DOI:
10.1039/D4TA05592F
(Paper)
J. Mater. Chem. A, 2024,
12, 30340-30350
Exploring reaction mechanisms for CO2 reduction on carbides†
Received
9th August 2024
, Accepted 11th October 2024
First published on 11th October 2024
Abstract
The electrocatalytic conversion of carbon dioxide (CO2) into valuable fuels offers immense promise in pursuing sustainable energy solutions. Transition metal carbides (TMCs) with their robustness and intriguing electronic properties have traditionally emerged as captivating contenders in the quest for efficient catalysts for the CO2 reduction reaction (CO2RR). The presence of carbon atoms in TMC structures unlocks a unique reaction mechanism for the CO2RR, namely the Mars–van Krevelen (MVK) mechanism, facilitating CO2 capture and more efficient conversion to high-value-added chemicals. This work is the first report on the use of TMCs for the CO2RR where comprehensive reaction pathways for different product formations are investigated. This theoretical study delves into the electronic intricacies of TMCs, unraveling their potential to drive the transformative journey toward a greener tomorrow. Here, we analyzed 11 TMCs to explore the reactivity trends toward CO, formic acid, methane, methanediol, and methanol formation. VC is the best candidate explored to produce formic acid at 0 V onset potential. In addition, WC is the best candidate explored to produce methanol at an onset potential of −0.36 V. These results demonstrate that our studied TMCs as electrocatalysts are more promising than previously studied materials (metals and oxides) for application in the CO2RR, and thus require more attention and investigation.
1. Introduction
Since the start of the 20th century, the world climate has been changing more noticeably. The balance in nature is lost due to dramatically increasing CO2 emissions due to burning of carbonaceous fuels which results in global warming. An urgent effort is needed to overcome this problem to reduce the ratio of CO2 and turn it into fuel and other useful chemical products. On the other hand, the large-scale use of unsustainable fossil fuels for energy has led to an energy crisis in the world and increased the amount of CO2 in the atmosphere. Due to this, the world climate is changing day by day, and scientific effort is needed to overcome the emission of CO2 in the air by converting the already present CO2 into green energy fuels.1–3 Previous experimental and theoretical research concluded that conversion of CO2 into valuable products can be achievable, but not yet efficient for commercialization. In the whole process of the CO2 reduction reaction (CO2RR), the catalyst plays a key role in capturing CO2 and converting it into valuable chemical products and fuels. CO2 is considered a chemically stable molecule, which presents challenges when attempting to reduce it electrochemically due to the high reduction overpotential, poor product selectivity, and low current efficiency. To tackle this challenge, researchers and scientists have been investigating innovative catalyst materials that can effectively overcome these obstacles and enhance this electrochemical process.4–8
In the previous decades, many experimental studies have been done for the electrochemical CO2RR on different metal catalysts i.e. pure metals of Cu, Ag, Au, and Pt have been studied as catalysts for the CO2RR.9–12 It is well known that Cu is considered a widely used catalyst for the reduction of CO2 to CH4 but its overpotential is high (∼0.9 V) and moreover it produces 15 different carbon-containing products which require a large amount of energy to separate them. In addition to pure Cu, polycrystalline Cu was also examined for application in the CO2RR, but this has the same problem as pure Cu.1,6,13–16
In addition to pure metal electrocatalysts, metal oxide catalysts i.e. TiO2 studied by the DFT method were considered impressive and more efficient for CO2 conversion to CH3OH and CH4 products.17 In another study, RhO2 was predicted to be a novel candidate for the formation of formic acid at −0.20 V onset potential.18 However, the stability and electron conductivity of oxides as cathode materials are insufficient, which limits their application in the CO2RR.19,20 There are numerous studies conducted to explore novel catalyst materials for the CO2RR such as graphene-based materials, metals, metal oxides, zeolites, sulfides, and metal–organic frameworks, but yet activity and efficiency are not optimal for the commercialization purposes and the utilization of catalysts for large-scale implementation is still a challenge and more study is required to overcome this problem.21–30 Traditional catalysts may have limitations in terms of selectivity, efficiency, or cost. By searching for other materials, researchers aim to identify catalysts that can enhance the performance of CO2RR processes, making it more economically viable and environmentally friendly. The surface chemistry of transition metal carbides (TMCs) is comparable to that of precious Pt-group metals, which was already described by Levy and Boudart.31,32 Some of the TMCs were investigated and showed promise for reactions such as CO oxidation,33 hydrogenation,34 methane dry reforming,35 desulfurization,36 conversation of methane to gas and water gas shift reaction.37,38 TMCs demonstrate significant efficacy in facilitating various transformations and are regarded as attractive candidates for catalytic applications.39 Consequently, they are considered a potential alternative to precious metal catalysts owing to their high chemical and thermal stability40 high mechanical stability,41 and low cost.42 Hence these materials have received great attention for application in catalysis, but there is no comprehensive investigation of TMCs for the CO2RR. Utilizing advanced quantum mechanical simulations and computational models, we explored the binding energies and different reaction pathways of 11 TMCs (CrC, HfC, MoC, YC, ScC, NbC, ZrC, TaC, TiC, WC, and VC), with a focus on their suitability for catalyzing CO2 reduction. Here we are doing screening studies on 11 different TMCs to find out which one is more favourable and promising for the CO2RR thermodynamically for more in-depth investigation in the future. These data provide valuable information about the activity of these TMCs. The difference in the PDS values associated with different products can be an indication of selectivity to some extent, however, kinetic barrier calculations are needed to determine product selectivity which is not the scope of the present work.
2. Computational method
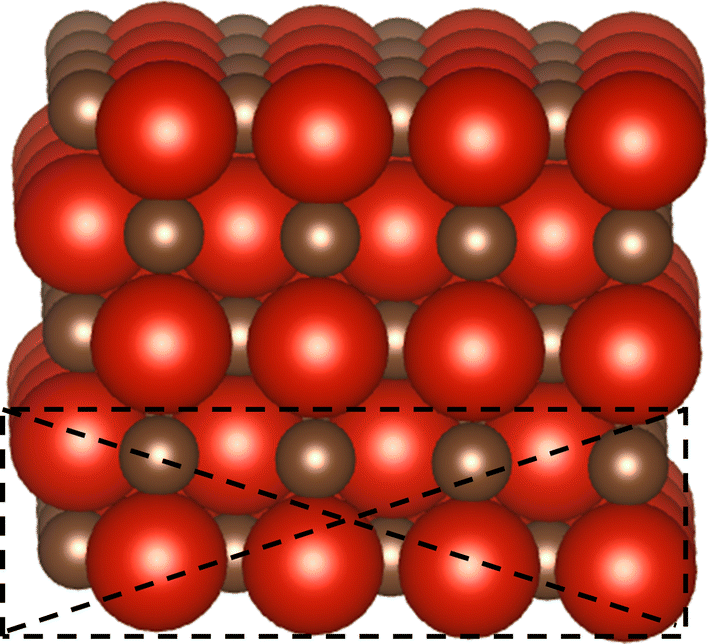
The interaction of CO2 with TMC surfaces and the formation of different intermediates were studied by DFT calculations with Generalized Gradient Approximation (GGA) using the RPBE exchange–correlation functional.43 The Vienna ab initio simulation package (VASP) was used for the modeling, with a cutoff energy of 450 eV and a 4 × 4 × 1 Monkhorst–Pack K-point.44 The Projector augmented wave (PAW) method was implemented due to the full wavefunction by utilizing computationally efficient pseudopotentials, significantly reducing the computation time.45 The TMC surface was considered in the rocksalt (RS) crystallographic structure with the texture orientation of (100). The electrodes modeled consisted of 5 layers with each layer made of 4 atoms of carbon and 4 atoms of metal, 40 atoms in total in the unit cell of each TMC structure. The boundary condition was periodic with a 20 Å vacuum along the z direction to avoid self-interaction of neighboring layers. The bottom two layers were fixed, and the top 3 layers were allowed to fully relax together with any adsorbates on the surface as shown in Fig. 1.
 |
| | Fig. 1 Schematic representation of a TMC, the dark brown color shows carbon atoms and the red color shows metal atoms. The dotted line shows that the bottom two layers are fixed. | |
The binding energy of an adsorbate was calculated according to eqn (1):46
| | | ΔEads = E(adsorbate/TMC slab) − E(TMC slab) − E(adsorbate) | (1) |
Here
E(adsorbate/TMC slab) is the total energy of the system with an adsorbate on the TMC slab,
E(TMC slab) is the total energy of the pristine TMC slab, and
E(adsorbate) is the total energy of the adsorbate. The negative Δ
Eads corresponds to the exothermic adsorption of species. The computational hydrogen electrode (CHE) model as proposed by Nørskov
et al.47 was used to calculate the free energy diagram for the CO
2RR along different paths at certain applied potentials. According to this method, at room temperature, the kinetic barrier is negligible for the proton-coupled electron step and the potential limiting step is the most positive energy difference between the two adjacent steps. The reaction free energy of an adsorbate for the CO
2RR at any arbitrary potential U
vs. RHE and any arbitrary pH was calculated using the equations:
| | | ΔG(URHE) = ΔG(URHE=0) + neURHE | (2) |
| | | eURHE = eURHE + 2.3kbTpH | (3) |
where
URHE is the applied potential referred to the standard hydrogen electrode (SHE),
n is the number of electrons,
e is the charge of electrons,
kb is the Boltzmann constant and
T is the temperature. By putting
eqn (2) into
eqn (3) we will get:
| | | ΔG(URHE) = ΔG(URHE=0) + n(eURHE = eURHE + 2.3kbTpH) | (4) |
Generally, overpotential is independent of the pH of the electrolyte so in this study we will consider pH = 0. Hence for each elementary step, ΔG (U = 0) is calculated by
| | | ΔG(U=0) = ΔEDFT + ΔEZPE − TΔS + ΔE0 K→T | (5) |
where Δ
G is the Gibbs free energy, Δ
EDFT is the electronic energy calculated by DFT, Δ
EZPE is the zero-point energy correction and
TΔ
S is entropy differences between the gas phase and adsorbed species calculated by vibrational frequencies for the adsorbed species. Δ
E0 K→T is the change in internal energy because of temperature and its value is

where
Cp(
T′) represents specific heat at constant pressure and the integral of this equation contributes negligibly at room temperature. The value of the gas phase is taken from the thermodynamic tables from textbooks.
48
A crucial step towards a rational design of new catalysts that are selective in reducing CO2 to specific hydrocarbons and alcohols is to determine the detailed reaction mechanism for the process. To accomplish this, a detailed description of the electrochemical solid–liquid interface is also required. It has been observed that a detailed description of the electrochemical solid–liquid interface model is successful in capturing the experimental trends of product distribution for the CO2RR on pure metals.49,50 Although the more approximated thermochemical model (TCM) and the implicit computational hydrogen electrode (CHE) cannot completely capture these trends, they do capture the onset potentials for the CO2RR and other electrochemical reactions quite well.50–54 The molecular level structure of water on noble transition metals is qualitatively well known and therefore realistic model systems can be used. Unfortunately, for transition metal carbide (TMC) surfaces and especially for the (100) facets of rocksalt structures, the molecular scale structure of water is still controversial, and hence, realistic model systems have not been fully developed yet. Therefore, most of the analyses in this work are based on the calculated thermodynamics of adsorbed intermediates on the TMC surfaces, and the TCM and CHE models are used to capture the onset potentials for the CO2RR. For pure metals, activation energies can be calculated for each of the proton–electron transfer steps (toward different intermediates and products) by setting up a charged double-layer model including a solid electrode and an aqueous electrolyte solution as has been done for the metal–liquid interface for the CO2RR49,55 There, the charge difference in the double layer sets up the applied electric potential explicitly and therefore the proton–electron transfer barriers can be calculated as a function of explicitly varied potential. This approach is helpful in reproducing the experimentally observed product distribution as a function of both the metal type and the applied potential. However, for these TMCs and as elaborated above, there is a lack of understanding on the water layer structure in modeling plus the fact that there are not many experimental reports on these TMCs to compare the computational results and trends with. Furthermore, previous reports on Cu pure metal, in the (111) facets of hexagonal structures, suggest stabilization energies of 0.1–0.25 eV for CO* and CHO* when water layers are included in the model. This correction has not been included in our analysis, as we are conducting a screening study focused on identifying trends across 11 different surfaces based on thermodynamic data. Readers interested in this work are strongly advised to pay attention to the fact that the conclusion made in this paper is mainly based on thermodynamics where the authors tried to provide some hints and understanding on the possibility of using TMCs, opening the path for further investigation of their utilization for the CO2RR.
3. Results and discussion
The thermochemical model50 was employed to predict favorable reaction pathways and onset potentials for the CO2RR on the (100) facets of the RS structure of TMCs. A number of studies have been carried out using this model successfully i.e., hydrogen evolution reaction (HER),56,57 and nitrogen reduction reaction (NRR)58–61 and here we have employed the same method. From the earlier transition metals, 11 carbides including CrC, HfC, MoC, YC, ScC, NbC, ZrC, TaC, TiC, WC, and VC were selected for this investigation, as the later transition metals form carbides in structures other than the RS.62,63 The electronic structure of these TMCs has been reported in our previous publication.64 In the initial phase, the adsorption-free energy of all species was systematically investigated on the metal, bridge, and carbon sites of each TMC. This investigation revealed that the carbon sites are particularly more active for adsorption compared to the metal sites. We have undertaken a comprehensive exploration involving the absorption of various species to analyze their poisoning effects on TMCs, as illustrated in Fig. 2.
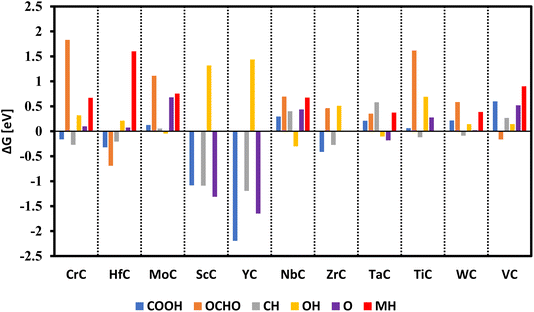 |
| | Fig. 2 Comparison of adsorption energies of different species onto the clean surface of TMCs, and CH and MH represent the adsorption of protons on carbon and metal sites of TMCs. For some of the carbides (ScC, YC, ZrC, TiC), the MH adsorption is not shown as it is not feasible for protons to adsorb on the metal site. | |
The findings reveal positive binding energies of H, O, and OH species for most TMCs rendering a lower likelihood of poisoning, with the exceptions being ScC and YC where O binds exergonically. Consequently, these materials (ScC and YC) have been selected only for investigation within the framework of methane formation via the Mars–van Krevelen (MvK) mechanism. Adsorption of other key species such as COOH and OCHO was found to be exergonic for most of these surfaces. A unique property of TMC is to unlock a special reaction mechanism, namely the MvK. In this mechanism, the carbon atom already incorporated in the catalyst structure reacts with protons and converts to CH4, releasing from the surface spontaneously and leaving a carbon vacancy on the surface which should be refilled with atmospheric CO2 to sustain the catalytic cycle of methane formation. This MvK does not work on most of the other catalyst materials and thus TMCs offer an advantage. The other most common and possible mechanism on all the other catalyst surfaces is the conventional surface mechanism where CO binds the clean surface in the form of COOH or OCHO. We investigate both of these mechanisms here.
3.1. CO2RR pathways via the MvK mechanism
In actual experiments, it is important to note that TMCs might exhibit some defects in the form of carbon vacancies, primarily due to the inherent difficulty in always achieving complete stoichiometry. Another possible reason for the formation of vacancies would be coming from the electrochemical conditions where the catalytic cycle starts with surface protonation until a surface carbon atom is reduced to methane (CH4) and released. The carbon vacancy is then formed which should be filled with atmospheric CO2 where the C atom of CO2 fills the vacancy of TMCs, and two oxygen atoms are further reduced to two water molecules as mentioned in eqn (6) and (7) followed by the Kröger–Vink notation:65| | | * + 4(H+ + e−) → CH4(g) + CVac | (6) |
| | | CVac + CO2 + 4(H+ + e−) → 2H2O + * | (7) |
To consider the effect of proton coverage on the surface, we investigated surface protonation by adding protons one after the other and explored all the possible adsorption sites, which are the metal site, the bridge site, and the carbon site. It was found that all the protons tend to bind the carbon site only to make CH4 (as shown in Fig. 3). Thus, no proton coverage builds on the surface and thus it is an indication of unfavourable HER on these surfaces.
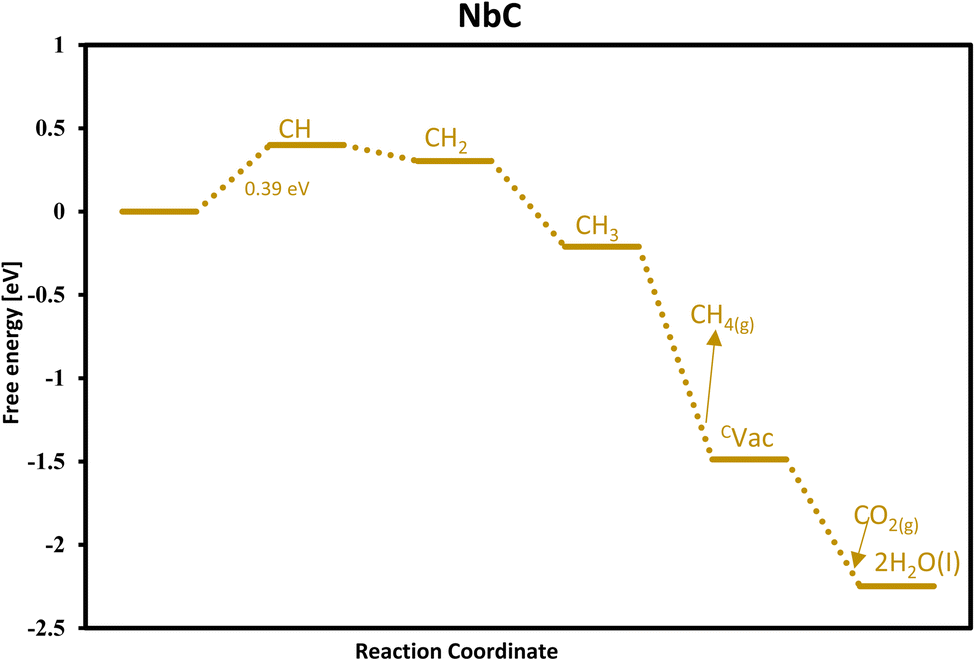
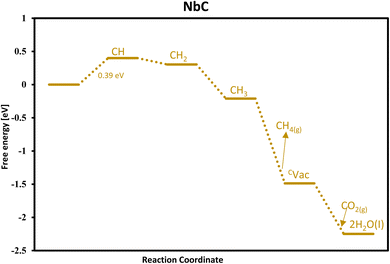 |
| | Fig. 3 Free energy diagram for methane formation via the MvK mechanism on the surface of NbC. | |
Our findings conclude NbC as the optimal candidate for CH4 formation via the MvK with a predicted onset potential of −0.39 V and its free energy diagram is shown in Fig. 3.
The other promising candidates for CH4 formation via the MvK mechanism are WC, MoC, VC, and TiC with onset potentials of −0.41, −0.44, −0.44, and −0.46 V, respectively, as shown in the free energy diagram of Fig. S1.† Further reduction of oxygen atoms of CO2 is an exergonic process for HfC, WC, ZrC, TaC, TiC, and VC, except for MoC, CrC, ScC and YC where more potential is needed for water release. The onset potential required for methane formation on the surface of ScC and YC is −1.24 and −1.68 V, respectively. Due to these high onset potential values and energy steps, we have excluded ScC and YC for further investigations.
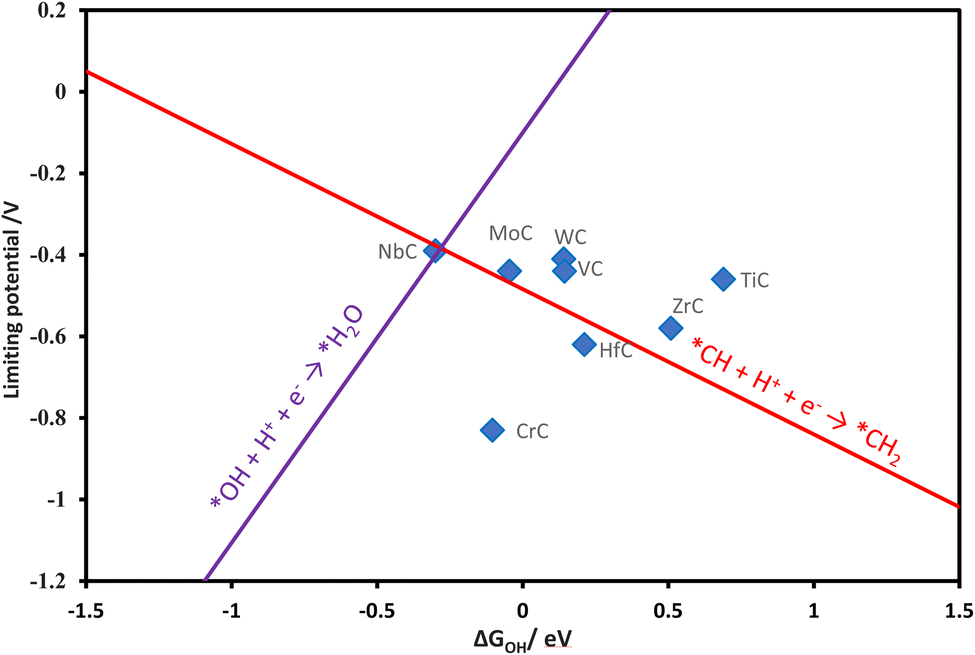
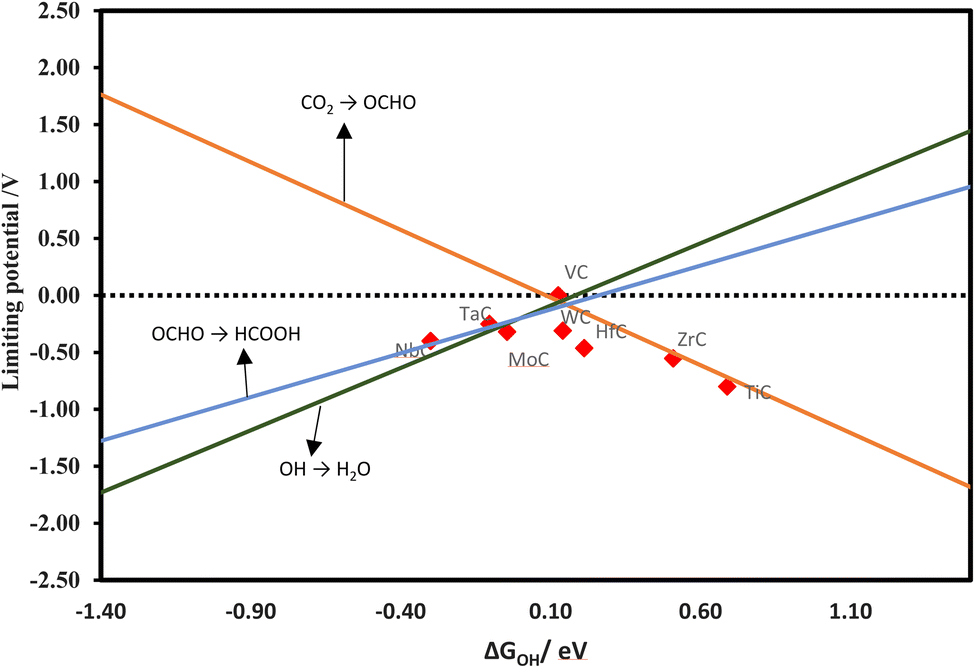
A volcano plot has been generated by using the linear relations of various reaction steps. The Gibbs free energy change of the OH (ΔGOH) has been found as a reliable descriptor in previous research,18,66 for investigating the CO2RR on transition metal oxide surfaces and also found reliable in our work. The theoretical volcano plot for the formation of methane via the MvK mechanism was constructed from the scaling relations and is shown in Fig. 4. NbC is located on top of the volcano as the most promising catalyst among the TMCs studied. The left leg of the volcano is related to the reaction between OH and water, while the right leg depicts the formation of CH2 after the formation of the CH intermediate.
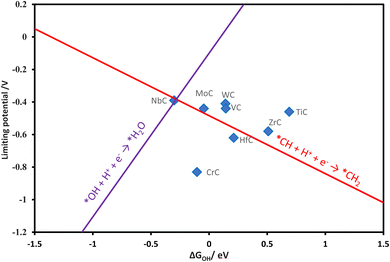 |
| | Fig. 4 Theoretical volcano for the formation of methane via the MvK mechanism. | |
3.1.1. Stability against decomposition via vacancy migration.
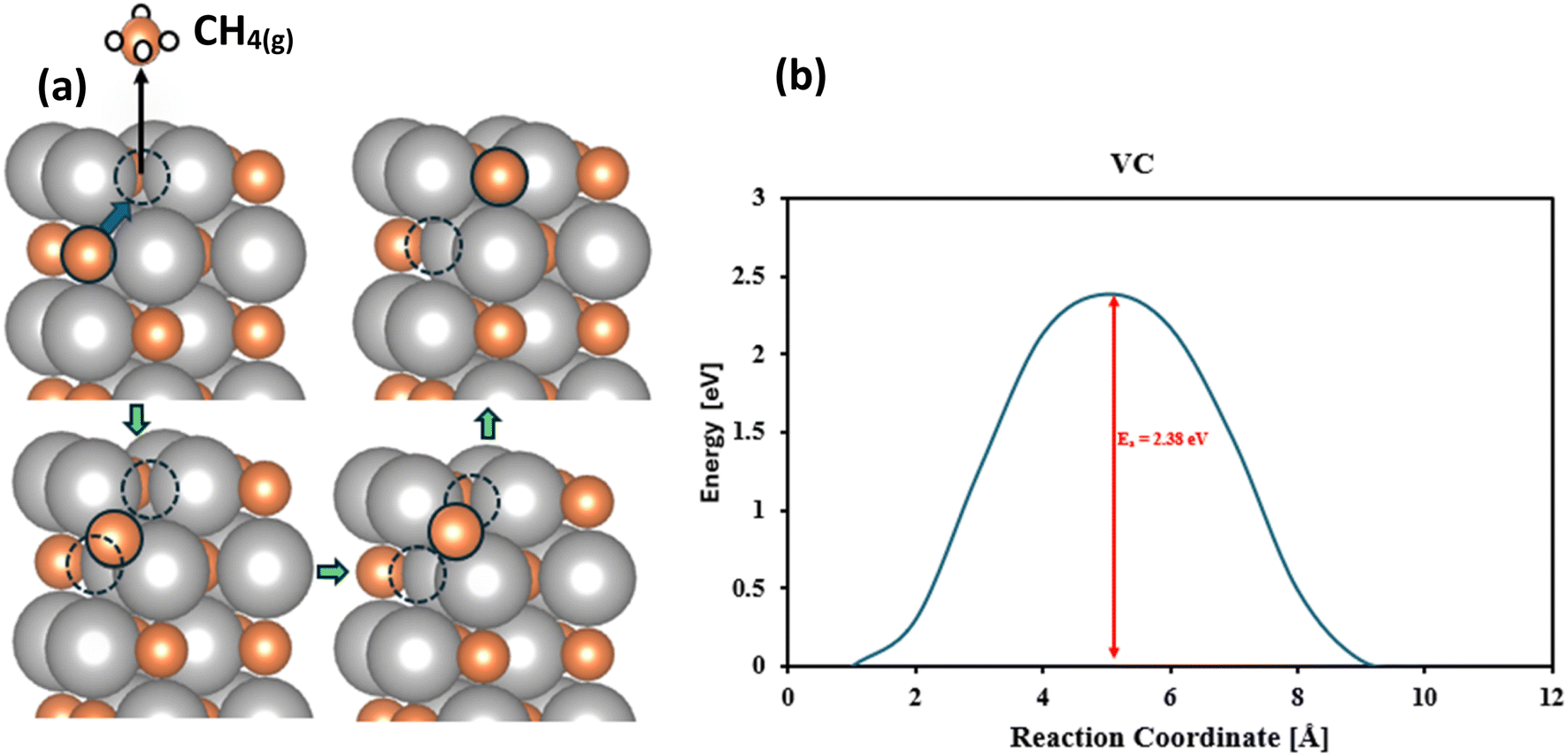
For most of these surfaces, it is relatively facile to form methane and then fill the vacancy with atmospheric CO2 and complete the catalytic cycle. In the MvK, there is a possibility of migration of the surface vacancy into the bulk which means migration of the C atoms from the bulk to the surface. If this process is facile (thermodynamically and kinetically) it can contribute to further reduction of C atoms of the material and conversion of the carbide to its pure metal, rendering decomposition and material instability. Therefore, it is crucial to investigate this phenomenon carefully. Using the Climbing Image Nudged Elastic Band (CI-NEB) method we have investigated the diffusion barrier of migration of carbon atoms from the sublayer to the surface. For our promising candidate, VC, the carbon migration barrier is 2.38 eV as shown in Fig. 5 which means that this process should be very slow under ambient conditions and this catalyst should be stable. Similarly, for our other investigated candidates, CrC, HfC, MoC, NbC, TaC, TiC, WC, and ZrC the diffusion barrier for carbon migration is calculated to be 1.77, 3.87, 1.87, 2.69, 3.55, 2.96, 1.91, and 3.30 eV, respectively, and the conclusion is that these TMCs should be very stable against decomposition to parental metals.
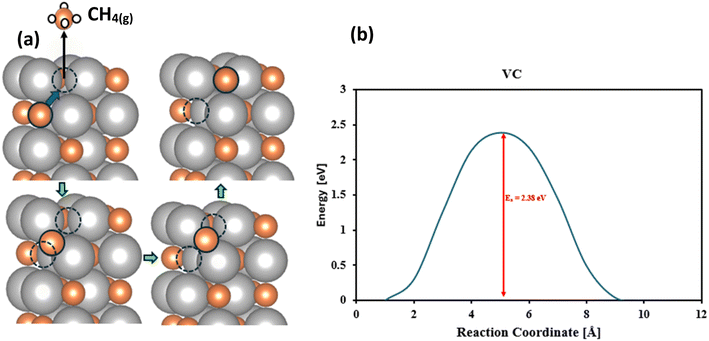 |
| | Fig. 5 Schematic diagram for migration of carbon atoms from the sublayer to surface vacancies. (a) Represents the visualization of the motion of carbon atoms from the sublayer to the surface, orange color indicates the carbon atoms, silver color indicates the metal atoms and white color indicates the proton. (b) Represents the minimum energy path for the migration of carbon atoms from the sublayer to the surface. | |
3.2. CO2RR pathways via the conventional method
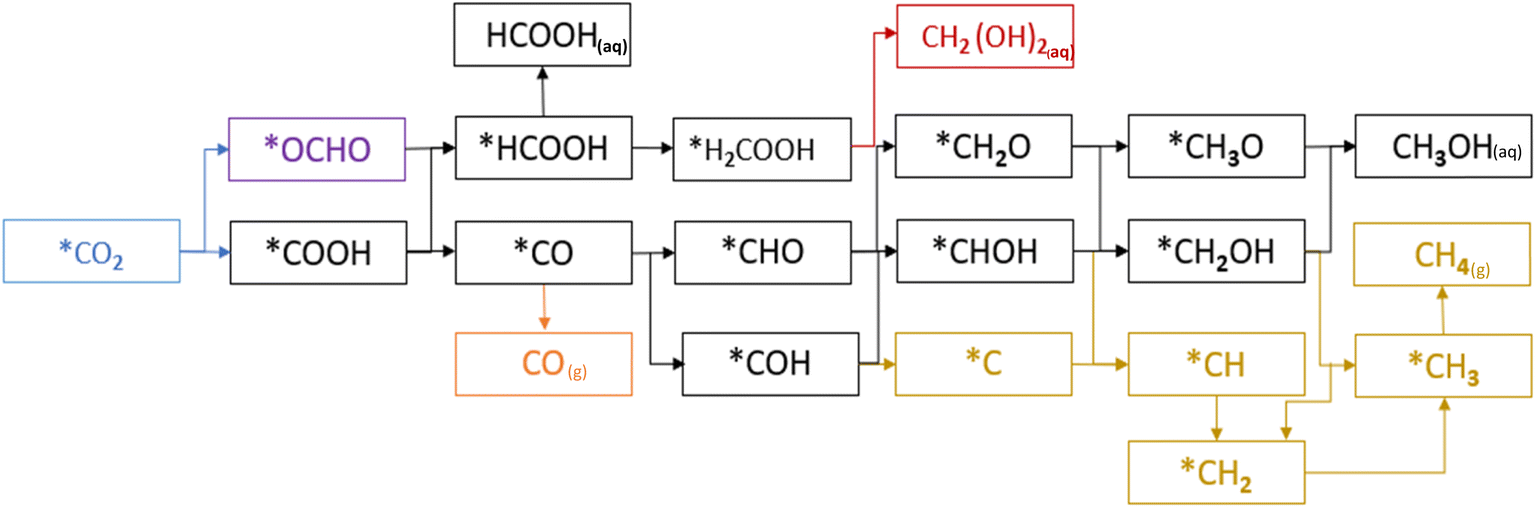
In the first step of the CO2RR two possible intermediates, OCHO and COOH, can be formed by the first electron–proton transfer step, and further protonation on OCHO and COOH continues until the product(s) is(are) formed as shown in Fig. 6. Our analysis concludes that the carbon sites of TMCs exhibit the highest level of activity and the carbon of COOH and OCHO attaches with the carbon of TMCs as shown in the schematic of the adsorption of COOH and OCHO in Fig. S3.† Additionally, in the majority of situations, the adsorption of COOH is more energetically favorable compared to OCHO, as illustrated in Fig. 2. However, OCHO adsorption is favorable on HfC and VC only. Regarding HfC, the progression through OCHO after a few protonation steps exhibits endergonic characteristics. This is why we have chosen to analyze the COOH pathway, which displays the lowest energy values on the free energy diagram as shown in Fig. S1.†
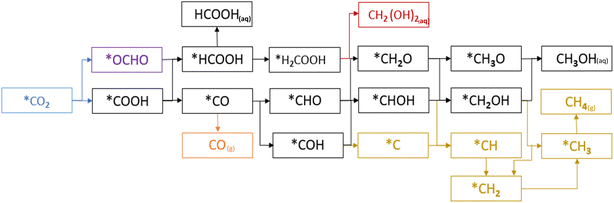 |
| | Fig. 6 Proposed mechanism of the CO2 reduction reaction via OCHO and COOH. | |
CO2 adsorption is the bottleneck in the CO2RR and the majority of the materials explored and reported so far cannot bind it favorably, and thus researchers working with DFT usually investigate the protonation form of CO2 (COOH or OCHO) as well67–69 and as such we followed the same approach. However, we looked into the direct adsorption of CO2 on the clean surface of these TMCs, and we found out that only HfC, ScC, YC, and ZrC bind CO2 exergonically. Conducting protonation of CO2 on these 4 surfaces, the PDS was found to be high for ScC and YC and thus we have not considered ScC and YC for further investigation. For HfC and ZrC, however, COOH was found to be more exergonic than direct adsorption of CO2.
3.2.1. Selectivity between CO2RR versus HER.
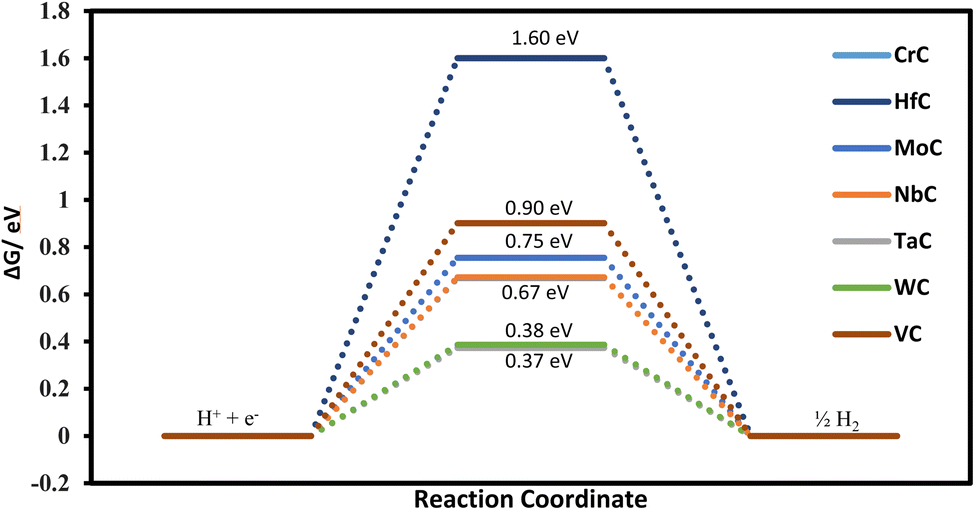
The first step of protonation provides information on selectivity towards the CO2RR or HER. Our calculated free energies for all the TMCs suggest that the HER is of no concern as COOH always binds the surface stronger than H. For example, the calculated Gibbs free energies of H adsorption for TaC and WC are 0.37 and 0.38 eV, respectively. Although these free energies are considered small and thus HER-susceptible, the COOH binding free energies for TaC and WC are only 0.21 and 0.22 eV, respectively. This indicates that the active site should be occupied by the COOH rather than H and thus more selectivity towards the CO2RR is expected. Similarly, (COOH or OCHO) adsorption vs. hydrogen adsorption for CrC, HfC, MoC, NbC, ZrC, TiC, and VC shows more tendency towards the CO2RR rather than the HER (refer to Fig. 2 and 7). Fig. 7 shows that our predicted catalyst systems possess weaker adsorption energy for atomic H adsorption, suggesting poor HER activity/selectivity. Therefore, the impediment against the HER indicates that the TMC system can act as an efficient catalyst for the CO2RR.
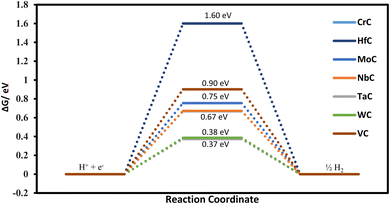 |
| | Fig. 7 Free energy diagram for H adsorption on the metal site as an indication of the HER. | |
3.2.2. CO2RR pathways towards the formation of formic acid (HCOOH) and CO.
In the second protonation step, formic acid and CO are the main products of the CO2RR before the formation of methane, methanol, and methanediol. The reactions follow the protonation steps as:| | | *COOH or *OCHO + H+ + e− → *HCOOH | (8) |
| | | *COOH or *OCHO + H+ + e− → *CO + H2O | (9) |
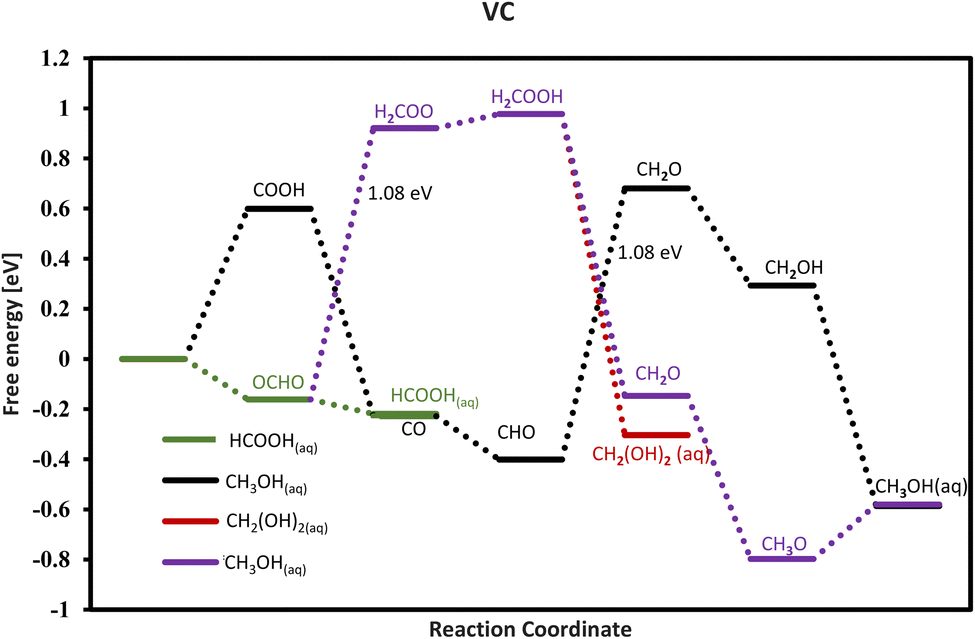
Our investigation reveals that only VC does not bind the *HCOOH intermediate on its surface and this catalyst has the potential to produce aqueous formic acid with 0 V onset potential as shown in the free energy diagram (Fig. 8), and thus this candidate is the best TMC to produce formic acid. The VC needs a relatively large onset potential of around −1.08 V to form methanol and methanol. Therefore, the selectivity is easily tunable for VC by tuning the onset potential applied and only producing formic acid.
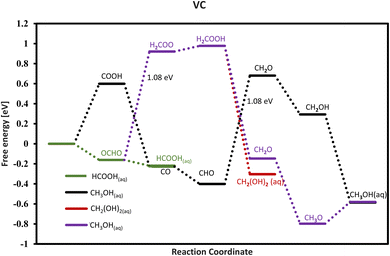 |
| | Fig. 8 Free energy for VC and the green line indicates the formation of formic acid at 0 V. CO and HCOOH are formed via different paths and not together via the same path. The HCOOH formation is followed by the green path which comes from OCHO which is an exergonic step, while the CO formation is followed by the black path which comes from COOH and is endergonic. | |
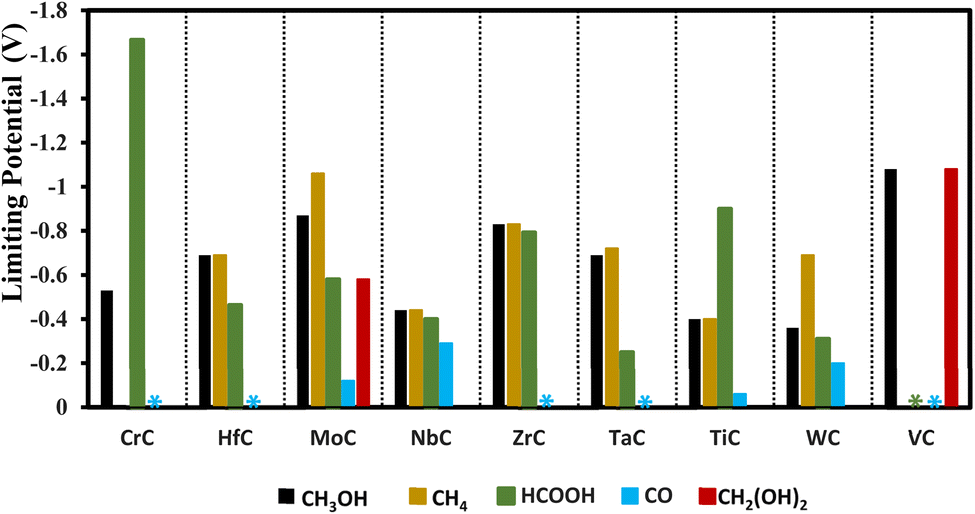
In addition to VC, other carbides like TaC, WC, NbC, and HfC are explored as good candidates to produce formic acid with small onset potentials of −0.25, −0.31, −0.40, and −0.46 V, respectively as shown in Fig. 9.
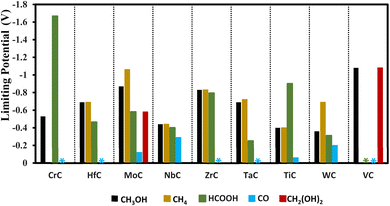 |
| | Fig. 9 Onset potential for the formation of methanol, methane, formic acid, CO, and methanediol. The representation of the steric effects indicates that the mentioned products in specific colors are formed with 0 V onset potential. | |
In addition, we have developed a volcano diagram for the production of formic acid using scaling relations. Fig. 10 illustrates the model, where each line represents a reaction involved in the production of formic acid. We have utilized OH as a descriptor, which is analogous to the volcano plot used for methane creation via the MvK mechanism. The results indicate VC as a promising candidate on the top of the volcano. The CrC, HfC, ZrC, TaC, and VC are expected to convert CO2 to CO spontaneously. These materials are promising for CO formation at 0 V. MoC, NbC, TiC, and WC are also good candidates for the formation of CO with an appropriate onset potential of −0.12, −0.29, −0.06, and −0.20 V, respectively.
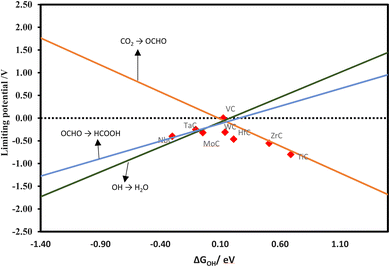 |
| | Fig. 10 Theoretical volcano for the formation of HCOOH. | |
3.2.3. CO2RR pathways toward the formation of methanol.
The CO2RR towards methanol formation involves 6 proton–electron transfer steps as shown in Table 1. In this work, we have investigated two routes to produce methanol, one route is *COOH to *HCOOH and another route is *COOH to *CO + H2O.
Table 1 Possible pathways for methanol formation
| Pathways |
(e + H+) steps |
| 1 |
2 |
3 |
4 |
5 |
6 |
| A |
*COOH |
*HCOOH |
*H2COOH |
*CH2O + H2O |
*CH2OH |
CH3OH (aq) |
| B |
*OCHO |
*HCOOH |
*H2COOH |
*CH2OH + OH |
CH3OH (aq) + *OH |
H2O(l) |
| C |
*COOH |
*HCOOH |
*H2COOH |
*CH2OH + OH |
*CH2OH + H2O(l) |
CH3OH (aq) |
| D |
*COOH |
*HCOOH |
*H2COOH |
*CH3O + OH |
*CH3O + H2O(l) |
CH3OH (aq) |
| E |
*COOH |
*CO + H2O(l) |
*COH or *CHO |
*CHOH or *CH2O |
*CH2OH or *CH3O |
CH3OH (aq) |
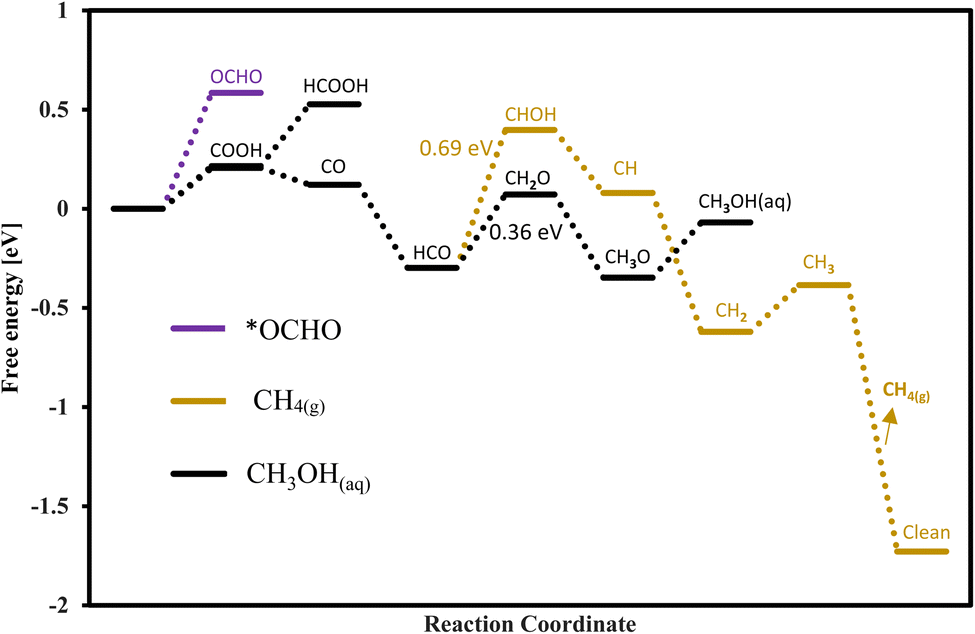
The route followed through COOH to CO + H2O is more promising as compared to the other one because in this route we have required less onset potential as shown in the free energy diagrams of Fig. S2.† For seven TMCs (CrC, HfC, MoC, NbC, ZrC, Tic, WC) the COOH adsorption is favorable, while for TaC and VC the OCHO adsorption is more favorable for methanol formation. The mechanisms for methanol formation differ among various TMCs. To simplify the representation of these reactions, we focus on the most energetically favorable stages in the minimal free energy pathways, disregarding other reaction steps. The surfaces of HfC, WC, MoC, ZrC, TiC, and CrC followed path E, NbC followed path C, TaC followed path B, and VC followed path A as mentioned in Table 1 for the formation of methanol. Our analysis reveals that WC is the best candidate for methanol formation with the potential determining step of 0.36 eV via the black pathway. The free energy diagram for the WC is shown in Fig. 11 for both methane and methanol formation and the adsorption configuration of each intermediate is shown in Fig. S4.† In addition to WC, the studied TiC, NbC, and CrC are also good candidates for methanol formation with corresponding onset potentials of −0.40 V, (−0.44 V, −0.67 V) and −0.53 V, respectively. The scaling relations for methanol formation were investigated by considering several possible descriptors, but the correlation was not good mainly due to the fact that each TMC follows a different reaction path for the formation of methanol and also the PDS for each TMC is not associated with a similar step. Thus, TMC showed to deviate from the scaling relations.
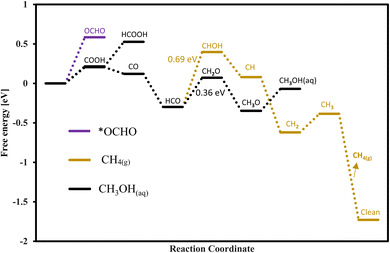 |
| | Fig. 11 Free energy diagram for the methanol formation on WC. | |
The potential determining step values for formic acid, CO, methanediol methanol, and methane formation on each TMC are shown in Table 2.
Table 2 Onset potential (V) for the CO2RR towards the various products and intermediates (╳ represents these products are not formed on these catalysts)
| Catalyst |
Formic acid |
CO |
Methanol |
Methanediol |
Methane |
HER on the metal site (MH) |
| CrC |
−1.67 |
0.00 |
−0.53 |
╳ |
Unstable |
−0.66 |
| HfC |
−0.46 |
0.00 |
−0.69 |
╳ |
−0.52 |
−1.60 |
| MoC |
−0.58 |
−0.12 |
−0.87 |
−0.58 |
−0.87 |
−0.75 |
| NbC |
−0.40 |
−0.29 |
−0.44 |
╳ |
Unstable |
−0.67 |
| ZrC |
−0.79 |
0.00 |
−0.83 |
╳ |
−0.83 |
— |
| TaC |
−0.25 |
╳ |
−0.69 |
╳ |
−0.69 |
−0.37 |
| TiC |
−0.90 |
−0.06 |
−0.4 |
╳ |
−0.9 |
— |
| WC |
−0.31 |
−0.20 |
−0.36 |
╳ |
−0.69 |
−0.38 |
| VC |
0.00 |
0.00 |
−1.08 |
−1.08 |
╳ |
−0.90 |
3.2.4. CO2RR pathways towards the formation of methanediol and methane.
Among the TMCs explored here, only MoC and VC are found to catalyze methanediol formation with the onset potentials of −0.58 V and −1.08 V, respectively. The path followed for methanediol consists of 4 steps as COOH/OCHO → HCOOH → H2COOH → CH2(OH)2(aq). On all the other TMCs, further protonation of H2COOH breaks it into other intermediates and thus methanediol formation cannot be achieved.
Through the conventional mechanism, TiC and NbC are found suitable candidates for the formation of methane with the onset potentials of −0.40 and −0.44 V, respectively, and interestingly these candidates have the same onset potential for the formation of methanol, meaning that both of these products are expected to be produced at these applied potentials. In addition, HfC, MoC, ZrC, TaC, and WC are also suitable for the formation of methane with the required onset potential values of −0.69, −1.06, −0.83, −0.72, and −0.69 V, respectively, but these materials are more selective towards formic acid formation as compared to methane with onset potentials of −0.46, −0.58, −0.79, −0.25, and −0.41 V, respectively. Only the pathways with minimum free energy are used and shown for methane formation in the free energy diagram for each candidate in Fig. S2.†
3.3. Comparison of CO2RR onset potentials for TMCs studied in this work with some other metal-based catalysts previously reported
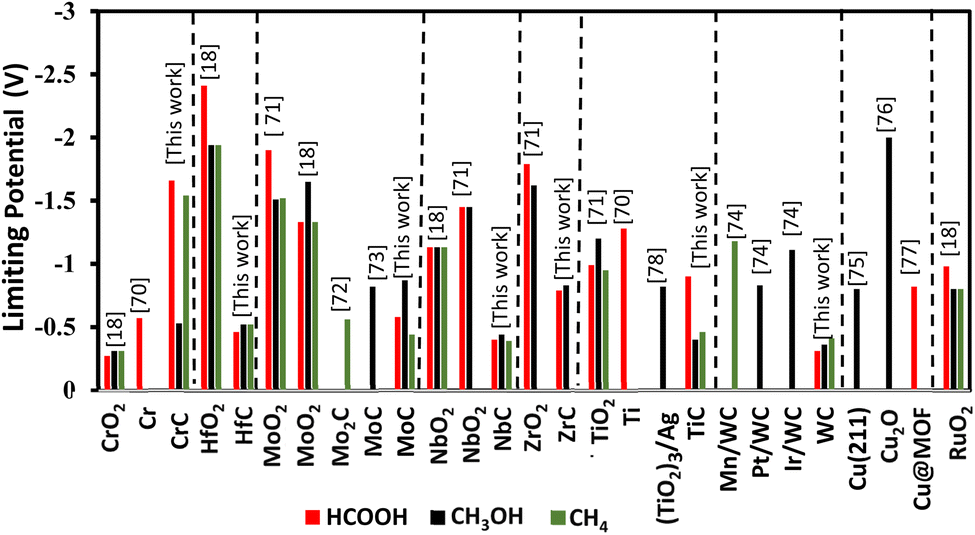
We have compared these results with previously studied metal-based catalysts as shown in Fig. 12. In this research work except CrC other TMCs show small onset potentials as compared to previously studied TMOs, pristine metals, and dopants. HfC, MoC, NbC, and TiC offer selectivity towards formic acid, methanol, and methane and are better than previously studied oxides of the same parental metal (HfO2, NbO2, TiO2, RuO2 and (TiO2)3/Ag and pristine Ti). The conclusion drawn from Fig. 12 shows that our findings from the TMCs for the CO2RR are more novel than previously studied metals.
 |
| | Fig. 12 Comparison of CO2RR onset potentials for different product formations on TMCs studied in this work with similar metal-based catalysts previously reported for formic acid, methanol, and methane.18,70–78 | |
4. Conclusion
Our comprehensive theoretical analysis reveals that specific TMCs exhibit remarkable catalytic activity for CO2 reduction, surpassing the performance of conventional catalysts. The electronic structure of these TMCs plays a pivotal role in facilitating the conversion of CO2 into valuable chemical products, offering a glimpse into the potential applications of these materials in renewable energy technologies. Our novel findings concluded that the presence of carbon atoms in the TMCs is fruitful for the CO2RR. Exploring the performance of a range of TMCs, we found out that the VC is the best candidate for the formation of formic acid with 0 V required onset potential, the WC is the best candidate to produce methanol with −0.36 V onset potential, and the MoC is the best candidate to produce methanediol with −0.58 V onset potential. Through an extensive literature survey, we conducted a thorough comparison of our results with those of transition metal oxides and pristine metals studied in the past. This analysis revealed that the TMCs demonstrated a smaller onset potential, rendering them ideal for CO2RR applications. This theoretical exploration of transition metal carbides for CO2 reduction reactions opens new frontiers in the quest for green energy solutions. As we stand at the cusp of a sustainable energy revolution, the theoretical novelties presented here pave the way for experimental endeavors to unlock the full potential of transition metal carbides in catalyzing a greener future.
Data availability
The calculations have been performed using the Vienna ab initio simulation package VASP (6.3.2 version).79 The data that support the article have been uploaded in the ESI† in the form of free energy diagrams.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The calculations were carried out utilizing the Icelandic high-performance computer cluster, Elja. The Technology & Development Fund has provided financial support under grant no. 2215496-0611.
References
- Y. Hori, Electrochemical CO2 reduction on metal electrodes, Mod. Aspects Electrochem., 2008, 89–189 CrossRef CAS PubMed.
- T. R. Karl and K. E. Trenberth, Modern global climate change, Science, 2003, 302(5651), 1719–1723 CrossRef CAS PubMed.
- E. V. Kondratenko,
et al., Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes, Energy Environ. Sci., 2013, 6(11), 3112–3135 RSC.
- S. D. Ebbesen and M. Mogensen, Electrolysis of carbon dioxide in solid oxide electrolysis cells, J. Power Sources, 2009, 193(1), 349–358 CrossRef CAS.
- Q. Fu,
et al., Syngas production via high-temperature steam/CO2 co-electrolysis: an economic assessment, Energy Environ. Sci., 2010, 3(10), 1382–1397 RSC.
- E. Bertheussen,
et al., Electroreduction of CO on polycrystalline copper at low overpotentials, ACS Energy Lett., 2018, 3(3), 634–640 CrossRef CAS.
- S. Tang,
et al., Synergistic effect of surface-terminated oxygen vacancy and single-atom catalysts on defective MXenes for efficient nitrogen fixation, J. Phys. Chem. Lett., 2020, 11(13), 5051–5058 CrossRef CAS PubMed.
- S. Tang,
et al., Metal-free boron nitride nanoribbon catalysts for electrochemical CO2 reduction: combining high activity and selectivity, ACS Appl. Mater. Interfaces, 2018, 11(1), 906–915 CrossRef PubMed.
- C. W. Li and M. W. Kanan, CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films, J. Am. Chem. Soc., 2012, 134(17), 7231–7234 CrossRef CAS PubMed.
- N. Hoshi, M. Kato and Y. Hori, Electrochemical reduction of CO2 on single crystal electrodes of silver Ag (111), Ag (100) and Ag (110), J. Electroanal. Chem., 1997, 440(1–2), 283–286 CrossRef CAS.
- Y. Chen, C. W. Li and M. W. Kanan, Aqueous CO2 reduction at very low overpotential on oxide-derived Au nanoparticles, J. Am. Chem. Soc., 2012, 134(49), 19969–19972 CrossRef CAS PubMed.
- A. Rodes, E. Pastor and T. Iwasita, Structural effects on CO2 reduction at Pt single-crystal electrodes: Part 1. The Pt (110) surface, J. Electroanal. Chem., 1994, 369(1–2), 183–191 CrossRef CAS.
- K. P. Kuhl,
et al., New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059 RSC.
- K. Schouten,
et al., A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes, Chem. Sci., 2011, 2(10), 1902–1909 RSC.
- T. Cheng, H. Xiao and W. A. Goddard III, Reaction mechanisms for the electrochemical reduction of CO2 to CO and formate on the Cu (100) surface at 298 K from quantum mechanics free energy calculations with explicit water, J. Am. Chem. Soc., 2016, 138(42), 13802–13805 CrossRef CAS PubMed.
- E. Bertheussen,
et al., Quantification of liquid products from the electroreduction of CO2 and CO using static headspace-gas chromatography and nuclear magnetic resonance spectroscopy, Catal. Today, 2017, 288, 54–62 CrossRef CAS.
- G. K. Ramesha, J. F. Brennecke and P. V. Kamat, Origin of catalytic effect in the reduction of CO2 at nanostructured TiO2 films, ACS Catal., 2014, 4(9), 3249–3254 CrossRef CAS.
- E. Tayyebi,
et al., Trends of electrochemical CO2 reduction reaction on transition metal oxide catalysts, J. Phys. Chem. C, 2018, 122(18), 10078–10087 CrossRef CAS.
- L. Zhai,
et al., Titania-modified silver electrocatalyst for selective CO2 reduction to CH3OH and CH4 from DFT study, J. Phys. Chem. C, 2017, 121(30), 16275–16282 CrossRef CAS.
- J.-J. s. Velasco-Vélez,
et al., On the activity/selectivity and phase stability of thermally grown copper oxides during the electrocatalytic reduction of CO2, ACS Catal., 2020, 10(19), 11510–11518 CrossRef PubMed.
- L. Chico-Mesa, E. Herrero and R. M. Arán-Ais, Tuning carbon dioxide electroreduction through selective facet exposure, Curr. Opin. Chem. Eng., 2024, 43, 100997 CrossRef.
- Z. Liu,
et al., Electrochemical CO2-to-CO conversion: A comprehensive review of recent developments and emerging trends, Sep. Purif. Technol., 2024, 330, 125177 CrossRef CAS.
- S. Du,
et al., Catalysts and electrolyzers for the electrochemical CO2 reduction reaction: from laboratory to industrial applications, Chem. Commun., 2024, 60(10), 1207–1221 RSC.
- S. González, F. Illas and J. L. Fierro, Evidence for spontaneous CO2 activation on cobalt surfaces, Chem. Phys. Lett., 2008, 454(4–6), 262–268 Search PubMed.
- M. A. Petersen, J.-A. van den Berg and W. J. van Rensburg, Role of step sites and surface vacancies in the adsorption and activation of CO on χ-Fe5C2 surfaces, J. Phys. Chem. C, 2010, 114(17), 7863–7879 CrossRef CAS.
- D. C. Sorescu, W. A. Al-Saidi and K. D. Jordan, CO2 adsorption on TiO2 (101) anatase: A dispersion-corrected density functional theory study, J. Chem. Phys., 2011, 135(12) DOI:10.1063/1.3638181.
- Y. Jiao,
et al., A density functional theory study on CO2 capture and activation by graphene-like boron nitride with boron vacancy, Catal. Today, 2011, 175(1), 271–275 CrossRef CAS.
- N. Y. Dzade, A. Roldan and N. H. de Leeuw, Activation and dissociation of CO2 on the (001), (011), and (111) surfaces of mackinawite (FeS): A dispersion-corrected DFT study, J. Chem. Phys., 2015, 143(9) DOI:10.1063/1.4929470.
- D. Smykowski, B. Szyja and J. Szczygieł, DFT modeling of CO2 adsorption on Cu, Zn, Ni, Pd/DOH zeolite, J. Mol. Graphics Modell., 2013, 41, 89–96 CrossRef CAS PubMed.
- L. Valenzano,
et al., Computational and Experimental Studies on the Adsorption of CO, N2, and CO2 on Mg-MOF-74, J. Phys. Chem. C, 2010, 114(25), 11185–11191 CrossRef CAS.
- R. Levy and M. Boudart, Platinum-like behavior of tungsten carbide in surface catalysis, Science, 1973, 181(4099), 547–549 CrossRef CAS PubMed.
- H. H. Hwu and J. G. Chen, Surface chemistry of transition metal carbides, Chem. Rev., 2005, 105(1), 185–212 CrossRef CAS PubMed.
- L. K. Ono and B. Roldan-Cuenya, Effect of interparticle interaction on the low temperature oxidation of CO over size-selected Au nanocatalysts supported on ultrathin TiC films, Catal. Lett., 2007, 113, 86–94 CrossRef CAS.
- P. M. Patterson, T. K. Das and B. H. Davis, Carbon monoxide hydrogenation over molybdenum and tungsten carbides, Appl. Catal., A, 2003, 251(2), 449–455 CrossRef CAS.
- A. J. Brungs, A. P. York and M. L. Green, Comparison of the group V and VI transition metal carbides for methane dry reforming and thermodynamic prediction of their relative stabilities, Catal. Lett., 1999, 57(1–2), 65–69 CrossRef CAS.
- J. Rodriguez,
et al., Adsorption and decomposition of SO2 on TiC (0 0 1): An experimental and theoretical study, Surf. Sci., 2003, 543(1–3), L675–L682 CrossRef CAS.
- J. B. Claridge,
et al., New catalysts for the conversion of methane to synthesis gas: molybdenum and tungsten carbide, J. Catal., 1998, 180(1), 85–100 CrossRef CAS.
- F. Viñes,
et al., Catalyst size matters: Tuning the molecular mechanism of the water–gas shift reaction on titanium carbide based compounds, J. Catal., 2008, 260(1), 103–112 CrossRef.
- M. M. Sullivan, C.-J. Chen and A. Bhan, Catalytic deoxygenation on transition metal carbide catalysts, Catal. Sci. Technol., 2016, 6(3), 602–616 RSC.
- Y. N. Regmi,
et al., Electrocatalytic activity and stability enhancement through preferential deposition of phosphide on carbide, ChemCatChem, 2017, 9(6), 1054–1061 CrossRef CAS.
- T. J. Harrington,
et al., Phase stability and mechanical properties of novel high entropy transition metal carbides, Acta Mater., 2019, 166, 271–280 CrossRef CAS.
- Y. C. Kimmel,
et al., Trends in electrochemical stability of transition metal carbides and their potential use as supports for low-cost electrocatalysts, ACS Catal., 2014, 4(5), 1558–1562 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(11), 7413 CrossRef.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953 CrossRef PubMed.
- G. Li,
et al., Role of dissociation of phenol in its selective hydrogenation on Pt (111) and Pd (111), ACS Catal., 2015, 5(3), 2009–2016 CrossRef CAS.
- J. K. Nørskov,
et al., Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108(46), 17886–17892 CrossRef.
-
P. W. Atkins, J. De Paula and J. Keeler, Atkins' Physical Chemistry, Oxford University Press, 2023 Search PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, Calculations of product selectivity in electrochemical CO2 reduction, ACS Catal., 2018, 8(6), 5240–5249 CrossRef CAS.
- A. A. Peterson,
et al., How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels, Energy Environ. Sci., 2010, 3(9), 1311–1315 RSC.
- K. P. Kuhl,
et al., Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces, J. Am. Chem. Soc., 2014, 136(40), 14107–14113 CrossRef CAS PubMed.
- A. A. Peterson and J. K. Nørskov, Activity descriptors for CO2 electroreduction to methane on transition-metal catalysts, J. Phys. Chem. Lett., 2012, 3(2), 251–258 CrossRef CAS.
- J. Rossmeisl,
et al., Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607(1–2), 83–89 CrossRef CAS.
- P. M. Gíslason and E. Skúlason, Catalytic trends of nitrogen doped carbon nanotubes for oxygen reduction reaction, Nanoscale, 2019, 11(40), 18683–18690 RSC.
- J. Hussain, H. Jónsson and E. Skúlason, Faraday efficiency and mechanism of electrochemical surface reactions: CO2 reduction and H2 formation on Pt (111), Faraday Discuss., 2016, 195, 619–636 RSC.
- Y. Abghoui and E. Skúlason, Hydrogen evolution reaction catalyzed by transition-metal nitrides, J. Phys. Chem. C, 2017, 121(43), 24036–24045 CrossRef CAS.
- Y. Abghoui, Superiority of the (100) over the (111) facets of the nitrides for hydrogen evolution reaction, Top. Catal., 2022, 65(1), 262–269 CrossRef CAS.
- A. Iqbal, E. Skúlason and Y. Abghoui, Understanding the Mechanistic Pathways of N2 Reduction to Ammonia on (110) Facets of Transition Metal Carbides, Crystals, 2024, 14(9), 770 CrossRef CAS.
- A. Iqbal, E. Skúlason and Y. Abghoui, Catalytic Nitrogen Reduction on the Transition Metal Carbonitride (110) Facet: DFT Predictions and Mechanistic Insights, J. Phys. Chem. C, 2024 DOI:10.1021/acs.jpcc.4c02148.
- A. Iqbal, E. Skúlason and Y. Abghoui, Are (100) facets of transition metal carbonitrides suitable as electrocatalysts for nitrogen reduction to ammonia at ambient conditions?, Int. J. Hydrogen Energy, 2024, 64, 744–753 CrossRef CAS.
- A. Iqbal, E. Skulason and Y. Abghoui, Electrochemical Nitrogen Reduction to Ammonia at Ambient Condition on the (111) Facets of Transition Metal Carbonitrides, ChemPhysChem, 2024, e202300991 CrossRef CAS PubMed.
- K. Korir,
et al., First-principle calculations of the bulk properties of 4d transition metal carbides and nitrides in the rocksalt, zincblende and wurtzite structures, Diamond Relat. Mater., 2011, 20(2), 157–164 CrossRef CAS.
- N. Szymanski,
et al., Unconventional superconductivity in 3d rocksalt transition metal carbides, J. Mater. Chem. C, 2019, 7(40), 12619–12632 RSC.
- V. Ellingsson,
et al., Nitrogen reduction reaction to ammonia on transition metal carbide catalysts, ChemSusChem, 2023, 16(22), e202300947 CrossRef CAS PubMed.
-
F. Kröger and H. Vink, Relations between the concentrations of imperfections in crystalline solids, in Solid State Physics, Elsevier, 1956, pp. 307–435 Search PubMed.
- A. Bhowmik, T. Vegge and H. A. Hansen, Descriptors and thermodynamic limitations of electrocatalytic carbon dioxide reduction on rutile oxide surfaces, ChemSusChem, 2016, 9(22), 3230–3243 CrossRef CAS PubMed.
- N. Atrak, E. Tayyebi and E. Skúlason, Electrochemical CO2 reduction towards formic acid and methanol on transition metal oxide surfaces as a function of CO coverage, Catal. Sci. Technol., 2023, 13(11), 3321–3336 RSC.
- B. Kang,
et al., Computational evaluation of CO2 conversion into formic acid via a novel adsorption mechanism on metal-free B4C12, J. Colloid Interface Sci., 2024, 654, 371–378 CrossRef CAS PubMed.
- J. Wang,
et al., Tackling the activity and selectivity challenges of electrocatalysts toward the CO2RR via biatom catalysts on the 2D extended phthalocyanines, Int. J. Quantum Chem., 2024, 124(1), e27308 CrossRef CAS.
- F. Li, H. Ai, C. Shi, K. H. Lo and H. Pan, Single transition metal atom catalysts on Ti2CN2 for efficient CO2 reduction reaction, Int. J. Hydrogen Energy, 2021, 46(24), 12886–12896 CrossRef CAS.
- N. Atrak, E. Tayyebi and E. Skúlason, Effect of co-adsorbed water on electrochemical CO2 reduction reaction on transition metal oxide catalysts, Appl. Surf. Sci., 2021, 570, 151031 CrossRef CAS.
- S. K. Kim, Y. J. Zhang, H. Bergstrom, R. Michalsky and A. Peterson, Understanding the low-overpotential production of CH4 from CO2 on Mo2C catalysts, ACS Catal., 2016, 6(3), 2003–2013 CrossRef CAS.
- H. Li and K. Reuter, Active-site computational screening: role of structural and compositional diversity for the electrochemical CO2 reduction at Mo carbide catalysts, ACS Catal., 2020, 10(20), 11814–11821 CrossRef CAS.
- S. Wannakao, N. Artrith, J. Limtrakul and A. M. Kolpak, Catalytic activity and product selectivity trends for carbon dioxide electroreduction on transition metal-coated tungsten carbides, J. Phys. Chem. C, 2017, 121(37), 20306–20314 CrossRef CAS.
- S. Back, H. Kim and Y. Jung, Selective heterogeneous CO2 electroreduction to methanol, ACS Catal., 2015, 5(2), 965–971 CrossRef CAS.
- J. Hazarika and M. S. Manna, Electrochemical reduction of CO2 to methanol with synthesized Cu2O nanocatalyst: Study of the selectivity, Electrochim. Acta, 2019, 328, 135053 CrossRef CAS.
- Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi and J. Tang, Nanoengineering metal–organic framework-based materials for use in electrochemical CO2 reduction reactions, Small, 2021, 17(16), 2006590 CrossRef CAS PubMed.
- L. Zhai, C. Cui, Y. Zhao, X. Zhu, J. Han, H. Wang and Q. Ge, Titania-modified silver electrocatalyst for selective CO2 reduction to CH3OH and CH4 from DFT study, J. Phys. Chem. C, 2017, 121(30), 16275–16282 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Atef
Iqbal
,
Atef
Iqbal
 where Cp(T′) represents specific heat at constant pressure and the integral of this equation contributes negligibly at room temperature. The value of the gas phase is taken from the thermodynamic tables from textbooks.48
where Cp(T′) represents specific heat at constant pressure and the integral of this equation contributes negligibly at room temperature. The value of the gas phase is taken from the thermodynamic tables from textbooks.48