 15–21 CrossRef CAS
- R. V. Siriwardane, C. Robinson, M. Shen and T. Simonyi, Energy Fuels, 2007, 21, 2088–2097 CrossRef CAS
- Z. Yin, K. Wang, P. Zhao and X. Tang, Ind. Eng. Chem. Res., 2016, 55, 1142–1146 CrossRef CAS
- N. Almagro-Herrera, S. Lozano-Calvo, A. Palma, J. C. García and M. J. Díaz, Fuel, 2024, 367, 131501 CrossRef CAS
- Y. Nishimura, D. Yokogawa and S. Irle, Chem. Phys. Lett., 2014, 603, 7–12 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00224e |
| This journal is © The Royal Society of Chemistry 2024 |