
Multi-electron bipolar-type organic molecules for high-capacity dual-ion zinc batteries†
Chengmin
Hu
a,
Xiaozhe
Yang
a,
Pingxuan
Liu
a,
Ziyang
Song
a,
Yaokang
Lv
 b,
Ling
Miao
*a,
Mingxian
Liu
*a and
Lihua
Gan
*a
b,
Ling
Miao
*a,
Mingxian
Liu
*a and
Lihua
Gan
*a
aShanghai Key Laboratory of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, P. R. China. E-mail: 22169@tongji.edu.cn; ganlh@tongji.edu.cn; liumx@tongji.edu.cn
bZhejiang College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China
First published on 13th March 2024
Abstract
Organic materials featuring element sustainability and functional tunability have currently sprung up as viable cathode alternatives for zinc ion batteries. Nevertheless, organic materials generally suffer from performance deficiencies such as limited capacity and low stability. Here, a bipolar-type organic molecule indanthrone (IDT) is demonstrated as the cathode, with n-type (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and p-type (N–H) redox sites involved in multi-electron transfer processes that store both cations and anions, resulting in a significantly enhanced capacity. Meanwhile, the decoration of p-type N–H bridges into the n-type anthraquinone network expands the conjugate system and upgrades the high-voltage durability, maintaining 88.7% of its initial capacity after 3000 cycles at 10 A g−1. The as-designed Zn//IDT battery provides a competitive specific capacity of 238 mA h g−1 at 0.2 A g−1 and an energy density of 159.1 W h kg−1. In addition, bipolar-type N–H and CO moieties are confirmed to involve a mixed energy-storage mechanism with multi-electron participation through various ex situ characterization studies and electrochemical tests. The Zn2+ ion is the redox-related cation of CO, while the CF3SO3− anion is involved in the conversion between NH+ and N–H during the storage process. This work provides a new approach for designing high-performance organic cathodes for advanced zinc batteries.
O) and p-type (N–H) redox sites involved in multi-electron transfer processes that store both cations and anions, resulting in a significantly enhanced capacity. Meanwhile, the decoration of p-type N–H bridges into the n-type anthraquinone network expands the conjugate system and upgrades the high-voltage durability, maintaining 88.7% of its initial capacity after 3000 cycles at 10 A g−1. The as-designed Zn//IDT battery provides a competitive specific capacity of 238 mA h g−1 at 0.2 A g−1 and an energy density of 159.1 W h kg−1. In addition, bipolar-type N–H and CO moieties are confirmed to involve a mixed energy-storage mechanism with multi-electron participation through various ex situ characterization studies and electrochemical tests. The Zn2+ ion is the redox-related cation of CO, while the CF3SO3− anion is involved in the conversion between NH+ and N–H during the storage process. This work provides a new approach for designing high-performance organic cathodes for advanced zinc batteries.
Introduction
A variety of aqueous rechargeable batteries have been widely explored benefiting from their non-flammability and excellent environmental friendliness.1–4 Multivalent ion storage systems may enable multi-electron transfer compared to monovalent ion storage systems. In particular, aqueous divalent zinc ion batteries (ZIBs) have emerged as a promising storage family due to the notably high theoretical capacity (819 mA h g−1), outstanding hydrophilicity and ample abundance of zinc metal.5–9 ZIBs are generally constructed from commercial zinc foil as the anode, cathode materials, and aqueous zinc salt electrolytes. The quality of cathode materials plays a crucial role in the overall performance of ZIBs. Until now, a series of inorganic cathode materials have been developed for ZIBs.10–14 However, during the charging/discharging process, the zinc ion exists as the large-sized hydrated Zn(H2O)62+ in the electrolyte, which causes an undesirable phase/structure transition within inorganic cathode materials, resulting in unsatisfactory electrochemical performance.15–18 In contrast, the storage mechanism of organic materials only involves charge-state evolution of redox-active functionalities without structural changes. Very recently, organic materials featuring element sustainability and functional tunability have sprung up as viable cathode alternatives for ZIBs.19,20Based on energy storage mechanisms, organic materials are classified into n-type, p-type, and bipolar-type.21–23 n-Type organics receive electrons to be reduced and converted into ionic compounds and then combined with cations in electrolytes.24–28 Typically, n-type organics often possess highly electroactive functional groups (e.g., CO, CN and –NO2) that provide high capacity.29–31 However, the inevitable solubility of n-type organic molecules at the discharge state in electrolytes leads to poor cycling capacity.32,33 p-Type organics undergo the oxidized process and lose electrons and then change into ionic compounds to combine with anions.34,35 p-Type organics show faster reaction kinetics benefiting from no bond rearrangement during energy storage. Nevertheless, p-type organics tend to show limited capacity because of excessive redox-inactive compositions, failing to deliver high-energy density for practical applications. Bipolar-type organics consisting of n-type and p-type functional sites are considered emerging electrode materials. The bipolar characteristics can maximize their positively/negatively charged states to combine with anions/cations, which endow them with capacity-stability growth.36–39 Therefore, to remedy the performance deficiency aroused by a single type of functional site, molecular-level design and mechanism exploration of bipolar-type organics deserve much more effort towards high-performance ZIBs.
Here, a bipolar-type molecule IDT is reported as an organic cathode material. The bipolar IDT molecule participates in a multi-electron transfer process by combining n-type and p-type active motifs to produce an enhanced specific capacity. Besides, the introduction of N–H bridges increases the conjugated level and enhances the structural stability of IDT, enabling IDT to maintain 88.7% of its initial capacity after 3000 cycles at a current of 10 A g−1. As envisaged, the Zn//IDT batteries provide excellent electrochemical performance, with a high capacity (238 mA h g−1) and a high energy density (159.1 W h kg−1). Excellent electrochemical performance contributes to the mixed energy storage mechanism. Zn2+ is the redox-related cation of CO, while the OTF− (OTFCF3SO3) anion is involved in the conversion between NH+ and N–H during the storage process. This work provides a new way to design high performance organic cathodes for ZIBs.
Results and discussion
Structural characterization
The quinone-based indanthrone (IDT) is composed of two N–H bridges coupling with anthraquinones (AQ).40 Fourier transform infrared spectroscopy (FTIR) was used to characterize the molecular structures of IDT and AQ (Fig. S1†). The characteristic peaks at around 1500 and 3434 cm−1 represent N–H groups exclusively in the IDT molecule, and the peak near 1686 cm−1 is attributed to the coexistent CO bond.41 The FTIR results indicate a bipolar-type IDT molecule with multiple active sites simultaneously combined with n-type carbonyl and p-type imide groups. Compared with AQ, IDT coupling of p-type N–H bridges with the n-type AQ network presents an expanded conjugate structure, which could facilitate electron migration as well as high-voltage durability characteristic of p-type organics. Electronic structures of IDT and AQ were calculated and studied by using the density functional theory (DFT). The HOMO/LUMO energy levels are shown in Fig. 1a and b. Compared to the high LUMO energy (−2.79 eV) of AQ, IDT with a lower LUMO energy (−2.90 eV) corresponds to stronger electron affinity and higher reduction potential.42 As shown in Fig. 1c, the HOMO/LUMO energy gaps of IDT and AQ are 2.16 and 4.20 eV, respectively. The narrow HOMO/LUMO energy gap of IDT indicates convenient electron transfer with a low energy barrier according to the molecular orbital theory. Besides, the solid-state UV-vis spectra of IDT and AQ are shown in Fig. S2.† According to the Tauc plots, the optical energy gaps (Eg) were calculated to be 3.48 eV for AQ and 2.25 eV for IDT. The narrow Eg value endows IDT with higher charge transfer efficiency.43,44 Conductivity measurements were also carried out using a four-probe tester system, with conductivities of 9.2 × 10−9 and 5.2 × 10−11 S cm−1 (1 MPa) for IDT and AQ, respectively (Fig. S3†). As given above, IDT has a smaller HOMO/LUMO energy gap (2.16 eV) and lower LUMO energy level (−2.90 eV), which indicates that the introduced N–H groups expand the conjugated structure of IDT, resulting in a better electronic conductivity.45
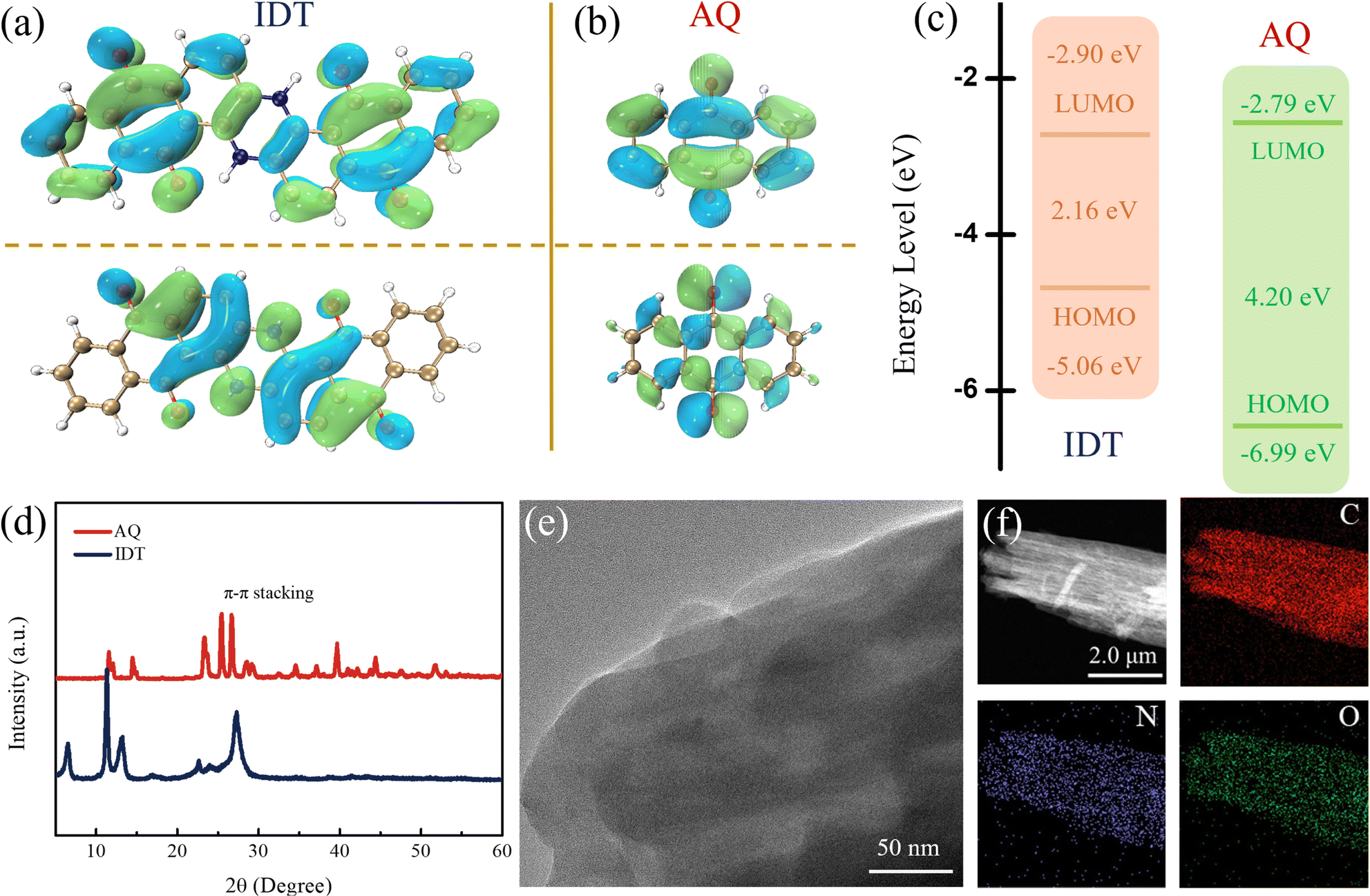 | ||
| Fig. 1 (a–c) LUMO/HOMO diagrams and energy levels. (d) XRD patterns. (e) HRTEM image of IDT. (f) EDS mapping images of IDT. | ||
Additionally, an extended conjugate system consolidates the IDT structure owing to the π–π stacking interaction, which could further endure efficient electron transfer and high working potential. XRD patterns show that IDT and AQ have sharp characteristic π–π stacking peaks at around 27° (100 crystal planes), which are attributed to the ordered stacking by the conjugation of molecular planes (Fig. 1d).46,47 This is also evident from the SEM images, where both IDT and AQ maintain a blocky structure (Fig. S4†). The three-dimensional stacking phenomenon caused by planar conjugation can be clearly observed from high-resolution transmission electron microscopy (HRTEM) (Fig. 1e). From the corresponding EDS mapping, the uniform and dense distribution of the N/O elements representing the highly active groups in IDT can be observed (Fig. 1f). The thermogravimetric analysis (TGA) results related to structure stability are shown in Fig. S5.† AQ began to decompose at 243 °C, while the initial decomposition temperature of IDT is 541 °C due to the conjugation expansion. The coupling of N–H bridges with AQ molecules realizes the integration of n-type (CO) and p-type (N–H) redox sites, and the correspondingly extended conjugated system allows electron delocalization throughout the molecule, accelerating the transport of charged ions and providing fast charge kinetics.
Electrochemical performance
The unique structure with n-type (CO) and p-type (N–H) redox sites empowers the bipolar-type IDT to be potential cathode material for ZIBs. ZIBs were constructed in 2 M Zn(OTF)2 aqueous electrolyte, utilizing IDT as the cathode and commercial zinc foil as the anode. The electrochemical performance was initially evaluated by the galvanostatic charge/discharge (GCD) test. GCD curves at different current densities are shown in Fig. 2a. At current densities of 0.2, 0.5, 1, 2, 3, 4, and 5 A g−1, the bipolar-type IDT cathode can provide a capacity of 238, 223, 208, 199, 183, 167 and 154 mA h g−1, respectively. In contrast, n-type AQ exhibits smaller capacities at current densities of 0.2, 0.5, 1, 2, 3, 4, and 5 A g−1 for 139, 110, 89, 72, 60, 48, and 36 mA h g−1, respectively (Fig. S6†). Even when the current density increases to 10 A g−1, the IDT cathode maintains a capacity retention of 55.0% to provide an excellent capacity of 131 mA h g−1, while AQ can only provide 27 mA h g−1 with a capacity retention of 19.4% (Fig. S7†).
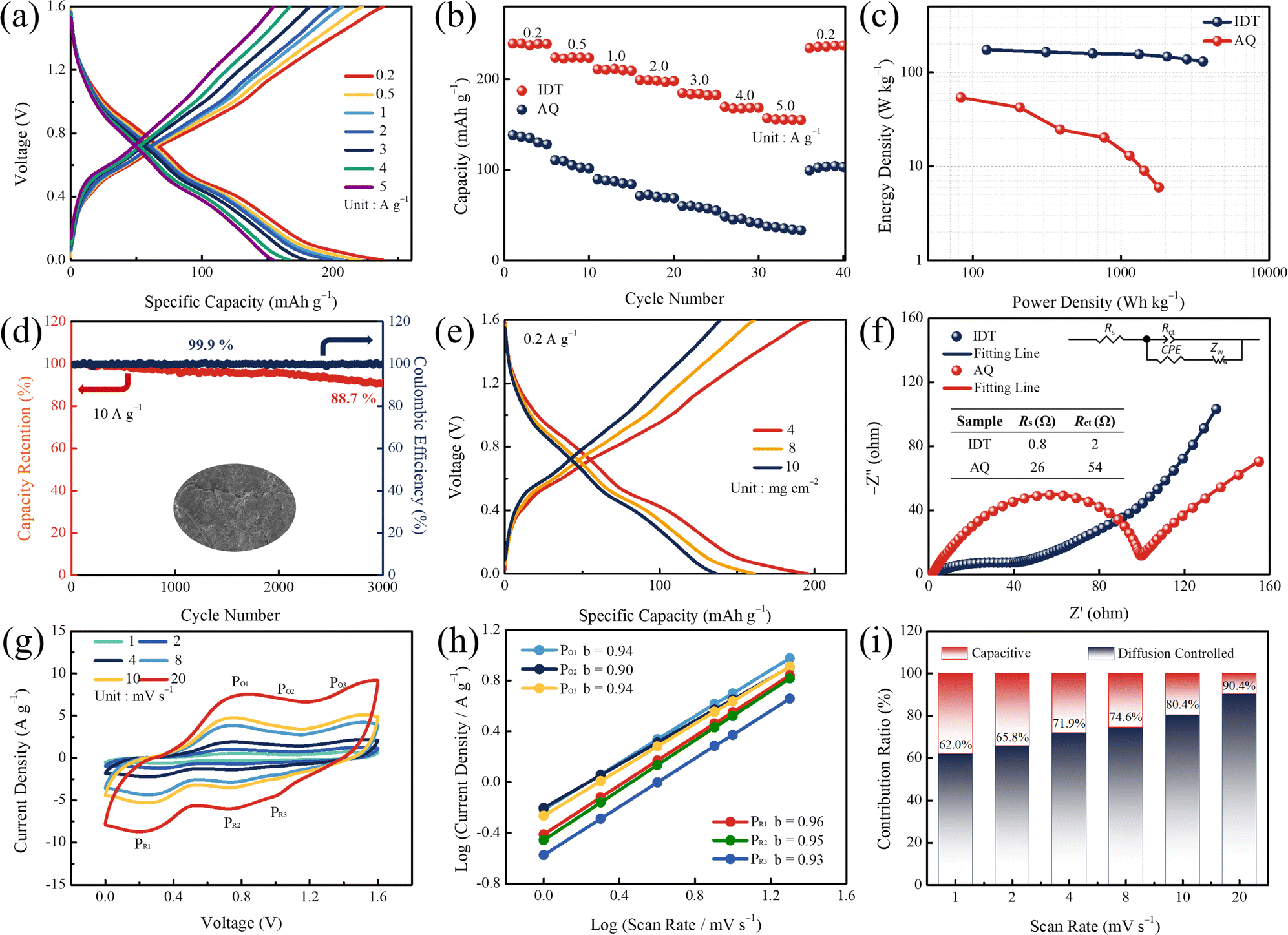 | ||
| Fig. 2 (a) GCD curves of the IDT cathode at different current densities. (b) Rate performance tests of IDT and AQ. (c) Ragone plot. (d) Cyclability of the Zn//IDT battery (inset shows SEM images after cycling). (e) Specific capacities of the IDT cathode under different mass loadings. (f) EIS plots. (g) CV curves. (h) Calculated b values. (i) Normalized capacitive and diffusion-controlled contribution ratios at various scan rates of the Zn//IDT battery. | ||
Rate performance tests at different current densities are shown in Fig. 2b. When the current density increases from 0.2 to 5 A g−1 and then rapidly switches to 0.2 A g−1, the IDT cathode returns to the initial capacity quickly. However, the AQ cathode shows an irreversible trend of capacity decline. The high capacity of IDT benefits from its high-density functional groups and dual-ion storage mechanism at bipolar-type redox sites. The Ragone plots are shown in Fig. 2c. The IDT cathode can provide a maximum energy density of 159.1 W h kg−1 at a power density of 142.7 W kg−1 and a maximum power density of 4126.5 W kg−1 at an energy density of 115.6 W h kg−1. After 3000 high-intensity cycles at high current densities of 10 A g−1, the Zn//IDT battery maintains 88.7% capacity retention and a coulombic efficiency close to 100% (Fig. 2d). On the other hand, Zn//AQ only shows a 52.2% capacity retention after 3000 cycles (Fig. S8†). In contrast with the original smooth Zn anode, the cycled Zn anode exhibits a rough surface structure (Fig. S9†), which comes from the repeated deposition/stripping process of the Zn anode during electrochemistry. Compared to the current reported organic cathodes, the Zn//IDT battery shows excellent energy storage performance (Table S1†). The performance at high-mass loads is a critical step in driving large-scale applications. Specifically, the IDT cathodes have capacities of 198, 169 and 135 mA h g−1 with mass loadings of 4, 8, and 10 mg cm−2 (Fig. 2e). The highly accessible surface group shortens the ion transport path and reduces the volume change during the reversible coordination of Zn2+.48–50 The excellent zinc ion storage performance of the IDT cathode is attributed to the stability of the expanded conjugate structure and the high-density bipolar redox sites. Strong structural stability ensures high-potential tolerance and long cycle-life, and the high density of active groups ensures high ion accessibility guaranteeing excellent capacity availability.51,52 An equivalent circuit was adopted for fitting the Nyquist plots (Fig. 2f), including the equivalent series resistances (Rs), the charge transfer resistance (Rct), the constant phase angle element (CPE), and Warburg resistance (Zw). Electrochemical impedance spectroscopy (EIS) experiments reveal an Rs of 0.8, 2 Ω and a corresponding Rct of 26, 54 Ω for IDT-based and AQ-based ZIBs, respectively (Fig. 2f). IDT-based ZIBs show lower Rs and Rct, which are attributed to the increase of active functional groups for enhanced interfacial compatibility and the expanded conjugate structure for reduced transport hindrance. CV profiles at scan rates from 1 to 20 mV s−1 show three pairs of redox peaks (marked PO1/PR1, PO2/PR2 and PO3/PR3, Fig. 2g). The shape of CV curves remains stable at increasing scan rates, due to excellent structural stability and lower ion transport resistance.53,54 Charge storage kinetics are investigated based on Dunn's method. The relationship between current (i) and scan rate (v) can be expressed as i = kvb, where k is a constant.55,56 Plotting log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i from logv, the values of b for PO1/PR1, PO2/PR2 and PO3/PR3 were 0.94/0.96, 0.90/0.95 and 0.94/0.93, respectively (Fig. 2h), which are close to the ideal surface management process (b = 1).57–59 Further derivation shows that the capacity contribution of diffusion control gradually increases from 62% to 90.4% when the scan rate is increased from 1 to 20 mV s−1 (Fig. 2i). The AQ cathode was only with 56% of diffusion control at a scan rate of 20 mV s−1 (Fig. S10†). The unique conjugated structure of IDT enables rapid ion migration with low energy hindrance, and the high-density bipolar redox sites reduce the ion diffusion distance to achieve high-speed reaction kinetics.
i from logv, the values of b for PO1/PR1, PO2/PR2 and PO3/PR3 were 0.94/0.96, 0.90/0.95 and 0.94/0.93, respectively (Fig. 2h), which are close to the ideal surface management process (b = 1).57–59 Further derivation shows that the capacity contribution of diffusion control gradually increases from 62% to 90.4% when the scan rate is increased from 1 to 20 mV s−1 (Fig. 2i). The AQ cathode was only with 56% of diffusion control at a scan rate of 20 mV s−1 (Fig. S10†). The unique conjugated structure of IDT enables rapid ion migration with low energy hindrance, and the high-density bipolar redox sites reduce the ion diffusion distance to achieve high-speed reaction kinetics.
Charge storage mechanism
The cyclic voltammetry curves of IDT show three pairs of redox peaks at 1 mV s−1, and only one pair of redox peaks can be observed for the CV curve of AQ (Fig. S11†). According to CV curves of IDT and AQ, the energy storage mechanism involving multi-electron transfer is initially inferred. Theoretically, bipolar-type IDT can store anions and cations simultaneously within a single charging/discharging cycle. During the discharging process, the CO group accepts electrons and then converts into C–O− to combine with Zn2+ cations. This corresponds to redox peaks centered at 0.33/0.78 V and 0.81/1.18 V in CV curves. During the charging process, the N–H group can be oxidized to NH+ and then combines with OTF− anions, which corresponds to the redox peak of 1.26/1.42 V in the CV curve. To demonstrate the dual-ion storage mechanism, a series of ex situ characterization studies were used to monitor the structural changes of the IDT cathode during the charging and discharging process. Characterization studies were carried out based on the different states of the IDT cathode in a single charge/discharge turn (Fig. 3a), where A, B and C refer to 1.6, 0.7 and 0 V during the discharge process and D and E refer to 1.1 and 1.6 V during the charging process. Ex situ XPS spectra are shown in Fig. S12.† In the XPS contour plot of Zn 2p, it is seen that the Zn 2p signal belonging to Zn2+ coordination with active sites on the cathode surface is gradually enhanced with the discharge voltage from 1.6 to 0 V and then weakened after the increase of the charging voltage from 0 to 1.6 V (Fig. 3b). In contrast, S 2s and 2p signals experience a continuous decrease during the discharge process and a gradual enhancement during the charging process (Fig. 3c). The high-resolution XPS spectra of Zn 2p, S 2s and 2p in different states reveal the corresponding redox trends. The uptake of Zn2+ ions occurs in the discharge process, while the uptake of OTF− occurs during the charging process. The high-resolution O 1s spectra in different states are shown in Fig. 3d. During the discharging process, Zn2+ combines with the active CO group to form a C–O–Zn bond, resulting in a decrease in the intensity of the CO peak. When recharging to 1.6 V, the CO intensity increases again, indicating the desorption of Zn2+ ions. Like the N species, the high-resolution N 1s XPS spectra are shown in Fig. 3e. When discharging at 0–1.6 V, N–H content increases while NH+ content decreases. When charging to 1.6 V, the contents of N–H and NH+ show an opposite trend, indicating that OTF− adorbs during the charging process.60 CV curves of IDT in ZnSO4/H2O electrolytes also show three pairs of redox peaks, which is similar to that in Zn(OTF)2/H2O electrolytes (Fig. S13†). The XPS results and CV curves further verify the dual-ion storage mechanism of IDT. XRD patterns in different states are shown in Fig. S14.† When discharging to 0 V, the characteristic peak attributed to Zn(OTF)2[Zn(OH)2]3·xH2O61 appeared at around 32.8° and completely disappeared again during the subsequent charging process. The formation of Zn(OTF)2[Zn(OH)2]3·xH2O is also confirmed by the emerging flocculated nanosheets in SEM images at state C and elemental mapping images of Zn and S species distribution (Fig. 3f). GCD tests in 0.3 M Zn(OTF)2/acetonitrile show that IDT could deliver a capacity of 195 mA h g−1, which is less than that in Zn(OTF)2/H2O (Fig. S15†). In HOTF electrolyte with different pH, the capacity of IDT increased with the number of protons in the electrolyte (Fig. S16†), validating the role of protons as auxiliaries in the charging and discharging process. These structural characterization studies and electrochemical tests strongly demonstrate the bipolar-type charge storage mechanism within the IDT cathode, where Zn2+ combines with n-type CO sites at low potential and the CF3SO3− anion is involved in the conversion of p-type N–H moieties at high potential.
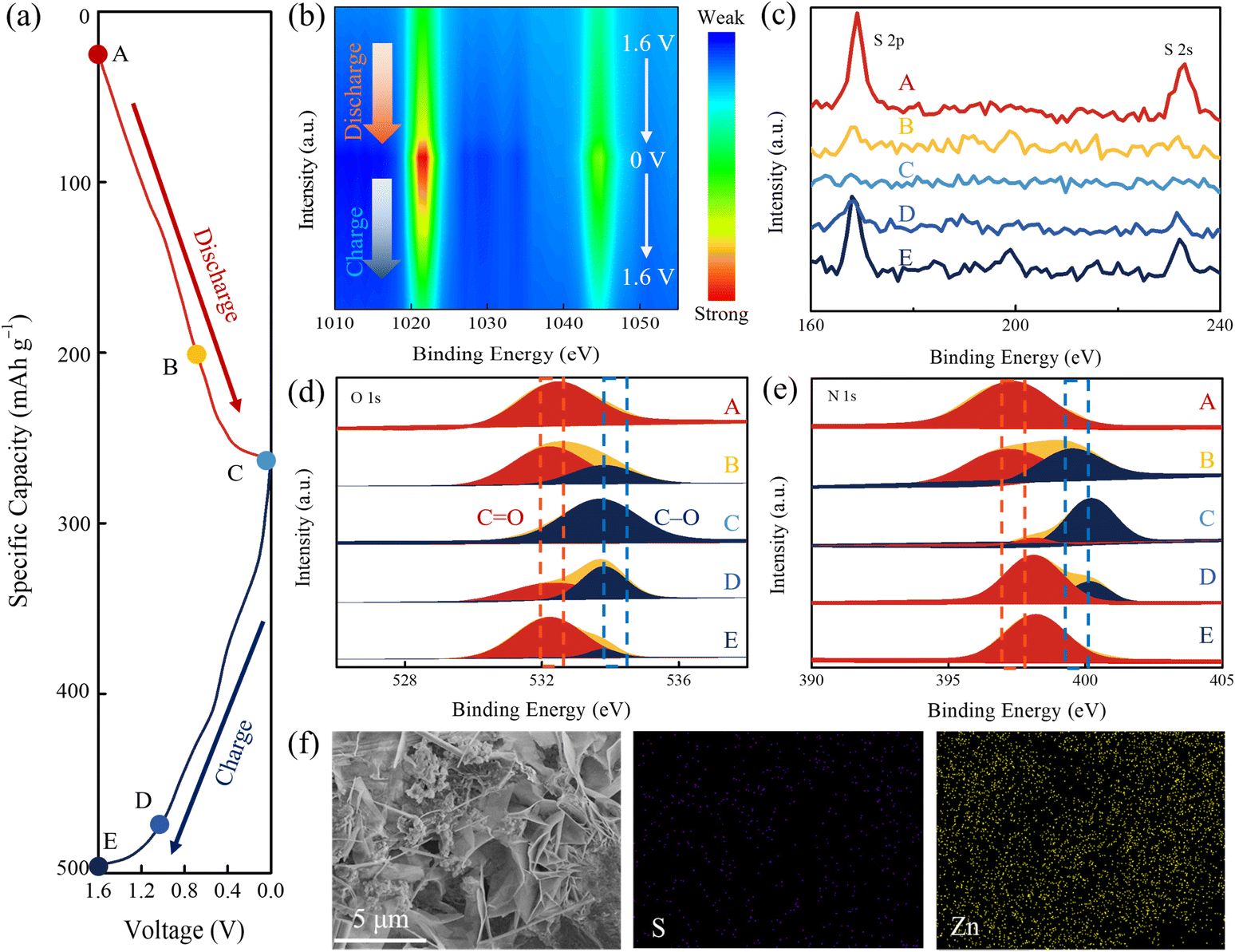 | ||
| Fig. 3 (a) A GCD profile. Ex situ XPS spectra: (b) Zn 2p, (c) S 2p, (d) O 1s, and (e) N 1s at various discharged/charged states. (f) SEM image. | ||
Density functional theory calculations
Molecular electrostatic potential (ESP) simulation was used to characterize the active sites of bipolar-type IDT and n-type AQ. The red region represented by the carbonyl group indicates a better environment for cation adsorption during discharging, while the the blue region where the imine group is located shows an easier position for anion adsorption during charging (Fig. S17†). The ESP plot of the bipolar-type IDT molecule shows two types of adsorption sites for both the cation and anion, while AQ shows the cation adsorption site characteristic of the n-type cathode. According to ESP plots, it can be seen that functional groups at positions A, B and C have symmetrical potential distributions, respectively. Combined with the three pairs of redox peaks in the CV curve of IDT (Fig. 3g), it is initially inferred that the redox process of charge storage involves six electron transfers including three steps (Fig. 4a). The storage mechanism was de-validated using DFT calculations on the electronic configuration of IDT combined with Zn2+ and OTF− ions during the charging and discharging process. During discharging, two CO sites of the IDT cathode first coordinate with a Zn2+ ion (Fig. 4a), corresponding to the redox peak at 0.81/1.18 V in CV curves (Fig. 2g); then the residual two CO sites continue to combine with a Zn2+ ion, attributing to the redox peak at 0.33/0.78 V. During charging, two N–H sites couple with two OTF− ions, which correspond to the redox peak at 1.26/1.42 V. The electronic configurations of IDT bonded to different numbers of Zn2+ and OTF− ions were named IDT-Znx and IDT-OTFx, respectively (x represents the different positions, x = A, B, C). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy gaps of the electronic conformations of IDT combined with Zn2+/OTF− were obtained using DFT calculations. During the discharge process, the energy band gaps of the electronic conformations formed by IDT combined with Zn2+ ions at different positions (IDT-ZnA and IDT-ZnAB) are 1.42 and 1.23 eV, respectively (Fig. 4b). IDT-ZnAB possesses a lower energy band gap, indicating a stable molecular structure. HOMO diagrams give the qualitative acceptance of electron information, and it can be seen that the electron cloud arrangement on the HOMOs of IDT-ZnAB tends to be saturated, which reinforces the validation of the full utilization of its four CO active sites. Similarly, it can be found that IDT-OTFC has a lower energy gap than IDT indicating that all its N–H bonds can be used as active groups to store OTF− ions. ESP plots are shown in Fig. 4c. The uncoordinated CO in IDT-ZnA and IDT-ZnAB maintains the same trend of increasing electronegativity concentration, which facilitates further binding with Zn2+ ions. Upon acceptance of four Zn2+ ions, the electronegativity concentration decreases significantly, which indicates that its electron arrangement has reached the highest reduced level and does not allow further acceptance of Zn2+. The electropositive concentration of N–H groups in IDT remains weakly electropositive upon binding to OTF− ions, indicating full utilization of the dual N–H active site. Gibbs free energies (G) of the coordination products were counted to further determine the most stable electronically arranged conformations.24 As depicted in Fig. 4c, the Gibbs free energies of IDT-ZnA and IDT-ZnAB are −1.10 and −2.06 eV, respectively. The Gibbs free energies remain negative and lower than those of the double active sites when all four CO active sites are bound to Zn2+ ions, which further demonstrates the utilization of the four active sites. The Gibbs free energy of IDT-OTFC is 2.50 eV, indicating that the adsorption of anions is a heat-absorbing process. The results of theoretical calculations indicate the six-electron transfer process of the two types of active sites in IDT and further confirm the optimal coordination process of Zn2+ and OTF− at n-type and p-type redox sites during electrochemical processes.
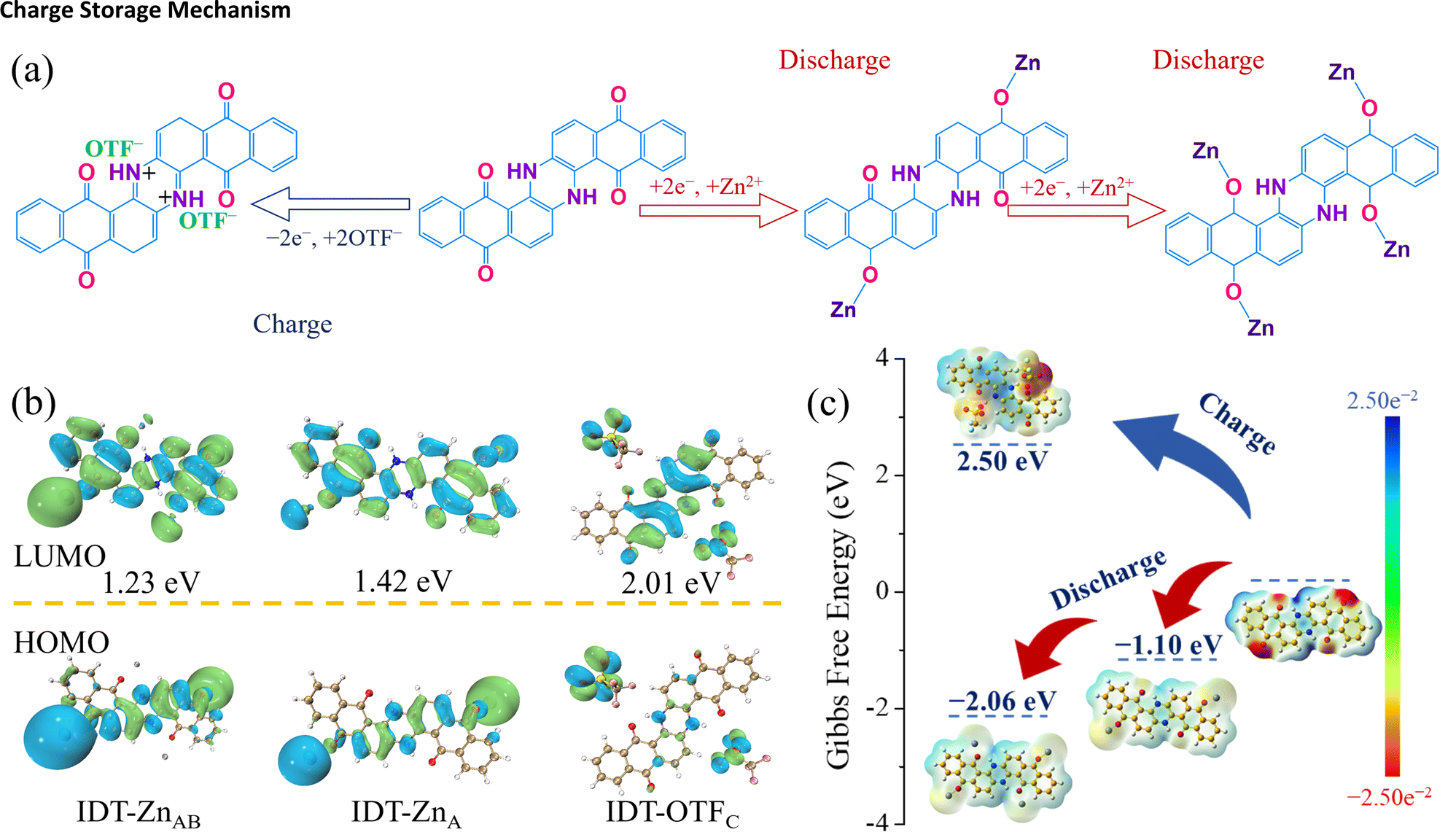 | ||
| Fig. 4 (a) Mechanism illustration of the Zn//IDT battery with 2 M Zn(OTF)2 electrolyte. (b) The calculated energy level of IDT associated with different amounts of Zn2+ and OTF−. (c) Molecular ESP simulation and Gibbs free energy. | ||
Conclusions
In summary, we demonstrate a novel bipolar-type conjugated structure of the indanthrone (IDT) molecule composed of p-type N–H bridges with n-type anthraquinone units as an organic cathode material. This novel bipolar conjugated structure greatly increases the conductivity, improves the stability of the structure, and ensures the high utilization of high-density active sites. A narrow Eg of IDT is 2.16 eV obtained using DFT calculations and solid-state UV-vis spectroscopy, and its unique molecular structure provides fast reaction kinetics and a strong electrochemical response. As expected, the Zn//IDT battery has a high capacity of 238 mA h g−1 at 0.2 A g−1. It maintains 88.7% of its initial capacity after 3000 cycles at 10 A g−1. Ex situ characterization studies and electrochemical tests confirm its excellent electrochemical performance due to the hybrid energy storage mechanism of multi-electron participation involving bipolar active carbonyl (CO) and amine (N–H) groups. Zn2+ is a redox-related cation for CO, while the OTF− anion participates in the conversion of CO between NH+ and N–H during storage. The bipolar ionic storage mechanism offers promising battery chemistry, and such a molecular design concept provides a new idea for the design of high-performance organic electrodes for AZIBs.
Author contributions
Chengmin Hu: project administration, conceptualization, methodology, formal analysis, data curation, writing – original draft. Xiaozhe Yang: conceptualization, investigation. Pingxuan Liu: investigation, visualization. Ziyang Song: methodology, investigation, visualization, writing – review & editing. Yaokang Lv: investigation. Ling Miao: visualization, formal analysis, investigation, writing – review & editing. Mingxian Liu: funding acquisition, methodology, supervision, writing – review & editing. Lihua Gan: funding acquisition, supervision, project administration, visualization, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22272118, 22172111, 21905207, and 22309134), the Science and Technology Commission of Shanghai Municipality, China (No. 22ZR1464100, 20ZR1460300, and 19DZ2271500), the China Postdoctoral Science Foundation (2022M712402), the Shanghai Raising-Star Program (23YF1449200), the Zhejiang Provincial Science and Technology Project (2022C01182), and the Fundamental Research Funds for the Central Universities (22120210529 and 2023-3-YB-07).Notes and references
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS.
- D. Zhang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Chem. Sci., 2024, 15, 4322–4330 RSC.
- X. Jia, C. Liu, Z. G. Neale, J. Yang and G. Cao, Chem. Rev., 2020, 120, 7795–7866 CrossRef CAS PubMed.
- W. Zhou, D. Zhu, J. He, J. Li, H. Chen, Y. Chen and D. Chao, Energy Environ. Sci., 2020, 13, 4157–4167 RSC.
- W. Xin, L. Miao, L. Zhang, H. Peng, Z. Yan and Z. Zhu, ACS Mater. Lett., 2021, 3, 1819–1825 CrossRef CAS.
- P. Ruan, S. Liang, B. Lu, H. J. Fan and J. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202200598 CrossRef CAS PubMed.
- L. Zhang, L. Miao, W. Xin, H. Peng, Z. Yan and Z. Zhu, Energy Storage Mater., 2022, 44, 408–415 CrossRef.
- H. Li, C. Guo, T. Zhang, P. Xue, R. Zhao, W. Zhou, W. Li, A. Elzatahry, D. Zhao and D. Chao, Nano Lett., 2022, 22, 4223–4231 CrossRef CAS PubMed.
- X. Chen, W. Li, D. Reed, X. Li and X. Liu, Electrochem. Energy Rev., 2023, 6, 33 CrossRef CAS.
- L. Ma, S. Chen, C. Long, X. Li, Y. Zhao, Z. Liu, Z. Huang, B. Dong, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1902446 CrossRef CAS.
- K. Yang, Y. Hu, T. Zhang, B. Wang, J. Qin, N. Li, Z. Zhao, J. Zhao and D. Chao, Adv. Energy Mater., 2022, 12, 2202671 CrossRef CAS.
- F. Mo, G. Liang, Q. Meng, Z. Liu, H. Li, J. Fan and C. Zhi, Energy Environ. Sci., 2019, 12, 706–715 RSC.
- Y. Zhang, F. Wan, S. Huang, S. Wang, Z. Niu and J. Chen, Nat. Commun., 2020, 11, 2199 CrossRef CAS PubMed.
- T. Bashir, S. Zhou, S. Yang, S. A. Ismail, T. Ali, H. Wang, J. Zhao and L. Gao, Electrochem. Energy Rev., 2023, 6, 5 CrossRef CAS.
- M. R. Tuttle, S. T. Davis and S. Zhang, ACS Energy Lett., 2021, 6, 643–649 CrossRef CAS.
- W. Jian, W. Zhang, X. Wei, B. Wu, W. Liang, Y. Wu, J. Yin, K. Lu, Y. Chen, H. N. Alshareef and X. Qiu, Adv. Funct. Mater., 2022, 32, 2209914 CrossRef CAS.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- M. Tang, C. Jiang, S. Liu, X. Li, Y. Chen, Y. Wu, J. Ma and C. Wang, Energy Storage Mater., 2020, 27, 35–42 CrossRef.
- H. Zhang, S. Xie, Z. Cao, D. Xu, L. Wang, H. Fang, J. Shen and M. Ye, ACS Appl. Energy Mater., 2021, 4, 655–661 CrossRef CAS.
- K. Qin, J. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 RSC.
- C. Chen, C. S. Lee and Y. Tang, Nano-Micro Lett., 2023, 15, 121 CrossRef PubMed.
- Z. Cheng, J. Le, J. Wang, W. Sun, L. Zheng, L. Cai, D. Lu, Y. Shen, J. Wu, F. Fu and H. Chen, Adv. Energy Mater., 2021, 12, 2101930 CrossRef.
- Y. Xu, G. Zhang, J. Liu, J. Zhang, X. Wang, X. Pu, J. Wang, C. Yan, Y. Cao, H. Yang, W. Li and X. Li, Energy Environ. Mater., 2023, 6, e12575 CrossRef CAS.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- R. Zhang, H. Xu, D. Luo, J. Chi, Z. Fan, H. Dou and X. Zhang, Chem. Eng. J., 2023, 458, 141336 CrossRef CAS.
- G. Sun, B. Yang, X. Chen, Y. Wei, G. Yin, H. Zhang and Q. Liu, Chem. Eng. J., 2022, 431, 134253 CrossRef CAS.
- K. W. Nam, H. Kim, Y. Beldjoudi, T. W. Kwon, D. J. Kim and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2541–2548 CrossRef CAS PubMed.
- Y. Zhang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202316835 CrossRef CAS PubMed.
- F. Ye, Q. Liu, H. Dong, K. Guan, Z. Chen, N. Ju and L. Hu, Angew. Chem., Int. Ed., 2022, 61, e202214244 CrossRef CAS PubMed.
- S. Bian, Y. Yang, S. Liu, F. Ye, H. Tang, Y. Wu and L. Hu, Chem.–Eur. J., 2024, e202303917 CrossRef CAS PubMed.
- F. Ye, Q. Liu, C. J. Lu, F. Q. Meng, T. Lin, H. L. Dong, L. Gu, Y. P. Wu, Z. L. Tang and L. F. Hu, Energy Storage Mater., 2022, 52, 675–684 CrossRef.
- D. Kundu, P. Oberholzer, C. Glaros, A. Bouzid, E. Tervoort, A. Pasquarello and M. Niederberger, Chem. Mater., 2018, 30, 3874–3881 CrossRef CAS.
- Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Adv. Sci., 2024, 11, 2310319 CrossRef PubMed.
- H. Glatz, E. Lizundia, F. Pacifico and D. Kundu, ACS Appl. Energy Mater., 2019, 2, 1288–1294 CrossRef CAS.
- Y. Luo, F. Zheng, L. Liu, K. Lei, X. Hou, G. Xu, H. Meng, J. Shi and F. Li, ChemSusChem, 2020, 13, 2239–2244 CrossRef CAS PubMed.
- S. Gu, J. Chen, R. Hao, X. Chen, Z. Wang, I. Hussain, G. Liu, K. Liu, Q. Gan, Z. Li, H. Guo, Y. Li, H. Huang, K. Liao, K. Zhang and Z. Lu, Chem. Eng. J., 2023, 454, 139877 CrossRef CAS.
- F. A. Obrezkov, A. F. Shestakov, S. G. Vasil'ev, K. J. Stevenson and P. A. Troshin, J. Mater. Chem. A, 2021, 9, 2864–2871 RSC.
- Y. Zhao, N. Xu, M. Ni, Z. Wang, J. Zhu, J. Liu, R. Zhao, H. Zhang, Y. Ma, C. Li and Y. Chen, Adv. Mater., 2023, 35, e2211152 CrossRef PubMed.
- Z. Li, J. Tan, Y. Wang, C. Gao, Y. Wang, M. Ye and J. Shen, Energy Environ. Sci., 2023, 16, 2398–2431 RSC.
- B. Pan, L. Bai, C. M. Hu, X. Wang, W. S. Li and F. G. Zhao, Adv. Energy Mater., 2020, 10, 2000181 CrossRef CAS.
- T. Shi, G. Li, Y. Han, Y. Gao, F. Wang, Z. Hu, T. Cai, J. Chu and Z. Song, Energy Storage Mater., 2022, 50, 265–273 CrossRef.
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330 RSC.
- Z. Song, L. Miao, H. Duan, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202401049 Search PubMed.
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, L. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219136 CrossRef CAS PubMed.
- L. Lin, Z. Lin, J. Zhu, K. Wang, W. Wu, T. Qiu and X. Sun, Energy Environ. Sci., 2023, 16, 89–96 RSC.
- H. Peng, S. Huang, V. Montes-Garcia, D. Pakulski, H. Guo, F. Richard, X. Zhuang, P. Samori and A. Ciesielski, Angew. Chem., Int. Ed., 2023, 62, e202216136 CrossRef CAS PubMed.
- Z. Li, J. Tan, X. Zhu, S. Xie, H. Fang, M. Ye and J. Shen, Energy Storage Mater., 2022, 51, 294–305 CrossRef.
- T. Sun, W. Zhang, Z. Zha, M. Cheng, D. Li and Z. Tao, Energy Storage Mater., 2023, 59, 102778 CrossRef.
- W. Du, L. Miao, Z. Song, X. Zheng, C. Hu, Y. Lv, L. Gan and M. Liu, Chem. Eng. J., 2024, 484, 149535 CrossRef CAS.
- J. Zhang, W. Li, J. Wang, X. Pu, G. Zhang, S. Wang, N. Wang and X. Li, Angew. Chem., Int. Ed., 2023, 62, e202215654 CrossRef CAS PubMed.
- J. Li, N. Luo, L. Kang, F. Zhao, Y. Jiao, T. J. Macdonald, M. Wang, I. P. Parkin, P. R. Shearing, D. J. L. Brett, G. Chai and G. He, Adv. Energy Mater., 2022, 12, 2201840 CrossRef CAS.
- Q. Huang, L. Huang, Y. Jin, Y. Sun, Z. Song and F. Xie, Chem. Eng. J., 2024, 482, 148912 CrossRef CAS.
- Z. Zhou, X. Zhou, M. Zhang, S. Mu, Q. Liu and Y. Tang, Small, 2020, 16, e2003174 CrossRef PubMed.
- T. Shi, Z. Song, Y. Lv, D. Zhu, L. Miao, L. Gan and M. Liu, Chin. Chem. Lett., 2024, 35, 109559 CrossRef.
- Y. Kong, C. Tang, X. Huang, A. K. Nanjundan, J. Zou, A. Du and C. Yu, Adv. Funct. Mater., 2021, 31, 2010569 CrossRef CAS.
- Z. Tie, Y. Zhang, J. Zhu, S. Bi and Z. Niu, J. Am. Chem. Soc., 2022, 144, 10301–10308 CrossRef CAS PubMed.
- P. Liu, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Small, 2024, 20, 2400774 Search PubMed.
- Q. Huang, Y. Jin, L. Huang, Y. Cong and Z. Xu, J. Mater. Chem. A, 2023, 11, 12297–12307 RSC.
- C. Hu, Y. Qin, Z. Song, P. Liu, L. Miao, H. Duan, Y. Lv, L. Xie, M. Liu and L. Gan, J. Colloid Interface Sci., 2024, 658, 856–864 CrossRef CAS PubMed.
- Q. Tang, Z. Liang, J. Liu, J. Xu and Q. Miao, Chem. Commun., 2010, 46, 2977–2979 RSC.
- Y. Dai, J. Li, L. Chen, K. Le, Z. Cai, Q. An, L. Zhang and L. Mai, ACS Energy Lett., 2021, 6, 684–686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00476k |
| This journal is © The Royal Society of Chemistry 2024 |