
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of cellular membrane-mimicking glycopolymer interfaces by a solvent-assisted method on QCM-D sensor chips and their molecular recognition†
Masanori
Nagao
 *a,
Tsukuru
Masuda
b,
Madoka
Takai
b and
Yoshiko
Miura
*a
*a,
Tsukuru
Masuda
b,
Madoka
Takai
b and
Yoshiko
Miura
*a
aDepartment of Chemical Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: nagaom@chem-eng.kyushu-u.ac.jp; miuray@chem-eng.kyushu-u.ac.jp
bDepartment of Bioengineering, The University of Tokyo, Tokyo 113-8656, Japan
First published on 30th January 2024
Abstract
Carbohydrate-based membranes that show molecular recognition ability are interesting mimics of biointerfaces. Herein, we prepared glycopolymer membranes on QCM-D sensor chips using a solvent-assisted method and investigated their interactions with a target lectin. The membrane containing the glycopolymer with a random arrangement of the carbohydrate units adsorbed more lectin than that containing the glycopolymer with an organized block of carbohydrate units.
Carbohydrates play important roles in biological activities (e.g., pathogen infection, immune response, and cell-to-cell communication) through molecular recognition on cell surfaces, where carbohydrates selectively interact with corresponding proteins (lectins).1–4 Since monovalent interaction between a carbohydrate unit and a lectin binding site is generally weak, multivalent binding is important to strengthen carbohydrate–lectin interactions.5,6 Glycoconjugates exist as clusters on cell surfaces and exhibit multivalent binding with multiple lectin binding sites. A glycopolymer contains multiple carbohydrate units in a single molecule.7–10 Glycopolymers exhibit multivalent interactions with target lectins and have been used as pathogen inhibitors and biosensors.11–13
Since the molecular recognition of carbohydrates occurs on cell surfaces, material interfaces modified with carbohydrates have attracted much attention as biointerface mimics and analysis platforms.14–17 Carbohydrates can be immobilized by thiol–gold interactions on gold surfaces14,16 and silane coupling reactions on glass substrates.18,19 Locally dense immobilized carbohydrates on solid surfaces can form strong multivalent interactions with the target lectin. For example, Percec and co-workers fabricated a glycodendrimer surface with a precisely controlled arrangement of carbohydrate units.20 The well-defined distance between the carbohydrate units determined the multivalency and strengthened the interaction of the glycodendrimer surface with lectins. Furthermore, while not related to carbohydrate interactions, there have been reports indicating that the method of immobilizing enzymes on the surface can impact their activity.21–23 These examples demonstrate the substantial impact that the immobilization method for functional groups can have on the properties of biointerface interactions.
Molecular recognition of fluid membranes formed through non-covalent interactions is attracting increasing attention. For example, Ritcher and co-workers reported highly selective molecular recognition between adamantane units and cyclodextrin polymers on a fluid lipid bilayer supported on a substrate.24 Such fluid lipid bilayers are formed by the liposome shaping and the Langmuir–Blodgett method.25,26 In addition to these traditional methods, the solvent-assisted method was reported by Cho and co-workers.27–29 This method enabled the facile preparation of a uniform lipid bilayer membrane. Furthermore, Meier and co-workers demonstrated that the solvent-assisted method can be applied to synthetic polymer membranes.30 In this pioneering work, the authors also demonstrated the molecular recognition of the formed polymer membrane.
In this context, we hypothesized that the solvent-assisted method would be useful for obtaining a glycopolymer membrane-based molecular recognition platform. However, the applicability of the solvent-assisted method for this purpose and the effects of polymer structures on surface molecular recognition remain unclear. Herein, we investigate the bottom-up formation of glycopolymer membranes on a quartz crystal microbalance with dissipation (QCM-D) sensor chips using the solvent-assisted method and the molecular recognition behavior of the glycopolymer membranes toward a target lectin (Fig. 1). QCM-D is an excellent tool for analyzing interactions between biomolecules.31 While there have been several reports on the interaction between glycopolymer interfaces and proteins using QCM-D,32–34 this work focuses on observing the in situ formation of non-covalent polymer membranes using the solvent-assisted method and their interactions with proteins. In this work, we use the term “membrane” to describe the “membrane-like” solid-supported polymer assemblies for simplicity.30
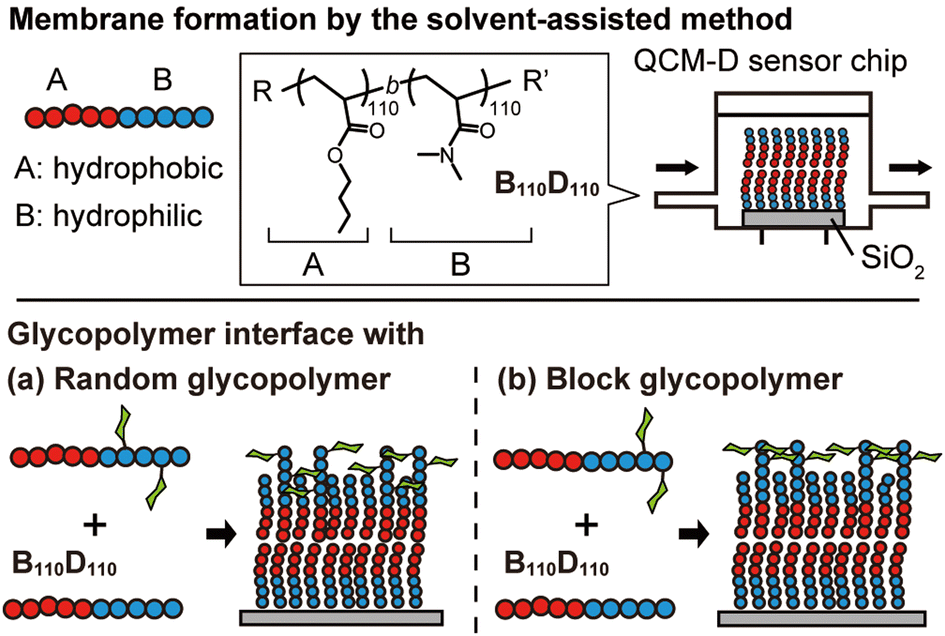 | ||
| Fig. 1 Schematic illustration of polymer membrane formation on a quartz crystal microbalance with a dissipation (QCM-D) sensor chip by the solvent-assisted method. B is butyl acrylate and D is N,N-dimethyl acrylamide. In the lower half, membrane formation using glycopolymers with a random arrangement (a) and a block arrangement (b). | ||
First, we investigated the amphiphilic polymer structures required to form polymer membranes. The block copolymers were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization, which is a type of living radical polymerization. Hydrophobic and hydrophilic blocks were prepared with butyl acrylate (BA) and N,N-dimethylacrylamide (DMA) monomers, respectively. The target degree of polymerization for each block was varied (Tables S1 and S2, ESI†). The compositions of the obtained polymers were determined by 1H NMR (Fig. S1–S9, ESI†). Size exclusion chromatography (SEC) analysis demonstrated that the peaks of the hydrophobic BA polymers (polyBAm, m = 50, 100, and 200) shifted to higher molecular weight (Mn) after polymerization with the hydrophilic DMA (polyBAm-b-DMAn, BmDn), confirming successful chain extension (Fig. S10 and S11, ESI†).
The self-assembly behaviour of the synthesized BmDn diblock copolymers was evaluated by dynamic light scattering (DLS) measurements. The DLS measurements of B58D24, B58D64, B110D55, and B110D110 polymer solutions in phosphate-buffered saline (PBS) solution (0.1 g L−1) showed that the copolymers had diameters larger than 30 nm, indicating the formation of self-assembled structures such as micelles in aqueous solution (Table 1 and Fig. S12, ESI†). B200D100 and B200D200 did not completely dissolve in ethanol and were excluded from subsequent experiments.
| Polymer | M n (g mol−1) | M w/Mna | D h (nm) | Surface coveragec (%) |
|---|---|---|---|---|
| a Molecular weight (Mn) and dispersity (Mw/Mn) were determined by SEC analysis (N,N-dimethylformamide with 10 mM LiBr as an eluent) calibrated with a polymethylmethacrylate standard. b Hydrodynamic diameter was determined by dynamic light scattering (0.1 g L−1 in PBS at 25 °C). Measurements were repeated three times. c Coverage of the QCM-D sensor surface was determined by: 100% × (1 − ΔFBSA on polymer membrane/ΔFBSA on bare glass substrate), where ΔFBSA is the frequency change caused by the addition of bovine serum albumin. | ||||
| B58D24 | 8300 | 1.36 | 48 ± 5 | 26 |
| B58D64 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
1.21 | 32 ± 1 | 43 |
| B110D55 | 16100 |
1.33 | 48 ± 8 | 39 |
| B110D110 | 22100 |
1.20 | 34 ± 4 | 53 |
To determine the appropriate polymer structure for the solvent-assisted method, the synthesized diblock copolymers were applied to form polymer membranes on QCM-D substrates. As a representative example, the QCM-D frequency profile of B110D110 during membrane formation is shown in Fig. 2 (profiles for the other polymers are shown in Fig. S13–S15, ESI†). A silica-coated QCM-D sensor chip was set in the module, and the signal was stabilized with a flow of PBS solution, followed by ethanol and polymer solution in ethanol (10 mg L−1). Then, the solvent was switched from the polymer solution to PBS, and the frequency change (ΔF) was observed (step 4 in Fig. 2). The negative ΔF indicated polymer adsorption on the QCM-D substrate. To evaluate the polymer membrane coverage of the sensor surface, the solvent was sequentially changed to bovine serum albumin (BSA) solution in PBS (0.5 g L−1), which led to BSA adsorption on the uncovered area of the sensor surface. The surface coverage of the polymer membrane was calculated using eqn (1):
| Surface coverage = 100% × (1 − ΔFBSA on polymer membrane/ΔFBSA on bare glass substrate) | (1) |
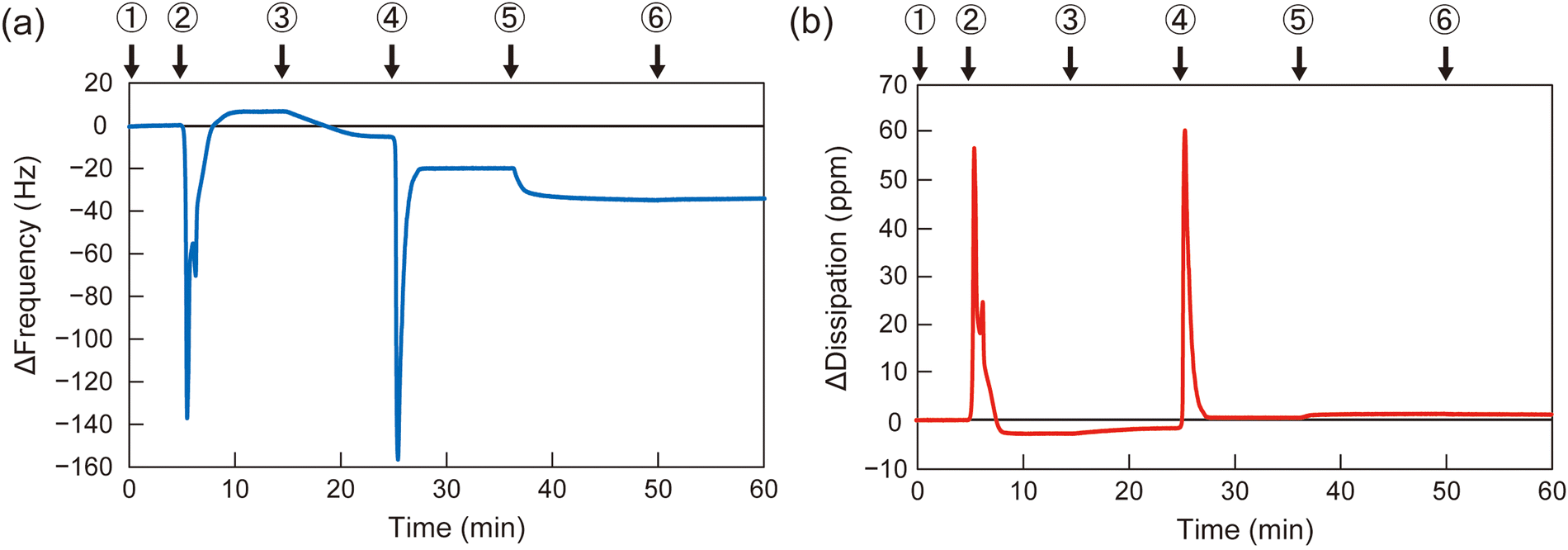 | ||
| Fig. 2 QCM-D frequency (a) and dissipation (b) profiles for polymer membrane formation with B110D110 upon addition of PBS (1), ethanol (2), polymer solution in ethanol (3), PBS (4), BSA solution in PBS (5), and PBS (6). | ||
Next, amphiphilic glycopolymers were synthesized based on the structure of B110D110. Mannose units were introduced into the hydrophilic block as side chains in two types of sequence: random or block. The target degree of mannose acrylamide was 10 and copolymerized with DMA for the random glycopolymer poly(BA110-b-(DMA-r-Man)103) (RG). Mannose acrylamide was polymerized with B110D110 as the macro-RAFT agent to produce block glycopolymer poly(BA110-b-DMA110-b-Man7) (BG). The number of mannose units in each RG and BG was determined to be nine and seven, respectively, by 1H NMR (Fig. 3 and Fig. S17, S18, ESI†). Both RG and BG showed similar molecular weights (Fig. S19, ESI†). These results indicated that RG and BG have comparable structures, except for their sequence.
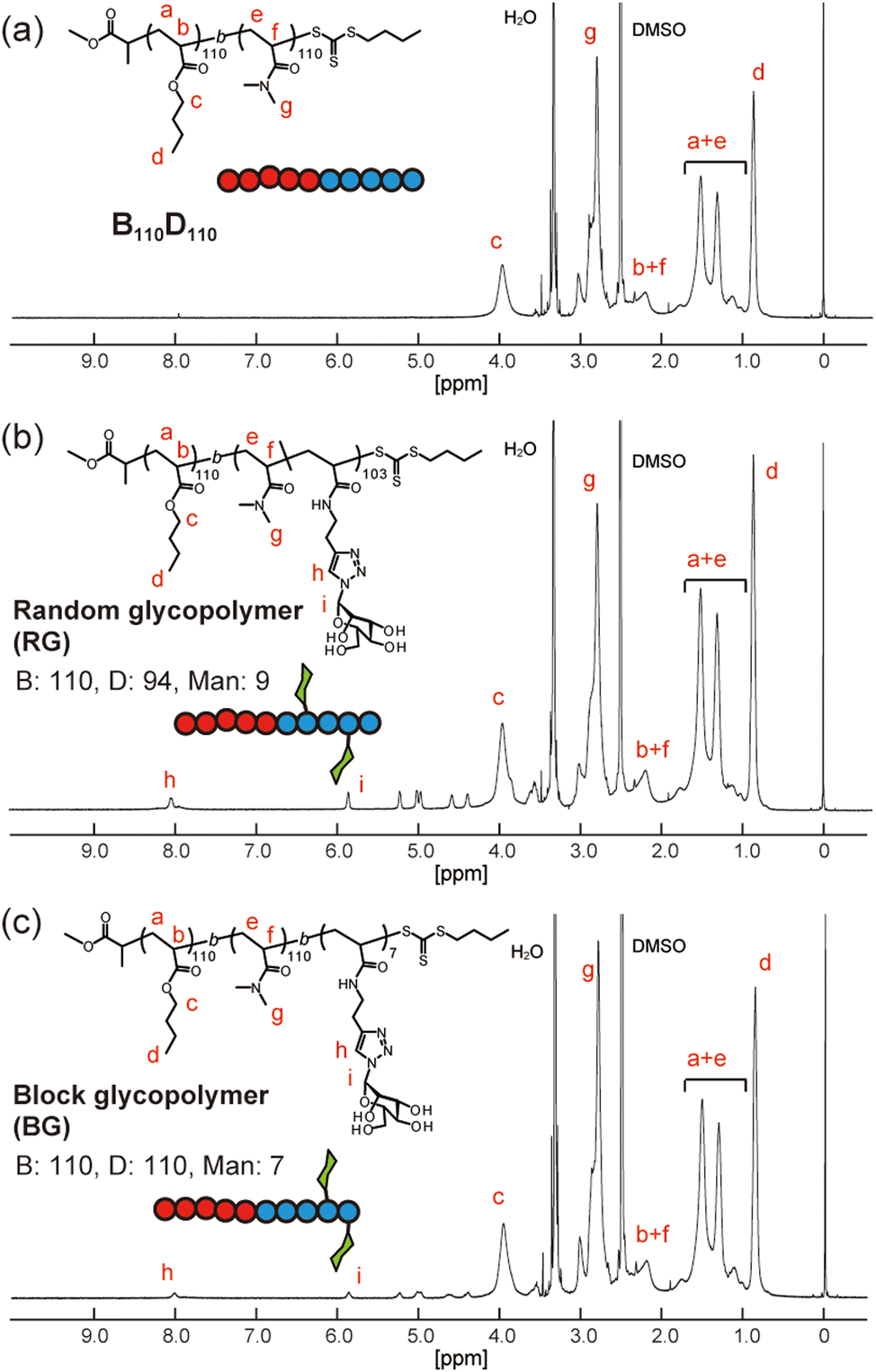 | ||
| Fig. 3 1H NMR spectra of (a) B110D110, (b) random glycopolymer (RG), and (c) block glycopolymer (BG) in d6-dimethylsulfoxide. | ||
Then, polymer membranes containing the glycopolymers were formed using the solvent-assisted method. Polymer solutions were prepared with either RG or BG (10 or 50 wt%, respectively) and B110D110 (90 or 50 wt%) in ethanol. As in the control experiment with only B110D110, the mixtures of B110D110 and glycopolymers also showed decreased ΔF after solvent exchange (step 4 in Fig. 4a and b). The surface coverage of the glycopolymer membranes was less than 30%, which was lower than that of B110D110 (Table 2). The decreased surface coverage was attributed to the hydrophilic–hydrophobic balance of the polymers being affected by the presence of mannose units. The glycopolymer membranes on the QCM-D sensor chips were observed by atomic force microscopy (AFM) after drying; no vesicle structure was observed (Fig. S20, ESI†). The AFM results further confirmed the formation of glycopolymer membranes on the QCM-D sensor surface using the solvent-assisted method.
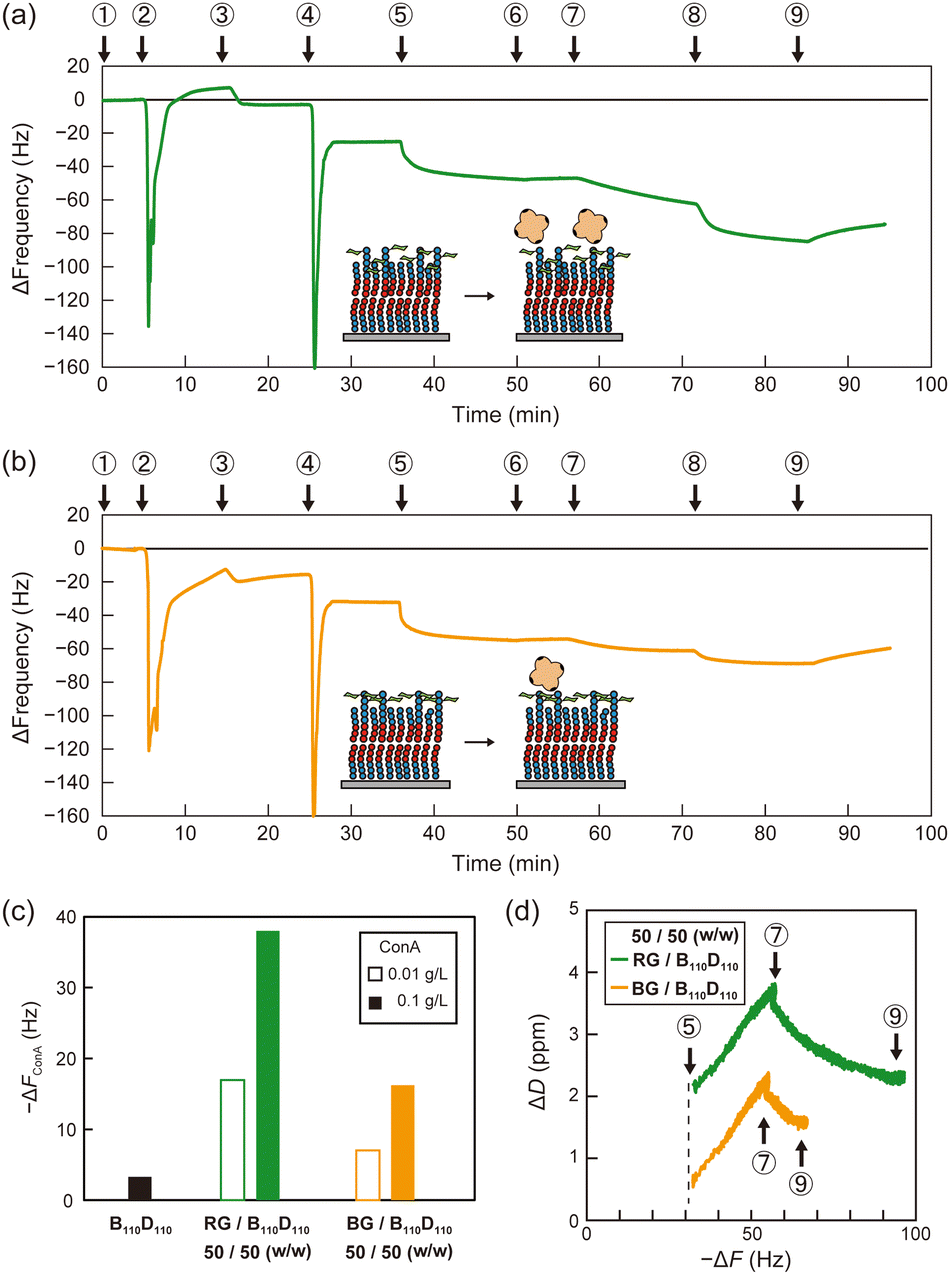 | ||
| Fig. 4 QCM-D frequency profiles for polymer membrane formation with (a) B110D110:RG = 50:50 (w/w) and (b) B110D110:RG = 50:50 (w/w). In the profiles, PBS (1), ethanol (2), polymer solution in ethanol (3), PBS (4), BSA solution in PBS (5), PBS(+) (6), ConA solution in PBS(+) (C = 0.01 g L−1) (7), ConA solution in PBS(+) (C = 0.1 g L−1) (8), and PBS (+) (9). (c) The frequency change caused by ConA adsorption for each polymer membrane. (d) ΔD–ΔF plots of ConA adsorption for each glycopolymer membrane (step 5 to step 9). | ||
| Glyco polymera | Glycopolymer/B110D110 (w/w) | Surface coverageb (%) | [ConA] (g L−1) | −ΔFConA (Hz) |
|---|---|---|---|---|
| a RG and BG are poly(BA110-b-(DMA-r-Man)103) and poly(BA110-b-DMA110-b-Man7), respectively. b Surface coverage of QCM-D sensor surface was determined by: 100% × (ΔFBSA on polymer membrane/ΔFBSA on bare glass substrate). | ||||
| RG | 10:90 |
24 | 0.01 | 4 |
| 0.1 | 12 | |||
| 50:50 |
28 | 0.01 | 18 | |
| 0.1 | 36 | |||
| BG | 10:90 |
26 | 0.01 | 2 |
| 0.1 | 7 | |||
| 50:50 |
27 | 0.01 | 7 | |
| 0.1 | 17 | |||
Molecular recognition by the glycopolymer membranes was then evaluated using concanavalin A (ConA). ConA is a tetrameric lectin that specifically binds to mannose units in the presence of calcium ions.4 After blocking the uncovered sensor surface with BSA, the membranes were exposed to ConA in PBS with calcium ion (PBS(+)) solution (0.01 g L−1), which caused a ΔF decrease (step 7 in Fig. 4a and b). The sensor frequency changes caused by ConA (ΔFConA) were −18 and −7 Hz for the membranes with 50 wt% RG and BG, respectively (Fig. 4c and Table 2). The concentration of ConA solution was sequentially increased to 0.1 g L−1 (step 8 in Fig. 4a and b), which caused ΔFConA to further decrease to −36 and −17 Hz for RG and BG, respectively (Fig. 4c). Stated values are the averages of triplicate experiments (Table S3, ESI†). The dissipation profiles of the membranes are shown in Fig. S21 (ESI†). In contrast, peanut agglutinin (PNA), a non-mannose binding lectin did not induce frequency changes, indicating the retained selectivity of the mannose units on the glycopolymer surfaces (Table S4 and Fig. S22, S23, ESI†). Membranes with a lower ratio of glycopolymers to B110D110 of 10 wt% adsorbed less ConA than those with a higher glycopolymer: B110D110 ratio (Table 2 and Fig. S24, S25, ESI†). The polymer membrane with only B110D110 (no glycopolymer) showed negligible ConA adsorption (Fig. 4c). These results demonstrated that using BSA to block uncovered sensor surfaces worked well to avoid nonspecific adsorption of ConA, even at low surface coverage, and that ConA adsorption on the QCM-D surface was derived from specific molecular recognition by mannose units of the glycopolymers. Interestingly, the amount of ConA adsorbed on the sensor surface was larger for the membrane with RG than that with BG, even though they had the same surface coverage (Table 2). This indicates that the glycopolymer membrane containing mannose units with a random arrangement interacted more strongly with ConA or had larger capacity for ConA binding. It is noteworthy that our previous works also reported that the strongest binding of glycopolymers to lectins was not achieved at the highest carbohydrate unit density.35,36 Alexander-Katz and co-workers reported that polymers featuring the functional group in an alternating fashion demonstrate an advantage in multivalent interactions with the target compared to those with a block structure, particularly under high target concentrations. This phenomenon arises from the competition for multiple binding sites on the target.37
The slope of the dissipation change (ΔD)–ΔF plots obtained from QCM-D measurements provides information about the viscoelasticity of the sensor surface in adsorption experiments. A higher slope indicates that a surface has a relatively relaxed structure and high viscosity.38Fig. 4d shows that the slopes of the ΔD–ΔF plots for ConA adsorption were the same for the membranes with RG and BG. This indicates that the glycopolymer membranes had comparable elasticity, and the amount of ConA adsorbed on the membrane was affected by the statistical arrangement of carbohydrate units on the surface, and not by dynamic properties such as fluidity.
To demonstrate the advantage of membrane formation by the solvent-assisted method, we prepared a glycopolymer membrane on a QCM-D sensor chip by spin coating using the same polymer solution (B110D110:RG = 50:50, 10 mg L−1 in ethanol). The spin-coated surface displayed ΔFConA of −7 Hz upon exposure to ConA solution (0.1 g L−1), which was smaller than that of the equivalent membrane prepared by the solvent-assisted method (Fig. S26 and S27, ESI†). The deference on the adsorption amount of ConA was attributed to the varying amounts of glycopolymers on the sensor surface. At step 2 in Fig. S26 (ESI†), the frequency change due to the adsorption of BSA on the spin-coated polymer was as large as that on a bare glass surface of the QCM sensor, indicating that the polymer is sparsely deposited on the sensor surface. Considering that the adsorption of PNA on the polymer membranes prepared by the solvent-assisted method was below 3 Hz (Table S4, ESI†), the adsorption of ConA on the spin-coated surface is likely specific to mannose units, implying the presence of a certain amount of polymer on the sensor surface. This experiment aimed to showcase the effects of different membrane formation methods at the same polymer concentration, highlighting the solvent-assisted method's efficiency in forming a membrane at lower concentrations. A high polymer concentration in the spin-coating method could potentially result in more polymer deposition on the sensor surface.
In conclusion, we prepared synthetic polymer membranes on QCM-D sensor surfaces using the solvent-assisted method. The formation of these membranes relies on the self-assembly of amphiphilic polymers, where the hydrophobic blocks aggregate in the aqueous solvent, as observed upon replacing the polymer solution in ethanol to PBS solution. Among the amphiphilic copolymers tested, B110D110, with a diblock structure featuring higher molecular weight and hydrophobic unit ratio, yielded the membrane with the highest coverage of the QCM-D sensor chip. Introduction of glycopolymers containing mannose units, arranged either randomly or in organized blocks, into the B110D110 polymer membranes revealed that the arrangement of carbohydrate units at the membrane surface influences molecular recognition ability. Specifically, the membrane with randomly arranged mannose units adsorbed more ConA than its counterpart with organized blocks of the mannose units, suggesting competition for multiple binding sites on ConA. Controlled polymerization techniques enable the precise design of synthetic polymer structures, paving the way for the development of highly functional interfaces based on synthetic polymers in the future. Furthermore, the polymer membrane formed via the solvent-assisted method exhibited superior ConA adsorption compared to the spin-coating method, underscoring the advantages of the solvent-assisted approach. Moving forward, applying this method to create non-covalently attached membranes on surfaces with amphiphilic polymers featuring different glass transition temperatures of hydrophobic blocks could unveil the correlation between polymer membrane fluidity and functionality.
Author contributions
The experiments were planned and conducted by M. Nagao and T. Masuda. The manuscript was written by M. Nagao. Y. Miura and M. Takai provided advice to improve the quality of the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a JSPS Grant-in-Aid (JP22K14728, JP22H05430, JP22H05048, JP23H02015) and by funding from Fukuoka University (Grant No. 221045).References
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- J. Poole, C. J. Day, M. Von Itzstein, J. C. Paton and M. P. Jennings, Nat. Rev. Microbiol., 2018, 16, 440–452 CrossRef CAS PubMed.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
- J. J. Lundquist and E. J. Toone, Chem. Rev., 2002, 102, 555–578 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed.
- G. Yilmaz and C. R. Becer, Macromol. Chem. Phys., 2020, 221, 2000006 CrossRef CAS.
- L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC.
- M. Nagao, H. Matsumoto and Y. Miura, Chem. – Asian J., 2023, 18, e202300643 CrossRef CAS PubMed.
- T. Masuda and M. Takai, J. Mater. Chem. B, 2022, 10, 1473–1485 RSC.
- J. Li, X. Y. Tian, L. P. Zong, Q. Zhang, X. J. Zhang, R. Marks, S. Cosnier and D. Shan, ACS Appl. Mater. Interfaces, 2019, 11, 32366–32372 CrossRef CAS PubMed.
- A. Singh, J. C. Arango, A. Shi, J. B. d’Aliberti and S. A. Claridge, J. Am. Chem. Soc., 2023, 145, 1668–1677 CrossRef CAS PubMed.
- K. R. Love and P. H. Seeberger, Angew. Chem., Int. Ed., 2002, 41, 3583–3586 CrossRef CAS PubMed.
- J. Shi, T. Yang, S. Kataoka, Y. Zhang, A. J. Diaz and P. S. Cremer, J. Am. Chem. Soc., 2007, 129, 5954–5961 CrossRef CAS PubMed.
- S. Park, J. C. Gildersleeve, O. Blixt and I. Shin, Chem. Soc. Rev., 2013, 42, 4310–4326 RSC.
- C. D. Rillahan, E. Schwartz, R. McBride, V. V. Fokin and J. C. Paulson, Angew. Chem., Int. Ed., 2012, 51, 11014–11018 CrossRef CAS PubMed.
- G. T. Carroll, D. Wang, N. J. Turro and J. T. Koberstein, Langmuir, 2006, 22, 2899–2905 CrossRef CAS PubMed.
- H. Seto, S. Kamba, T. Kondo, M. Hasegawa, S. Nashima, Y. Ehara, Y. Ogawa, Y. Hoshino and Y. Miura, ACS Appl. Mater. Interfaces, 2014, 6, 13234–13241 CrossRef CAS PubMed.
- N. Yu. Kostina, D. Söder, T. Haraszti, Q. Xiao, K. Rahimi, B. E. Partridge, M. L. Klein, V. Percec and C. Rodriguez-Emmenegger, Angew. Chem., Int. Ed., 2021, 60, 8352–8360 CrossRef CAS PubMed.
- J. Guo, L. Yang, Z. Gao, C. Zhao, Y. Mei and Y.-Y. Song, ACS Catal., 2020, 10, 5949–5958 CrossRef CAS.
- J. Guo, L. Yang, C. Zhao, Z. Gao, Y.-Y. Song and P. Schmuki, J. Mater. Chem. A, 2021, 9, 14911–14919 RSC.
- J. Xu, Y. Xue, X. Jian, Y. Zhao, Z. Dai, J. Xu, Z. Gao, Y. Mei and Y.-Y. Song, Chem. Sci., 2022, 13, 6550–6557 RSC.
- G. V. Dubacheva, T. Curk, D. Frenkel and R. P. Richter, J. Am. Chem. Soc., 2019, 141, 2577–2588 CrossRef CAS PubMed.
- R. P. Richter, R. Bérat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- C. Draghici, V. Mikhalevich, G. Gunkel-Grabole, J. Kowal, W. Meier and C. G. Palivan, Langmuir, 2018, 34, 9015–9024 CrossRef CAS PubMed.
- A. R. Ferhan, B. K. Yoon, S. Park, T. N. Sut, H. Chin, J. H. Park, J. A. Jackman and N. J. Cho, Nat. Protoc., 2019, 14, 2091–2118 CrossRef CAS PubMed.
- S. R. Tabaei, J. A. Jackman, S. O. Kim, B. Liedberg, W. Knoll, A. N. Parikh and N. J. Cho, Langmuir, 2014, 30, 13345–13352 CrossRef CAS PubMed.
- J. J. J. Gillissen, S. R. Tabaei and N. J. Cho, Phys. Chem. Chem. Phys., 2016, 18, 24157–24163 RSC.
- S. Di Leone, J. Vallapurackal, S. Yorulmaz Avsar, M. Kyropolou, T. R. Ward, C. G. Palivan and W. Meier, Biomacromolecules, 2021, 22, 3005–3016 CrossRef CAS PubMed.
- J. Wei, Y. Shao, S. Qiao, A. Li, S. Hou and W.-B. Zhang, Anal. Chem., 2023, 95, 16435–16446 CrossRef CAS PubMed.
- Y. Wang, R. Narain and Y. Liu, Langmuir, 2014, 30, 7377–7387 CrossRef CAS PubMed.
- J. Liu, C. Fu, S. Wang, L. Tao, L. Yan, D. M. Haddleton, G. Chen and Y. Wei, Macromolecules, 2014, 47, 4676–4683 CrossRef.
- Y. Chen, M. S. Lord, A. Piloni and M. H. Stenzel, Macromolecules, 2015, 48, 346–357 CrossRef CAS.
- M. Nagao, Y. Fujiwara, T. Matsubara, Y. Hoshino, T. Sato and Y. Miura, Biomacromolecules, 2017, 18, 4385–4392 CrossRef CAS PubMed.
- T. Ishida, M. Nagao, T. Oh, T. Mori, Y. Hoshino and Y. Miura, Chem. Lett., 2022, 51, 308–311 CrossRef CAS.
- E. Zumbro and A. Alexander-Katz, ACS Omega, 2020, 5, 10774–10781 CrossRef CAS PubMed.
- M. Tagaya, Polym. J., 2015, 47, 599–608 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02663a |
| This journal is © The Royal Society of Chemistry 2024 |