
Collagen fiber-reinforced, tough and adaptive conductive organohydrogel e-skin for multimodal sensing applications†
Zhen
He
ab,
Jialu
Shen
 ab,
Maohua
Lan
ab and
Haibin
Gu
*ab
ab,
Maohua
Lan
ab and
Haibin
Gu
*ab
aKey Laboratory of Leather Chemistry and Engineering of Ministry of Education, Sichuan University, Chengdu 610065, China. E-mail: guhaibinkong@126.com
bNational Engineering Laboratory for Clean Technology of Leather Manufacture, Sichuan University, Chengdu 610065, China
First published on 13th June 2024
Abstract
Conductive hydrogels (CHs) with high sensitivity and multifunctional property are considered as excellent materials for wearable devices and flexible electronics. Surface synapses and internal multilayered structures are key factors for highly sensitive pressure sensors. Nevertheless, current CHs lack environmental adaptability, multifunctional perception, and instrument portability, which seriously hinders their application as sensors. Here, waste collagen fibers (buffing dust of leather), polyvinyl alcohol (PVA) and gelatin (Gel) were used as the basic framework of the hydrogel, loaded with a conductive material (silver nanoparticles (BD-CQDs@AgNPs)) and an anti-freezing moisturizer (glycerol (Gly)), resulting in a multifunctional conductive organohydrogel (BPGC-Gly). As a temperature and humidity sensor, it demonstrated an excellent temperature response range (−20–60 °C) and was capable of rapid response (2.4 s) and recovery (1.6 s) to human breathing. As a strain/pressure sensor, it allowed real-time monitoring of human movement and had a high low-pressure sensitivity (S = 4.26 kPa−1, 0–12.5 kPa). Interestingly, BPGC-Gly could also be used as a portable bioelectrode or the acquisition, monitoring and analysis of EMG/ECG signals. In this work, BPGC-Gly was assembled with wireless transmission to achieve multimodal heath detection, which opens new avenues for multi-responsive CHs, comprehensive human health monitoring and next-generation wearable electronic skin (e-skin).
Introduction
Nowadays, with the development of human health monitoring devices and the rise of the internet of everything (IOE), flexible sensor devices (FSDs) that can simultaneously detect strain/pressure, temperature/humidity, and bioelectric signals are attracting widespread attention for application in wearable devices,1 human–computer interaction systems,2 soft robotics,3 and electronic skin.4 They can simulate the stretchability and tactile sensitivity of human skin, realize the perception of the external environment, and convert mechanical deformation into electrical signals for real-time monitoring.5,6 However, most of FSDs have shortcomings such as poor fatigue resistance, single functionality, poor portability, and contradiction between mechanical strength and toughness, resulting in reduced accuracy and stability of the sensor, and greatly shortened service life.7–9 In recent years, conductive hydrogels (CHs) have become ideal materials for manufacturing FSDs due to their excellent mechanical flexibility, conductivity, wearability, and multi-responsiveness.10,11 However, in practical applications, hydrogel-based flexible sensors inevitably have some limitations. On the one hand, most of the reported hydrogels consist of synthetic precursors with single function and low sensitivity.12 On the other hand, with CHs being hydrophilic gels with a three-dimensional (3D) network structure, their free water can be easily evaporated or frozen, resulting in their poor environmental stability, which hinders the stability and long-term use of FSDs.13,14 Therefore, it is particularly important to develop and prepare low-cost CHs with ultra-sensitivity, multifunctionality, good biocompatibility and environmental stability.Firstly, sensitivity is one of the most critical indicators of CHs. To enhance their sensitivity, current researchers have focused on engineering CHs’ surface structures by techniques such as micro-pattern processing (micro-dome, micro-pyramid arrays and others),15 3D printing,16 pattern photolithography,17 and self-wrinkling.18 For example, Meng et al. used polypyrrole (PPy) and conductive wood to prepare a micro-pattern anti-expansion hydrogel for underwater detection of stethoscope.19 This carefully designed outer surface improves the sensitivity of CHs, but the methods are costly and require specialized equipment. Nevertheless, this approach, constructing CHs with “external protrusions”, is valuable for enhancing external perception. The natural animal biomass materials have attracted the attention of researchers due to their unique structures, such as synapses, natural fibers, pores, etc.20–22 For instance, Wei et al. prepared piezoresistive sensors with oriented multilayer structures from multilevel fiber structures contained in tannery solid wastes (TSWs).23 A unique material, buffing dust (BD), has attracted our attention; it is a leather waste with a composition similar to human skin, with abundant production, collagen-containing fiber structure and low-cost characteristics.24,25
Secondly, conductivity and antimicrobial properties need to be considered when preparing FSDs. Carbon quantum dots (CQDs), as a zero-dimensional carbon material with excellent electrical conductivity, water solubility and biocompatibility, have been widely reported in the fields of medical imaging, environmental monitoring and chemical analysis.26,27 In this work, BD-CQDs were prepared from Tara tannin, a vegetable tanning agent containing many phenolic hydroxyl structures, and BD. These BD-CQDs with a large amount of amine and catechol groups have enhanced reducing ability.28 Therefore, BD-CQDs were used as a reducing agent and stabilizer to prepare BD-CQDs silver nanoparticles (BD-CQDs@AgNPs), endowing the hydrogel with good conductivity and antibacterial properties, thereby realizing its long-term use possibility. Thirdly, to address the adaptability of CHs in different environments (low temperature and dry), strategies like hydrophobic coatings,29 salts,30 and polyols have been proposed.31 Among them, glycerin (Gly) has been widely used in food, cosmetics and medical industries because of its low cost and good moisturizing properties.32 Therefore, on the one hand, the introduction of Gly can form a strong hydrogen bond with water molecules and inhibit the evaporation of water, thus conferring anti-freezing and moisturizing properties to the hydrogel.33 On the other hand, the introduction of Gly is conducive to the improvement of hygroscopicity as well as the ability of sensing humidity change of CHs.34
In this paper, highly sensitive and multifunctional BPGC-Gly organohydrogels with “surface synapses” and “internal fibers” structures were prepared by using BD as the functional raw material for hydrogels, and by using polyvinyl alcohol (PVA), gelatin (Gel), BD-CQDs@AgNPs and Gly. The obtained BPGC-Gly organohydrogel could be used as a multi-functional sensor integrating temperature, humidity, strain, pressure and bioelectric detection. It could sense temperature (−20–60 °C) and humidity (97–46.7% RH (relative humidity)) over a wide range, and quickly respond (2.4 s) and recover (1.6 s) to different frequencies of breathing. As a strain/pressure sensor, the prepared sensor could monitor the human movement status in real time. Also, it enabled the monitoring of position and weight of varying magnitudes and exhibited high pressure sensitivity (S = 4.26 kPa−1) even under low-pressure conditions (0–12.5 kPa). In addition, owing to the high sensitivity of the BPGC-Gly bioelectrode, it could accurately collect human electrophysiological signals (electromyography (EMG) and electrocardiogram (ECG)), and further obtained more comprehensive human movement health data. These natural animal biomass-based organohydrogels show great potential in electronic skin, health monitoring, and biological electrodes.
Experimental section
Materials
The materials used, characterization instruments and details of experimental characterization methods are described in the ESI,† and informed consent was obtained for the experiments involving human participants.Preparation of BD-CQDs
The preparation was done according to the previously reported literature.35 The mixed solution with 150 mg of BD, 50 mg of Tara tannin and 30 mL of deionized (DI) water was reacted at 180 °C for 8 h. And then, the reaction product was cooled to room temperature (RT, 25 °C) and filtered (0.22 μm filter membrane) to obtain BD-CQDs.Preparation of sliver nanoparticles (BD-CQDs@AgNPs)
Firstly, the pH value of 5 mL of BD-CQDs was adjusted to 8. Then, the mixed solution was placed in a water bath at 40 °C, and silver nitrate solution (0.06 M, 0.2 mL) was slowly added and stirred for 10 min to obtain BD-CQDs@AgNPs. To study the effect of pH on AgNP synthesis, sodium hydroxide was added to bring pH in the range of 6–10 (Fig. S1, S2 and Table S1, ESI†). As shown in Fig. S3, S4 and Table S2 (ESI†), the influence of silver nitrate concentration on the particle size of AgNPs was explored.Preparation of BPGC and BPGC-Gly
Firstly, PVA particles were stirred at 90 °C for 2 h to obtain the PVA solution. And then, different amounts of gelatin and BD were thoroughly mixed in BD-CQDs@AgNPs (0.5 mL) and then added to the PVA solution and stirred for 30 min at 60 °C to obtain the pre-hydrogels. Finally, the pre-hydrogels were subjected to three freeze–thaw operations (freezing condition: −21 °C/6 h, thawing condition: RT/6 h) to obtain the BPGC hydrogel. The composition of BxPGyC hydrogels is shown in Table 1, where x and y represent the amount of BD and Gel, respectively.| Sample | PVA (g) | Gelatin (mg) | BD (mg) | BD-CQDs@AgNPs (mL) | H2O (mL) | Gly (mL) |
|---|---|---|---|---|---|---|
| PVA | 0.1 | 0 | 0 | 0 | 0.9 | |
| PC | 0.1 | 0 | 0 | 0.6 | 0.9 | |
| B20PG | 0.1 | 20 | 20 | 0 | 0.9 | |
| B20PC | 0.1 | 0 | 20 | 0.6 | 0.9 | |
| B20PG10C | 0.1 | 10 | 20 | 0.6 | 0.9 | |
| B20PG20C | 0.1 | 20 | 20 | 0.6 | 0.9 | |
| B20PG30C | 0.1 | 30 | 20 | 0.6 | 0.9 | |
| PG20C | 0.1 | 30 | 0 | 0.6 | 0.9 | |
| B10PG20C | 0.1 | 30 | 10 | 0.6 | 0.9 | |
| B30PG20C | 0.1 | 30 | 30 | 0.6 | 0.9 | |
| BPGC-Gly3% | 0.1 | 20 | 20 | 0.6 | 0.75 | 0.05 |
| BPGC-Gly7% | 0.1 | 20 | 20 | 0.6 | 0.80 | 0.10 |
| BPGC-Gly10% | 0.1 | 20 | 20 | 0.6 | 0.75 | 0.15 |
| BPGC-Gly13% | 0.1 | 20 | 20 | 0.6 | 0.70 | 0.20 |
Similarly, under the same pre-preparation conditions, the pre-hydrogels were mixed well with different volumes of Gly, and then the mixed system was subjected to three repeated freezing–thawing cycles to obtain the BPGC-Gly organohydrogel. The composition of the BPGC-Glyz hydrogel is shown in Table 1, where z denotes the percentage of Gly in the total volume.
Results and discussion
Preparation and characterization of BD-CQDs and BD-CQDs@AgNPs
The preparation process of BD-CQDs@AgNPs is shown in Fig. S5a (ESI†). Firstly, BD-CQDs were prepared by hydrothermal reaction using BD as a N source and Tara tannin as a C source. Secondly, silver nitrate (AgNO3) was reduced by BD-CQDs in an alkaline environment (pH = 8) to obtain BD-CQDs@AgNPs with antimicrobial and conductive properties. Finally, the microstructures and physicochemical properties of BD-CQDs and BD-CQDs@AgNPs were analyzed in detail (ESI†).Preparation and mechanical characterization of the BPGC hydrogel
In general, Fig. 1 briefly illustrates the preparation of the BPGC hydrogel. Firstly, a certain amount of Gel was dissolved in a pre-prepared PVA solution (10 wt%) and stirred for 15 min. Then, BD-CQDs@AgNPs and BD were added to the mixed solution and stirred for 30 min to obtain the pre-hydrogel (pre-BPGC). Finally, the BPGC hydrogel was prepared by freezing (6 h, −21 °C) and thawing (6 h, 25 °C) the pre-hydrogel three times. In particular, the organic hydrogel (BPGC-Gly) was obtained by replacing part of DI water with Gly and repeating the above experimental procedure. The crosslinking network of the BPGC hydrogel was mainly composed of various hydrogen bonds (between PVA and Gel, PVA and BD, Gel and BD) and the crystal domain of the PVA chain. Meanwhile, BD-CQDs@AgNPs enhanced the electrical conductivity of the BPGC hydrogel, while their small particle size endowed the BPGC hydrogel with antibacterial properties. Notably, collagen fibers provided by BD could serve as a multi-network support structure for the BPGC hydrogel, contributing to improvement of its physical and mechanical properties. The anti-freezing and moisturizing properties of BPGC-Gly were conferred by Gly, and Gly could also form strong hydrogen bonds with PVA, BD and Gel, which is conducive to the improvement of mechanical properties.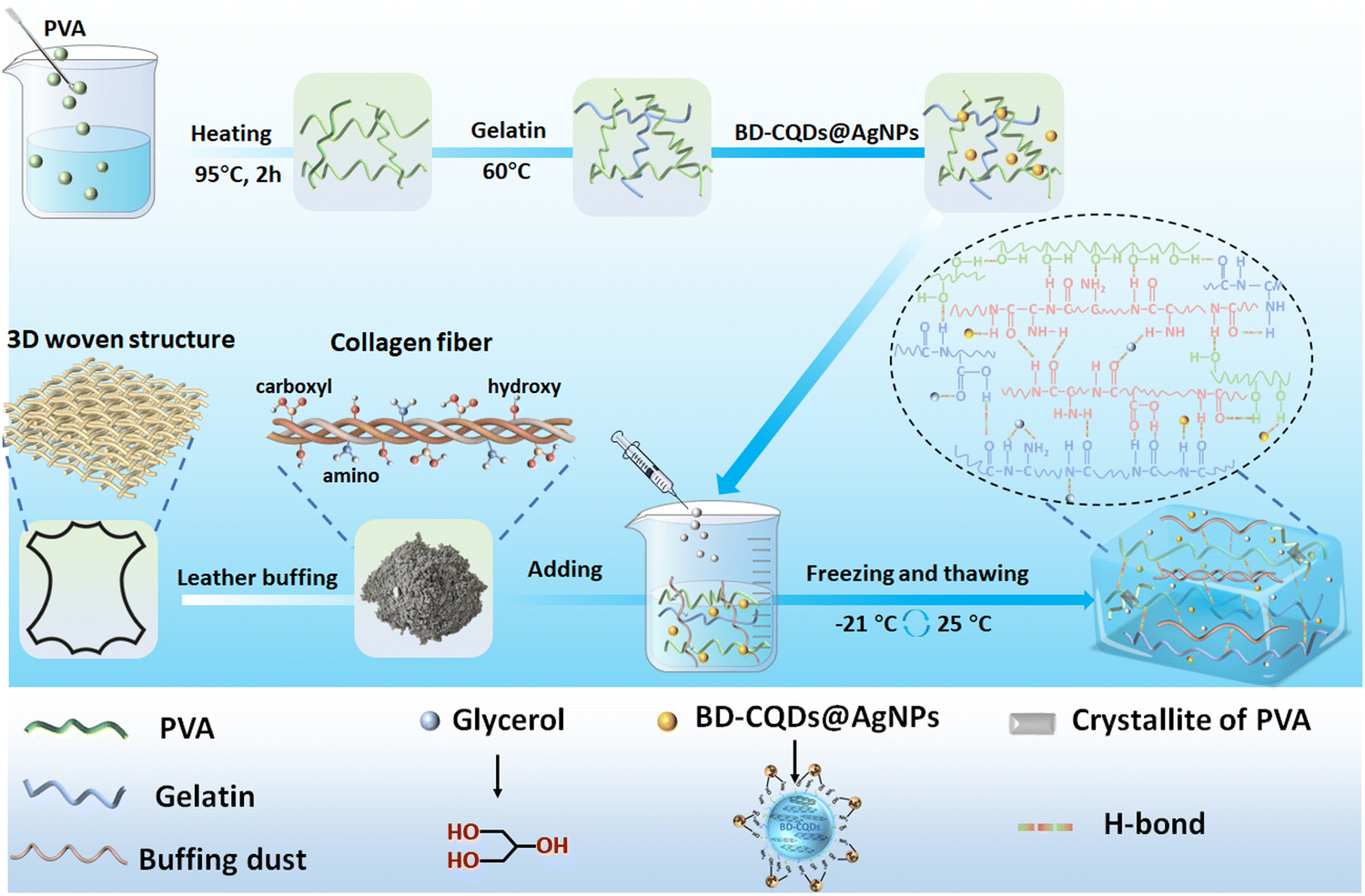 | ||
| Fig. 1 Schematic fabrication route for the BPGC-Gly organohydrogel. | ||
Firstly, using the mechanical strength of BPGC as a reference, we optimized the Gel and BD dosages through stretching and compression experiments. As depicted in Fig. 2a, with increasing Gel content, the hydrogel's tensile strength and strain both increased, from 0.16 MPa and 200% (PVA) to 0.5 MPa and 280% (B20PG20C). The main reason is that the addition of Gel enhanced the hydrogen bonding with PVA and BD, and could prevent fracture through effective energy dissipation during tensile deformation.36 However, further increasing the Gel dosage (B20PG30C) did not significantly improve tensile strength or elongation, probably due to the saturation of hydrogen bonding sites in PVA and BD. When comparing hydrogels' toughness and conductivity (Fig. 2b), BD-CQDs@AgNPs endowed the hydrogel with electrical conductivity, with values above 0.70 S m−1 compared to 0.28 S m−1 of the pure PVA hydrogel. Meanwhile, the change in toughness was in line with the stress strength, which increased with the increase in the amount of Gel (Fig. 2b). Notably, as shown in Fig. 2c, by comparing the compressive stress changes of hydrogels at the same compressive strain (50%), it could be found that the compressive strains of PVA, B20PC, B20PG10C, B20PG20C and B20PG30C were 0.042, 0.060, 0.072, 0.085 and 0.095 MPa, respectively. In comparison, B20PG20C and B20PG30C exhibited better compressive properties, which was consistent with the results of the stress–strain experiments. In general, considering the mechanical properties, toughness, conductivity and energy saving performance of hydrogels, we chose the amount of Gel in B20PG20C for subsequent experiments.
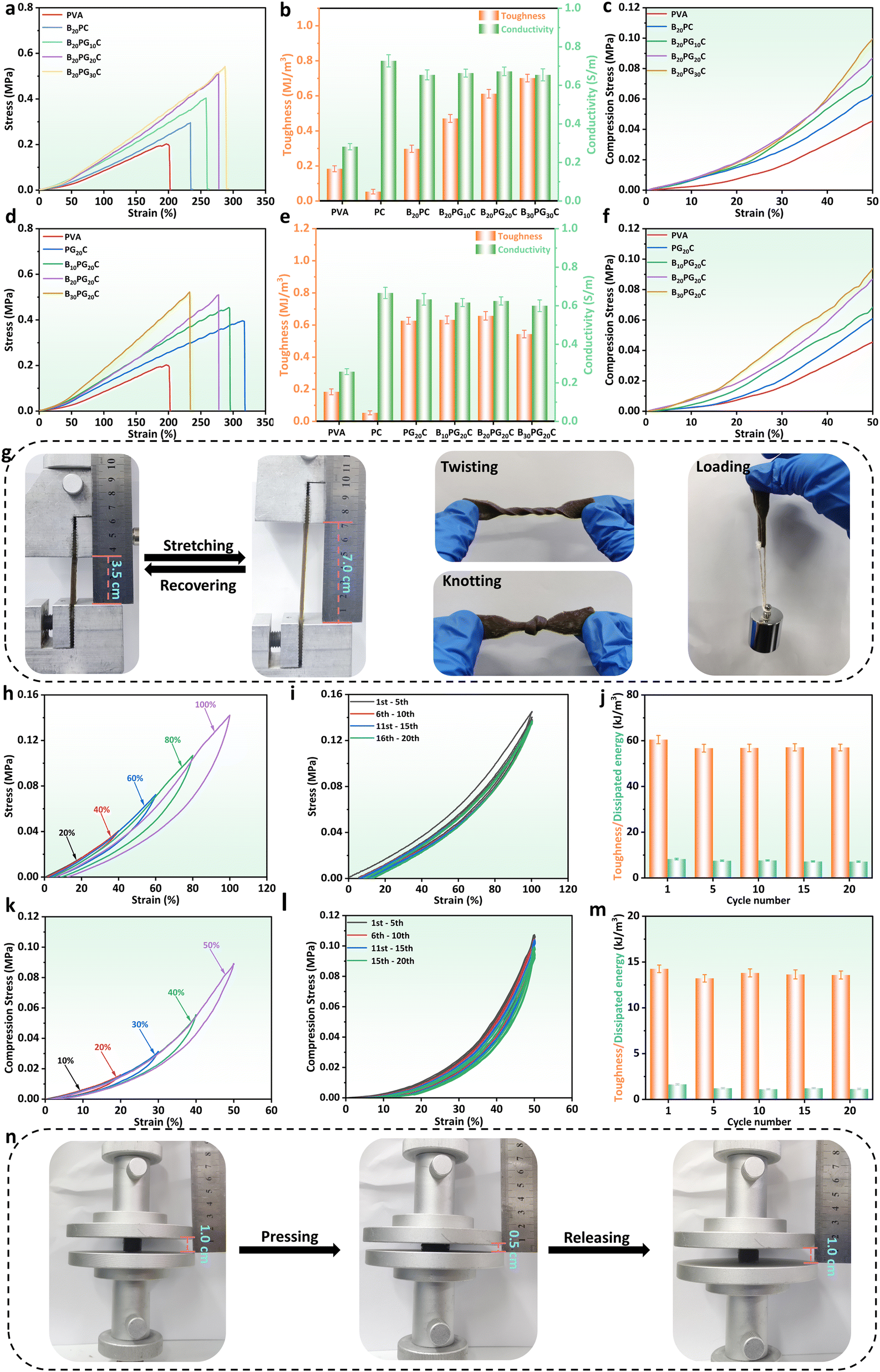 | ||
| Fig. 2 Optimization and mechanical properties of the BPGC hydrogel. Stress–stain curves (a), toughness and conductivity (b), and compression stress–stain curves (c) of BPGC hydrogels with different contents of Gel. Stress–stain curves (d), toughness and conductivity (e), and compression stress–stain curves (f) of BPGC hydrogels with different contents of BD. (g) Pictures of the BPGC hydrogel after stretching-recovery, twisting, knotting and hanging of 200 g weight. (h) Stress–strain cycle curves of the BPGC hydrogel under different strains (20%, 40%, 60%, 80% and 100%). Cyclic stress–strain curves (20 cycles) (i), and toughness and dissipated energy (j) of the BPGC hydrogel at 100% strain. (k) Compression stress–strain cycle curves of the BPGC hydrogel under different strains (10%, 20%, 30%, 40% and 50%). Cyclic compression stress–strain curves (20 cycles) (l), and toughness and dissipated energy (m) of the BPGC hydrogel at 50% strain. (n) Pictures of BPGC hydrogel after compression-recovery. | ||
Similarly, the amount of BD was optimized in terms of tensile and compressive properties. As shown in Fig. 2d, with the increase of the amount of BD, the fracture strength of the hydrogel gradually increased, while the breaking elongation gradually decreased. Specifically, the active groups (amino, hydroxyl, carboxyl) contained in BD could form hydrogen bonds with the side-chain groups of PVA and Gel, thereby enhancing the tensile strength of the hydrogel.37 As displayed in Fig. 2e the toughness of P20GC, B10PG20C, B20PG20C and B30PG20C was up to 0.60 MJ m−3 and above. Simultaneously, compressive stress–strain experiments were carried out on hydrogels with different amounts of BD. Under the same compressive strain (50%), both B20PG20C and B30PG20C could reach the compressive strength of 0.085 MPa (Fig. 2f). Considering the tensile strength, compressive strength, strain, and toughness of hydrogels, the B20PG20C hydrogel (referred to as the BPGC hydrogel) was chosen to further investigate the mechanical properties of the hydrogel.
The BPGC hydrogel could be stretched to twice its original length without damage, even in the twisted and knotted state, indicating that it has good mechanical strength (Fig. 2g). Meanwhile, as depicted in Fig. 2h, the recovery ability of BPGC was characterized by tensile loading–unloading experiments under different tensile strains. Obviously, BPGC almost returned to the original state after unloading, and only a small hysteresis loop was produced, indicating its excellent recovery performance. As shown in Fig. S7a and b (ESI†), the dissipated energy was strain-dependent. As BPGC's strain increased from 20% to 100% (20% per increment), the dissipated energy corresponded to 0.35, 0.93, 2.93, 5.60, and 9.88 kJ m−3, respectively, which was due to the fact that more of the dynamic hydrogen bonds in the BPGC hydrogel were broken to release more mechanical energy with the increase of external tension.36 Meanwhile, the fatigue resistance of BPGC was studied by 20 stretching cycles under 100% strain. As depicted in Fig. 2i, only the first stretching cycle curve showed a slight hysteresis loop, which was similar to that of most hydrogels, suggesting that some of the dynamic hydrogen bonds in the BPGC hydrogel were broken.36 In the subsequent 19 tensile cycle experiments, negligible irrecoverable deformation of BPGC could be found, proving its excellent fatigue resistance. Subsequently, the dissipated energy and toughness were calculated for the 1st, 5th, 10th, 15th, and 20th cycles of the cycling process. As shown in Fig. 2j, the toughness values of the 5 groups under 100% strain were close, ranging from 56.82 to 60.50 kJ m−3. Meanwhile, the dissipated energy corresponding to the 5 cycles ranged from 7.12 to 8.25 kJ m−3, accounting for 12.50–13.50% of the total work. All these results indicated that the BPGC hydrogel has superior fatigue resistance property.
Similarly, compression experiments revealed BPGC's mechanical properties. As shown in Fig. 2k, compression-unloading experiments were performed on the BPGC hydrogel at different strains (10%, 20%, 30%, 40% and 50%). Findings consistent with the tensile-unloading experiments were observed, where the hysteresis loop of BPGC increased with increasing compressive strain (dissipation energy ranged from 0.07 to 2.35 kJ m−3, Fig. S7b, ESI†). In addition, BPGC was subjected to 20 compression-unloading cycles at 50% strain (Fig. 2l). As expected, the 20-cycle curves could almost overlap and BPGC could automatically recover to the original state without any damage, indicating its excellent compressive fatigue resistance and rebound ability. At the same time, the dissipated energy and toughness of the 1st, 5th, 10th, 15th, and 20th cycles of the cycling process are shown in Fig. 2m, and the toughness values are very close to each other, around 14.00 kJ m−3. And the corresponding dissipated energy accounted for 8.08–11.50% of the total work of each cycle. In addition, Fig. 2n displays that after 20 compression cycles (50% strain), BPGC recovered to its original state without damage. Based on the above tensile and compressive results, the exceptional fatigue resistance and recovery capacity of BPGC has proven to be the basis for subsequent multifunctional sensors and flexible bioelectrodes.
Structural characterization of the BPGC-Gly organohydrogel
Various characterization methods were used to further study the surface morphology as well as the internal interaction of the BPGC-Gly organohydrogel. SEM was applied to characterize the cross-sectional microstructure of PVA, PGC, BPGC and BPGC-Gly at different scales. As illustrated in Fig. 3, all the four hydrogels exhibited a honeycomb-like 3D porous structure. Obviously, the BPGC-Gly hydrogel (1000×) exhibited a denser 3D network than the other three hydrogels due to the increased intramolecular and intermolecular hydrogen bonds among BD, PVA, Gel, Gly and BD-CQDs@AgNPs (Fig. 3a–d). As illustrated in Fig. 3c, a significant collagen fiber network could be seen in the high-magnification (5000×) SEM images of BPGC, and the diameter of the collagen fiber bundle in BPGC was about 3.38 μm, demonstrating the successful introduction of BD as a filler.38 Notably, collagen fibers with a diameter of 100 nm were observed in the further enlarged SEM image (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) of BPGC and the D-Period (65 nm) belonging to the collagen fibers was identified.39 Therefore, the SEM images from micro to nano scale had fully proved that BPGC retained the 3D woven network of collagen fibers, which was conducive to improving the mechanical properties of the hydrogel and the stability of the skeleton. In contrast, no D-Period was found in BPGC-Gly (Fig. 3d), which may be because the microstructure of the collagen fiber bundles was wrapped after the adsorption and permeation of Gly. Furthermore, the top cross-section of BPGC-Gly was observed using SEM. As shown in Fig. 3e, the 3D SEM image of BPGC-Gly reveals its multilevel and multi-pore structure, which mainly was attributed to the introduction of BD that provided a fiber support structure of varying size. Upon further magnification (200×) of the top surface, BPGC-Gly revealed a distinct collagen fiber-Gel-PVA hybrid synapse, where the collagen fibers were clearly distributed across the synapse. Additionally, BPGC-Gly displayed a high synapse structure, with vertical height ranging from 50 to 100 μm, which greatly contributes to the sensitivity enhancement of BPGC-Gly. Therefore, through a simple and low-cost preparation method, imparting this “external synapse” and internal multilevel network structure to the hydrogel significantly improved its pressure sensitivity and mechanical strength. In addition, the energy-dispersive X-ray spectroscopy (EDS) spectrum of BPGC-Gly revealed a uniform distribution of Ag elements throughout the hydrogel (Fig. S8, ESI†). These results indicated that BD was uniformly embedded in the hydrogel, functioning as its internal supportive skeleton. Additionally, the successful incorporation of BD-CQDs@AgNPs into the BPGC hydrogel has been achieved.
000×) of BPGC and the D-Period (65 nm) belonging to the collagen fibers was identified.39 Therefore, the SEM images from micro to nano scale had fully proved that BPGC retained the 3D woven network of collagen fibers, which was conducive to improving the mechanical properties of the hydrogel and the stability of the skeleton. In contrast, no D-Period was found in BPGC-Gly (Fig. 3d), which may be because the microstructure of the collagen fiber bundles was wrapped after the adsorption and permeation of Gly. Furthermore, the top cross-section of BPGC-Gly was observed using SEM. As shown in Fig. 3e, the 3D SEM image of BPGC-Gly reveals its multilevel and multi-pore structure, which mainly was attributed to the introduction of BD that provided a fiber support structure of varying size. Upon further magnification (200×) of the top surface, BPGC-Gly revealed a distinct collagen fiber-Gel-PVA hybrid synapse, where the collagen fibers were clearly distributed across the synapse. Additionally, BPGC-Gly displayed a high synapse structure, with vertical height ranging from 50 to 100 μm, which greatly contributes to the sensitivity enhancement of BPGC-Gly. Therefore, through a simple and low-cost preparation method, imparting this “external synapse” and internal multilevel network structure to the hydrogel significantly improved its pressure sensitivity and mechanical strength. In addition, the energy-dispersive X-ray spectroscopy (EDS) spectrum of BPGC-Gly revealed a uniform distribution of Ag elements throughout the hydrogel (Fig. S8, ESI†). These results indicated that BD was uniformly embedded in the hydrogel, functioning as its internal supportive skeleton. Additionally, the successful incorporation of BD-CQDs@AgNPs into the BPGC hydrogel has been achieved.
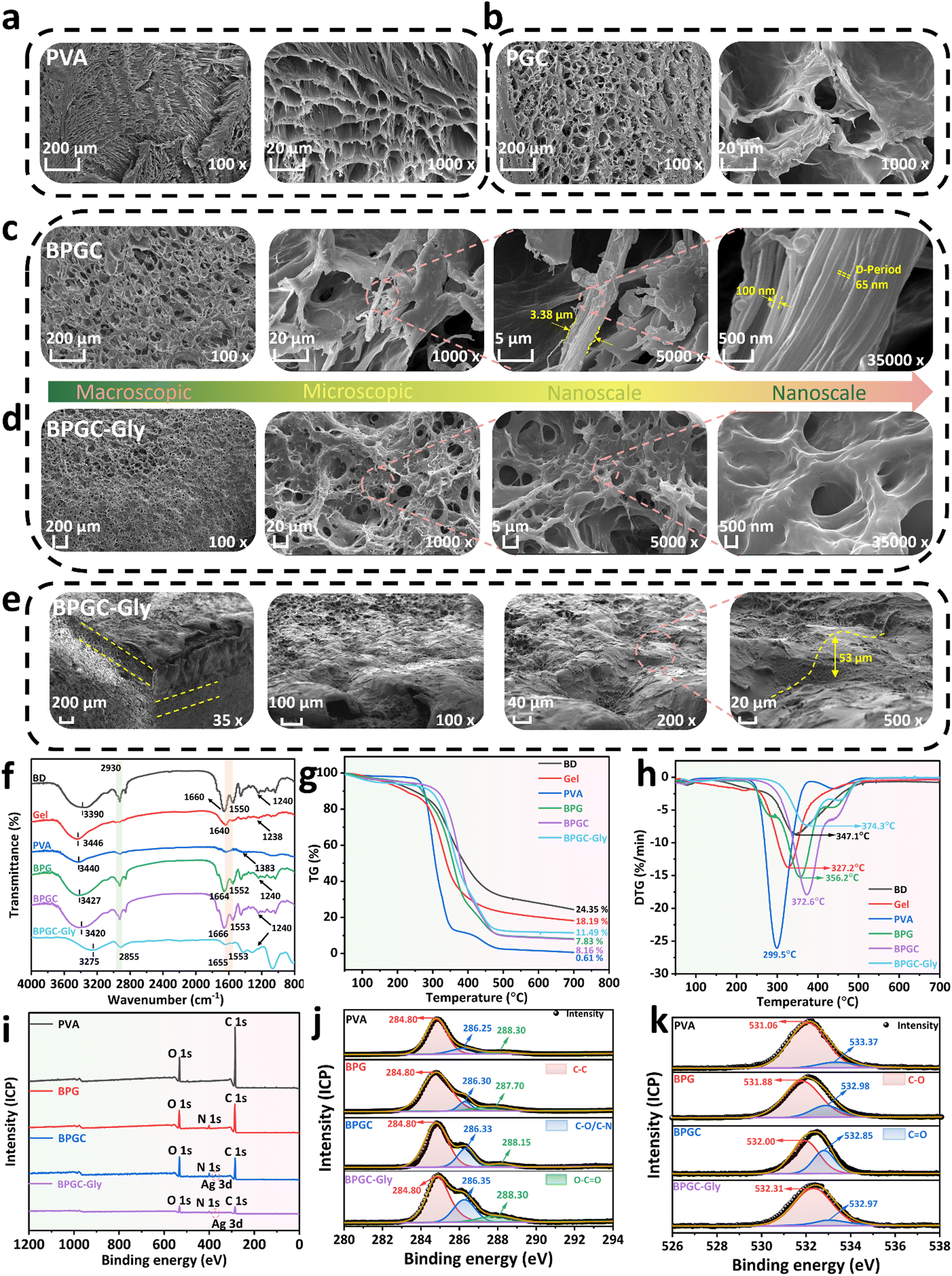 | ||
| Fig. 3 Structural characterization of the BPGC-Gly organohydrogel. SEM images of PVA (a), PGC (b), BPGC (c), and BPGC-Gly (d) at different magnifications. (e) SEM images 3D stereo cross-section and top cross-section of BPGC-Gly. FTIR spectra (f), TG (g) and DTG (h) curves of BD, PVA, Gel, BPG, BPGC and BPGC-Gly. XPS full spectra (i), high-resolution C 1s spectra (j), and O 1s spectra (k) of PVA, BPG, BPGC and BPGC-Gly. | ||
The physical and chemical interactions within BPGC-Gly were further explored by using Fourier transform infrared (FTIR) spectroscopy. As illustrated in Fig. 3f, the O–H stretching vibration peak at 3340 cm−1 could be found in the FTIR spectrum of PVA, which belonged to the intramolecular or intermolecular hydrogen bond of PVA, while the stretching vibration absorption peak corresponding to the primary alcohol C–O could be observed at 1383 cm−1.28 Obviously, the major groups and structures of collagen (methylene, carboxyl and amino) could be observed in the FTIR spectra of B, G, BPG, BPGC and BPGC-Gly. Specifically, the stretching vibration peaks of N–H and O–H could be seen near 3420 cm−1, and the asymmetric and symmetric stretching vibration absorption peaks of –CH2– could be observed at 2930 and 2855 cm−1, respectively.40 The peak near 1640–1666 cm−1 corresponded to the amide I band, which was the characteristic peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration in the carboxyl group.37 The characteristic peaks of C–N stretching vibration and N–H bending vibration (amide II band) appeared at 1553 cm−1.41 The C–N bond stretching vibration and in-plane N–H bending of the protein amide bond were recorded near 1238 cm−1 (amide III band).40 Notably, the stretching vibration peaks of O–H of these five samples were significantly deviated, and a right shift of peaks was found from the PVA hydrogel (3440 cm−1) to BPGC (3420 cm−1), indicating that a number of hydrogen bonds were formed among BD, PVA and Gel.42 After Gly was added, a blue shift of the peak appeared at 3275 cm−1 for BPGC-Gly, suggesting the further enhancement of hydrogen bonding.
O stretching vibration in the carboxyl group.37 The characteristic peaks of C–N stretching vibration and N–H bending vibration (amide II band) appeared at 1553 cm−1.41 The C–N bond stretching vibration and in-plane N–H bending of the protein amide bond were recorded near 1238 cm−1 (amide III band).40 Notably, the stretching vibration peaks of O–H of these five samples were significantly deviated, and a right shift of peaks was found from the PVA hydrogel (3440 cm−1) to BPGC (3420 cm−1), indicating that a number of hydrogen bonds were formed among BD, PVA and Gel.42 After Gly was added, a blue shift of the peak appeared at 3275 cm−1 for BPGC-Gly, suggesting the further enhancement of hydrogen bonding.
Thermogravimetry (TG) and derivative thermogravimetry (DTG) were used for thermal stability testing of hydrogels and some raw materials. Firstly, as indicated in Fig. 3g, the initial partial mass loss occurring between 50 and 250 °C was attributed to the significant loss of free water. Additionally, the prominent weight loss stage observed in the range of 250–450 °C was due to the decomposition of the biomass skeleton and the breaking of intermolecular hydrogen bonds.43 Finally, at the 450–700 °C stage, it is evident that the residual mass of the hydrogels increased gradually from PVA (0.16%) to BPG (7.83%), BPGC (8.16%), and BPGC-Gly (11.49%). This indicated that the introduction of BD, Gel, BD-CQDs@AgNPs, and Gly enhanced the thermal stability of the hydrogels. Furthermore, compared to the pure PVA hydrogel, the decomposition temperature of BPG was increased by 56.7 °C, which was attributed to the introduction of BD and Gel materials which enhanced the intramolecular and intermolecular hydrogen bonding in the BPG hydrogel, resulting in the formation of a dense cross-linked network (Fig. 3h). Simultaneously, due to the incorporation of BD@CQDs@AgNPs and Gly, the decomposition temperature of BPGC (372.6 °C) and BPGC-Gly (374.3 °C) was higher than that of the BPG hydrogel (356.2 °C), demonstrating that BD@CQDs@AgNPs and Gly could also form a significant number of hydrogen bonds within the BPG structure, thereby enhancing the cross-linked network and thermal stability of the hydrogel. This finding was consistent with the results obtained from SEM.
Fig. 3i presents the X-ray photoelectron spectroscopy (XPS) full spectra of PVA, BPG, BPGC and BPGC-Gly. The characteristic peak of Ag 3d appeared in the XPS spectra of BPGC and BPGC-Gly, indicating the successful introduction of BD-CQDs@AgNPs. This finding is consistent with the EDS mapping results based on SEM. Clearly, the ratio of O/C in the PVA hydrogel is 0.20, while in BPGG and BPGC-Gly, the O/C ratio increased to 0.36 and 0.38, respectively. These findings indicated that the introduction of BD, Gel, Gly, and BD-CQDs@AgNPs enhanced the presence of polar functional groups on the surface of BPGC. To demonstrate the multiple hydrogen bonding interactions between BD, Gel, Gly and PVA, we further analyzed and compared the XPS high-resolution spectra of PVA, BPG, BPGC and BPGC-Gly. The C 1s peak of the PVA sample could be deconvoluted into three peaks, the C–C–H peak (284.80 eV), the C–OH peak (286.25 eV), and the O–CO peak (288.30 eV), where the presence of the O–CO bond may be a proof for the residue of the ethyl acetate group.44 However, after the addition of BD and Gel, the high-resolution C 1s spectrum of the BPG hydrogel exhibited three characteristic peaks with higher intensities (Fig. 3j). Particularly, the C–OH peak had shifted to a higher binding energy (BE), indicating that this subpeak may be composed of C–O and C–N bonds, which was consistent with the results obtained from FTIR. Furthermore, in the spectra of BPGC and BPGC-Gly, significant shifts could be found in the C–O/C–N and O–CO peaks, indicating that the introduction of BD-CQDs@AgNPs and Gly also enhanced the hydrogen bonding interactions among the various components. As illustrated in Fig. 3k, it could be observed that the high-resolution O 1s spectrum of the PVA sample contained two single peaks corresponding to C–O (531.06 eV) and CO (533.37 eV) bonds, whereas an increase in the binding energy of the C–O bond by 0.72 eV could be found in the BPG sample.45 Additionally, the BE of the two peaks in BPGC and BPGC-Gly also experienced displacement compared to BPG. These results collectively demonstrated the successful incorporation of BD, Gel, BD-CQDs@AgNPs and Gly into the BPGC hydrogel, and the hydrogen bonding among the various components was the main interaction (mainly relying on amino, hydroxyl, and carboxyl groups).
Anti-freezing and moisturizing properties of BPGC-Gly
It is well known that hydrogels, as a 3D network structure material with ultra-high-water content, may have limited applications in low temperature environments. The introduction of Gly in this work was expected to confer good anti-freezing and moisturizing properties to the BPGC hydrogel. In order to investigate the anti-freezing and moisturizing properties of BPGC-Gly, the amount of Gly was optimized. As shown in Fig. S9a (ESI†) with the increase in the amount of Gly, the crystallization peak of BPGC-Gly also decreased. Also, it could be observed that the DSC curve of BPGC exhibited a crystallisation peak at 1.5 °C, while the lowest crystallisation peak of BPGC-Gly could reach −40.3 °C, which was due to the fact that the hydrogen bond formed between Gly and H2O molecules prevented the formation of ice crystals, thus significantly lowering the freezing point of water in BPGC-Gly.34 Notably, BPGC-Gly exhibited better tensile stress and compression properties compared to BPGC (Fig. S9b, ESI†). Specifically, the fracture strength of BPGC-Gly3%, BPGC-Gly7%, BPGC-Gly10% and BPGC-Gly13% increased to 0.63, 0.65, 0.72 and 0.75 MPa, respectively, while the corresponding tensile elongation dropped to 300%, 292%, 270% and 245%, respectively. As illustrated in Fig. S9c (ESI†), the toughness of the organic hydrogel generally exhibited an increasing trend with increasing Gly content, reaching a peak of 0.86 MJ m−3 (BPGC-Gly10%). Meanwhile, with the increase of Gly content, the compressive property of BPGC hydrogels presented a similar behavior to the tensile performance (Fig. S9d, ESI†). This may be due to the fact that the hydroxyl groups contained in Gly are easy to form hydrogen bonds with the active groups in BPGC, thus enhancing the intermolecular force and winding tightness, resulting in enhanced mechanical properties. However, the conductivity gradually decreased, which may be due to the introduction of hydrophilic organic solvent hindering the electron movement in the BPGC hydrogel.32 Therefore, in order to balance the mechanical properties, electrical conductivity, and anti-freezing behavior of BPGC-Gly, BPGC-Gly10% (referred to as BPGC-Gly) was finally selected for the subsequent experiments. In order to observe the freeze-resistance and electrical conductivity of the obtained hydrogels more visually, BPGC and BPGC-Gly were placed at −21 °C for 24 h and compared. As illustrated in Fig. S9e (ESI†), the two hydrogels exhibited considerably different phenomena. BPGC turned into a brown-green hard lump, lacking flexibility and conductive properties, while BPGC-Gly could be easily twisted and was able to light up an LED lamp. In summary, these results provide preliminary evidence that BPGC-Gly has excellent freezing resistance and electrical conductivity at low temperatures.In addition, the weight changes of BPGC and BPGC-Gly after being placed at 20 °C for 5 d were compared. As depicted in Fig. S9f (ESI†), BPGC had a residual weight of about 12% and did not have a moisturizing effect. In contrast, the weight change rate of BPGC-Gly was no more than 30%, demonstrating its long-term moisturizing properties. Next, we tested the mechanical properties of BPGC-Gly at different placement time (0–5 d). As shown in Fig. S9g (ESI†), with increasing time, the tensile strain of BPGC-Gly decreased while its tensile stress increased. Similarly, under the same strain conditions, the compressive strength of BPGC-Gly increased (Fig. S9h, ESI†). These phenomena could be attributed to the partial evaporation of free water within BPGC-Gly, leading to partial collapse of the internal 3D network, which was consistent with the observed facts.46 Furthermore, the mechanical properties of BPGC-Gly at different temperatures (−20 °C, −10 °C, 0 °C, 10 °C, and 20 °C) were compared (Fig. S9i–k, ESI†). It was found that the tensile and compressive performance of BPGC-Gly remained almost consistent at different temperatures. These experimental phenomena indicated the importance of anti-freezing and moisture retention properties of BPGC-Gly in maintaining its good tensile and compressive strength. It overcame the dependency on the environment seen in conventional conductive hydrogels and is beneficial for monitoring human movement in low-temperature environments.
Antibacterial and cytotoxicity properties of BPGC-Gly
It is well known that porous hydrogels with high water content provide an ideal living environment for bacterial growth. If the hydrogel is not promptly treated with antibacterial agents in time, its service life will be significantly shortened. In this work, BD-CQDs@AgNPs with long-lasting antibacterial capacity were introduced into the BPGC hydrogel in order to obtain good antibacterial effects. To verify this aim, the classical Gram-positive (S. aureus) and Gram-negative bacteria (E. coli) were selected as experimental strains to investigate the antibacterial properties of PVA, BPG, BPGC, and BPGC-Gly hydrogels, and the inoculated media without the sample were used as blank control groups. The size of the inhibition zone around the hydrogel was observed by the classical disc diffusion method.40 As illustrated in Fig. 4a, the presence of strains of bacteria at the edges of the PVA hydrogel indicates that it has no antibacterial properties against S. aureus and E. coli. BPG demonstrated a small inhibition circle (d = 17 mm). Meanwhile, the other two groups of hydrogels containing BD-CQDs@AgNPs showed an obvious antibacterial effect, especially the inhibition area of BPGC-Gly against E. coli and S. aureus was 8.41 cm2 and 5.30 cm2, respectively, which might be caused by the synergistic effect of Gly and BD-CQDs@AgNPs (Fig. 4b). In addition, in order to explore the antibacterial mechanism of BD-CQD@AgNPs, Zeta potentials of bacteria were further measured, and it was found that both E. coli (−17.5 mV) and S. aureus (−17.7 mV) were negatively charged. Since the average Zeta potential of BD-CQD@AgNPs was −39.2 mV, it is difficult for negative bacteria to come into contact with negatively charged BD-CQD@AgNPs by electrostatic adsorption, and the reaction between bacteria and silver nanoparticles may be driven by intermolecular forces.47 Meanwhile, Gram-negative bacteria (E. coli) have a thinner cell wall compared to Gram-positive bacteria (S. aureus) and are therefore more susceptible to BD-CQD@AgNPs.48 In general, BD-CQDs@AgNPs have a particle size of nanometers (3.80 nm), and can enter the cell membrane of bacteria at low concentration, thus interfering with cell wall synthesis and inhibiting bacterial growth.37,40,48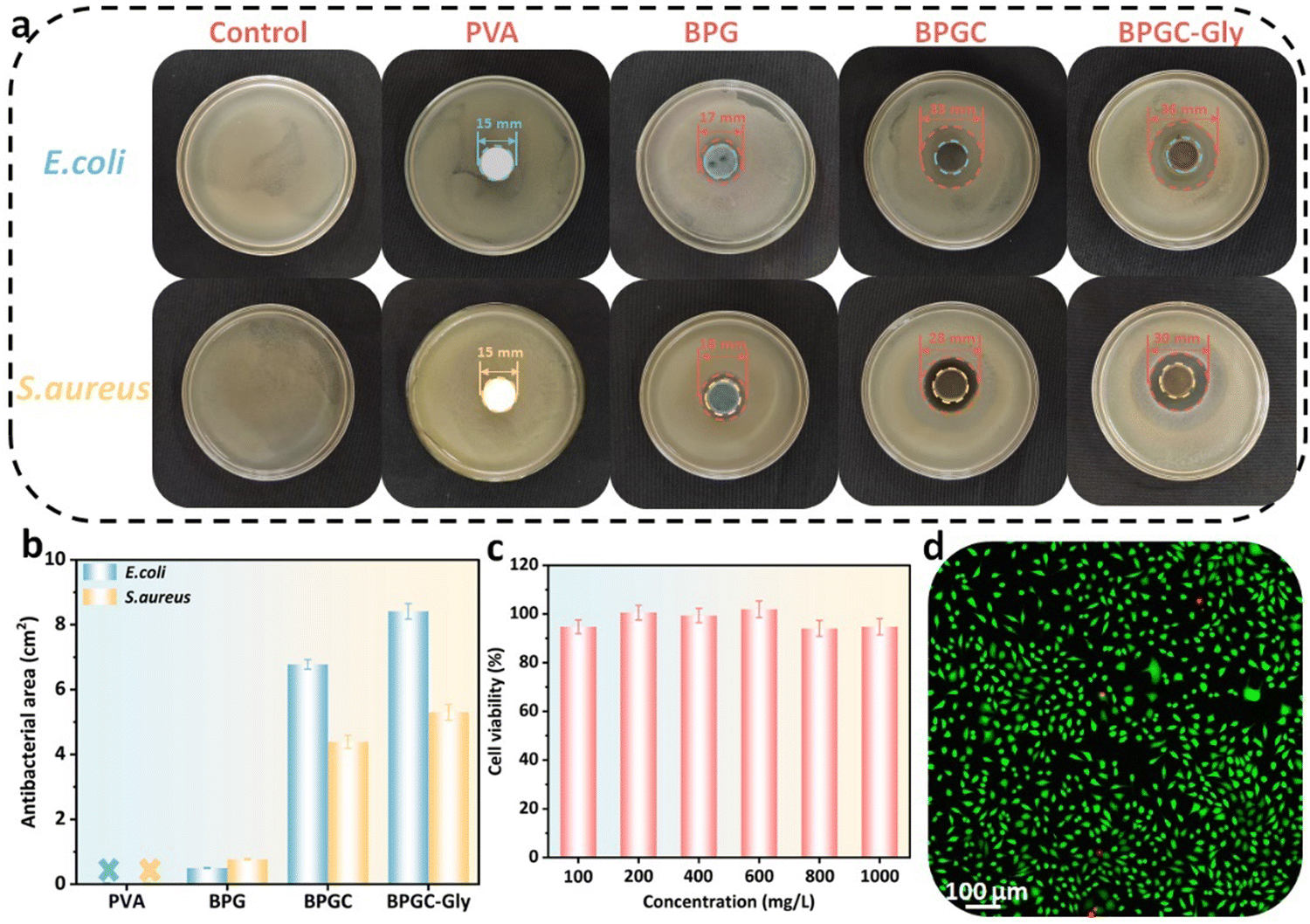 | ||
| Fig. 4 Antibacterial and cytotoxicity properties of BPGC-Gly. Antibacterial pictures (a) and statistics of the antibacterial area (b) of PVA, BPG, BPGC, and BPGC-Gly against E. coli and S. aureus. (c) Cell viability of BPGC-Gly at different concentrations. (d) CLSM image of L929 cells after culturing for 24 h with 1000 mg mL−1 BPGC-Gly extract (living cells: green fluorescence, dead cells: red fluorescence). | ||
To further illustrate the biocompatibility of BPGC-Gly, common CCK-8 cytotoxicity experiments were performed.43 L929 cells were cultured with different concentrations (0.1–1.0 mg mL−1) of the BPGC-Gly extract and cell viability was recorded. As shown in Fig. 4c, the survival rates of L929 cells at all concentrations exceeded 94%, proving that BPGC-Gly was non-toxic to L929 cells. Meanwhile, LIVE/DEAD cell staining was performed on the L929 cells treated with the highest concentration (1 mg mL−1) to further evaluate the cytocompatibility of BPGC-Gly. As illustrated in Fig. 4d, only a very small number of L929 cells showed red fluorescence (dead cells) and the majority of cells exhibited green fluorescence (living cells), further demonstrating the good biocompatibility of BPGC-Gly.
Electrical and sensing properties of BPGC-Gly
With the introduction of BD-CQDs@AgNPs, BPGC-Gly exhibited excellent conductivity, providing a solid foundation for its use as a flexible sensor. As illustrated in Fig. 5a, using BPGC-Gly as a conductor, an LED light could be successfully illuminated. Furthermore, the light intensity gradually decreased with the increase of the strain rate, indicating the strain-sensitivity of BPGC-Gly. Additionally, we have fabricated BPGC-Gly into a rod-shaped touchscreen pen, allowing touchscreen interaction and drawing on a tablet (Fig. S10, ESI†). Both of these phenomena prove that BPGC-Gly possesses good conductivity and holds significant potential for electronic applications.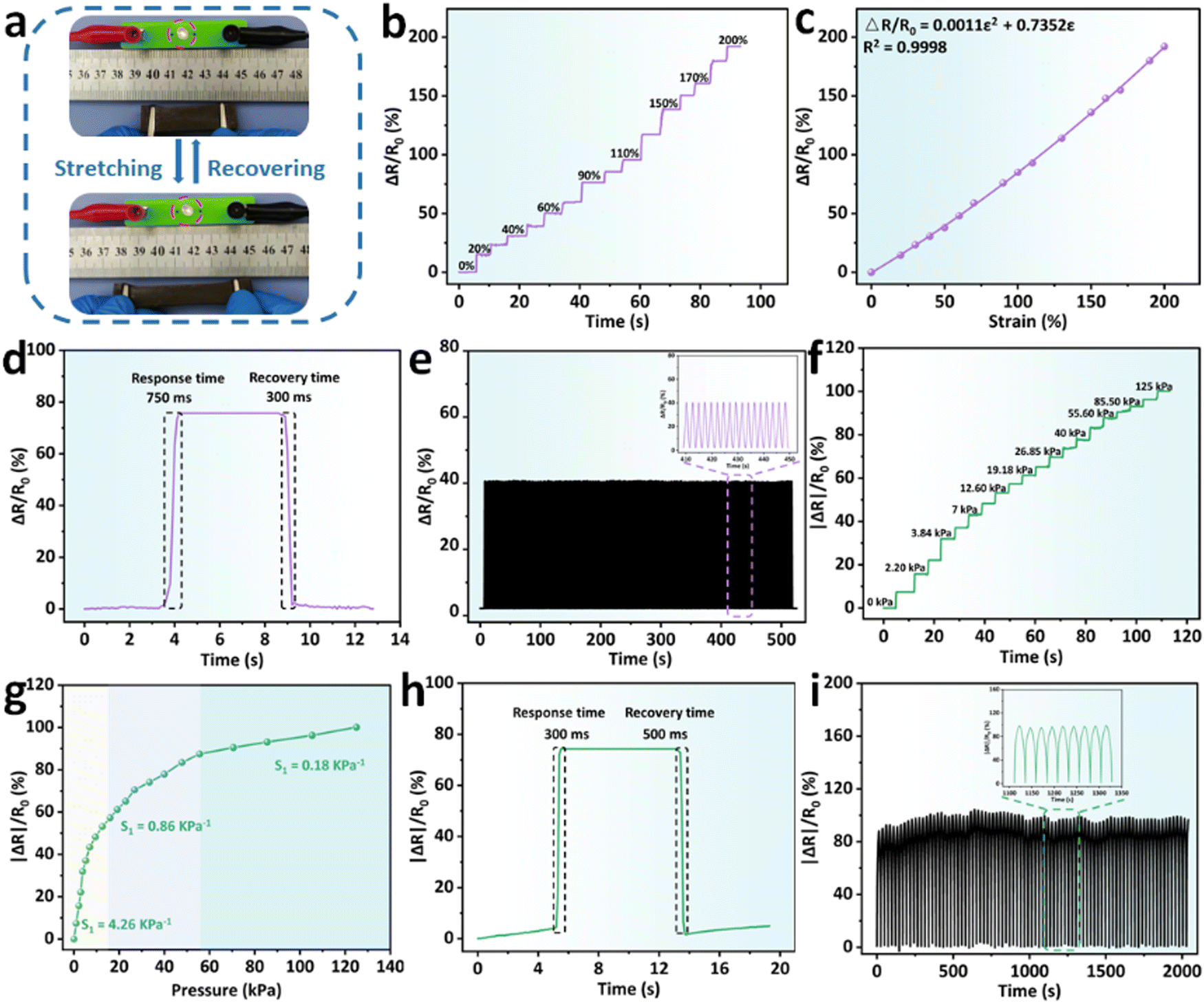 | ||
| Fig. 5 Electrical and sensing properties of BPGC-Gly. (a) Pictures of an LED bulb changing brightness as BPGC-Gly undergoes stretching-recovering. (b) ΔR/R0vs. time curve of BPGC-Gly at different strains (0–200%). (c) Quadratic fitting curve of ΔR/R0vs. strain of BPGC-Gly. (d) Tensile response and recovery time of BPGC-Gly. (e) Tensile cycling curve of ΔR/R0vs. time of BPGC-Gly at 50% tensile strain (500 s, inset: a local magnification of 410–450 s). (f) ΔR/R0vs. time curve of BPGC-Gly at different pressure (0–125 kPa). (g) Quadratic fitting curve of ΔR/R0vs. pressure of BPGC-Gly. (h) Compression response and recovery time of BPGC-Gly. (i) Compression cycling curve of ΔR/R0vs. time of BPGC-Gly at 50% compression strain (2000 s, inset: a local magnification of 1100–1350 s). | ||
In addition, the stability and sensitivity of external force feedback are important indices of flexible sensor devices. As illustrated in Fig. 5b, the change in the relative resistance of BPGC-Gly at different tensile strains (0–200% with 20%) was recorded. Specifically, BPGC-Gly could last 2–4 s for each strain gradient, with a strong stable signal output. Furthermore, by calculating the gauge factors (GF) as reported in the literature,40 a curve with the equation ΔR/R0 = 0.0011ε2 + 0.7352ε could be fitted, where ε denoted the tensile strain. Here, the quadratic curve was differentiated, and the calculation formula of GF was obtained (GF = 0.0022ε + 0.7352), indicating that BPGC-Gly had strain sensitivity and offered the possibility of human motion detection (Fig. 5c). Meanwhile, the pressure sensitivity (S) was further determined to quantify the compression sensitivity of BPGC-Gly. As indicated in Fig. 5f, the absolute change in the relative resistance of BPGC-Gly under different pressure (0–125 kPa) was tested and documented. As shown in Fig. 5g, the S could be divided into three regions. A pressure sensitivity of 4.26 kPa−1 was observed in the pressure range of 0–12.5 kPa. In the moderate pressure range (12.5–55.5 kPa), the pressure sensitivity was reduced to 0.86 kPa−1. As the pressure increased to 55.5–125 kPa, the S dropped to 0.18 kPa−1. These results indicated that BPGC-Gly was highly sensitive to pressure changes in the low-pressure range. Compared to other pressure-sensitive hydrogels, it exhibited a wider range of strain detection, demonstrating its potential as a pressure sensor (Table S3, ESI†).
Furthermore, the rapid recovery capability is another crucial indicator for strain/pressure sensors. We performed stretching-release and compression-release experiments on BPGC-Gly to determine its response time. As shown in Fig. 5d and h, BPGC-Gly demonstrated swift response and recovery to both stretching and compression. Specifically, it had a response time of 750 ms and a recovery time of 300 ms during the stretching experiment. Furthermore, in the compression experiment, it maintained a difference in response and recovery time of no more than 200 ms. These results demonstrated the excellent responsiveness of BPGC-Gly to stress/pressure. Lastly, the cyclic stability and fatigue resistance of BPGC-Gly as a strain/pressure sensor were investigated. As depicted in Fig. 5e and i, the stretching/compression cycling tests were conducted at 50% strain for 500 s/2000 s. Throughout the testing process, there was very little performance loss observed, and the magnified images of the curves revealed nearly identical signals. These results suggested that BPGC-Gly possessed outstanding durability under stretching and compression, making it suitable as a flexible sensor for long-term motion detection and heavy load monitoring.
Temperature-sensing and humidity-sensing properties of the BPGC-Gly sensor
Due to the rich hydrated ion network in the prepared BPGC-Gly organohydrogel, it has the ability to adsorb and release water molecules based on environmental humidity. Therefore, we utilized BPGC-Gly as a transducing material to perceive the changes in the external environment. Firstly, copper wires were connected to both ends of BPGC-Gly to create a dual-electrode chemical resistor configuration for temperature and humidity sensing. Secondly, a fixed bias voltage of 1.5 V was applied to BPGC-Gly to measure the changes in conductivity under varying temperature and humidity conditions. It is worth noting that electronic migration is influenced by temperature changes. As the temperature increases, the ionic motion speeds up, which leads to an increase in the ionic conductivity of BPGC-Gly. As shown in Fig. 6a, with increasing temperature (from −20 °C to 60 °C), the conductivity of the BPGC-Gly sensor also increased (0.17 to 1.39 S m−1). Therefore, the real-time conductivity of the BPGC-Gly sensor could effectively reflect the real-time temperature. Meanwhile, as illustrated in the inset of Fig. 6a, the conductivity of the BPGC-Gly sensor remained relatively stable even after being placed at −21 °C for 7 d, indicating that it possessed low-temperature conductive stability. Additionally, 3M tape was used to seal the BPGC-Gly sensor for temperature sensing tests. The response is defined as the change in relative conductivity (ΔG/G0), where G0 represents the initial conductivity. As shown in Fig. 6b, when the BPGC-Gly sensor was exposed to hot (45 °C) and cold water (0 °C) from room temperature (20 °C), the dynamic response curves varied due to the changes in relative conductance. The relative conductance of the BPGC-Gly temperature sensor was studied over time during the temperature cycling from 0 °C to 45 °C. As seen in Fig. 6c, the dynamic response curves demonstrated repeatable signals, suggesting the stability and reliability of the BPGC-Gly temperature sensor. Notably, the response time and recovery time of the BPGC-Gly temperature sensor were 193.3 s and 345.4 s, respectively (Fig. 6d). These findings indicated that the BPGC-Gly sensor could achieve rapid temperature responsiveness, which will be beneficial for monitoring overheating or fever-related conditions in the human body.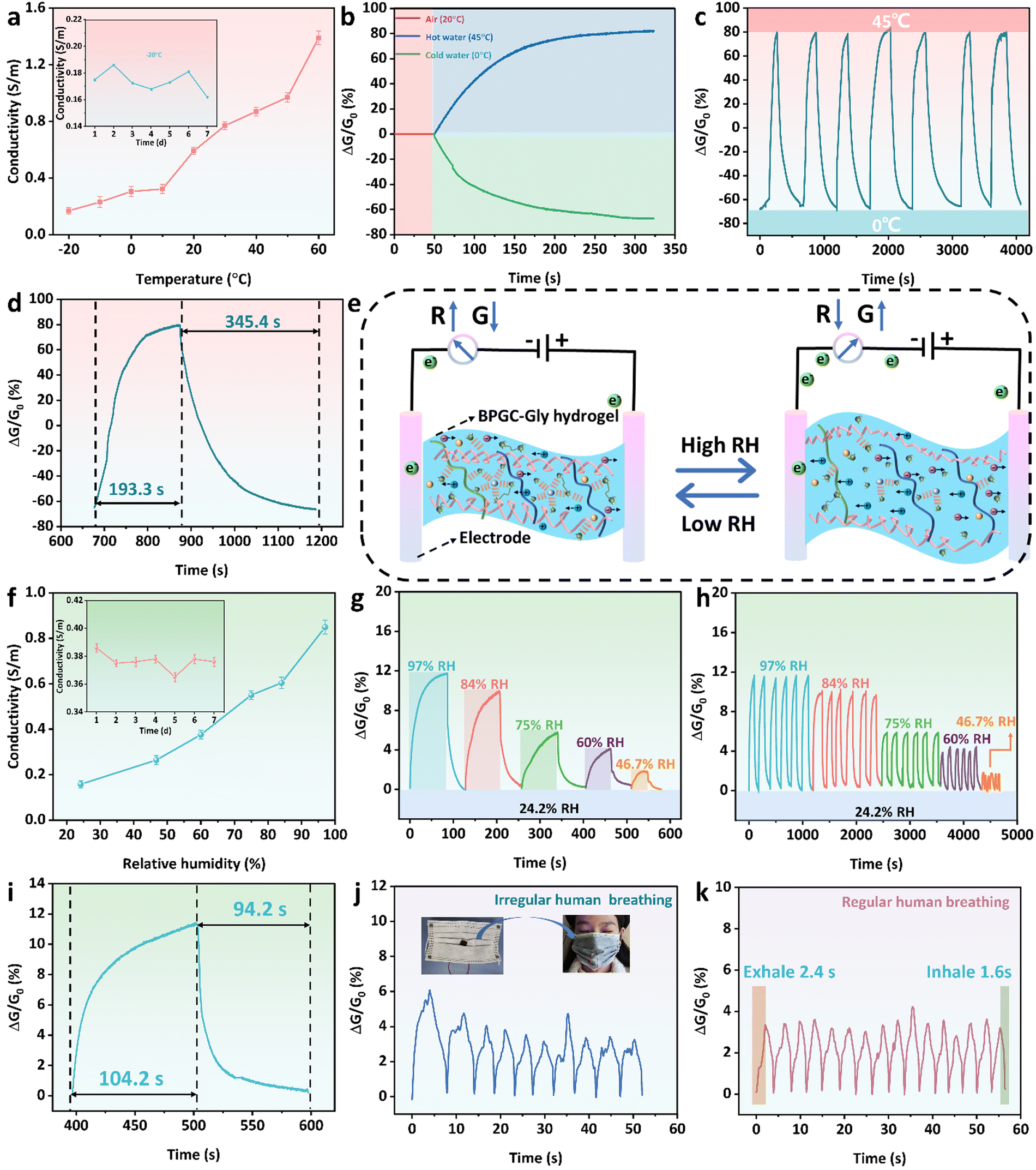 | ||
| Fig. 6 Temperature and humidity sensing properties of the BPGC-Gly sensor. (a) Conductivity of the BPGC-Gly sensor at different temperatures (inset: conductivity of the BPGC-Gly sensor after 7 days). (b) Real-time conductance change curve of the BPGC-Gly sensor at different water temperatures (0 °C, 20 °C, and 45 °C). The 10-cycle (c) and amplified 2nd cycle (d) curves of real-time conductance of the BPGC-Gly sensor in response to 0 °C and 45 °C. (e) Schematic of the humidity sensing mechanism of the BPGC-Gly sensor. (f) Conductivity of the BPGC-Gly sensor at different humidity (inset: conductivity of the BPGC-Gly sensor after 7 days at 20 °C with RH 60%). (g) Real-time conductance response of the BPGC-Gly sensor to RH ranging from 97% to 46.7%. The 30-cycle (h) and amplified 2nd cycle (i) curves of real-time conductance of the BPGC-Gly sensor in response to RH ranging from 97% to 46.7%. Real-time response of the BPGC-Gly sensor to irregular (j) and regular (k) human breathing. | ||
Further systematic investigations were conducted on the performance of the BPGC-Gly sensor in relative humidity detection. The detailed working principle of the BPGC-Gly humidity sensor is presented in Fig. 6e. In low relative humidity (RH) environments, the low vapor pressure induces the evaporation of some free water from BPGC-Gly, impeding the migration of internal ions and leading to the increased resistance. However, the presence of Gly in BPGC-Gly locks the moisture, preventing excessive water loss and maintaining stability in low RH environments. In high RH environments, due to the highly hydrophilic groups (amino, carboxyl, and hydroxyl groups) within BPGC-Gly, water molecules adsorb and condense on the hydrogel through chemical and physical means, causing a slight increase in the volume of the organohydrogel.49 Overall, the BPGC-Gly humidity sensor utilizes its moisture absorption and release properties to detect the RH changes. The conductance variation reflected the humidity level, allowing for accurate humidity sensing. Similarly, the RH changes could be observed through the change of electrical conductivity. As illustrated in Fig. 6f, with the increase of environmental RH, the conductivity of the BPGC-Gly humidity sensor also increased. Specifically, the conductivity increased from 0.15 S m−1 at 24.2% RH to 0.85 S m−1 at 97% RH. Furthermore, as shown in the inset of Fig. 6f, the conductivity of the BPGC-Gly humidity sensor was also effectively maintained in an open environment (21 °C, 60% RH). In addition, the conductance changes under different RH conditions were studied to evaluate the humidity sensing performance of the BPGC-Gly sensor. As illustrated in Fig. 6g, starting with an initial 24.2% RH, the BPGC-Gly sensor was placed in chambers with different RH levels (97%, 84%, 75%, 60%, and 46.7% RH) until the conductance no longer changed, and then transferred back to the initial environment until it recovered to the initial conductance value. Notably, the response ranges of the BPGC-Gly sensor were 12% and 2% in environments with 97% RH and 46.7% RH, respectively, demonstrating a wide humidity response range. Additionally, the humidity cycling stability of the BPGC-Gly sensor was also analyzed. As depicted in Fig. 6h, the standard deviation of the response curves for various humidity changes within 30 cycles did not exceed 3%, proving the reliability and stability of the humidity response of the BPGC-Gly humidity sensor. Especially, the response time and recovery time of the BPGC-Gly humidity sensor from RH 24.2% to RH 84% and then returning to the RH 24.2% environment were 104.2 s and 94.2 s, respectively, indicating that the BPGC-Gly sensor has the ability to quickly sense the humidity change (Fig. 6i).
To further explore the practical application potential of the BPGC-Gly sensor, it was fixed onto a mask to monitor real-time human respiration. As shown in Fig. 6j and k, the changes of relative conductance with time were clear and repeatable for different breathing patterns (irregular and regular modes), which proved that the BPGC-Gly sensor has the ability to recognize breath environments with different lengths and intensities. Meanwhile, due to the small size of the humidity chamber within the mask, the airflow generated by human respiration easily changes the RH around the sensor. Therefore, compared to regular chamber humidity tests, the time required for mask respiration detection was shorter. As depicted in Fig. 6k, under the regular human breathing mode, the response time of exhaled gas was 2.4 s, and the response time of inhaled gas was 1.6 s, indicating that BPGC-Gly possesses fast respiratory responsiveness. In conclusion, these results demonstrated that the BPGC-Gly sensor, with its dual sensing capabilities for temperature and humidity, has potential applications in diagnosing respiratory system diseases and sleep monitoring.
Strain/pressure sensing properties of the BPGC-Gly sensor
Based on its excellent mechanical properties, electrical conductivity, anti-freezing properties, biocompatibility and sensing stability, the BPGC-Gly organohydrogel could be used as a strain/pressure sensor. When BPGC-Gly is applied with a voltage, its internal porous structure provides channels for the directional movement of ions, thus forming an electric current. As shown in Fig. S11 (ESI†), when BPGC-Gly is stretched, the change in the cross-sectional area and length causes the internal channels to change, making it more difficult for the ions to move, which is reflected in an increase in BPGC-Gly's resistance and a decrease in the current. Similarly, when BPGC-Gly is compressed, the cross-sectional area increases and the length decreases, making the internal channels tighter, and the connection is tighter despite the reduced space for ion movement, which may make ion movement easier, resulting in a decrease in resistance and an increase in current. As shown in Fig. 7, the BPGC-Gly sensor could be used to monitor different movements of the human body (small, medium and large movements). As depicted in Fig. 7a, when the BPGC-Gly sensor was utilized to monitor the pronunciation of volunteers, the sensor could obtain the highly repetitive and reliable electrical signals when word and sentence (“Hi”, “Hello” and “How are you”) were repeatedly said. These results indicated that the BPGC-Gly sensor not only could detect small movements of the human body, but also had certain speech recognition capability. Meanwhile, when the BPGC-Gly sensor was attached to the volunteer's face skin, medium movements of smiling (Fig. 7b) and swallowing (Fig. 7c) could be detected and a continuous and stable signal output was obtained. Notably, the relative resistance signals in real time at different tensile frequencies and different bending angles could also be monitored by using the BPGC-Gly sensor. As shown in Fig. 7d and e, repeated movements of knee bending and walking at different frequencies could be clearly captured by the BPGC-Gly sensor and the changing frequency did not affect the shape of the peaks, proving that the sensor has stable and accurate performance for human monitoring. In addition, a series of human movements at different angles such as finger bending, wrist bending and elbow bending could also be clearly and completely recorded by the BPGC-Gly sensor (Fig. 7f–h). Thus, through the peak height, width and peak shape of the electrical signal, we could obtain the feedback on the health of human joint movement. In addition, the motion detection performance of the BPGC-Gly sensor in different environments was also investigated. As depicted in Fig. S11 (ESI†), the BPGC-Gly sensor was placed in −21 °C/49% RH, 20 °C/97% RH, and 20 °C/24.2% RH environments, respectively. After 60 h, the BPGC-Gly sensor was used to monitor the relative resistance variations over time during different human movements, producing stable characteristic peaks similar to those at room temperature. These results indicated that the BPGC-Gly sensor has good environmental adaptability and could be used for monitoring human motion in different temperature and humidity environments.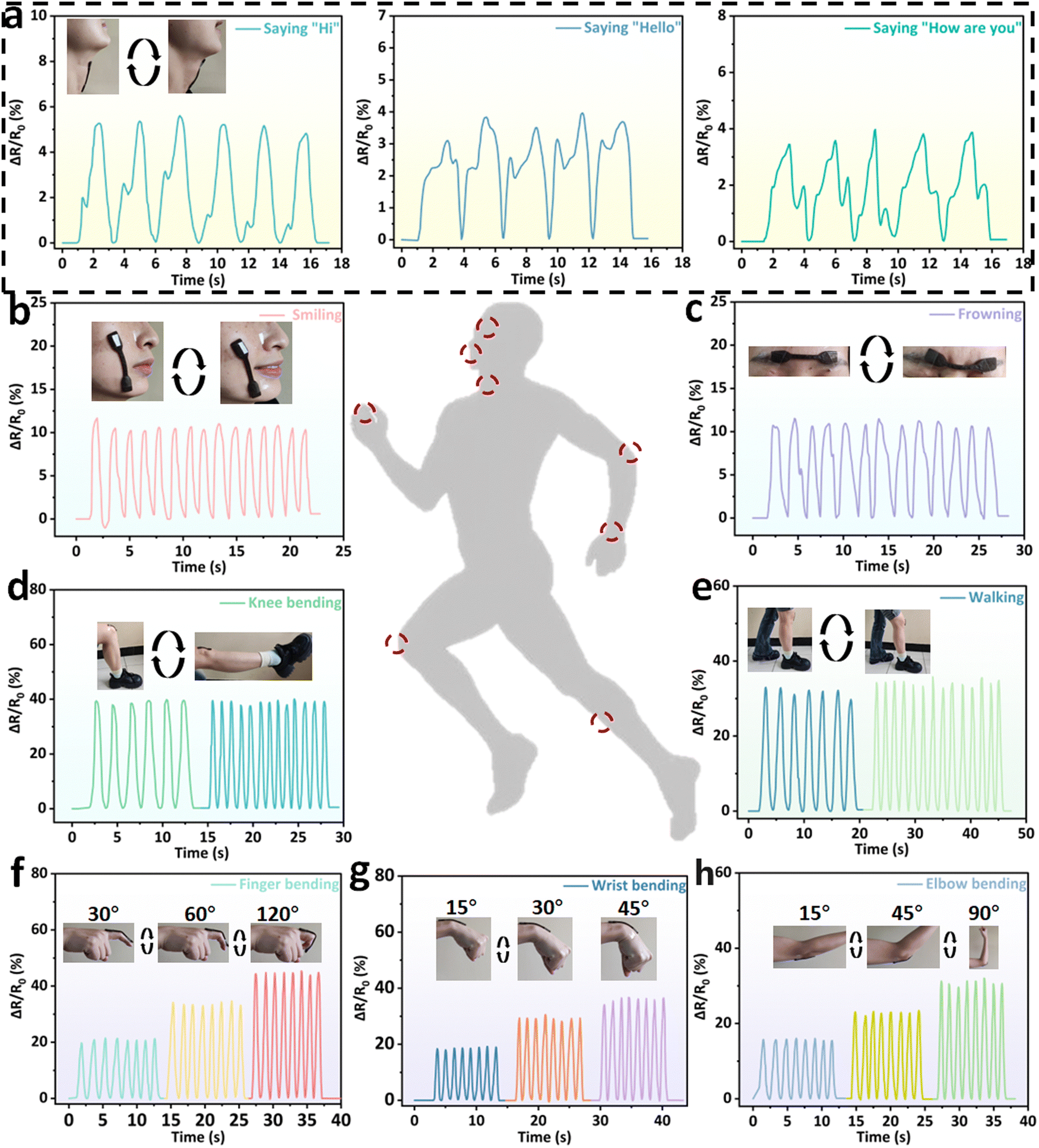 | ||
| Fig. 7 Strain sensing property of the BPGC-Gly sensor. Real-time ΔR/R0 change curves of BPGC-Gly as strain sensors to monitor various human movements, including human pronunciation (a), smiling (b) and frowning (c), knee bending (d) and walking (e) at different frequencies and finger (f), wrist (g) and elbow (h) bending at different angles. | ||
Based on the previous experiments, we know that BPGC-Gly exhibited pressure hypersensitivity, rapid responsiveness, and long-term stability. To further explore the potential of BPGC-Gly as a flexible pressure electronic device, we conducted a detailed analysis of its rapid responsiveness and stable feedback. As illustrated in Fig. 8a, under different frequencies of pressure stimulation, the peak heights of the curves of |ΔR|/R0 changing over time remained unchanged. Moreover, with increasing frequency, the response and recovery time of the signal peak also become shorter, indicating the reproducible and rapid feedback properties of BPGC-Gly at different frequencies. Additionally, during the continuous testing process under different pressures, almost no performance loss or interruption of continuous feedback was observed (Fig. 8b). BPGC-Gly with a highly porous structure exhibited a wide range of pressure (8.5–125 kPa) durability and stable feedback during compression. Its rapid responsiveness was demonstrated by compressing it from 0% to 50% and restoring it to its original state. As shown in Fig. 8c, BPGC-Gly could be almost restored to the original state and produce a fast response curve under rapid and sustained pressure. The aforementioned tests, including varying compression frequency, pressure, and compression-recovery tests, further confirm that BPGC-Gly possesses remarkable sensitivity and stability, making it a promising candidate for various applications in flexible pressure electronic devices.
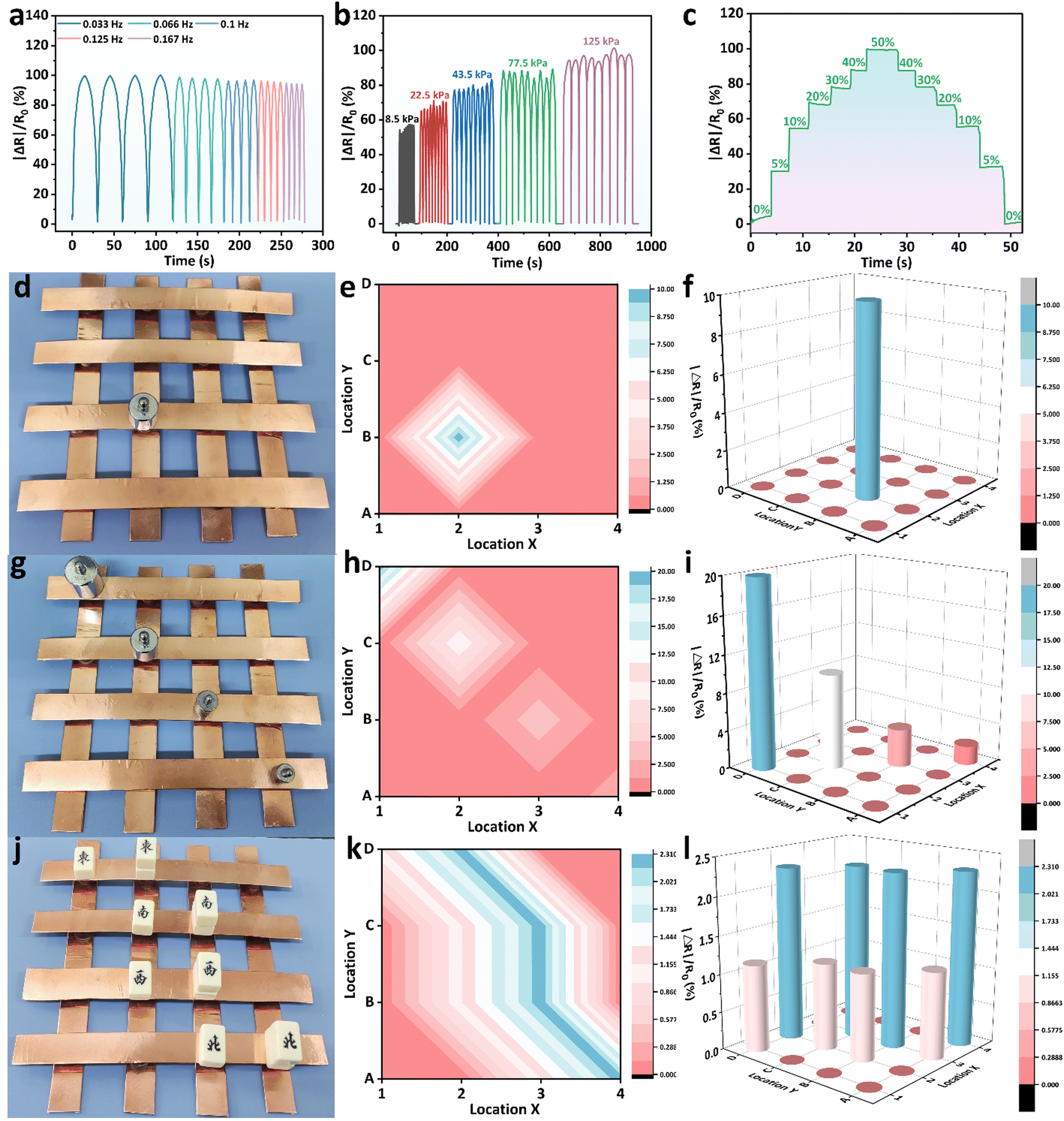 | ||
| Fig. 8 Pressure sensing property of the BPGC-Gly sensor. (a) The resistance change curves of the BPGC-Gly sensor at different pressure frequencies. (b) The resistance change curves of five compressing and releasing cycle tests of the BPGC-Gly sensor under different pressures. (c) The resistance change curves of the BPGC-Gly sensor compressing from 0% to 50% and releasing to the original state. The digital picture of 50 g weight (d) placed on the BPGC-Gly sensor array, and the corresponding 2D-mapped pressure distribution (e), and 3D-mapped response graph (f). The digital picture of different weights (g) placed on the BPGC-Gly sensor array, and the corresponding 2D-mapped pressure distribution (h), and 3D-mapped response graph (i). The digital picture of different numbers of rectangular blocks (j) randomly arranged on the BPGC-Gly sensor array, and the corresponding 2D-mapped pressure distribution (k), and 3D-mapped response graph (l). | ||
BPGC-Gly pieces were coupled with an electrochemical workstation to create a 4 × 4 matrix pressure sensor for detecting pressure distribution in space. The working principle was based on the compression of BPGC-Gly under external pressure, which increases the contact area and shortens the ion flow path, resulting in a change in BPGC-Gly's conductivity.50 Each BPGC-Gly sensor was shaped as a cylindrical patch (15 × 15 × 10 mm) for individual operation. When a 50 g weight was placed on a single unit of the array sensor (Fig. 8d), the 2D mapping pressure distribution (Fig. 8e) and the 3D pressure signal distribution (Fig. 8f) could be clearly observed. Similarly, by placing different weights (100 g, 50 g, 20 g, and 10 g) on the BPGC-Gly pressure array, the response signal decreased as the weight decreased. Notably, the BPGC-Gly array sensor could also accurately identify different external intensity pressures and integrate into 2D and 3D mapping images. As shown in Fig. 8g–i, by placing different rectangular blocks of varying quantities on the pressure sensor unit, accurate pressure position distribution and relative resistance intensity could be obtained graphically. The visualization application of the BPGC-Gly pressure array sensor has potential applications in the human–computer interaction system of multi-touch devices.
Electrophysiological monitoring property of the BPGC-Gly sensor
Currently, hydrogel sensors face challenges such as the need for an external power supply and difficulty in monitoring subtle physiological indicators like heartbeat and muscle contractions.51 Therefore, the development of a portable power device and sensitive wearable sensing equipment is particularly important. Due to its good biocompatibility, mechanical properties, electrical conductivity, and sensitivity, BPGC-Gly provided the possibility of detecting the human physiological index. In our work, BPGC-Gly was used as an electrode patch and bioelectrical signals of the human body were output in a microcomputer via Bluetooth transmission module in order to solve the above problem (Fig. S13, ESI†). Specifically, BPGC-Gly was made into an electrode pad (10 mm diameter and 1 mm thick) for detecting electromyography (EMG) and electrocardiogram (ECG) signals of volunteers.Fig. 9a illustrates the mechanism of using BPGC-Gly as a flexible electrode for bioelectric detection. Let's take muscle cells as an example. It is well known that muscle contraction begins in the central nervous system, where alpha motor neurons generate muscle contraction signals (neurons are mainly composed of dendrites, cell bodies, and axons).52 As shown in the left picture of Fig. 9a, when the signal has not reached the cell body, the neuron exhibits a resting potential (approximately −70 mV due to the outflow of K+ ions from the cell membrane). The neuron is in a polarized state at this time. However, when an organism is stimulated by a stimulus, such as a nerve impulse or chemical signal, the organism generates an excitatory or inhibitory electric signal along the motor neuron. When the electrical signal reaches the cell body, the balance of the polarized state is broken, and a significant influx of Na+ from outside the cell membrane causes an increase in the membrane potential of the muscle fiber (about +40 mV), known as depolarization (in the right picture of Fig. 9a). Lastly, the potential at this time is called the action potential, and its generation causes a change in the transmembrane potential difference, which prompts the electrons in the BPGC-Gly flexible electrode attached to the skin to move directionally.37,52 Therefore, the electrochemical signal curve of the generated bioelectricity can be obtained based on the potential difference between the reference and working electrodes.
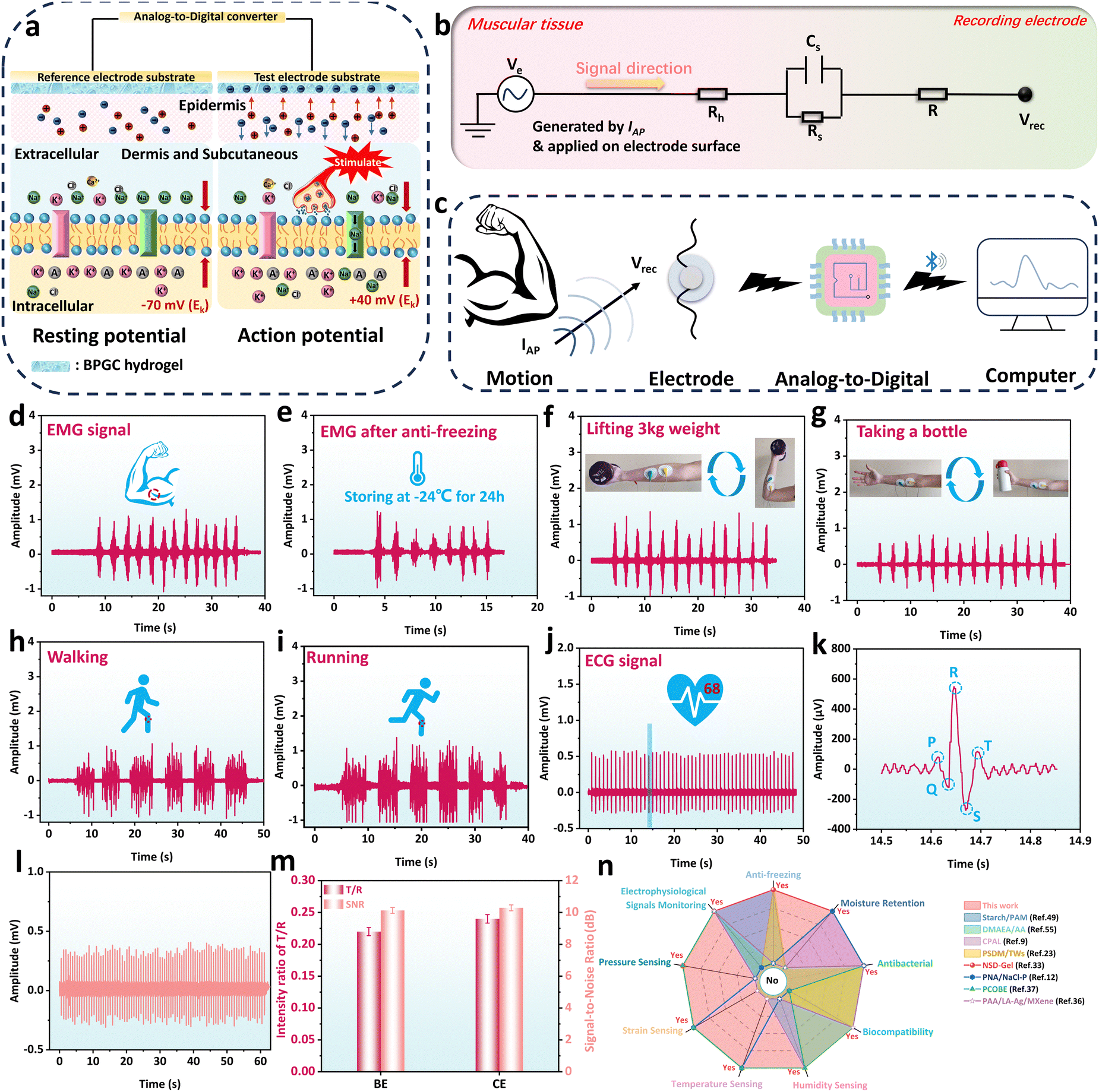 | ||
| Fig. 9 Electrophysiological monitoring property of the BPGC-Gly sensor. Diagram of the sensing mechanism of bioelectric signals (a), equivalent circuit model of bioelectric signals (b) and schematic diagram of the BPGC-Gly sensor for acquisition, monitoring and analysis of bioelectricity (c). EMG signals of spreading arm movements at room temperature (d) and low temperature (e). Real-time EMG signals for various movements, including lifting weight (f), taking a bottle (g), walking (h) and running (i). ECG signals (j) and amplification of 14.5–14.9 s ECG signals (k) monitored using BPGC-Gly. (l) The signals of ECG using a commercial electrode (CE). (m) The intensity ratio of T/R and SNR of the BPGC-Gly electrode (BE) and CE. (n) Comprehensive performance comparison of BPGC-Gly with previously reported multifunctional conductive hydrogels. | ||
Fig. 9b depicts the hydrogel-skin tissue equivalent model for bioelectrical recording. Generally, human skin tissue cells consist of stratum corneum, epidermal cells, and dermal tissue.53 These three structures could generally be regarded as body impedance and skin impedance. The skin impedance is composed of the equivalent resistance Rs (which has a higher impedance and can reach resistivity values of 100–1000 KΩ m) of the stratum corneum and the equivalent capacitance Cs between the epidermis and dermis.54,55 Since the dermis and subcutaneous tissue mainly consist of cells and a large amount of extracellular matrix, their equivalent impedance is smaller and can be represented as the resistance Rh.37,52 Additionally, Fig. 9c demonstrates the process of bioelectrical signal acquisition, detection, and analysis. Specifically, the electrical signals generated by movement are conducted through neural and muscular tissues, causing electron migration within the BPGC-Gly flexible electrode, which is in contact with the human epidermis. Subsequently, bioelectric signals are acquired by collecting the potential difference between the working and reference electrodes. And then, these signals were conveyed to the core control circuitry through the analog-to-digital conversion circuitry, and transmitted to a computer terminal in real-time via Bluetooth for real-time data detection and analysis. Compared to traditional bioelectrodes, the BPGC-Gly electrode exhibits excellent sensitivity, flexibility, stability, reusability, and low impedance. These characteristics are advantageous for maintaining a high signal-to-noise ratio in flexible electrodes, thereby enhancing the clarity and reliability of bioelectrical signals.
As illustrated in Fig. 9d and e, the bioelectric acquisition device based on BPGC-Gly was used for muscle signal detection at room temperature (25 °C) and low temperature (−20 °C). It could be found that prolonged elbow flexion training showed stable and repetitive EMG signals, demonstrating the ability of the BPGC-Gly flexible electrode to monitor subtle muscle signals of the human body, as well as its tolerance to low-temperature environments. Based on the portability and sensitivity of the BPGC-Gly bioelectrode acquisition device, we have attempted to apply it to motion monitoring in other parts of the human body. As depicted in Fig. 9f and g, BPGC-Gly could adhere completely to the arm skin for signal transmission. We placed the reference electrode adjacent to the working electrode and grounded the ground electrode on the arm bone. By performing repetitive weightlifting and cup-grasping motions, stable motion signal curves were obtained. Through comparison, it could be observed that different motion activities of the same muscle resulted in varying peak signal intensities. This phenomenon allowed us to identify different actions on the same body part based on peak intensity. Additionally, we further utilized BPGC-Gly for monitoring large-scale body movements such as walking and running. As shown in Fig. 9h and i, the bioelectrical signal intensity and waveform obtained during fast walking and fast running (with consistent stride length) are essentially the same. However, due to the higher frequency of running movements, the corresponding electrical signals displayed a faster frequency state. Notably, the ECG signals of volunteers could also be clearly and completely detected, and the heart rate of the volunteers could be predicted by detecting the signal peak, which was compared with the heart rate of the smart bracelet. It was found that they both showed 68 beats per minute (Fig. 9j). Meanwhile, the magnified ECG could display characteristic peaks of the heartbeat, including P-wave, QRS complex and T-wave, which are key indicators of symptoms such as arrhythmia and myocarditis (Fig. 9k). Due to the importance of the T/R ratio and signal-to-noise ratio (SNR) as key indicators of accuracy in ECG detection devices, a comparison was made between the BPGC-Gly electrode (BE) and commercial electrode pads (CE) in terms of these two parameters. As shown in Fig. 9l, the ECG waveform detected by CE was consistent in shape with that of BE. Additionally, the T/R ratio for BE and CE was measured to be 0.22 and 0.24, respectively, while the SNR was reported as 10.29 and 10.13, respectively (Fig. 9m). These results indicated that BE exhibited comparable monitoring accuracy to commercial electrodes, making it suitable for ECG/EMG monitoring during muscle movements. The above findings showed that BPGC-Gly has excellent micro signal sensing performance and is expected to be used as a portable bioelectrode and for human–computer interaction, human motion monitoring, and heart disease monitoring through a complete data acquisition, reception, and analysis process. Finally, to showcase the versatility of the BPGC-Gly sensor, it was qualitatively and quantitatively compared with previously reported hydrogel sensors (Fig. 9n and Table S4, ESI†). The results indicated that the BPGC-Gly sensor possessed excellent properties, including anti-freezing, moisturizing, antibacterial, and biocompatibility. Additionally, BPGC-Gly exhibited a diverse range of multifunctional sensing capabilities, encompassing temperature, humidity, strain sensing, pressure sensing, and biological signals. This brings new possibilities to the field of multifunctional conductive hydrogels.
Conclusions
In summary, a multifunctional conductive organohydrogel BPGC-Gly was designed and fabricated, utilizing waste collagen fibers (BD), Gel, and PVA as the basic framework, loaded with a conductive antibacterial material (silver nanoparticles (BD-CQDs@AgNPs)) and the anti-freezing moisturizer Gly. The resulting BPGC-Gly displayed a natural fiber-like structure, boasting exceptional mechanical properties, conductivity, anti-freezing properties, and moisturizing properties. In terms of structure, the introduction of BD endowed BPGC-Gly with a “surface synapse” and an internal multilayered fiber network, enabling high sensitivity with an S value of up to 4.26 kPa−1 under low pressure conditions (0–12.5kPa). In terms of performance, the unique multilevel fiber skeleton structure of BPGC-Gly, coupled with the hydrogen bonding interactions among different substances, served as a dissipative pathway. Consequently, BPGC-Gly possessed the advantages of “three highs” (high tensile strength (0.72 MPa), high elongation (270%), and high toughness (0.86 MJ m−3)). In terms of applications, based on its outstanding properties, this leather solid waste-derived multifunctional organohydrogel exhibited diverse modal sensing capabilities, including temperature, humidity, strain, pressure, and bioelectrical signals. As a temperature and humidity sensor, it demonstrated an excellent temperature response range (−20–60 °C) and a wide humidity detection range (24.2–97% RH), with rapid response (2.4 s) and recovery (1.6 s) to human respiration. As a strain/pressure sensor, the fabricated BPGC-Gly sensor has a wide response range and high sensitivity, enabling real-time monitoring of human movements. Notably, BPGC-Gly could also be used as a portable bioelectrode for collecting and analyzing bioelectrical signals (such as ECG and EMG) and could serve as a supplementary means for motion monitoring. In conclusion, this multidimensional and multimodal human health monitoring based on the multifunctional BPGC-Gly can provide comprehensive and complementary health monitoring data. Therefore, BPGC-Gly holds great potential in the fields of human health monitoring devices, wearable electronic devices, and human–machine interactions.Author contributions
Zhen He: conceptualization, methodology, investigation, writing – original draft, writing – review & editing. Jialu Shen: conceptualization, methodology, writing – review & editing. Manhua Lan: conceptualization, methodology, writing – review & editing. Haibin Gu: conceptualization, methodology, supervision, writing – review & editing, project administration, resources, funding acquisition.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21978180) is gratefully acknowledged. The authors appreciate the valuable help of Dr Zhonghui Wang from the College of Biomass Science and Engineering of Sichuan University and Hui Wang from the Analytical & Testing Center of Sichuan University. We thank eceshi (www.eceshi.com) for the TEM, XPS, and ICP test.References
- Y. Y. Lu, G. Yang, S. Q. Wang, Y. Q. Zhang, Y. H. Jian, L. He, T. Yu, H. Y. Luo, D. P. Kong, Y. L. Xianyu, B. Liang, T. Liu, X. P. Ouyang, J. C. Yu, X. Y. Hu, H. Y. Yang, Z. Gu, W. Huang and K. C. Xu, Nat. Electron., 2024, 7, 51–65 CrossRef CAS
.
- R. Y. Zhang, M. Hummelgård, J. Örtegren, M. Olsen, H. Andersson, Y. Yang, H. Olin and Z. L. Wang, Nano Energy, 2022, 100, 107503 CrossRef CAS
- G. M. Whitesides, Angew. Chem., Int. Ed., 2018, 57, 4258–4273 CrossRef CAS
- S. Shu, Z. M. Wang, P. F. Chen, J. W. Zhong, W. Tang and Z. L. Wang, Adv. Mater., 2023, 35, 2211385 CrossRef CAS
- F. Chen, Q. N. Zhuang, Y. C. Ding, C. Zhang, X. Song, Z. J. Chen, Y. K. Zhang, Q. J. Mei, X. Zhao, Q. Y. Huang and Z. J. Zheng, Adv. Mater., 2023, 35, 202305630 Search PubMed
- C. W. Zhi, S. Shi, S. Zhang, Y. Si, J. Q. Yang, S. Meng, B. Fei and J. L. Hu, Nano-Micro Lett., 2023, 15, 60 CrossRef
- I. Van Nieuwenhove, A. Salamon, K. Peters, G.-J. Graulus, J. C. Martins, D. Frankel, K. Kersemans, F. De Vos, S. Van Vlierberghe and P. Dubruel, Carbohydr. Polym., 2016, 152, 129–139 CrossRef CAS
- H. Q. Sun, S. Wang, F. Yang, M. Y. Tan, L. Bai, P. P. Wang, Y. Y. Feng, W. B. Liu, R. G. Wang and X. D. He, Mater. Horiz., 2023, 10, 5805–5821 RSC
- L. Tang, Y. W. Li, F. Liu, S. J. Wu, W. Wang, X. L. Sun, Z. H. Chen and J. X. Tang, ACS Appl. Electron. Mater., 2023, 5, 5651–5660 CrossRef CAS
- M. Wang, H. Chen, X. X. Li, G. Wang, C. Peng, W. Wang, F. Zhang, J. Q. Wang, H. H. Liu, G. Q. Yan and H. L. Qin, J. Mater. Chem. A, 2022, 10, 24096–24105 RSC
- L. H. Geng, W. Liu, B. B. Fan, J. M. Wu, S. Shi, A. Huang, J. L. Hu and X. F. Peng, Chem. Eng. J., 2023, 462, 142226 CrossRef CAS
- Q. Wang, Q. Zhang, G. Y. Wang, Y. R. Wang, X. Y. Ren and G. H. Gao, ACS Appl. Mater. Interfaces, 2021, 14, 1921–1928 CrossRef
- X. L. He, J. Dong, X. N. Zhang, X. Y. Bai, C. Zhang and D. S. Wei, Chem. Eng. J., 2022, 435, 135168 CrossRef CAS
- Q. Pang, H. T. Hu, H. Q. Zhang, B. B. Qiao and L. Ma, ACS Appl. Mater. Interfaces, 2022, 14, 26536–26547 CrossRef CAS PubMed
- K. N. Calahan, Y. Qi, K. G. Johannes, M. E. Rentschler and R. Long, Sci. Adv., 2022, 8, eabn2728 CrossRef CAS PubMed
- D. Oran, S. G. Rodriques, R. X. Gao, S. Asano, M. A. Skylar-Scott, F. Chen, P. W. Tillberg, A. H. Marblestone and E. S. Boyden, Science, 2018, 362, 1281–1285 CrossRef CAS PubMed
- S. Han, M. C. Lee, A. Rodríguez-delaRosa, J. Kim, M. Barroso-Zuppa, M. Pineda-Rosales, S. S. Kim, T. Hatanaka, I. K. Yazdi, N. Bassous, I. Sinha, O. Pourquié, S. Park and S. R. Shin, Adv. Funct. Mater., 2023, 34, 2304153 CrossRef PubMed
- J. H. Huang, G. Chen, T. H. Han, C. X. Yi, Y. Zhang, L. Ding, T. S. Sun, T. Jin and S. Zhou, Carbohydr. Polym., 2023, 319, 121196 CrossRef CAS PubMed
- Q. Y. Meng, Z. W. Ye, Y. Y. Wang, C. C. Liu, Q. F. Sun, J. L. Shamshina and X. P. Shen, Adv. Funct. Mater., 2023, 2304104 Search PubMed
- D. Masilamani, N. Ariram, B. Madhan and S. Palanivel, J. Cleaner Prod., 2023, 415, 137705 CrossRef CAS
- Y. Pei, S. Chu, Y. R. Zheng, J. H. Zhang, H. Liu, X. J. Zheng and K. Y. Tang, ACS Sustainable Chem. Eng., 2019, 7, 2530–2537 CrossRef CAS
- N. V. Salim, X. Jin, S. Mateti, H. Lin, V. Glattauer, B. Fox and J. A. M. Ramshaw, Mater. Today Adv., 2019, 1, 100005 CrossRef
- Y. Wei, B. Hao, Y. Wang, Y. Wang, H. Xiao, L. Li, X. Huang and B. Shi, J. Mater. Chem. C, 2022, 10, 8199–8205 RSC
- W. Hittini, A.-H. I. Mourad and B. Abu-Jdayil, J. Cleaner Prod., 2019, 236, 117603 CrossRef CAS
- P. M. Karmegam, P. Natarajan and S. Somasundaram, Chem. Eng. J., 2023, 451, 138553 CrossRef CAS
- L. Tian, Z. Li, P. Wang, X. H. Zhai, X. Wang and T. X. Li, J. Energy Chem., 2021, 55, 279–294 CrossRef CAS
- Y. Li, J. Gao, Y. X. Shi, Y. Z. Wang, M. Y. Li, A. Z. Pan, M. Y. Hu and G. J. Zhang, Carbon, 2024, 218, 118653 CrossRef CAS
- Z. He, J. C. Liu, X. Fan, B. Song and H. B. Gu, Ind. Eng. Chem. Res., 2022, 61, 17915–17929 CrossRef CAS
- P. Lee, A. Saura-Sanmartin and C. A. Schalley, Adv. Funct. Mater., 2023, 34, 2309140 CrossRef
- X. S. Zhang, H. W. Yan, C. Z. Xu, X. Dong, Y. Wang, A. P. Fu, H. Li, J. Y. Lee, S. Zhang, J. H. Ni, M. Gao, J. Wang, J. P. Yu, S. S. Ge, M. L. Jin, L. Wang and Y. Z. Xia, Nat. Commun., 2023, 14, 5010 CrossRef CAS PubMed
- R. R. Xue, Y. M. Zou, Z. Q. Wang, L. Mao, H. L. Wang, M. Zhang, A. Shao, J. C. Liu, N. Yao, Y. Y. Liu and Y. Ma, ACS Nano, 2023, 17, 17359–17371 CrossRef CAS PubMed
- B. Song, X. Fan and H. B. Gu, ACS Appl. Mater. Interfaces, 2022, 15, 2147–2162 CrossRef PubMed
- Z. X. Bai, X. C. Wang, M. H. Zheng, O. Y. Yue, M. C. Huang, X. L. Zou, B. Q. Cui, L. Xie, S. Y. Dong, J. J. Shang, G. D. Gong, A. M. Blocki, J. Guo and X. Liu, Adv. Funct. Mater., 2023, 33, 2212856 CrossRef CAS
- J. Wu, Z. X. Wu, H. H. Xu, Q. Wu, C. Liu, B. R. Yang, X. C. Gui, X. Xie, K. Tao, Y. Shen, J. M. Miao and L. K. Norford, Mater. Horiz., 2019, 6, 595–603 RSC
- Z. He, J. L. Shen, J. W. Zhang, W. Lin and H. B. Gu, ACS Sustainable Chem. Eng., 2023, 11, 13126–13141 CrossRef CAS
- L. Jiang, S. He, A. Liu, J. Zhang, J. Liu, S. R. He and W. Shao, Composites, Part B, 2022, 239, 109982 CrossRef CAS
- B. Song, X. Fan, J. L. Shen and H. B. Gu, Chem. Eng. J., 2023, 474, 145780 CrossRef CAS
- Y. H. Xiao, J. J. Zhou, C. H. Wang, J. W. Zhang, V. D. Radnaeva and W. Lin, Collagen Leather, 2023, 5, 2 CrossRef CAS
- Q. J. Chen, Y. Pei, K. Y. Tang and M. G. Albu-Kaya, Collagen Leather, 2023, 5, 20 CrossRef CAS
- X. Fan, T. Ke and H. B. Gu, Adv. Funct. Mater., 2023, 33, 2304015 CrossRef CAS
- L. Q. Peng, X. Zhang, L. J. Guo, J. H. Li, W. H. Zhang and B. Shi, J. Cleaner Prod., 2023, 414, 137653 CrossRef CAS
- L. L. Wang, X. Chen, S. H. Wang, J. R. Ma, X. X. Yang, H. L. Chen and J. X. Xiao, Adv. Healthcare Mater., 2024, 2302833 CrossRef CAS PubMed
- Q. Yang, D. H. Gao, R. Y. Miao, C. J. Qin, F. Song, W. J. Zhao, N. Zhao and H. Liu, J. Mater. Sci., 2023, 58, 1739–1751 CrossRef CAS
- O. F. Farag and E. Abdel-Fattah, J. Polym. Res., 2023, 30, 183 CrossRef CAS
- O. Duman, C. Ö. Diker, H. Uğurlu and S. Tunç, Carbohydr. Polym., 2022, 286, 119275 CrossRef CAS PubMed
- Q. Zhang, X. Liu, J. W. Zhang, L. J. Duan and G. H. Gao, J. Mater. Chem. A, 2021, 9, 22615–22625 RSC
- Y. Fang, C. Q. Hong, F. R. Chen, F. Z. Gui, Y. X. You and X. Guan,
et al.
, Inorg. Chem. Commun., 2021, 132, 108808 CrossRef CAS
- K. Saranya, G. C. Jayakumar, N. Usharani, A. Sundaramanickam and S. V. Kanth, J. Colloid Interface Sci., 2022, 608, 3141–3150 CrossRef PubMed
- S. Zeng, J. Y. Zhang, G. Q. Zu and J. Huang, Carbohydr. Polym., 2021, 267, 118198 CrossRef CAS PubMed
- M. Z. Chen, H. X. Wan, Y. Hu, F. Y. Zhao, X. N. An and A. Lu, Mater. Horiz., 2023, 10, 4510–4520 RSC
- H. Wang, Q. S. Ding, Y. B. Luo, Z. X. Wu, J. H. Yu, H. Z. Chen, Y. B. Zhou, H. Zhang, K. Tao, X. L. Chen, J. Fu and J. Wu, Adv. Mater., 2023, 2309868 Search PubMed
- H. Yuk, B. Y. Lu and X. H. Zhao, Chem. Soc. Rev., 2019, 48, 1642–1667 RSC
- N. R. Barros, H.-J. Kim, M. J. Gouidie, K. J. Lee, P. Bandaru, E. A. Banton, E. Sarikhani, W. J. Sun, S. M. Zhang, H.-J. Cho, M. C. Hartel, S. Ostrovidov, S. Ahadian, S. M. Hussain, N. Ashammakhi, M. R. Dokmeci, R. D. Herculano, J. M. Lee and A. Khademhosseini, Biofabrication, 2021, 13, 035030 CrossRef CAS PubMed
- R. Matsukawa, A. Miyamoto, T. Yokota and T. Someya, Adv. Healthcare Mater., 2020, 9, 2001322 CrossRef CAS PubMed
- R. J. Liu, J. T. Wang, G. F. Li, Z. Y. Fan, Q. Zhou, K. Wang and W. Huang, Adv. Funct. Mater., 2023, 33, 2214917 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00374h |
| This journal is © The Royal Society of Chemistry 2024 |