DOI:
10.1039/D4TA04399E
(Paper)
J. Mater. Chem. A, 2024,
12, 30329-30339
Regulating NO2 adsorption at ambient temperature by manipulating copper species as binding sites in copper-modified SSZ-13 zeolites†
Received
25th June 2024
, Accepted 6th October 2024
First published on 15th October 2024
Abstract
Atmospheric NO2 pollution poses significant risks to human health and the environment even at low concentrations, necessitating the development of efficient technologies for its removal under ambient conditions. In this study copper (Cu)-modified SSZ-13 zeolites (referred to as Cun+SSZ-13 where n represents the valence state of Cu) were developed for NO2 removal by adsorption. Cun+SSZ-13 zeolites containing Cu species with different valence states and proportions were prepared by reducing a Cu2+-exchanged SSZ-13 zeolite (Cu2+SSZ-13) using H2 at different temperatures. The Cun+SSZ-13 reduced at 190 °C showed the highest NO2 removal capacity (1.79 mmol g−1), outperforming pristine SSZ-13 and Cu2+SSZ-13 by 52.3% and 19.4%, respectively. The improvement was due to the increased amount of adsorption sites (Cu+ and H+) and the stronger affinity of Cu+ than Cu2+ for NO2, as confirmed by density functional theory (DFT) calculations. The generation of Cu0 nanoparticles and moisture in zeolites during reduction was undesirable for NO2 adsorption. However, this could be eliminated by lowering the reduction temperature and performing thermal activation, respectively. This work provides systematic methods for designing zeolite adsorbents for ambient NO2 removal and offers insights into the burgeoning field of air pollution control.
1. Introduction
Nitrogen oxides (NOx), including NO and NO2, are primarily emitted from mobile and stationary sources such as vehicles and industries. NOx have raised public concerns due to their detrimental effects on both human health and ecosystems.1–3 For example, NOx can interact with other air pollutants such as volatile organic compounds (VOCs) under sunlight, leading to the formation of secondary pollutants such as smog and highly irritating gases.4–6 Meanwhile, NOx themselves, as toxic components, can directly affect human health by decreasing lung function and causing respiratory symptoms, even at low concentrations.7 As NO2 is more active and around 30 times more toxic than NO,8 NO2 is categorized as a criteria pollutant representing NOx. Thus, atmospheric NO2 pollution and its control have attracted increased attention in recent decades, particularly in densely populated urban areas. For example, the concentration of roadside NO2 in Hong Kong remains at a harmful level (80 μg m−3),9 which is twice the World Health Organization (WHO) standard (40 μg m−3).10 The complex and congested nature of Hong Kong makes it challenging to dilute the roadside NO2. Therefore, efforts to abate atmospheric NO2 should focus on both reducing NO2 emissions in exhaust gases and removing NO2 from the ambient air.
De-NOx technologies, such as selective catalytic reduction (SCR) and selective non-catalytic reduction (SNCR), have been extensively developed and implemented, delivering high efficiency in NOx removal (99%).11–13 However, the operating temperature window (300–1100 °C) of these technologies makes their application prohibitive in low-temperature NOx (e.g., atmospheric NOx) abatement. Moreover, the generation of NH3 (as a reductant) by decomposing urea solution at low temperatures (<180 °C) poses a significant challenge for the SCR process.14 In addition, stricter regulations have increased the urgency for low-temperature NOx emission control. Adsorption is considered an alternative approach for NOx removal at low temperature. However, most adsorbents have only been evaluated for NOx adsorption at temperatures above 80 °C,15–17 omitting NO2 adsorption under ambient conditions. Thus, this study aims to develop effective NO2 adsorbents, with high NO2 adsorption capacity and selectivity, for atmospheric NO2 abatement at ambient temperature.
Adsorbents that exhibit a specific affinity for NO2 with moderate strength can afford high NO2 selectivity and capacity, making them desirable for NO2 adsorption. Given the presence of π bonds in the NO2 molecule, materials containing transition metal species are promising candidates for NO2 adsorption due to their unique interaction (π-complexation) with NO2, which enables π-backdonation from a d orbital metal center to the π* orbitals of the π-structured molecules.18 This interaction, with moderate strength, bridges the gap between physisorption and chemisorption, enabling the π-complexation adsorbents to exhibit both high selectivity and good reversibility.19 Copper (Cu), an affordable and abundant transition metal that has been extensively studied and used in high-temperature NOx SCR, has emerged as a promising candidate for NO2 adsorption.20–24 For instance, Cu-containing carbon showed a high NO2 removal capacity at room temperature, while the low thermal stability of carbon materials causes them to burn readily during regeneration at high temperatures.20 Cu nanoparticle (NP) loaded silica (KCC-1) has been reported to be an effective NO2 adsorbent, with the highly dispersed Cu NPs serving as effective adsorption sites for NO2.25 However, NO2 adsorption and dissociation on Cu2+ and Cu+ have not been studied. Cu-based metal–organic frameworks (MOFs) showed enhanced NO2 adsorption due to increased porosity and the reactive adsorption of NO2 on Cu.24 However, the high cost and low thermal/hydrothermal stability of MOFs limit their applications in real-word gas adsorption processes.
Small-pore zeolites, characterized by their three-dimensional porous structures, high thermal stability, and large surface areas, have extensive applications in gas adsorption and separation.26 Zeolites can also serve as unique ligands that tune the π-complexation between NO2 and Cu.27 Sierraalta et al. have demonstrated that Cu species-containing zeolites showed a stronger affinity for NO2 molecules than those containing other transition metals (Ag and Au).28 Onitsuka et al. reported that zeolites infused with Cu salts showed enhanced performance in adsorbing NOx at a low concentration of about 5 ppm.29 Note that extra-framework cations in zeolites are the dominant adsorption sites, whose properties dictate gas adsorption performance.30 For instance, the valence state of these cations can affect NO2 adsorption performance, particularly the affinity of the adsorbent for NO2. For specific types of cations, such as Cu, their valence state is directly related to the radius and the occupancy of the valence shell. Compared with Cu2+, Cu+ and Cu0 possess higher valence shell occupancies and fewer constraints (due to the larger radius) on the valence shell electrons, allowing more d-orbital electrons to be back-donated to the π* orbital of NO2. Consequently, the π-complexation occurring between NO2 and Cu ions at lower states (Cu+ and Cu0) is anticipated to improve gas–host interaction, leading to a higher affinity for NO2.
Herein, we prepared Cun+SSZ-13 zeolites (n = 0, 1, and 2) by reducing a Cu2+-exchanged SSZ-13 zeolite under a H2 atmosphere and systematically studied their NO2 removal performance and mechanisms. Thanks to the formation of Cu+ ions and the increased number of adsorption sites, Cun+SSZ-13 zeolites showed enhanced NO2 uptake and insignificant NO release. This work suggests that the tuning of Cu species as specific binding sites in zeolites could be an effective approach for regulating the affinity of zeolites for NO2 adsorption.
2. Experimental
2.1 Synthesis
2.1.1 Synthesis of H+SSZ-13 (Si/Al = 12).
The synthesis of H+SSZ-13 (Si/Al = 12) was conducted following a previously reported method.31 Specifically, 39.6 g of N,N,N-trimethyl-1-adamantanamine hydroxide (TMAdaOH, 25 wt%, Sachem Inc.) and 19.5 g of tetraethyl orthosilicate (TEOS, sigma 98%) were mixed with 4.05 g of deionized (DI) water. This mixture was stirred for 2 h at room temperature in a sealed polypropylene bottle. Afterwards, 1.05 g of aluminum ethoxide (Strem Chemical, 99%) was added to the mixture and stirred for another 1 h. The gel was then transferred into a 100 mL Teflon-lined autoclave and heated at 140 °C under static conditions for 6 days. After the crystallization, the product was filtered, thoroughly washed with DI water, and then dried at 80 °C overnight. Finally, the product was calcined in an air atmosphere at 550 °C for 8 h with a ramping rate of 2 °C min−1.
2.1.2 Synthesis of Cu2+SSZ-13.
The synthesis of Cu2+SSZ-13 involved a liquid-phase ion exchange process, a common approach for incorporating extra-framework cations into zeolites. Typically, 0.5 g of the as-synthesized H+SSZ-13 was dispersed in 50 mL of 0.5 M NH4NO3 and stirred for 12 h at 80 °C, with the process being repeated four times to ensure thorough ion exchange. The resulting NH4+SSZ-13 product was filtered, thoroughly washed with DI water, and dried at 80 °C overnight. Then, 0.5 g of the as-synthesized NH4+SSZ-13 was dispersed in 50 mL of 0.5 M Cu(NO3)2 solution and stirred for 12 h at 80 °C, again being repeated four times. The resulting Cu2+SSZ-13 was collected by filtration, washed extensively with DI water, and dried at 353 K overnight.
2.1.3 Synthesis of H2-reduced Cun+SSZ-13 samples.
A series of Cun+SSZ-13 samples were prepared by reducing Cu2+SSZ-13 under a H2 atmosphere at various temperatures. H2-temperature programmed reduction (TPR) was conducted on a Micromeritics ASAP 2950 instrument to identify optimal reduction conditions for the preparation of Cun+SSZ-13. The signal of H2 during the reduction process was detected using a thermal conductivity detector (TCD). Then, 0.1 g of Cu2+SSZ-13 was placed in a tube furnace and thermally treated under argon at 350 °C (ramping rate = 2 °C min−1) for 4 h to remove the pre-adsorbed impurities. After cooling to room temperature, 5% H2 (balanced with Ar, 20 cc min−1) was continuously purged and Cu2+SSZ-13 was heated at 100 °C to 700 °C for 3 h (ramping rate = 2 °C min−1). The resulting products are named Cun+SSZ-13-reducing temperature-R (e.g., Cun+SSZ-13-100-R).
2.2 Characterization
Synchrotron powder X-ray diffraction (PXRD) was measured using a Mythen-II detector at the PD beamline, Australian Synchrotron, ANSTO. To understand the valence state and the coordination environment of the Cu ions in Cun+SSZ-13 samples, Cu K-edge X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were collected at the XAS beamline, Australian Synchrotron, ANSTO. The morphologies of the samples were investigated using a scanning electron microscope (SEM) (EVO MA10, ZEISS, Germany). The specific surface area and pore volume of Cun+SSZ-13-190-R before and after NO2 dynamic column adsorption, as well as fresh H+SSZ-13, were determined using a 3Flex Surface Characterization Analyzer (Micromeritics Instrument Corp., USA) using N2 at −196 °C. Prior to measurement, the samples were degassed on the 3Flex Surface Characterization Analyzer at 300 °C for 3 h. The chemical composition (e.g., Si/Al and metal/Al ratios) of the samples was determined by Energy dispersive spectroscopy (EDX) using an EDX detector (Oxford Aztec Energy X-MAX 50) installed in a SEM (FEI Quanta 450 FEG). The in situ Fourier Transform Infrared (FTIR) spectra of Cun+SSZ-13-190-R were recorded on an IRAffinity-1 FTIR spectrophotometer (Shimadzu, Japan) at 25 °C, with an NO2 concentration of 1000 ppm and a flow rate of 60 mL min−1. Before the measurement, Cun+SSZ-13-190-R was thermally treated in the FTIR cell under a N2 atmosphere at 200 °C for 8 h to remove the pre-adsorbed gases and moisture.
2.3 Fixed-bed NO2/NO dynamic column adsorption at room temperature
The dynamic column breakthrough (DCB) of NOx was measured using a fixed-bed system (Fig. S1†) under ambient conditions (25 °C, 1 bar). The adsorbent (0.1 g) was well mixed with non-reactive glass beads (3 mm in diameter) to generate a homogeneous bed, and then packed into a stainless-steel tube. Unless otherwise specified, the samples were degassed at 300 °C under argon before NOx adsorption until the pre-adsorbed moisture was completely removed, as monitored by a Mass Spectrometer (Stanford Research Systems UGA300). Subsequently, NO2 or NO (both at 1000 ppm) in helium was purged through the column at a total inlet flow rate of 60 mL min−1 at 25 °C. The concentrations of NO2 and NO in the outlet gas were measured using an electrochemical sensor (RAE Systems, MultiRAE Plus PGM-50/5P).
2.4 Density functional theory (DFT) calculation
DFT calculations were performed using the QUANTUM ESPRESSO package.32–35 Ultrasoft pseudopotentials with the Perdew–Burke–Ernzerhof exchange–correlation functional were employed.36 The plane wave cutoffs were 30 Ry and 240 Ry for wavefunctions and the augmented charge density, respectively. The semiempirical Grimme's DFT-D3 scheme was used for van der Waals interaction correction.37 The SSZ-13 zeolite has a 1 × 1 × 1 supercell with a total trigonal P supercell size of 13.675 Å × 13.675 Å × 14.767 Å. Three Si atoms were replaced with Al atoms. To maintain the charge balance, hydrogen atoms were added near the Al atoms according to the charge of the inserted transition metal ions. The 6-membered ring (6MR) has been identified as the most favorable location for the extra-framework cations.38 Thus, we examined the local structures of the Cu ions in the 6MR, with 2 Si atoms being replaced by 2 Al atoms.
3. Results and discussion
3.1 Characterization of adsorbents
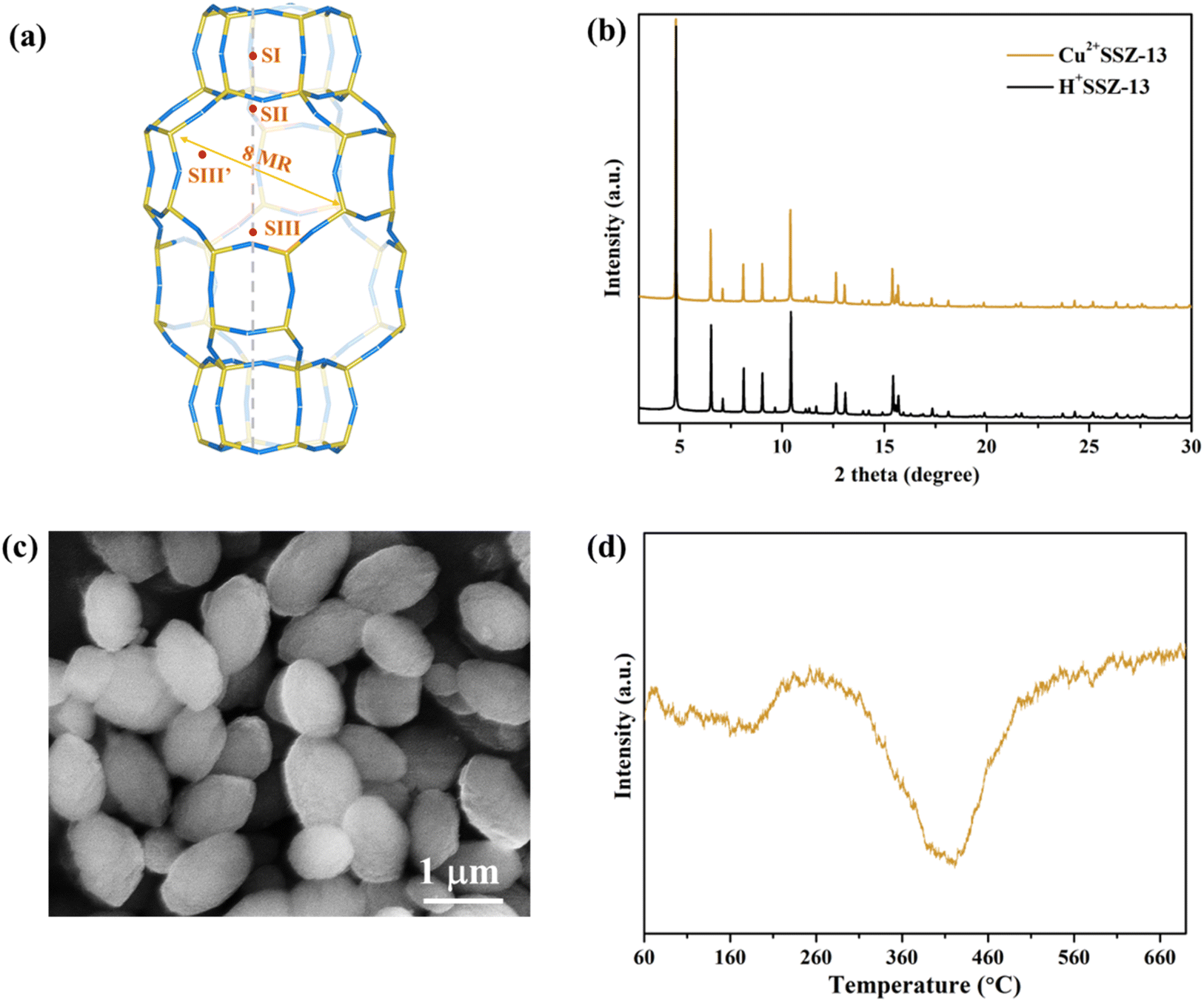
Cu2+SSZ-13 was prepared from H+SSZ-13 by an ion-exchange method, where Cu2+ substituted the extra-framework H+ cations (Fig. 1a). The synchrotron PXRD patterns of Cu2+SSZ-13 indicated the high purity and crystallinity characteristic of chabazite (CHA)-type SSZ-13 zeolite (Fig. 1b). As displayed in the SEM image (Fig. 1c), Cu2+SSZ-13 particles possess an elliptical shape and smooth exterior surfaces. Cun+SSZ-13 zeolites were then prepared by reducing Cu2+SSZ-13 under H2. Prior to the reduction, H2-TPR was conducted to determine the optimal reduction temperature (Fig. 1d). With the temperature increasing, two H2 consumption peaks appeared at around 200 and 400 °C, which were attributed to the reduction of Cu2+ at various locations within the zeolite.39 It is noteworthy that Cu2+ ions at different locations within the zeolite require different energy levels to be reduced. For example, the six-membered ring (6MR) in CHA is the most stable site for Cu2+, demanding more energy for reduction than those situated at an eight-membered ring (8MR).40,41 To achieve varying levels of Cu2+ reduction in SSZ-13 zeolite, both low (≤250 °C) and high temperature (≥390 °C) ranges were adopted.
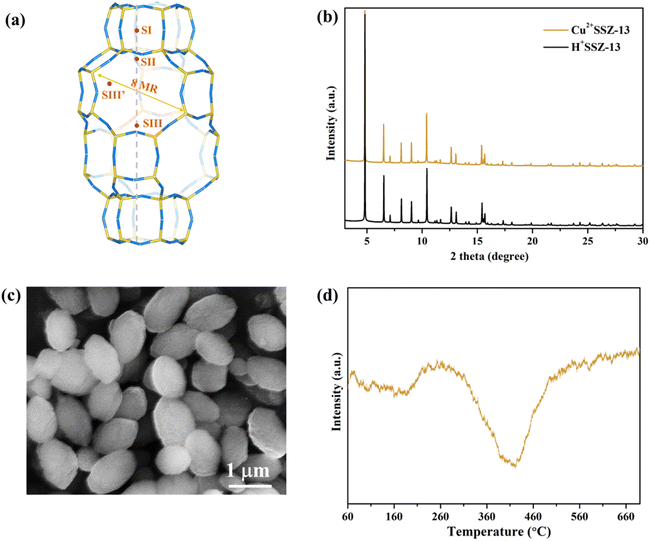 |
| | Fig. 1 (a) Illustration of the local crystal structure of chabazite (CHA) zeolites. SI–SIII’ are the possible positions of extra-framework cations; (b) synchrotron powder X-ray diffraction patterns of Cu2+SSZ-13 and pristine H+SSZ-13 (wavelength = 0.7735 Å); (c) SEM image of Cu2+SSZ-13; and (d) H2-TPR profile of Cu2+SSZ-13. | |
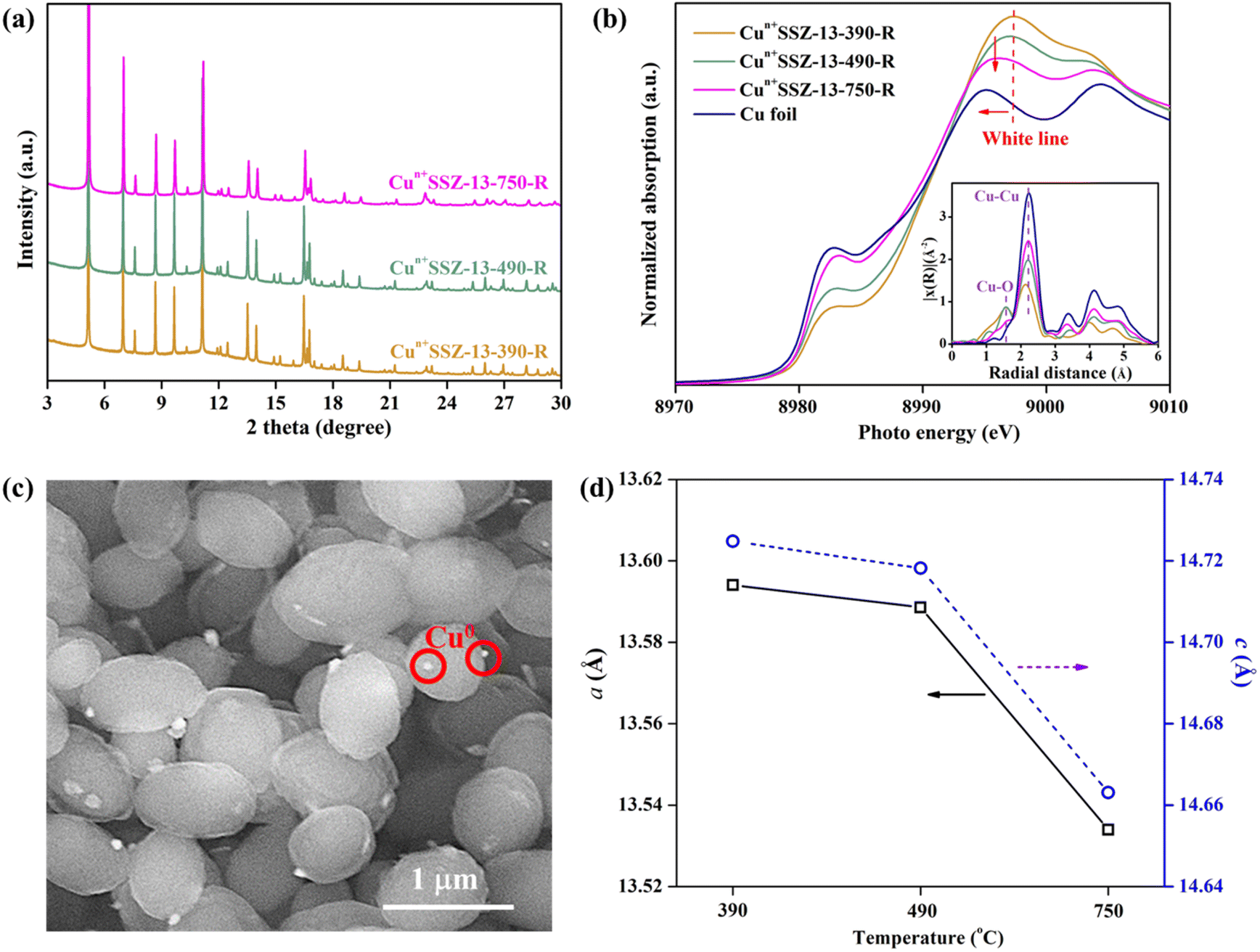
Upon high-temperature reduction (390, 490, and 750 °C), the structure of Cun+SSZ-13 samples remained intact, as evidenced by the unchanged PXRD peak locations of Cun+SSZ-13 and Cu2+SSZ-13 (Fig. 2a). However, the undesirable formation of Cu0 occurred on the external surface of the zeolite (Fig. 2c). Rietveld refinement of PXRD indicated that the crystallite size of these Cu0 particles (17, 23, and 25 nm at 390, 490, and 750 °C, respectively) increased with the increasing reduction temperature (Fig. S2†). The X-ray absorption spectroscopy (XAS) results showed that higher reduction temperatures resulted in a shift towards lower energy and a reduction in intensity of the white line peak (Fig. 2b), corresponding to decreases in the valence state and the occupancy of d orbitals of Cu ions.42 These observations suggest that during the reduction, Cu2+ migrated from the supercage to the external surface of zeolites and aggregated to form metallic Cu particles (Fig. 2c). This mitigation process can be explained by the R-plot EXAFS spectra (Fig. 2b, inset). Two peaks appearing at 1.7 Å and 2.2 Å were ascribed to the Cu–O and Cu–Cu bonds, respectively, arising from the coordination of Cu ions with lattice oxygen atoms in the zeolite (Cu–O) and Cu–Cu bonds in elementary Cu.43 The intensity of the Cu–O peak decreased while that of the Cu–Cu peak increased upon reduction at higher temperatures, indicating that more Cu2+ migrated from the cage to the external surface of zeolites to form elementary Cu. This result is consistent with the reduced lattice parameters of Cun+SSZ-13 at high reduction temperatures (Fig. 2d), which was attributed to the contraction of the zeolite lattice due to negative thermal expansion.44
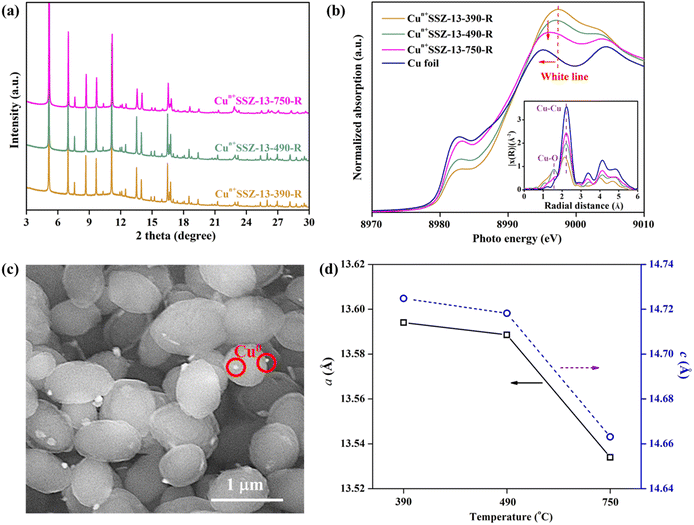 |
| | Fig. 2 (a) PXRD patterns (wavelength: 0.8275 Å) and (b) Cu K-edge XAS spectra of Cun+SSZ-13 reduced by H2 at 390, 490, and 750 °C (inset: Fourier transformed EXAFS modulation with phase correction in the k-range of 3.0–12.5 Å−1, k2-weighted); (c) SEM image of Cun+SSZ-13-390-R; and (d) variation of unit cell parameters (a and c) of Cun+SSZ-13 in relation to the H2-reduction temperature. | |
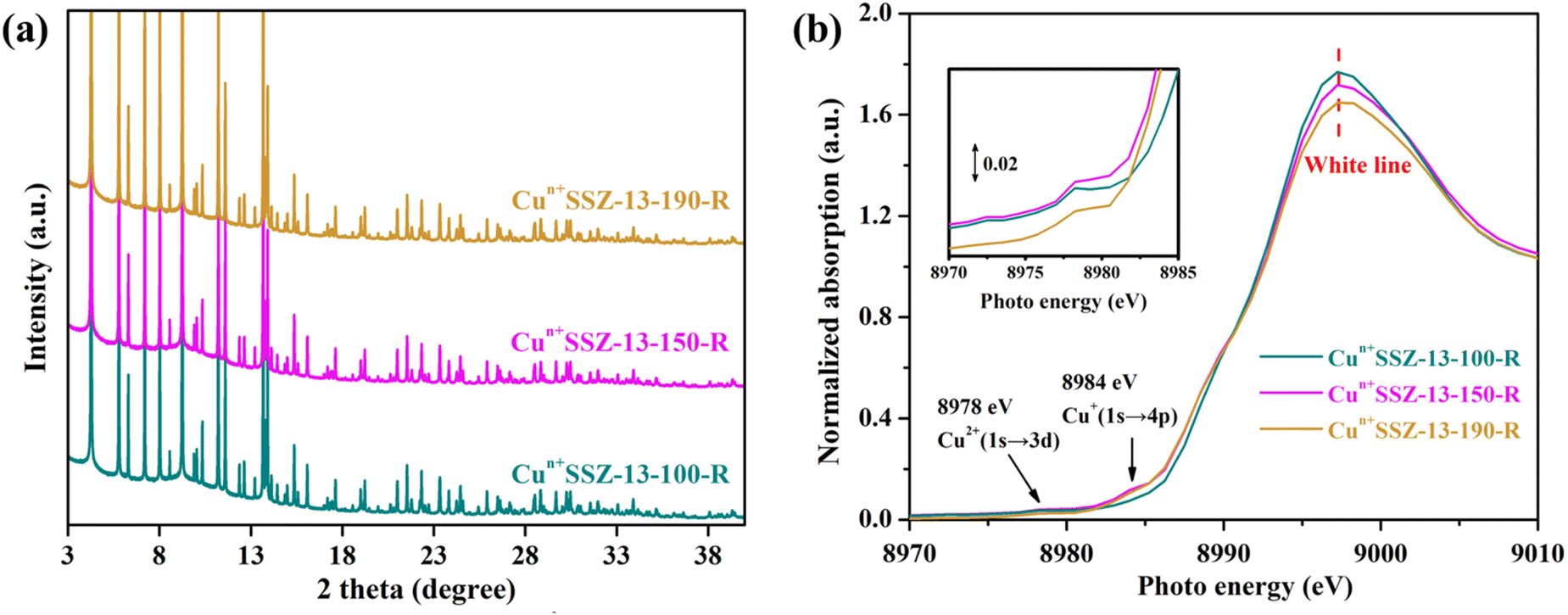
It is acknowledged that the well dispersed extra-framework cations in zeolites serve as the dominant active adsorption sites for gas molecules.27,45,46 Therefore, the presence of bulk Cu0 NPs on the external surface of zeolites should be minimized. Consequently, a further decrease in the reduction temperature was implemented. No characteristic peaks of elementary Cu were observed in the PXRD patterns of Cun+SSZ-13 samples reduced at 100, 150, 170 and 190 °C (Fig. 3a), indicating the absence of Cu0. The XAS results showed the changes in Cu2+/Cu+ levels within zeolites (Fig. 3b). The weak absorption peak ranging between 8975 and 8980 eV is the fingerprint of Cu2+, which is assigned to a dipole-forbidden 1s → 3d transition.47 The characteristic peak of Cu+ was observed at 8980–8985 eV, corresponding to the 1s → 4p transition.48 As the reduction temperature increased, the Cu2+ peak at 8978 eV diminished, while the Cu+ peak at 8984 eV gradually increased, suggesting that a higher reduction temperature facilitated the conversion of Cu2+ to Cu+. The edge peak at 8993–8997 eV, known as the ‘white line’ of Cu2+, indicates a higher oxidation state when the peak intensity is greater.48 As the reduction temperature increased, the intensity of the white line decreased, further suggesting a transformation of Cu2+ to Cu+.
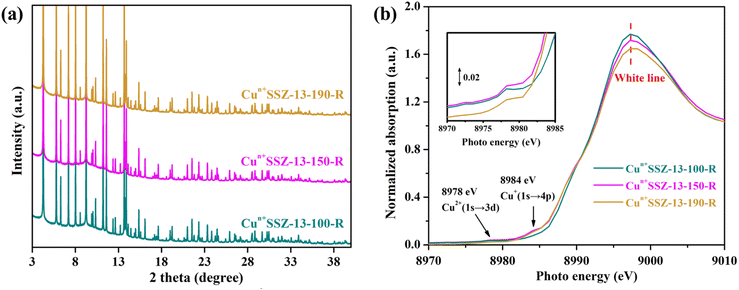 |
| | Fig. 3 (a) PXRD patterns (wavelength of 0.6880 Å) and (b) Cu K-edge XAS of Cun+SSZ-13 reduced by H2 at 100, 150, 170 and 190 °C. | |
3.2 NO2 adsorption
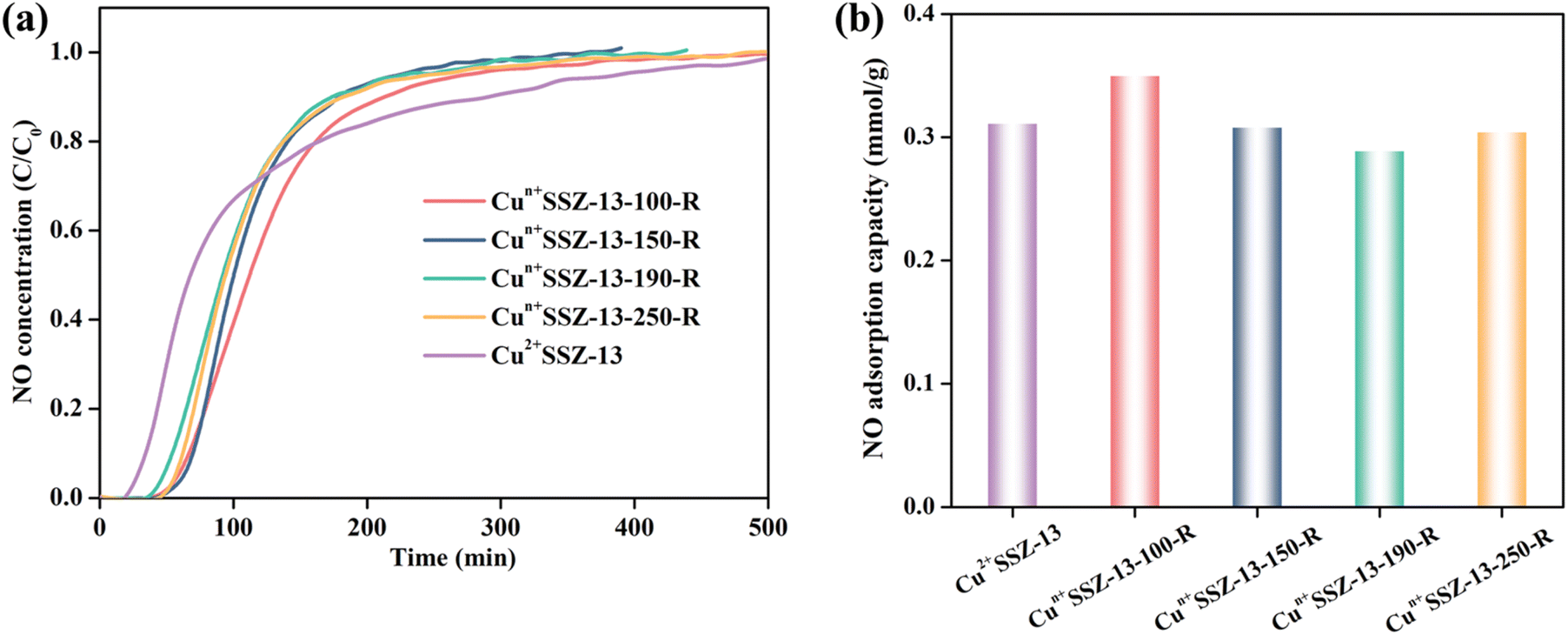
The NO2 adsorption performance of Cun+SSZ-13 reduced at different temperatures (i.e., 100, 150, 170, and 190 °C) was validated by NO2 dynamic adsorption at room temperature (Fig. 4a). Prior to NO2 adsorption, the adsorbents were activated at 300 °C under Ar to remove the pre-adsorbed impurities. The NO2 adsorption capacity of the Cun+SSZ-13 samples increased with the increasing reduction temperature (Fig. 4b), which is attributed to a higher proportion of Cu+ being present at a higher reduction temperature. The linear correlation between the NO2 capacity and the reduction temperature encouraged us to tentatively raise the reduction temperature to 250 °C, intending to further improve the NO2 adsorption capacity. However, the NO2 adsorption over Cun+SSZ-13-250-R was found to be even lower than that of the unreduced sample (Cu2+SSZ-13). This result was caused by the formation of Cu0 particles on the external surface of the zeolite (see the SEM image in Fig. S3†), which reduced the number of accessible active adsorption sites for NO2 molecules. The highest NO2 adsorption capacity, achieved by Cun+SSZ-13-190-R, was 1.79 mmol g−1 (Fig. 4b). This capacity demonstrated a significant increase of 19.4% and 52.3% with respect to that of Cu2+SSZ-13 and pristine H+SSZ-13, respectively, substantially surpassing the performance of the reported metal-modified zeolites (Table S1†). The above results clearly show that the H2 reduction of Cu2+SSZ-13 effectively improves the NO2 adsorption capacity, which is attributed to the increased affinity of binding sites for NO2.
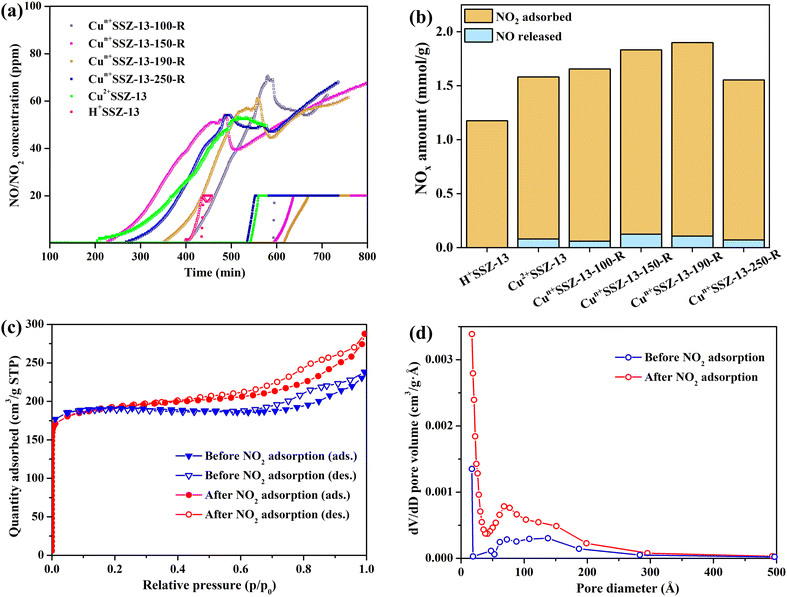 |
| | Fig. 4 (a) NO2 (1000 ppm) dynamic column adsorption at room temperature on H+SSZ-13, Cu2+SSZ-13 and Cun+SSZ-13 samples with thermal activation. The hollow (□) and solid (■) dots represent the concentration of NO and NO2, respectively. (b) The corresponding NO2 adsorption capacity and NO released amount. (c) N2 adsorption–desorption isotherms (−196 °C) and (d) pore size distributions of Cun+SSZ-13-190-R before and after NO2 (1000 ppm) dynamic column adsorption. | |
To assess the impact of NO2 adsorption on the specific surface area and pore volume of the Cu-modified zeolite, N2 adsorption–desorption isotherms were recorded for Cun+SSZ-13-190-R before and after NO2 dynamic adsorption (Fig. 4c, d and Table S2†). Post-adsorption, the BET surface area of Cun+SSZ-13-190-R remained largely unchanged, while the pore volume slightly increased from 0.34 to 0.40 cm3 g−1 which was probably due to the formation of additional interparticle stacking pores. Therefore, the porous structures of Cun+SSZ-13-190-R remained largely unaffected after a cycle of NO2 dynamic adsorption. It is anticipated that the lifespan and adsorption capacity of Cun+SSZ-13-190-R can be substantially preserved by regenerating it through a gentle heating process to drive off adsorbed water and NO2. Furthermore, in comparison with H+SSZ-13 without Cu modification, which featured micropores with a Brunauer–Emmett–Teller (BET) surface area of 771.69 m2 g−1 and a pore volume of 0.38 cm3 g−1 (Fig. S4†), Cun+SSZ-13-190-R exhibited a micro-mesoporous structure with a reduced surface area (557.97 m2 g−1) and pore volume (0.34 cm3 g−1). Despite the larger surface area and pore volume of the unmodified H+SSZ-13, the Cu-modified Cun+SSZ-13-190-R demonstrated a higher NO2 adsorption capacity. This finding suggests that the incorporation of Cu into the SSZ-13 zeolite significantly enhances the NO2 adsorption process, playing a more important role than the surface area and pore volume. Therefore, while surface area and pore volume are important factors in adsorption processes, the unique role of Cu within the SSZ-13 zeolite is the key determinant for the improved NO2 removal performance.
To evaluate the effect of moisture on NO2 adsorption, experiments were conducted on non-activated Cun+SSZ-13 samples (i.e., without thermal treatment prior to NO2 adsorption). Compared with Cu2+SSZ-13, the non-activated Cun+SSZ-13 samples showed a decreased NO2 capacity and an increased release of NO (Fig. S5†). The decline in capacity was due to the presence of moisture within zeolites generated during the H2 reduction process (eqn (1)). Such moisture would poison the active adsorption sites (Cu ions) by forming Cu hydrates and blocking sites intended for NO2 adsorption, thereby reducing the NO2 capacity. FTIR analysis (Fig. S6†) suggested that the moisture could facilitate NO2 transformation into NO3− and gaseous NO (eqn (2)).49 After a certain time, the amount of released NO decreased, and the NO release curves stabilized, suggesting the moisture was gradually consumed by NO2. Therefore, the thermal activation process is essential to mitigate the negative effects of moisture on NO2 adsorption.
| | | 2Cu(OH)+ + H2 → 2Cu+ + 2H2O | (1) |
| | | 3NO2 + H2O → 2HNO3 + NO(g) | (2) |
In addition to improving NO2 capacity, moisture removal also contributed to the decrease of NO release, which was evident from the longer NO retention time and the more gradual slopes of NO release curves (Fig. 4a). However, the correlation between the amount of NO released and the reduction temperature was inconsistent, which resulted from the different NO retention abilities of the samples. For example, Cun+SSZ-13-100-R showed the highest NO adsorption capacity (Fig. 5), aligning with the lowest amount of NO released (Fig. 4b). The NO adsorption capacity decreased with the increasing reduction temperature from 100 to 190 °C, while it increased when the reduction temperature was further raised to 250 °C.
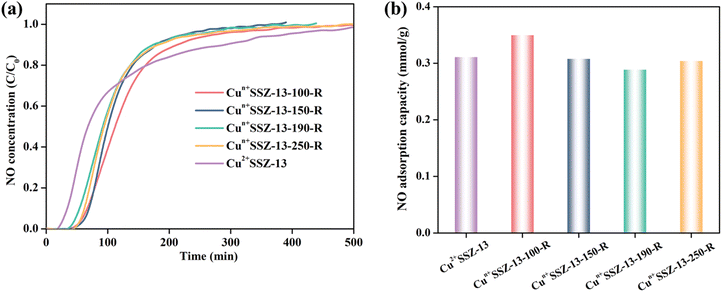 |
| | Fig. 5 (a) NO (1000 ppm) dynamic adsorption on Cun+SSZ-13 samples at room temperature; and (b) the corresponding NO adsorption capacity. | |
Our Cun+SSZ-13 adsorbent, which exhibits an enhanced capacity for ambient NO2 abatement yet sensitivity to moisture, can be effectively used for the reduction of NO2 emission from the engine pipeline when combined with desiccant materials or condensation systems that remove moisture from the exhaust gas. Additionally, our zeolite can be used in air purifiers or industrial settings to remove NO2, incorporating moisture control mechanisms such as heating elements or moisture-absorbing materials. Given the well-preserved specific surface area and pore volume after NO2 adsorption, as evidenced by porous property analysis, the Cun+SSZ-13-190-R adsorbent is expected to maintain its performance over multiple cycles of adsorption and thermal regeneration, thereby ensuring a long lifespan in the aforementioned practical applications.
3.3 Discussion of the mechanism
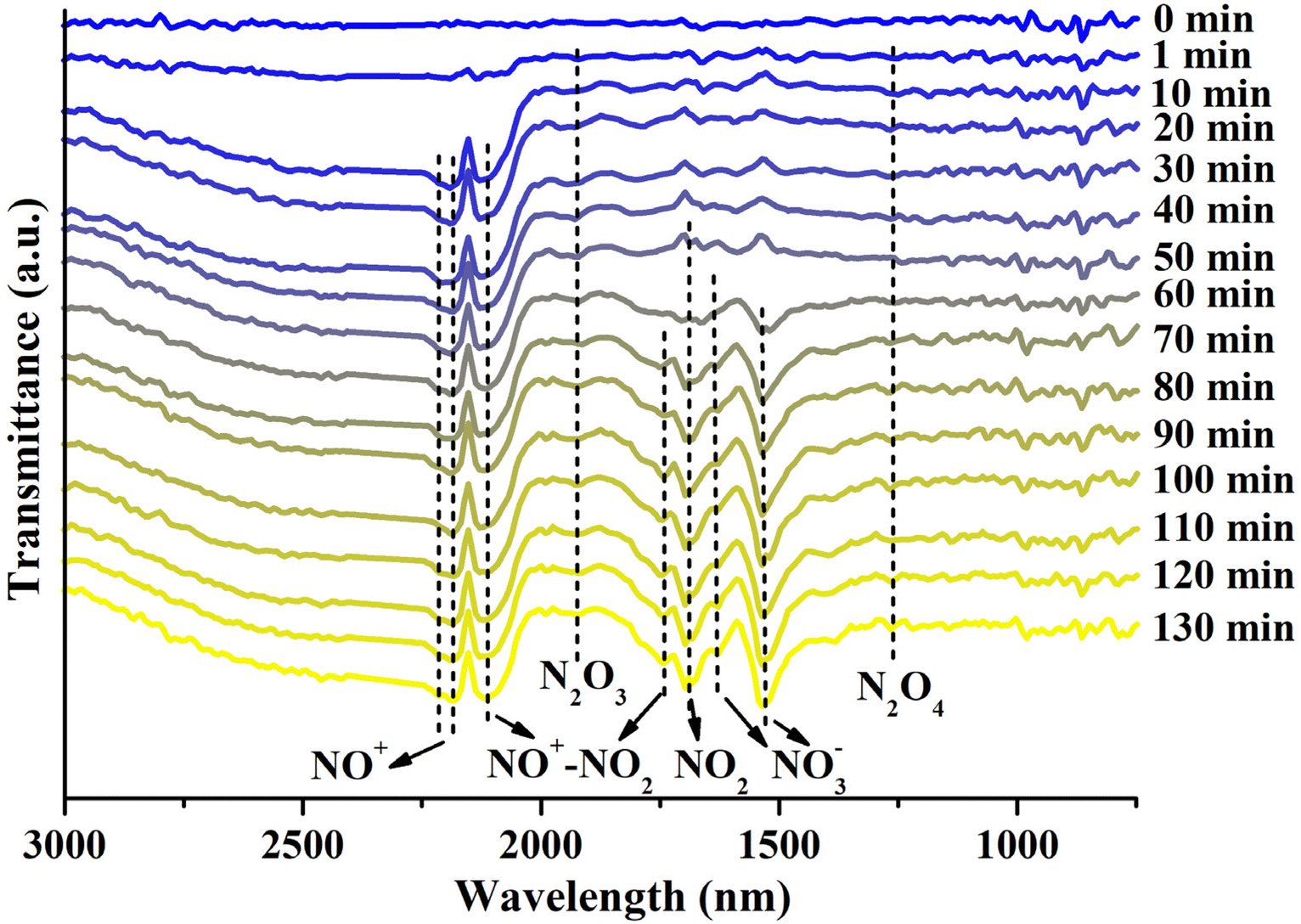
To elucidate the mechanism of NO2 adsorption on our Cu-modified zeolite, in situ FTIR analysis of the Cun+SSZ-13-190-R zeolite during the NO2 adsorption process was carried out (Fig. 6). The FTIR spectra revealed the presence of adsorbed NO2 at 1690 cm−1,50 which tended to form an N2O4 dimer (1261 cm−1)51 in small-pore zeolites due to the confinement effect.45 The adsorbed N2O4 can undergo ionization in the zeolite to form NO3− and NO+.52,53 As anticipated, the formation of nitrites was observed, including bridging NO3− (1630 cm−1) and monodentate NO3− (1534 cm−1).54,55 Meanwhile, the asymmetric band at around 2178 cm−1, corresponding to two NO+ stretches at 2214 and 2178 cm−1, is indicative of the generation of NO+ intermediates,56 which can serve as an adsorption site for NO2.45 The formation of NO+–NO2 complexes was confirmed by the presence of an N–O stretch at 1741 cm−1 accompanied by another NO stretch at 2109 cm−1,56 as well as a weak band at 1925 cm−1 assigned to N2O3,54 a product of NO2 adsorption on NO+.57 Although a small amount of NO was released during NO2 adsorption due to NO2 dissociation caused by oxygen vacancies, no distinct FTIR peaks indicative of adsorbed NO were observed. Based on these findings, the mechanism for NO2 adsorption on the Cun+SSZ-13-190-R zeolite during adsorption was proposed, as shown in eqn (3)–(6).| | | NO2 + zeolite → zeolite⋯NO2 | (3) |
| | | 2NO2 ⇌ N2O4 ⇌ NO3− + NO+ | (4) |
| | | 2NO+ + 2NO2 → NO+⋯NO2 + N2O3 | (5) |
| | | NO2 + Ovac → Ovac⋯O + NO (small amount) | (6) |
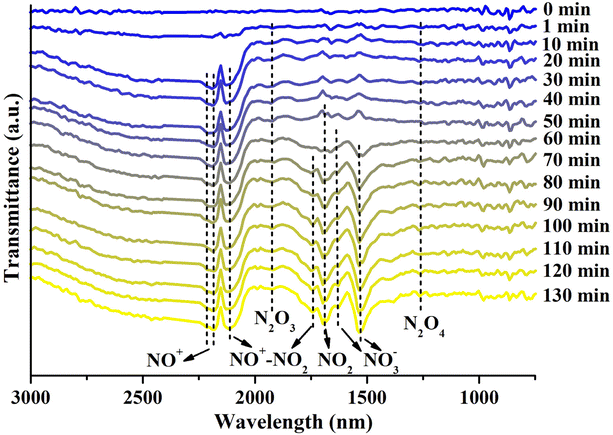 |
| | Fig. 6
In situ FTIR spectra of Cun+SSZ-13-190-R during NO2 adsorption, with the background of the fresh sample subtracted (conditions: NO2 concentration of 1000 ppm, flow rate of 60 mL min−1, 25 °C). | |
To understand the effects of copper valence states on NO2 adsorption in zeolites, DFT calculations were conducted to study the electronic and structural changes associated with Cu+ and Cu2+ ions upon NO2 adsorption. As shown in Fig. S7,† the number of cations (Cu+ and H+) in the zeolite after H2 reduction is increased. As the cations would serve as major adsorption sites, the increase in cation density would afford more binding sites for NO2 adsorption. Moreover, as shown in Table 1, the formation of Cu+ (binding energy of 20.06 kcal mol−1), which has a higher affinity for NO2 than Cu2+ (binding energy of 14.36 kcal mol−1), strengthens the interaction with NO2 through π-complexation. Thus, the increase in the density of adsorption sites with increasing binding strength enables greater NO2 uptake.
Table 1 The binding energy (kcal mol−1) of NO2 and NO molecules upon adsorption on Cu+ and Cu2+ in the SSZ-13 zeolite calculated by density functional theory (DFT)
|
|
NO2 |
NO |
| Cu+ |
−20.06 |
−30.31 |
| Cu2+ |
−14.36 |
−30.78 |
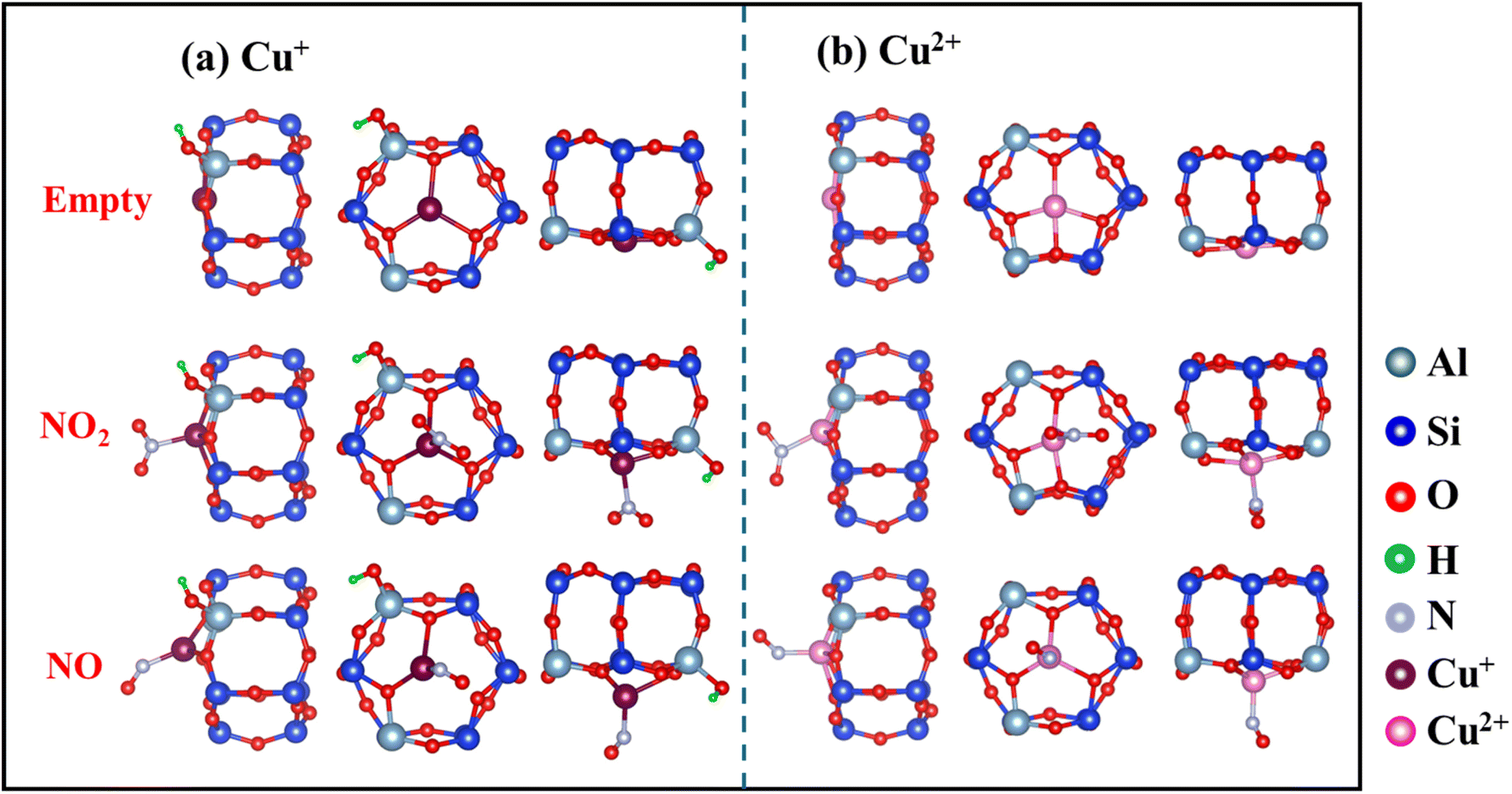
The optimized local structures of NO2/NO adsorbed on Cu+ and Cu2+ in SSZ-13 zeolites were analyzed (Fig. 7). When NO2 or NO is adsorbed, Cu–O bonds are partially disrupted, and the position of Cu ions shifts from a planar arrangement to a non-planar one. These changes in Cu–O coordination numbers and the displacement of Cu ion positions can indicate the intensity of interaction with NOx. Calculations showed reduced electron occupancies (ΔOc) in the 3d orbitals and increased ΔOc in the 4s and 4p orbitals (Table 2), suggesting σ-donation from NO2 and π-backdonation from Cu ions, respectively. In addition, Cu2+ (0.72 Å) has a smaller radius than Cu+ (0.96 Å) and attracts electrons more tightly.26 This constraint resulted in more stable electrons in the d orbitals of Cu2+, making it more difficult to enable backdonation to the π* orbital of NO2 and thus weakening the interaction with NO2. Thus, the higher affinity of Cu+ for NO2 than Cu2+, which is attributed to the more efficient π-complexation, facilitates NO2 adsorption by Cun+SSZ-13.
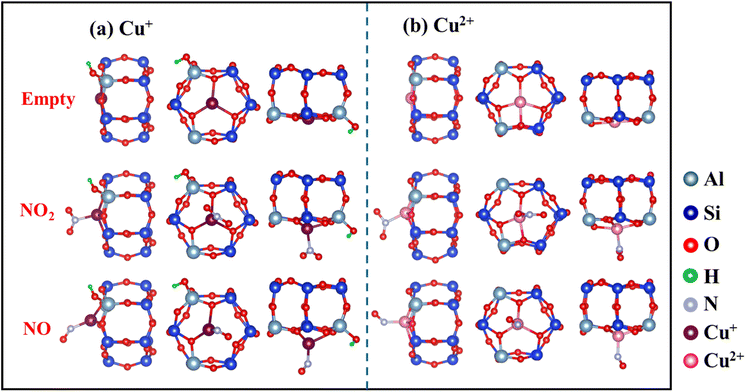 |
| | Fig. 7 Comparison of optimized local structures of NO2/NO adsorbed on Cu+ and Cu2+ cations in the SSZ-13 zeolite. The top, middle, and bottom images respectively depict the naked cation, and the cation interacting with NO2 and NO, all located at the 6 MR sites. | |
Table 2 Changes in electron occupancies (ΔOc) in the outer-shell orbitals of N, O and transition metal ions upon NO2 adsorption (Lowdin). Data was derived from the electron occupancies before and after NO2 and NO adsorption on Cu+SSZ-13 and Cu2+SSZ-13 zeolites (Table S3)
|
|
N-2s |
N-2pz |
N-2px |
N-2py |
O-2s |
O-2pz |
O-2px |
O-2py |
O-2s |
O-2pz |
O-2px |
O-2py |
Cu-3dz2 |
Cu-3dzx |
Cu-3dzy |
Cu-3dx2−y2 |
Cu-3dxy |
Cu-4s |
Cu-4pz |
Cu-4px |
Cu-4py |
∑3d |
∑4s + 4p |
| Cu+ + NO2 |
−0.09 |
0.32 |
−0.08 |
−0.16 |
−0.01 |
0.23 |
−0.12 |
−0.12 |
−0.02 |
0.18 |
−0.08 |
−0.12 |
−0.37 |
−0.05 |
−0.07 |
0.16 |
0.00 |
−0.01 |
0.16 |
0.01 |
0.01 |
−0.32 |
0.17 |
| Cu+ + NO |
−0.28 |
0.11 |
0.05 |
0.06 |
−0.01 |
0.10 |
−0.03 |
−0.05 |
0.00 |
0.00 |
0.00 |
0.00 |
−0.02 |
−0.18 |
−0.16 |
0.15 |
−0.06 |
0.05 |
0.16 |
−0.04 |
−0.01 |
−0.27 |
0.16 |
| Cu2+ + NO2 |
−0.10 |
0.31 |
−0.09 |
−0.15 |
−0.01 |
0.21 |
−0.13 |
−0.10 |
−0.02 |
0.19 |
−0.09 |
−0.14 |
−0.34 |
−0.09 |
−0.06 |
0.46 |
0.00 |
0.01 |
0.14 |
0.02 |
−0.01 |
−0.03 |
0.15 |
| Cu2+ + NO |
−0.23 |
−0.04 |
−0.03 |
0.06 |
−0.02 |
0.01 |
−0.07 |
−0.06 |
0.00 |
0.00 |
0.00 |
0.00 |
−0.17 |
−0.19 |
−0.17 |
0.47 |
−0.03 |
0.01 |
0.19 |
0.00 |
−0.01 |
−0.08 |
0.19 |
4. Conclusions
In this work, in-depth analysis of NO2 adsorption on Cun+SSZ-13 zeolites under ambient conditions was conducted, including evaluation of NO2 uptake, NO release, the affinity of adsorption sites for NO2, and the underlying adsorption mechanisms. Among the Cu-modified zeolites investigated, Cun+SSZ-13-190-R exhibited the highest NO2 adsorption capacity, outperforming both H+SSZ-13 and Cu2+SSZ-13. Our experimental and DFT studies demonstrated that the enhanced NO2 adsorption was attributed to the formation of abundant Cu+ ions within the zeolite, which have a higher affinity for NO2 than Cu2+, as well as the increased amount of adsorption sites. The elimination of moisture and Cu0 formed upon Cu2+SSZ-13 reduction was favorable for NO2 adsorption. The findings of this research could provide valuable insights for the development of adsorbents for NO2 removal at ambient temperature.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Science and Technology Innovation Commission of Shenzhen Municipality (Ref: JCYJ20210324134006019), the Research Grants Council of Hong Kong (Ref: CityU 11317722, 11310223), and the Innovation and Technology Commission — Innovation and Technology Fund (Ref: GHP/228/21GD). The authors would also like to acknowledge the research undertaken on the PD and XAS beamlines at the Australian Synchrotron, ANSTO. B. J. is supported by a Fellowship at the University of Wollongong, Australia.
References
- Z. Chen, J.-N. Wang, G.-X. Ma and Y.-S. Zhang, Lancet, 2013, 382, 1959 CrossRef PubMed.
- V. L. Feigin, G. A. Roth, M. Naghavi, P. Parmar, R. Krishnamurthi, S. Chugh, G. A. Mensah, B. Norrving, I. Shiue and M. Ng, Lancet Neurol., 2016, 15, 913–924 CrossRef PubMed.
- P. M. Edwards, S. S. Brown, J. M. Roberts, R. Ahmadov, R. M. Banta, J. A. Degouw, W. P. Dube, R. A. Field, J. H. Flynn and J. B. Gilman, Nature, 2014, 514, 351–354 CrossRef CAS PubMed.
- P. Georgopoulos, S. Arunachalam and S. Wang, J. Air Waste Manage. Assoc., 1997, 47, 838–850 CrossRef CAS PubMed.
- D. Pudasainee, B. Sapkota, M. L. Shrestha, A. Kaga, A. Kondo and Y. Inoue, Atmos. Environ., 2006, 40, 8081–8087 CrossRef CAS.
- M. Shao, Y. Zhang, L. Zeng, X. Tang, J. Zhang, L. Zhong and B. Wang, J. Environ. Manage., 2009, 90, 512–518 CrossRef CAS PubMed.
- G. B. Marks, W. Ezz, N. Aust, B. G. Toelle, W. Xuan, E. Belousova, C. Cosgrove, B. Jalaludin and W. T. Smith, Environ. Health Perspect., 2010, 118, 1476–1482 CrossRef CAS PubMed.
- R. Pastel and R. Sausa, Appl. Opt., 1996, 35, 4046–4052 CrossRef CAS PubMed.
-
The Government of the Hong Kong Special Administrative Region - Press Releases, EPD announces air quality monitoring results in 2019, 2020, https://www.info.gov.hk/gia/general/202001/20/P2020012000874.htm Search PubMed.
- Z. Hu, Z. Shen and J. C. Yu, Environ. Sci. Technol., 2017, 51, 7076–7083 CrossRef CAS PubMed.
- J. W. Girard, C. Montreuil, J. Kim, G. Cavataio and C. Lambert, SAE Int. J. Fuels Lubr., 2009, 1, 488–494 CrossRef CAS.
- C. Lambert, R. Hammerle, R. McGill, M. Khair and C. Sharp, SAE Trans., 2004, 580–589 Search PubMed.
- L. Muzio, G. Quartucy and J. Cichanowiczy, Int. J. Environ. Pollut., 2002, 17, 4–30 CrossRef CAS.
- V. Schmeisser, M. Weibel, L. S. Hernando, I. Nova, E. Tronconi and M. P. Ruggeri, SAE Int. J. Commer. Veh., 2013, 6, 190–199 CrossRef.
- H.-Y. Chen, J. E. Collier, D. Liu, L. Mantarosie, D. Durán-Martín, V. Novák, R. R. Rajaram and D. Thompsett, Catal. Lett., 2016, 146, 1706–1711 CrossRef CAS.
- C. Wan and J. Li, Carbohydr. Polym., 2015, 134, 144–150 CrossRef CAS PubMed.
- Y. Zheng, L. Kovarik, M. H. Engelhard, Y. Wang, Y. Wang, F. Gao and J. n. Szanyi, J. Phys. Chem. C, 2017, 121, 15793–15803 CrossRef CAS.
-
R. T. Yang, Adsorbents: Fundamentals and Applications, John Wiley & Sons, 2003 Search PubMed.
- A. Takahashi, R. T. Yang, C. L. Munson and D. Chinn, Langmuir, 2001, 17, 8405–8413 CrossRef CAS.
- M. Sun, A. Hanif, T. Wang, Q. Gu and J. Shang, Sep. Purif. Technol., 2023, 314, 123563 CrossRef CAS.
- Y. Tian, M. Kong, Z. Tao, C. Yang, S. Shang, Q. Gu, D. C. Tsang, L. Li and J. Shang, J. Hazard. Mater., 2023, 441, 129962 CrossRef CAS.
- M. Sun, A. Hanif, T. Wang, C. Yang, D. C. Tsang and J. Shang, J. Hazard. Mater., 2021, 418, 126400 CrossRef CAS PubMed.
- S. Shang, C. Yang, Y. Tian, Z. Tao, A. Hanif, M. Sun, H. H. S. Wong, C. Wang and J. Shang, ACS ES&T Eng., 2020, 1, 375–384 Search PubMed.
- S. Shang, C. Yang, C. Wang, J. Qin, Y. Li, Q. Gu and J. Shang, Angew. Chem., Int. Ed., 2020, 59, 19680–19683 CrossRef CAS PubMed.
- B. Levasseur, A. M. Ebrahim and T. J. Bandosz, J. Colloid Interface Sci., 2012, 377, 347–354 CrossRef CAS PubMed.
- M. Sun, Q. Gu, A. Hanif, T. Wang and J. Shang, Chem. Eng. J., 2019, 370, 1450–1458 CrossRef CAS.
- L. Rodriguez-Santiago, M. Sierka, V. Branchadell, M. Sodupe and J. Sauer, J. Am. Chem. Soc., 1998, 120, 1545–1551 CrossRef CAS.
- A. Sierraalta, R. Añez and M.-R. Brussin, J. Phys. Chem. A, 2002, 106, 6851–6856 CrossRef CAS.
-
S. Onitsuka, M. Ichiki and T. Watanabe, US Pat., 5158582, 1992 Search PubMed.
- D. Barthomeuf and B.-H. Ha, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 2158–2165 RSC.
- J. Yun and R. Lobo, Catal. Sci. Technol., 2015, 5, 264–273 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. d. Gironcoli, P. Delugas, J. R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- G. Paolo, B. Stefano, B. Nicola, C. Matteo, C. Roberto, C. Carlo, C. Davide, L. C. Guido, C. Matteo, D. Ismaila, C. Andrea Dal, G. Stefano de, F. Stefano, F. Guido, G. Ralph, G. Uwe, G. Christos, K. Anton, L. Michele, M.-S. Layla, M. Nicola, M. Francesco, M. Riccardo, P. Stefano, P. Alfredo, P. Lorenzo, S. Carlo, S. Sandro, S. Gabriele, P. S. Ari, S. Alexander, U. Paolo and M. W. Renata, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Zhang, J.-S. McEwen, M. r. Kollár, F. Gao, Y. Wang, J. Szanyi and C. H. Peden, ACS Catal., 2014, 4, 4093–4105 CrossRef CAS.
- Q. Guo, F. Fan, D. Ligthart, G. Li, Z. Feng, E. J. Hensen and C. Li, ChemCatChem, 2014, 6, 634–639 CrossRef CAS.
- A. G. Greenaway, I. Lezcano-Gonzalez, M. Agote-Aran, E. K. Gibson, Y. Odarchenko and A. M. Beale, Top. Catal., 2018, 61, 175–182 CrossRef CAS PubMed.
- J. H. Kwak, H. Zhu, J. H. Lee, C. H. Peden and J. Szanyi, Chem. Commun., 2012, 48, 4758–4760 RSC.
- G. Yalovega, V. Shmatko, A. Funik and M. Brzhezinskaya, Adv. Mater., 2016, 299–315 CAS.
- T. N. Huan, E. S. Andreiadis, J. Heidkamp, P. Simon, E. Derat, S. Cobo, G. Royal, A. Bergmann, P. Strasser and H. Dau, J. Mater. Chem. A, 2015, 3, 3901–3907 RSC.
- N. Shi, Y. Song, X. Xing and J. Chen, Coord. Chem. Rev., 2021, 449, 214204 CrossRef CAS.
- M. Sun, C. Ku, Z. Tao, T. Wang, C. Wen, A. Hanif, C. Wang, Q. Gu, P. Sit and J. Shang, Results Eng., 2023, 18, 101134 CrossRef CAS.
- C. Bian, D. Li, Q. Liu, S. Zhang, L. Pang, Z. Luo, Y. Guo, Z. Chen and T. Li, Chin. Chem. Lett., 2022, 33, 1169–1179 CrossRef CAS.
- R. C. Ambrosio and E. A. Ticianelli, J. Electroanal. Chem., 2005, 574, 251–260 CrossRef CAS.
- W. C. Gomes, A. d. O. W. Neto, P. M. Pimentel, D. M. de Araújo Melo and F. R. G. Silva, Colloids Surf., A, 2013, 426, 18–25 CrossRef CAS.
- F. Delachaux, C. Vallières, H. Monnier and M.-T. Lecler, Adsorption, 2019, 25, 95–103 CrossRef CAS.
- U. Bentrup, M. Richter and R. Fricke, Appl. Catal., B, 2005, 55, 213–220 CrossRef CAS.
- A. Mahmood, X. Wang, X. Xie and J. Sun, Colloids Surf., A, 2021, 626, 127058 CrossRef CAS.
- Z.-M. Wang, T. Arai and M. Kumagai, Ind. Eng. Chem. Res., 2001, 40, 1864–1871 CrossRef CAS.
- C. Sedlmair, B. Gil, K. Seshan, A. Jentys and J. A. Lercher, Phys. Chem. Chem. Phys., 2003, 5, 1897–1905 RSC.
- M. Kantcheva and E. Z. Ciftlikli, J. Phys. Chem. B, 2002, 106, 3941–3949 CrossRef CAS.
- K. Hadjiivanov, V. Avreyska, D. Klissurski and T. Marinova, Langmuir, 2002, 18, 1619–1625 CrossRef CAS.
- K. Khivantsev, N. R. Jaegers, H. A. Aleksandrov, L. Kovarik, M. A. Derewinski, Y. Wang, G. N. Vayssilov and J. Szanyi, Nat. Commun., 2021, 12, 6033 CrossRef CAS PubMed.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Calvin
Ku
a,
Aamir
Hanif
ab,
Calvin
Ku
a,
Aamir
Hanif