
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Materials derived from the human elastin-like polypeptide fusion with an antimicrobial peptide strongly promote cell adhesion†
Laura
Colomina-Alfaro
 a,
Paola
Sist
a,
Paola
D’Andrea
a,
Ranieri
Urbani
b,
Silvia
Marchesan
b,
Artemis
Stamboulis
c and
Antonella
Bandiera
*a
a,
Paola
Sist
a,
Paola
D’Andrea
a,
Ranieri
Urbani
b,
Silvia
Marchesan
b,
Artemis
Stamboulis
c and
Antonella
Bandiera
*a
aDepartment of Life Sciences, University of Trieste, 34127 Trieste, Italy. E-mail: abandiera.units.it
bDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, 34127 Trieste, Italy
cSchool of Metallurgy and Materials, Biomaterials Research Group, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 17th July 2024
Abstract
Protein and peptide materials have attracted great interest in recent years, especially for biological applications, in light of their possibility to easily encode bioactivity whilst maintaining cytocompatibility and biodegradability. Heterologous recombinant expression to produce antimicrobial peptides is increasingly considered a convenient alternative for the transition from conventional methods to more sustainable production systems. The human elastin-like polypeptide (HELP) has proven to be a valuable fusion carrier, and due to its cutting-edge properties, biomimetic materials with antimicrobial capacity have been successfully developed. In this work, we have taken advantage of this platform to produce a difficult-to-synthesise sequence as that of the human β-defensin 1 (hBD1), an amphipathic cationic peptide with structural folding constraints relevant to its bioactivity. In the design of the gene, highly specific endoproteinases recognition sites were introduced to release the active forms of hBD1. After the expression and purification of the new fusion construct, its biological activity was evaluated. It was found that both the fusion biopolymer and the released active forms can inhibit the growth of Escherichia coli in redox environments. Remarkably, 2D and 3D materials derived from the biopolymer showed a strong cell adhesion-promoting activity. These results suggest that HELP represents a multitasking platform that not only facilitates the production of bioactive domains and derived materials but could also pave the way for the development of new approaches to study biological interactions at the molecular level.
1. Introduction
Protein and peptide materials have become very popular over the last decade for a wide variety of biological uses and beyond, thanks to their inherent bioactivity, cytocompatibility, and biodegradability.1–3 In particular, those containing antimicrobial peptides (AMPs) are highly attractive in light of the emergence of antimicrobial resistance against traditional antibiotics, although their design entails a set of challenges to maintain their bioactivity.4,5 Recombinant fusion biotechnology has emerged as a promising platform to produce antimicrobial peptides. Standard chemical synthesis encounters several burdens that lead to increased production timelines and costs. These challenges include the hurdle of synthesising difficult sequences or lengthy peptides, inefficient purification processes due to the primary structure of the AMPs, and the need for the proper folding (e.g., the occurrence of disulfide bridges, post-translational modifications, etc.) to maintain the antimicrobial efficacy.6,7 In contrast, the recombinant production in Escherichia coli offers cost-effectiveness, scalability, and efficient expression systems, resulting in high yields and establishing the basis for large-scale protein production.8 To date, many fusion carriers have been developed and used for AMP production. Nevertheless, the elastin-like polypeptides (ELPs) are still the least exploited.9ELPs are a class of recombinant biopolymers inspired by elastin and therefore endowed with the inverse transition property, a thermo-responsive behaviour that allows these components to shift from the solvated form to the suspension state, depending on the temperature of the environment.10
The human elastin-like polypeptide (HELP) fusion carrier developed in our laboratory differs from most other ELPs described in the literature as it possesses the cross-linking domains characteristic of tropoelastin.11 These domains can be cross-linked by an enzymatic reaction, resulting in the formation of cytocompatible hydrogel matrices. This mild process does not affect cell viability, allowing its use for cell encapsulation and culture.12 Notably, the cross-linking domains are also the main target for elastase, an enzyme secreted by activated polymorphonuclear leukocytes during inflammation.13 Consequently, the presence of elastolytic proteases of different origins can trigger the release of any domain fused to HELP, as well as any compound embedded in the gel matrix.14 Recently, a HELP-AMP fusion with the antimicrobial peptide indolicidin has been described. In addition to the successful high-yield expression, the HELP-AMP fusion product retained both the HELP thermo-responsive properties and the AMP activity, demonstrating the advantage of using this carrier for the production of AMP domains.15
The human β-defensin 1 (hBD1) belongs to the defensins family, a class of small, cationic antimicrobial peptides with a well-defined tertiary structure stabilised by three specific disulfide bridges that are conserved among all β-defensins.16 hBD1 is constitutively expressed and produced by several types of epithelial cells, such as those of the urinary tract, pancreatic duct, respiratory tract, and intestine, as well as by keratinocytes.17 Recent evidence suggests that the members of the β-defensin family are multifunctional peptides that not only have functions related to host defence,18,19 but are also involved in cell proliferation and migration,20 wound healing, tissue regeneration,21,22 and tumour inhibition,23 positioning them as attractive components for translational applications. hBD1 plays a critical role in the innate immune system's defence against microbial infections and constitutes one of the body's first lines of defence against pathogens. It is well-known for its broad-spectrum antimicrobial activity against various bacteria, fungi, and viruses.24 However, the length of the sequence, the presence of six cysteine residues essential for the correct folding, as well as the need to increase hBD1 availability to produce multifunctional and biomimetic materials are the main challenges for its production by chemical synthesis. This has led us to employ the HELP carrier for the recombinant expression of hBD1 as an alternative and more sustainable route for the synthesis of this peptide.
This work describes the design, the production, the biochemical characterisation of the HELP-hBD1 fusion biopolymer (HhBD1) and the derived hBD1 peptides, as well as the biological assessment of their activity towards model microorganisms and cell lines. Derived materials such as coatings and matrices based on this new fusion biopolymer are evaluated for their interaction with biological systems.
2. Results and discussion
2.1 Production and characterisation of the HELP recombinant fusion with hBD1
hBD1 was employed to functionalise HELP to develop biomimetic components with antimicrobial properties. It has been described that this AMP is physiologically synthesised as a 68-residue prepropeptide (GenBank: CAA63405.1), the product of the DEFB1 gene.25 However, this product is further processed to originate functional domains ranging from 36 to 47 amino acids, which differ from each other by N-terminal truncation.26 Two major fragments endowed with antimicrobial activity have been described; one of 47 amino acids,27,28 and another one of 36 amino acids, which is described as the most active domain against bacterial cells.29,30 For this reason, the fragment of 47 amino acids was selected as the fusion partner at the C-terminus of the HELP carrier (Fig. 1). Remarkably, this last sequence does not contain any acidic residue.11 Thus, the unique aspartic acid at the beginning of the 36-residue hBD1 domain represents a specific target for the Asp-N endoproteinase, allowing the release of this active peptide from the recombinant fusion biopolymer. Following the same strategy, the addition of a triplet encoding a glutamic acid just before the open reading frame of the 47-residue hBD1 domain resulted in the presence of a second specific cleavage site, in this case for the Glu-C endoproteinase, as schematically shown in Fig. 1.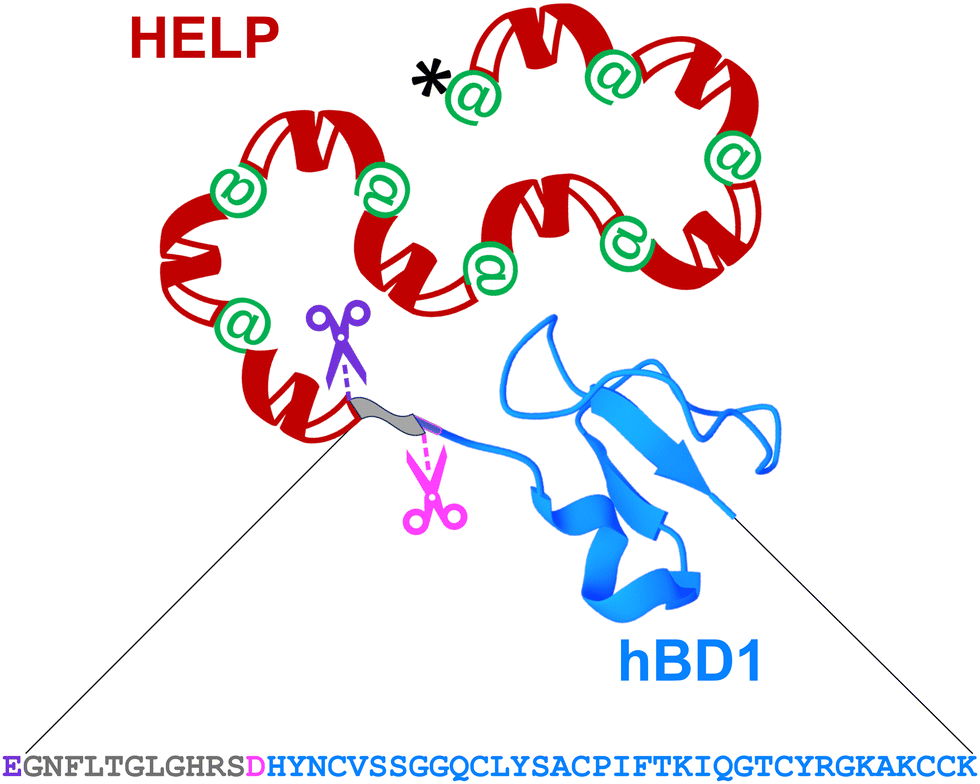 | ||
| Fig. 1 Schematic representation of the recombinant HhBD1 fusion biopolymer. The hBD1 sequence (grey-blue, 47 amino acids) is fused at the C-terminus of HELP (black, his-tag; green, cross-linking domains; red, hydrophobic domains of the human elastin). The hBD1 sequence is flanked by a glutamic acid residue, the specific proteolytic cleavage site for Glu-C (purple scissors). The 36 amino acids domain (blue) can be obtained by cleavage with the Asp-N specific protease (pink scissors). The image of the hBD1 fusion domain was generated with Mol*Viewer31 from the PDB entry 1E4S.32 | ||
After cloning, the fusion biopolymer, named HhBD1 (HELP-hBD1, MW 50694.45 Da), was expressed and produced in E. coli following a recombinant approach and purified by the inverse transition cycle, taking advantage of the thermo-responsive behaviour of the elastin domain, which is well described for the purification of ELP and ELP-based biopolymers (Fig. S1, ESI†).33,34 On average, more than 250 mg of pure HhBD1 were obtained per litre of bacterial culture using our lab-scale production system, which is in agreement with yields reported for other ELP-AMP fusions.35 Electrophoretic analysis of the pure HhBD1 (Fig. S1, lane 3, ESI†) showed the presence of additional bands that exhibited slower migration than that expected for the HhBD1 monomer, suggesting the presence of multimeric HhBD1 forms due to the formation of interchain disulfide bridges given that the primary structure of hBD1 contains six cysteine residues. The stability of the fusion protein at different pH values (see ESI,† Table S1) and the susceptibility to proteolysis by elastase were tested, and results are reported in Fig. S2 and S3 (ESI†). The HELP biopolymer was unaffected by both acidic and basic conditions. Intriguingly, prolonged incubation of HhBD1 in basic conditions resulted in the formation of a hydrogel-like layer on the bottom of the recipient with the consequent decrease in the concentration of HhBD1 in the supernatant.
As described above, the primary sequence of the HhBD1 biopolymer contains unique glutamic and aspartic acid residues, which are the recognition sites for highly specific endoproteinases. Therefore, the purified HhBD1 was treated with Glu-C and Asp-N to verify the release of the two hBD1 fragments (5069.42 Da and 3934.57 Da, respectively). The electrophoretic analysis (Fig. 2) showed that after the endoproteolytic reactions, most of the HhBD1 signal disappeared, accompanied by the formation of two new bands. The most prominent one migrated at the same level of the HELP band (about 50 kDa) and the other one ran along with the electrophoretic front (Fig. 2, boxed in purple), suggesting that the cleavage converted most of HhBD1 into HELP and hBD1. Both endoproteolytic reactions were performed on HELP as control and no effect on this biopolymer was observed, as expected (Fig. 2).
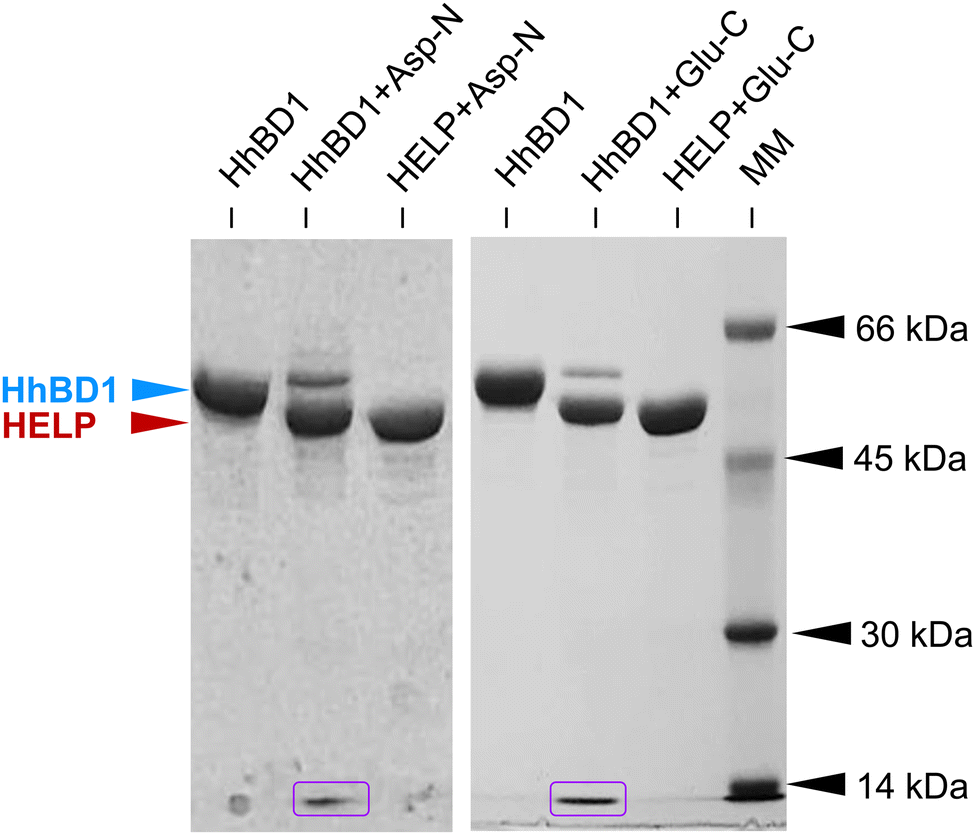 | ||
| Fig. 2 Representative image of the 10% SDS-PAGE analysis of Glu-C and Asp-N specific cleavage of HhBD1 and HELP biopolymers. The electrophoretic signals corresponding to HhBD1 (blue arrow), HELP (red arrow), and the released hBD1 domains (boxed in purple) are indicated. Molecular mass markers (MM): bovine serum albumin, 66 kDa; ovalbumin, 45 kDa; carbonic anhydrase, 30 kDa and lysozyme, 14 kDa. Coomassie blue staining. | ||
Turbidimetry analysis of the pure fusion biopolymer showed that, although no temperature-dependent transition was observed when the biopolymer was dissolved in buffer alone (Fig. 3, blue open symbols), in the presence of a nearly physiological concentration of NaCl, HhBD1 showed a sharper transition at a lower temperature (Tt 28 °C) compared to HELP under the same conditions (Tt 37.3 °C) (Fig. 3, compare red filled with blue filled symbols). This effect was already observed when HELP was fused with indolicidin.15 It has been described that the fused domains, when folded, may act providing a surface in close proximity to the carrier and that the surface properties, rather than the overall hydrophobicity of the domain, play a dominant role in modulating the Tt of elastin-like carriers.36 hBD1 exhibits an amphipathic conformation endowed with a predominantly apolar surface area (see ESI,† Table S2). This is consistent with the observed thermo-responsive behaviour of this HELP fusion biopolymer, which exhibits a lower Tt than the HELP carrier alone.
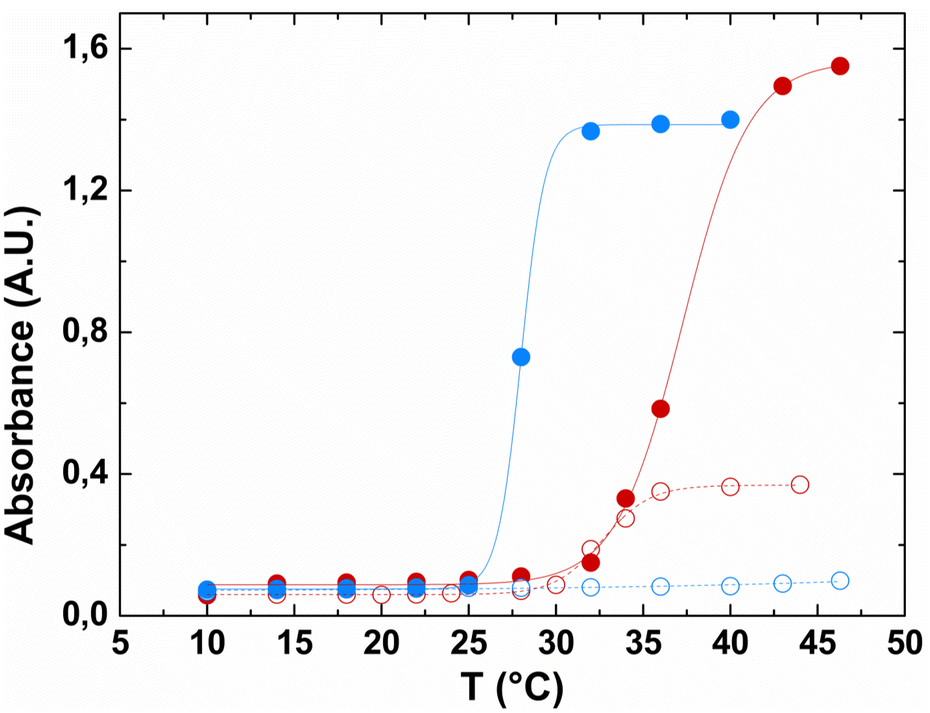 | ||
| Fig. 3 Turbidimetric analysis of the HELP and HhBD1 biopolymers at 2 mg mL−1 as a function of temperature. HELP (red symbols) and HhBD1 (blue symbols) were dissolved in 10 mM Tris (open symbols) and 10 mM Tris/0.15 mM NaCl (filled symbols) buffers, pH 8. | ||
These data showed the successful expression of the HhBD1 fusion biopolymer, which retained the thermo-responsive behaviour of HELP and displayed the expected cleavage pattern established in the design of the synthetic gene.
2.2 Biological activity evaluation of the HhBD1 biopolymer
After the production of purified HhBD1 exploiting the thermo-responsive behaviour of the HELP carrier, the first concern was to verify whether the fusion biopolymer retained the antimicrobial properties of the hBD1 domain. Although it was reported that the minimum inhibitory concentration of hBD1 (MIC) against Staphylococcus aureus and E. coli strains was 4–8 mg L−1 and 16–32 mg L−1, respectively,30 we could not detect any activity of HhBD1 following this method. Many reasons withstand behind this observation, especially the uncontrolled redox state of the fusion biopolymer in which the disulfide bridge formation is uncontrolled and unspecific and cannot be blocked due to the recombinant nature of HhBD1, and the impossibility of performing MIC assays adding reducing agents to the media, as these are toxic to the bacterial cells in solution. It has been described that the presence of cysteines is crucial for the antimicrobial activity of hBD1, especially the C-terminal cysteines, and that its antimicrobial capacity is closely related to the reduction of the disulfide bridges, and thus it can be evidenced in reducing environments.29 Following a modification of the radial diffusion assay described by these authors, in the presence of 2 mM DTT, we were able to detect the antimicrobial activity for the HhBD1 fusion biopolymer towards E. coli ATCC 25922 strain when 100 μg of HhBD1 were deposited (containing approximately 10 μg of hBD1). In contrast, no inhibition halos were observed in the absence of the reducing agent (see ESI,† Fig. S4). This activity was also retained after the cleavage of HhBD1 with Glu-C and Asp-N, and no significant differences were observed when comparing the diameters of the inhibition zone of the digestion mixtures with those of the fusion biopolymer (Fig. 4). HELP alone did not show any antimicrobial activity in any of the tested conditions (see ESI,† Fig. S1B and C), confirming that the antimicrobial activity of the HhBD1 fusion biopolymer is due to the presence of the hBD1 domain. | ||
| Fig. 4 Antimicrobial activity of HhBD1. (A) Radial diffusion assay performed in the presence of 2 mM DTT by incubating E. coli ATCC 25922 strain with the untreated HhBD1 biopolymer and with HhBD1 cleaved with Glu-C and Asp-N. Lysozyme was used as the positive control. (B) The diameter of the inhibition zones was measured and statistically analysed using a one-way ANOVA test. No significant differences (n.s.) were found with p < 0.05. | ||
The effect of HhBD1 on the viability of eukaryotic cells was tested using the WST-1 viability assay, in which the metabolic activity of osteoblastic (Fig. 5(A)) and fibroblastic (Fig. 5(B)) cells in the presence of different concentrations of HhBD1 and HELP was assessed. The viability assays did not evidence cytotoxicity on the tested cell lines. Rather, as already reported for HELP,37 they revealed a moderate pro-proliferative effect of both biopolymers.
 | ||
| Fig. 5 Viability of (A) MG-63 osteoblast and (B) NIH-3T3 fibroblast cell lines treated with HELP (red bars) and HhBD1 (blue bars) biopolymers at different concentrations. Values were normalised to the untreated control culture (black dotted lines). | ||
The evaluation of the biological activity of the HhBD1 biopolymer revealed that the fusion construct retained the antimicrobial activity, depending on the redox condition of the environment, as already described for hBD1. Furthermore, no cytotoxic effects on eukaryotic cells were detected at any of the concentrations tested (up to 500 μg mL−1).
Taken together, these data point to HhBD1 as a non-cytotoxic candidate component to produce biomaterials and composites endowed with antimicrobial activity that can be specifically triggered in a reducing environment.
2.3 HhBD1-based 3D matrix production and characterisation
The presence of cross-linking domains in the HELP carrier has been demonstrated as a valuable prerequisite for preparing hydrogel matrices endowed with biological functionality.15 Following an already described enzymatic method,12 the matrix-forming capacity of HhBD1 was explored and compared with that of HELP. 4% (w/v) aqueous solutions of HhBD1 in the presence of transglutaminase showed a capacity to form a porous, spongy, hydrogel-like matrix indistinguishable from that of HELP, as shown by scanning electron microscopy (SEM) analysis (Fig. 6(A)).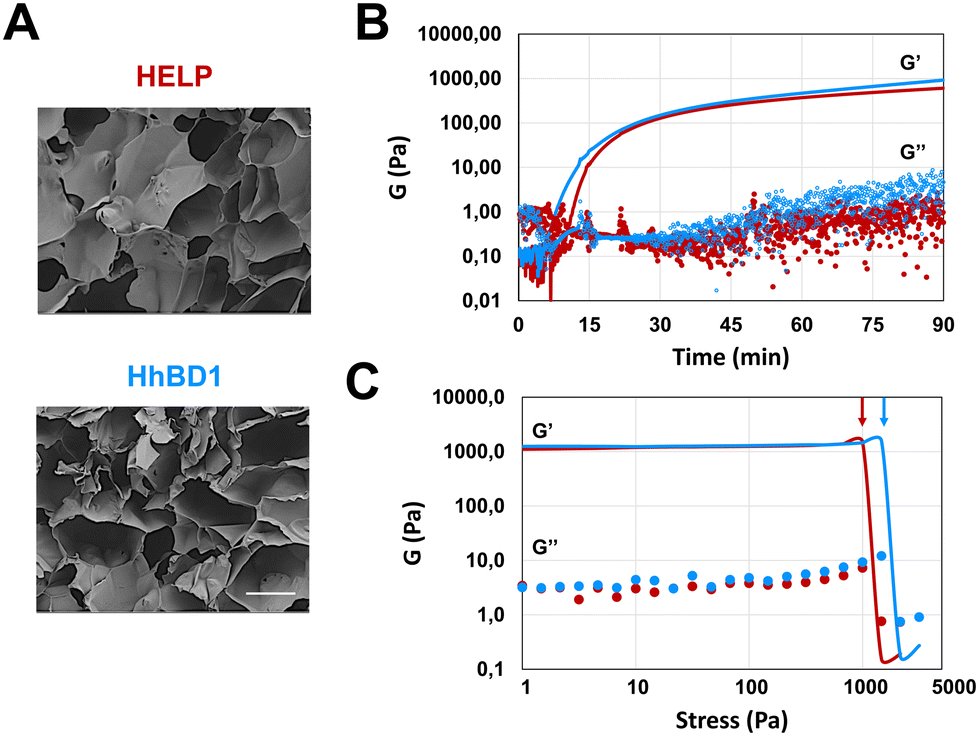 | ||
| Fig. 6 4% HELP and HhBD1 hydrogel matrices. (A) SEM analysis of the freeze-dried elastin-based hydrogel matrices. The bar is 40 μm. (B) Oscillatory rheological time sweep and (C) frequency sweep analyses of HELP (red) and HhBD1 (blue) matrices. G′, elastic or storage modulus; G′′, viscous or loss modulus. | ||
Oscillatory rheology was performed on 4% biopolymer solutions containing 2 μg mL−1 transglutaminase to follow the kinetics of the cross-linking reaction (Fig. 6). Although the gelation point is similar for the HELP and HhBD1 systems, the storage modulus of HhBD1 is anticipated compared to HELP, indicating faster enzyme kinetics (Fig. 6(B)). This could be due to the presence of additional glutamine and several lysine residues in the hBD1 domain which may be randomly involved in the cross-linking reaction, or higher solvent exposure of the cross-linking sites for HhBD1 relative to HELP. Both samples displayed elastic (G′) and viscous (G′′) moduli reaching a plateau after about 90 minutes, resulting in very similar storage moduli (1.0 ± 0.1 kPa) for both matrices. This fact indicates that the HELP cross-linking domains in both biopolymers are the main target of the transglutaminase rather than the hBD1 domain. It is worth noting that the presence of the hBD1 domain lowered the Tt of the HhBD1 fusion biopolymer (Fig. 2(B)), likely favouring the proximity of the HhBD1 chains and resulting in a more efficient cross-linking process, which is in agreement with the faster gelation kinetics. Both HELP and HhBD1 samples formed strong gels, with elastic moduli that were over two orders of magnitude higher than the viscous moduli. The stress sweep analyses supported the main findings of the time sweep tests, with both gels showing a remarkably wide linear viscoelastic range (up to nearly 1 kPa) and with the fracture point of the HELP matrix being slightly anticipated at 1.2 ± 0.3 kPa (Fig. 6(C), red arrow) compared to that of the HhBD1 gel at 1.8 ± 0.5 kPa (Fig. 6(C), blue arrow), suggesting more cross-linking points in the latter. The stability and nature of the hydrogels were further confirmed by frequency sweep analyses, which showed that the storage and loss moduli of both biopolymers were not affected by the applied frequency (see ESI,† Fig. S5). In summary, SEM and oscillatory rheology analyses evidenced that the HELP and HhBD1 matrices are comparable, although the addition of the antimicrobial domain slightly increased the cross-linking points and strength of the HhBD1 matrix.
The ability of HhBD1 to form a matrix was used as a convenient method to verify the specificity of the endoproteolytic cleavage by Glu-C and Asp-N. As described in the ESI,† each enzymatic reaction was performed on the matrix to cleave the fragments, which became soluble and were then recovered in the supernatant. The supernatant from each reaction was analysed by ESI-MS without further purification. The masses detected were consistent with those calculated for the two expected fragments, confirming the correctness of the amino acid sequence of the peptides and the high specificity of the reactions (see ESI,† Fig. S6A and B for Glu-C and Asp-N released fragments, respectively). Furthermore, these results confirmed the versatility of HELP fusion and the derived matrix to produce bioactive peptides.
2.4 Cell culture on HhBD1-derived substrates
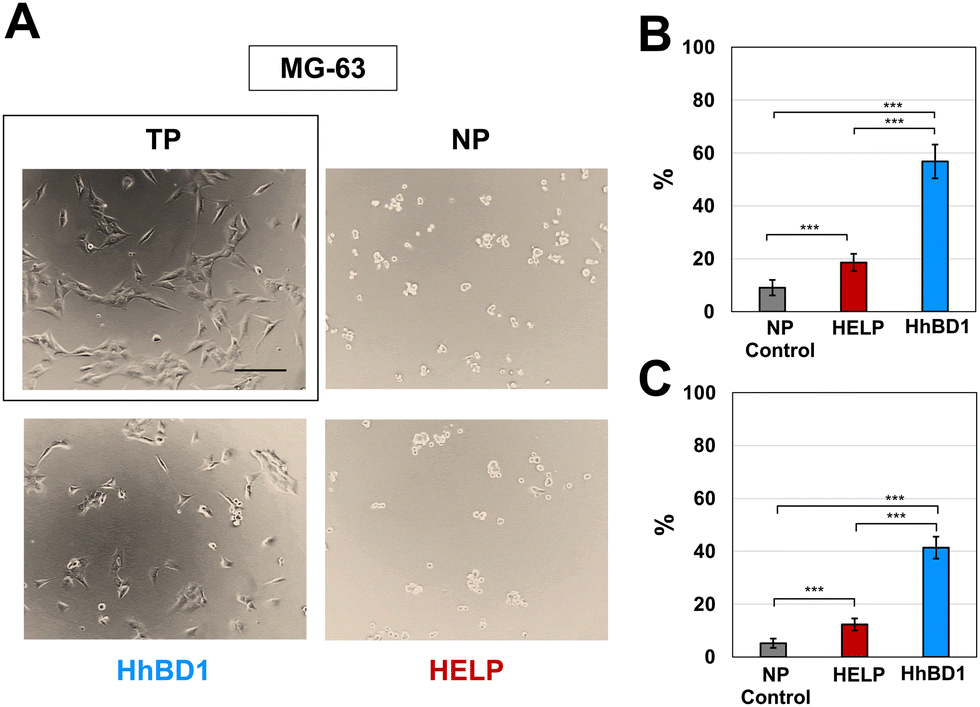 | ||
| Fig. 7 HhBD1 promoted the adhesion of osteoblastic cells. (A) Representative contrast phase microscopy images of MG-63 cultures on HELP- and HhBD1-coated NP surfaces. Cells cultured on untreated NP and TP (boxed) surfaces were the negative and positive controls for cell adhesion, respectively. (B) Adhesion assay using crystal violet staining and (C) viability assay of the attached cells. Values were normalised to the control cell culture on the TP surface and statistically evaluated using a one-way ANOVA test, ***p < 0.001. The bar is 200 nm. | ||
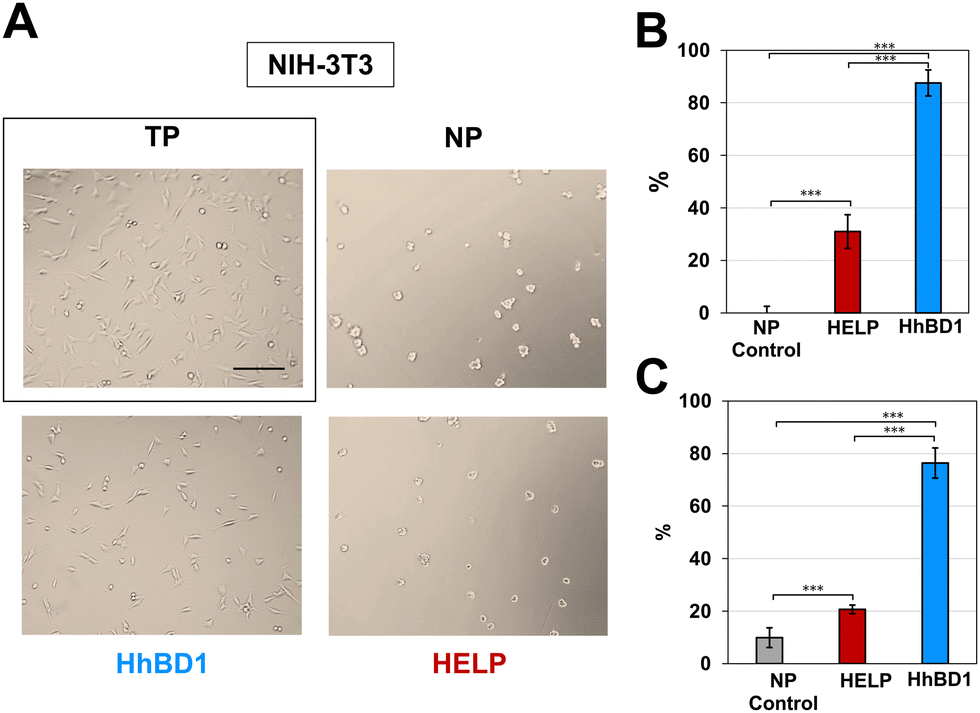 | ||
| Fig. 8 HhBD1 promotes the adhesion of fibroblastic cells. (A) Representative contrast phase microscopy images of NIH-3T3 cultures on HELP- and HhBD1-coated NP surfaces. Cells cultured on untreated NP and TP (boxed) surfaces were the negative and positive controls for cell adhesion, respectively. (B) Adhesion assay using crystal violet staining and (C) viability assay of the attached cells. Values were normalised to the control cell culture on the TP surface and statistically evaluated using a one-way ANOVA test, ***p < 0.001. The bar is 200 nm. | ||
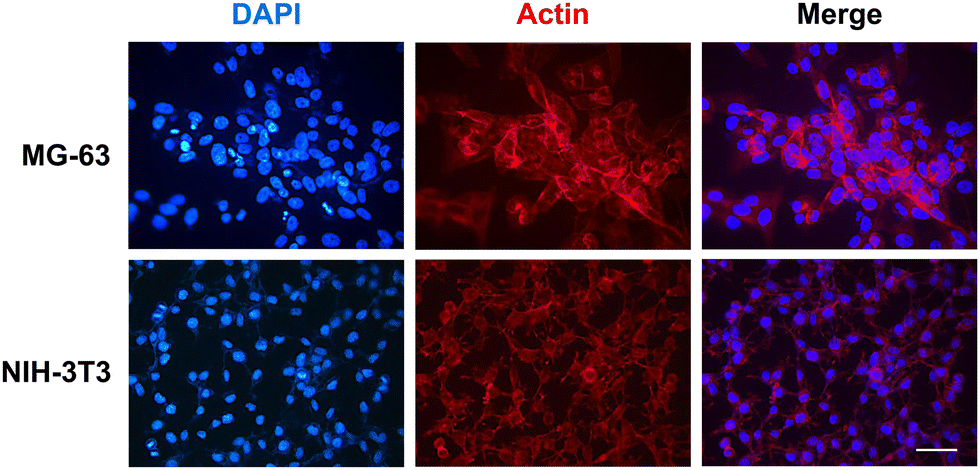
To further confirm these observations, thin-film coatings on NP were prepared with HELP and with HhBD1 to ensure a controlled coverage of the cell seeding surfaces with the two biopolymers (see Experimental Section 4.4). Fluorescence analysis was performed on the two cell lines cultured on the thin-films (Fig. 9(A) and (B)). To better highlight the pro-adhesive effect, cells were analysed 5 hours after seeding to prevent them from producing their extracellular matrix and starting to adhere to the substrate. Actin filaments were stained with Alexa Fluor 594 phalloidin, and nuclei were counterstained with DAPI. Consistently with previous results, only a few cells showed adhesion to the NP surface and to the HELP thin-film, whereas the HhBD1 thin-film coating strongly promoted cell adhesion to a level comparable to the culture on the TP control surface (Fig. 9(A) and (B)). DAPI staining further evidenced the difference between cultures on HELP and HhBD1 (see ESI,† Fig. S4). These results showed that the presence of the hBD1 domain on the surface has a robust pro-adhesive effect. In both cell lines, the morphology of the cells cultured on HhBD1 thin-films resembled those cultured on the control TP surface (Fig. 9). The cells cultured on the HhBD1 substrate showed highly organised actin meshwork. A slightly lower degree of spreading with respect to the cells cultured on control TP was observed. However, this could be related to irregularities on the surface of the thin-film as well as an uneven distribution of hBD1 on the surface. The cells cultured on HELP showed a different morphology, remaining rounded and showing a tendency to aggregate rather than spread, resembling the cultures on the untreated NP substrate. The DAPI staining emphasised the difference between the cultures on HELP and HhBD1 (see ESI,† Fig. S7).
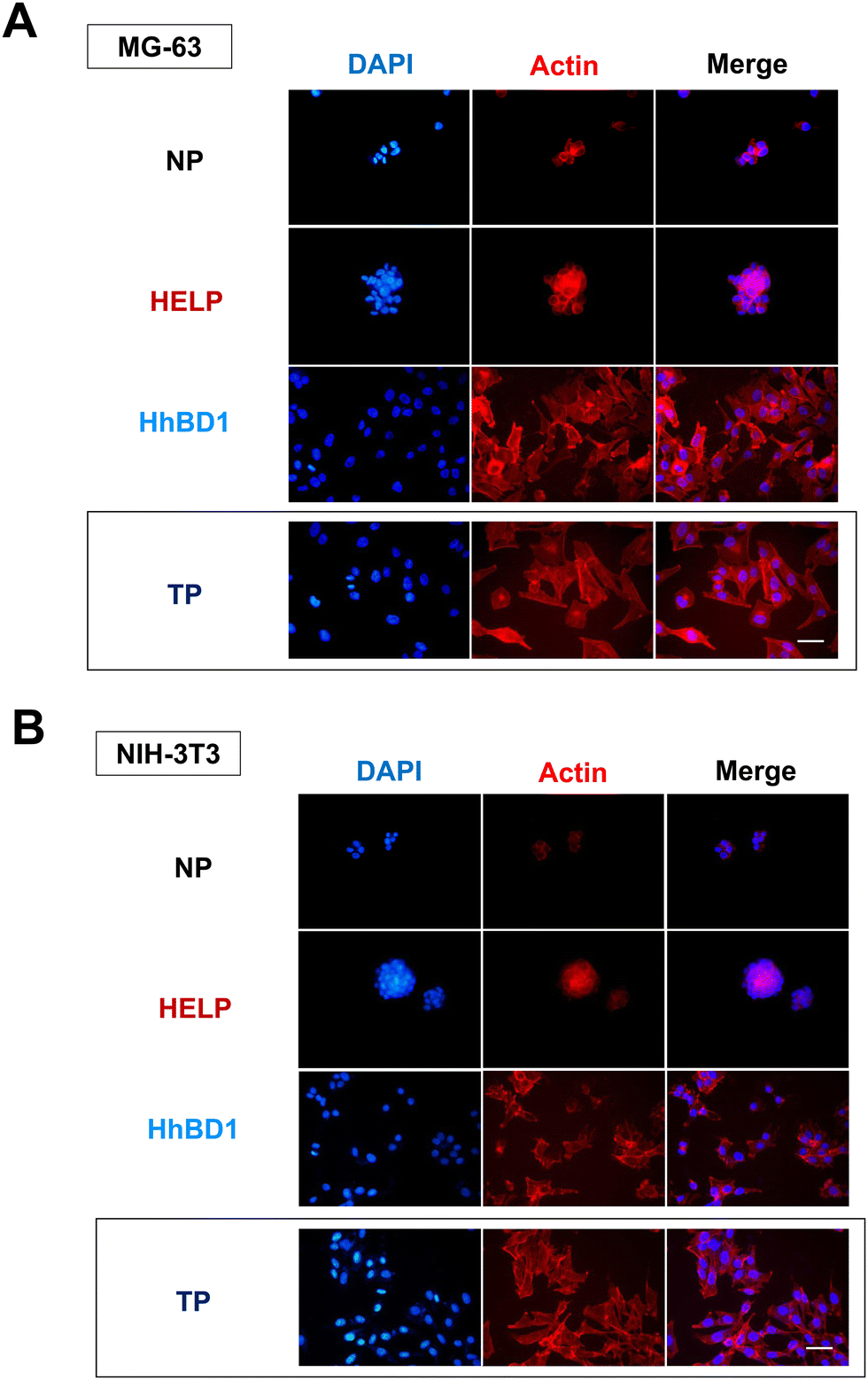 | ||
| Fig. 9 Fluorescence microscopy analysis of (A) MG-63 osteoblast and (B) NIH-3T3 fibroblast cell lines grown on HhBD1 and HELP thin-film NP coated surfaces. F-actin was labelled with Alexa Fluor 594 phalloidin, and nuclei were counterstained with DAPI. The bar is 50 μm. | ||
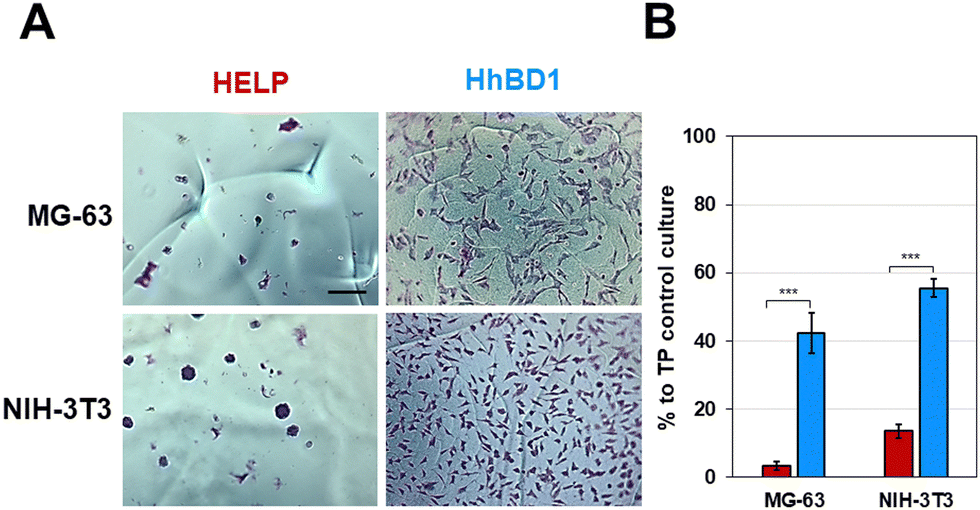 | ||
| Fig. 10 HhBD1 hydrogel matrices supported cell adhesion of osteoblast and fibroblast. (A) Representative contrast phase images of cells cultured on the hydrogel matrices and stained with toluidine blue. (B) Viability assay of cells cultured on the hydrogel matrices. Values were normalised to the TP cell growth control and statistically evaluated using a one-way ANOVA test, ***p < 0.001. The bar is 200 nm. | ||
Fluorescence microscopy analysis was carried out to examine the morphology and cytoarchitecture of the cells cultured on these matrices (Fig. 11 and Fig. S8, ESI†). Only few cell nuclei were visualised on the HELP matrix (Fig. S8B, ESI†), whereas on the HhBD1 matrix, the presence of a high number of stained nuclei was evident, indicating a robust cell adhesion of both osteoblast and fibroblast cells (Fig. S8B, ESI†). Both cell lines cultured on the HhBD1 matrices exhibited cells with a well-organised actin meshwork (Fig. 11), albeit cell dimensions appeared reduced to some extent compared to cells cultured on the standard TP surface (Fig. 9). This slight reduction in spreading may be due to the heterogeneous and irregular surface of the matrix as well as to the different surface stiffness respect that of the TP surface. However, cells were firmly attached and displayed a properly shaped cytoskeleton (Fig. 11).
 | ||
| Fig. 11 Fluorescence microscopy analysis of MG-63 and NIH-3T3 cells cultured on HhBD1 hydrogel matrices. F-actin was labelled with Alexa Fluor 594 phalloidin, and nuclei were counterstained with DAPI. The bar is 50 μm. | ||
These data, consistent with previous observations, confirmed that the presence of the hBD1 domain conferred to the HELP matrix a strong signalling effect for cell attachment towards both the tested lines. Interestingly, in contrast to the antimicrobial activity, the pro-adhesive capacity toward eukaryotic cells did not require reducing environmental conditions, suggesting that the peptide conformation may be a crucial factor in modulating its bioactivity. Coatings, thin-films, and hydrogel matrices derived from HhBD1 supported the osteoblasts and fibroblasts adhesion. Overall, these data revealed a pro-adhesive activity of the hBD1 domain, which, to our knowledge, is described for the first time. Although it is known that hBD1, like other β-defensins, is a multifunctional factor,24,41 a pro-adhesive activity has so far only been demonstrated for hBD5.42 Notably, it has recently been described that hBD1 can specifically bind to the pore region of the Kv1.3 potassium channel, leading to conformational changes while retaining the activation properties of the channel itself.43 There is now plenty of evidence that different classes of K+ channels play a role in integrin-dependent adhesion.44 It has been demonstrated that these channels can be physically associated with integrins, such that the conformational change of the channel in turn affects the structure of the adjacent integrin subunit, leading to activation of the latter and cell adhesion.45 Interestingly, these channels are reported to be involved in the regulation of cell size in non-excitable tissues.46 Our results are consistent with these findings and may even point to a possible unexplored mechanism related to cell adhesion.
3. Conclusions
Due to its length and conformation, the hBD1 is considered a difficult-to-synthesise sequence. The HELP carrier has been successfully employed as an alternative and more sustainable system to produce large amounts of this antimicrobial peptide with respect to chemical synthesis. The HhBD1 fusion biopolymer was produced and characterised, verifying the antimicrobial domain(s) release capacity achieved by the appropriate design of the synthetic gene. The analyses confirmed the potential of the new biopolymer, which retained both the thermo-responsive properties and the antimicrobial capacity, holding great promise to produce biocompatible and multifunctional materials. Furthermore, enzymatic crosslinking yielded remarkably strong and stable hydrogels from HhBD1. Unexpectedly, a strong cell adhesion-promoting activity was detected for the surfaces treated with the HhBD1 biopolymer as well as for the derived hydrogel matrix. This finding is consistent with recent data that indicate that hBD1 is a multifaceted factor with still unveiled biological functions and thus harbouring promise for a variety of clinical applications. Despite the development of countless strategies and materials for surface engineering, achieving efficient cell adhesion still represents a challenge. Our data indicates that the novel HhBD1 biopolymer is a versatile component for the fabrication of coatings and scaffolds that promote cell adhesion and that can be safely integrated into biological systems due to their biotic origin. In addition to being an alternative route to produce bioactive peptides and an effective way to obtain materials that directly integrate the functional domain, the HELP platform turned out to be a valuable system to study, by a reductionist approach, the biological interactions that are still unknown or not fully elucidated at the molecular level. The opportunity to embed a functional peptide in a biomimetic moiety, the availability of a suitable inactive control, as well as the possibility to choose between different configurations, such as the liquid solution and 2D or 3D material settings, are the main features of a powerful tool to decipher the complexity of biological mechanisms.4. Experimental
4.1 Production of the recombinant HhBD1 fusion biopolymer
4.2 Antimicrobial and biological evaluation of HhBD1
4.3 HhBD1 3D matrix production and characterisation
4.4 Cell culture on HhBD1-based substrates
Thin-film coatings (100 μg of biopolymer per cm2) were prepared depositing 10 μl (1.25 μg μL−1) of HhBD1 or HELP sterile aqueous solution (0.22 μm filtration) on the bottom (0.125 cm2) of the microwells of a μ-slide (15-Wells uncoated μ-Slides 3D, #81506, IBIDI, Grafelfing, Germany) and then air-dried under a sterile hood at room temperature.
All samples were washed three times with an excess of PBS, and cells were fixed by incubation with 4% (v/v) paraformaldehyde in PBS for 15 minutes at room temperature. Samples were then washed with PBS and blocked for 10 minutes with a solution containing 5% normal goat serum and 0.1% Triton X-100 in PBS. Then, 15 μL of blocking solution containing 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Missouri, USA) and Alexa Fluor 594 phalloidin (Molecular Probes, Eugene, USA) at a dilution of 100 ng mL−1 and 0.53 U mL−1, respectively were added to each sample and incubated at 4 °C for 2 hours. After washing three times for 10 minutes with 0.1% Triton X-100 in PBS, the μ-slide lid was sealed, and the coverslips were mounted on glass slides. Fluorescently labelled cells were visualised using a fluorescence microscope (Leica DMLS), and images were acquired with a Leica DFC450 C camera coupled to a Leica LAS v4.13 acquisition software (Leica Microsystems, Wetzlar, Germany). Image cropping, superimposition, and analysis were performed using ImageJ software.48
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Sabina Passamonti for her invaluable support and Dr Marco Stebel, Dr Davide Porrelli, and Dr Fabio Hollan for technical assistance. This work was supported by the H2020 Marie Sklodowska-Curie Action (AIMed project, grant no. 861138), the Horizon Europe STOP project, grant no. 101057961, and the iNEST (Interconnected North-Est Innovation Ecosystem) consortium funded by the European Union NextGenerationEU (PNRR). S.M. acknowledges funding from the Italian Ministry of University and Research through the PRIN program (SHAZAM grant no. 2022XEZK7K) funded by the European Union – Next Generation EU.References
- A. C. Conibear, A. Schmid, M. Kamalov, C. F. W. Becker and C. Bello, Curr. Med. Chem., 2020, 27, 1174–1205 CrossRef CAS PubMed.
- P. J. Jervis, C. Amorim, T. Pereira, J. A. Martins and P. M. T. Ferreira, Soft Matter, 2020, 16, 10001–10012 RSC.
- Y. Yuan, Y. Shi and H. S. Azevedo, Multifunctional Hydrogels for Biomedical Applications, 2022, pp. 97–126 Search PubMed.
- A. Rai, R. Ferrão, P. Palma, T. Patricio, P. Parreira, E. Anes, C. Tonda-Turo, M. C. L. Martins, N. Alves and L. Ferreira, J. Mater. Chem. B, 2022, 10, 2384–2429 RSC.
- A. Panjla, G. Kaul, S. Chopra, A. Titz and S. Verma, ACS Chem. Biol., 2021, 16, 2731–2745 CrossRef CAS PubMed.
- S. Deo, K. L. Turton, T. Kainth, A. Kumar and H. J. Wieden, Biotechnol. Adv., 2022, 59, 107968 CrossRef CAS PubMed.
- T. Vargues, G. J. Morrison, E. S. Seo, D. J. Clarke, H. L. Fielder, J. Bennani, U. Pathania, F. Kilanowski, J. R. Dorin, J. R. Govan, C. L. Mackay, D. Uhrín and D. J. Campopiano, Protein Pept. Lett., 2009, 16, 668–676 CrossRef CAS PubMed.
- Y. Li, Protein Expression Purif., 2011, 80, 260–267 CrossRef CAS PubMed.
- D. Wibowo and C. X. Zhao, Appl. Microbiol. Biotechnol., 2019, 103, 659–671 CrossRef CAS PubMed.
- R. Lin, S. Wang and W. Liu, Curr. Pharm. Des., 2018, 24, 3008–3013 CrossRef CAS PubMed.
- A. Bandiera, A. Taglienti, F. Micali, B. Pani, M. Tamaro, V. Crescenzi and G. Manzini, Biotechnol. Appl. Biochem., 2005, 42, 247–256 CrossRef CAS PubMed.
- A. Bandiera, Enzyme Microb. Technol., 2011, 49, 347–352 CrossRef CAS PubMed.
- A. Bandiera, A. Markulin, L. Corich, F. Vita and V. Borelli, Biomacromolecules, 2014, 15, 416–422 CrossRef CAS PubMed.
- L. Corich, M. Busetti, V. Petix, S. Passamonti and A. Bandiera, J. Biotechnol., 2017, 255, 57–65 CrossRef CAS PubMed.
- L. Colomina-Alfaro, P. Sist, S. Marchesan, R. Urbani, A. Stamboulis and A. Bandiera, Macromol. Biosci., 2023, e2300236 Search PubMed.
- D. M. Hoover, O. Chertov and J. Lubkowski, J. Biol. Chem., 2001, 276, 39021–39026 CrossRef CAS PubMed.
- E. Prado-Montes de Oca, Int. J. Biochem. Cell Biol., 2010, 42, 800–804 CrossRef CAS PubMed.
- F. Semple and J. R. Dorin, J. Innate Immun., 2012, 4, 337–348 CrossRef CAS PubMed.
- J. R. Shelley, D. J. Davidson and J. R. Dorin, Front. Immunol., 2020, 11, 1176 CrossRef CAS PubMed.
- H. Sugiwaki, M. Kotani, A. Fujita and S. Moriwaki, J. Cosmet., Dermatol., 2024, 23, 676–680 CrossRef PubMed.
- M. Takahashi, Y. Umehara, H. Yue, J. V. Trujillo-Paez, G. Peng, H. L. T. Nguyen, R. Ikutama, K. Okumura, H. Ogawa, S. Ikeda and F. Niyonsaba, Front. Immunol., 2021, 12, 712781 CrossRef CAS PubMed.
- G. Li, Q. Wang, J. Feng, J. Wang, Y. Wang, X. Huang, T. Shao, X. Deng, Y. Cao, M. Zhou and C. Zhao, Biomed. Pharmacother., 2022, 155, 113694 CrossRef CAS PubMed.
- C. Q. Sun, R. S. Arnold, C. L. Hsieh, J. R. Dorin, F. Lian, Z. Li and J. A. Petros, Cancer Biol. Ther., 2019, 20, 774–786 CrossRef CAS PubMed.
- Á. H. Álvarez, M. Martínez Velázquez and E. Prado Montes de Oca, Int. J. Biochem. Cell Biol., 2018, 104, 133–137 CrossRef PubMed.
- L. K. Ryan and G. Diamond, Viruses, 2017, 9, 153 CrossRef PubMed.
- E. V. Valore, C. H. Park, A. J. Quayle, K. R. Wiles, P. B. McCray, Jr. and T. Ganz, J. Clin. Invest., 1998, 101, 1633–1642 CrossRef CAS PubMed.
- K. Midorikawa, K. Ouhara, H. Komatsuzawa, T. Kawai, S. Yamada, T. Fujiwara, K. Yamazaki, K. Sayama, M. A. Taubman, H. Kurihara, K. Hashimoto and M. Sugai, Infect. Immun., 2003, 71, 3730–3739 CrossRef CAS PubMed.
- A. Bolatchiev, V. Baturin, I. Bazikov, A. Maltsev and E. Kunitsina, Fundam. Clin. Pharmacol., 2020, 34, 102–108 CrossRef CAS PubMed.
- B. O. Schroeder, Z. Wu, S. Nuding, S. Groscurth, M. Marcinowski, J. Beisner, J. Buchner, M. Schaller, E. F. Stange and J. Wehkamp, Nature, 2011, 469, 419–423 CrossRef CAS PubMed.
- A. Bolatchiev, PeerJ, 2020, 8, e10455 CrossRef PubMed.
- D. Sehnal, S. Bittrich, M. Deshpande, R. Svobodová, K. Berka, V. Bazgier, S. Velankar, S. K. Burley, J. Koča and A. S. Rose, Nucleic Acids Res., 2021, 49, W431–w437 CrossRef CAS PubMed.
- F. Bauer, K. Schweimer, E. Klüver, J. R. Conejo-Garcia, W. G. Forssmann, P. Rösch, K. Adermann and H. Sticht, Protein Sci., 2001, 10, 2470–2479 CAS.
- D. E. Meyer and A. Chilkoti, Nat. Biotechnol., 1999, 17, 1112–1115 CrossRef CAS PubMed.
- A. K. Varanko, J. C. Su and A. Chilkoti, Annu. Rev. Biomed. Eng., 2020, 22, 343–369 CrossRef CAS PubMed.
- L. Colomina-Alfaro, S. Marchesan, A. Stamboulis and A. Bandiera, Biotechnol. Bioeng., 2023, 120, 323–332 CrossRef CAS PubMed.
- K. Trabbic-Carlson, D. E. Meyer, L. Liu, R. Piervincenzi, N. Nath, T. LaBean and A. Chilkoti, Protein Eng., Des. Sel., 2004, 17, 57–66 CrossRef CAS PubMed.
- P. D’Andrea, D. Scaini, L. Ulloa Severino, V. Borelli, S. Passamonti, P. Lorenzon and A. Bandiera, Biomaterials, 2015, 67, 240–253 CrossRef PubMed.
- A. Bandiera, R. Urbani and P. Sist, Annu. Int. Conf. IEEE Eng. Med. Biol. Soc., 2010, 2010, 819–822 Search PubMed.
- A. Bandiera, L. Colomina-Alfaro, P. Sist, G. Gomez d’Ayala, F. Zuppardi, P. Cerruti, O. Catanzano, S. Passamonti and R. Urbani, Biomacromolecules, 2023, 24, 5277–5289 CrossRef CAS PubMed.
- P. D’Andrea, D. Civita, M. Cok, L. Ulloa Severino, F. Vita, D. Scaini, L. Casalis, P. Lorenzon, I. Donati and A. Bandiera, J. Appl. Biomater. Funct. Mater., 2017, 15, e43–e53 Search PubMed.
- S. K. Ghosh, T. S. McCormick and A. Weinberg, Front. Oncol., 2019, 9, 341 CrossRef PubMed.
- K. Howell and E. de Leeuw, Biochem. Biophys. Res. Commun., 2018, 502, 238–242 CrossRef CAS PubMed.
- J. Feng, Z. Xie, W. Yang, Y. Zhao, F. Xiang, Z. Cao, W. Li, Z. Chen and Y. Wu, Toxicon, 2016, 113, 1–6 CrossRef CAS PubMed.
- A. Becchetti, G. Petroni and A. Arcangeli, Trends Cell Biol., 2019, 29, 298–307 CrossRef CAS PubMed.
- M. Levite, L. Cahalon, A. Peretz, R. Hershkoviz, A. Sobko, A. Ariel, R. Desai, B. Attali and O. Lider, J. Exp. Med., 2000, 191, 1167–1176 CrossRef CAS PubMed.
- M. T. Pérez-García, P. Cidad and J. R. López-López, Am. J. Physiol.: Cell Physiol., 2018, 314, C27–c42 CrossRef PubMed.
- A. Bandiera, Prep. Biochem. Biotechnol., 2010, 40, 198–212 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HhBD1 cloning and purification; stability of HELP and HhBD1; hBD1 accessible surface area calculation, radial diffusion assays; oscillatory rheology analysis; mass spectrometry; fluorescence analyses of cell cultured on coatings and matrices. See DOI: https://doi.org/10.1039/d4tb00319e |
| This journal is © The Royal Society of Chemistry 2024 |