
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtracellular matrix mimetic supramolecular hydrogels reinforced with covalent crosslinked mesoporous silica nanoparticles†
Aygül
Zengin‡
,
Shahzad
Hafeez‡
,
Pamela
Habibovic
 ,
Matthew
Baker
and
Sabine
van Rijt
*
,
Matthew
Baker
and
Sabine
van Rijt
*
Department of Instructive Biomaterials Engineering, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, P. O. Box 616, 6200 MD Maastricht, The Netherlands. E-mail: s.vanrijt@maastrichtuniversity.nl
First published on 7th November 2024
Abstract
The extracellular matrix (ECM) is a dynamic environment that is primarily built up from fibrous proteins (e.g., elastins, fibronectins, collagens, and laminins) and plays a vital role in tissue regeneration processes. Therefore, the development of supramolecular hydrogels that can mimic the ECM's dynamicity and fibrous structure is of great interest in regenerative medicine. However, such hydrogels generally have weak mechanical properties and poor structural stability, which significantly limits their potential applications. To overcome this drawback, we developed a new type of hybrid network composed of supramolecular assemblies with covalent nanoparticle-based crosslinkers. The ECM mimetic hydrogels were created through UV-initiated thiol–ene crosslinking between norbornene functionalized benzene-1,3,5-tri carboxamide (NBTA) macromonomers and thiol functionalized mesoporous silica nanoparticles (MSN). We hypothesized that the MSN would improve the mechanical properties by crosslinking the NBTA supramolecular fibrous hydrogels. Notably, the covalent incorporation of MSNs did not disrupt the fibrous morphology of the resulting NBTA–MSN nanocomposites. Furthermore, these supramolecular nanocomposites demonstrated higher structural stability and elasticity compared to pristine NBTA hydrogels. Rheology studies showed that the mechanical properties of NBTA–MSN hydrogels could be tuned by adjusting MSN wt%. Interestingly, NBTA–MSN nanocomposites exhibited self-healing and injectability despite the covalent crosslinking of MSNs. In vitro studies confirmed that NBTA–MSN nanocomposites showed good cytocompatibility and maintained the viability of encapsulated MG63 cells. As a proof of concept, we also demonstrated that MSNs could act as ion reservoirs for calcium and phosphate within the hydrogel networks in addition to being covalent crosslinkers. Taken together, our work offers a promising strategy to create hybrid, biomimetic supramolecular nanocomposite materials for various applications such as injectable materials for bone tissue engineering, and reinforced bioinks for 3D printing applications.
1. Introduction
In native tissues, the extracellular matrix (ECM) dynamically supports tissue regeneration by providing biophysical and biochemical cues that control stem cell fate.1 The ECM consists of a fibrous three-dimensional (3D) supramolecular network where reversible non-covalent interactions facilitate its hierarchical assembly.2,3 The ECM dynamic environment regulates stem cell growth, differentiation, and enables communication within living tissues.4 Therefore, a key goal in the field of biomaterials is to develop synthetic ECMs that closely mimic the 3D structure and dynamic behaviour of natural ECMs, as this is crucial for controlling stem cell behaviour and the successful regeneration of complex living tissue. Supramolecular hydrogels are ideal materials since they allow the recreation of self-assembled structures that can mimic the fibrous structure, dynamicity, bioactivity, and complexity of the ECM. Supramolecular hydrogels are formed via the self-assembly of building blocks via noncovalent interactions such as hydrogen bonding, hydrophobic interactions, and π–π stacking. Reversibility, specificity, and directionality of noncovalent interactions enable tuning of structure, dynamicity, and mechanical and biological properties of synthetic hydrogel. Various ECM mimetic supramolecular hydrogelators have been developed such as peptide amphiphile,5 Ureido-pyrimidinone (UPy),6 bis-urea (BU),7 and self-assembling peptide hydrogels (SAPHs).8,9 ECM mimetic hydrogels have been shown to significantly influence stem cell fate, tissue formation, and organoid culture.10–12Benzene-1,3,5-tricarboxamide (BTA) is a promising supramolecular building block because it can self-assemble into one-dimensional (1D) nanofibres and has a tunable chemical structure.13,14 At high BTA concentrations, these nanofibres form mechanically tunable hydrogels. However, similar to other types of supramolecular hydrogels, the poor mechanical properties of BTA hydrogels significantly limit their applicability in stiffer tissues and organs.14–16 Therefore, strategies to improve the mechanical properties such as incorporating both non-covalent and covalent networks within supramolecular hydrogels are actively being explored.15,17–19
For example, the incorporation of nanoparticles into supramolecular hydrogels is a powerful approach to improve mechanical properties, and can also introduce additional functionalities (e.g. electrical and thermal conductivity, antibacterial properties, drug delivery) to the resulting hybrid hydrogels.20–24 Most of these studies incorporated diverse nanoparticles (carbon nanotubes, graphene oxide, cellulose, clay) as non-covalent crosslinkers or nanofillers within supramolecular networks. Among these, mesoporous silica nanoparticles (MSN) are promising nanomaterials to create organic/inorganic hybrid matrices. They can be easily functionalized to reinforce hydrogels, possess inherent bioactivity, and serve as efficient drug delivery vehicles through their uniform mesoporous structure.25 Previously, our work demonstrated that surface modification of MSNs with different chemical groups enables them to interact with polymer backbones, going beyond their roles as nanofillers. Specifically, employing MSNs as dynamic covalent crosslinkers within polyethylene glycol (PEG) hydrogels led to a remarkable increase in the storage modulus of PEG hydrogels up to 25-fold. The resulting nanocomposite was injectable and self-healing. We were also able to modulate the swelling, degradation, and drug release behaviour by adjusting MSN concentration within the network.26 In a separate study, we showed that MSNs as covalent crosslinkers in photocrosslinkable hydrogels through UV-initiated thiol–ene chemistry could also improve the mechanical properties.27 Additionally, other studies in the literature have indicated that MSNs can improve the mechanical properties of hydrogels such as PEG diacrylate (PEGDA), gelatin methacryloyl (GelMA), and chitosan.28–30 However, the impact of combining MSNs and fibrous hydrogels remains unexplored.
In this study, we integrated MSNs as covalent crosslinkers into ECM mimetic supramolecular hydrogels to enhance their mechanical properties. These nanocomposites (NBTA–MSN) were formed through thiol–ene crosslinking between norbornene-functionalized BTA macromonomers (NBTA) and thiol-functionalized MSNs upon UV exposure. To the best of our knowledge, this is the first study that incorporates nanoparticles as covalent crosslinkers into supramolecular networks through UV light-initiated thiol–ene chemistry. We systematically investigated the mechanical properties, self-healing behaviour, and injectability of the NBTA–MSN hydrogels. Moreover, the cytocompatibility of these nanocomposites was analyzed by encapsulating human osteoblasts-like cells (MG-63) within the hydrogel network. Finally, we evaluated the stability of designed NBTA–MSN hydrogels in a modified version of body-stimulated fluid (m-SBF).
2. Materials and methods
2.1 Materials
Tri-ethanolamine (TEA), tetraethyl orthosilicate (TEOS), 3-aminopropyl triethoxysilane (APTES), cetyltrimethylammonium chloride (CTAC), absolute ethanol, hydrochloric acid (HCl, 37%), ammonium fluoride (AF), ammonium nitrate (NH4NO3), and ATTO 647N-maleimide, 12-mercaptododecylphosphonic, Dulbecco's phosphate buffered saline, di-isoprpylethyl amine (DIPEA), dichloromethane (DCM), and diethyl ether was purchased from Sigma Aldrich. Polyethylene glycol (PEG) 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, 1,1 carbonyldiimidazole, and 1,12-diaminododecane were bought from Sigma Aldrich. Norbornene-2-methylamine (a mixture of isomers) was bought from TCI Chemicals.
000 g mol−1, 1,1 carbonyldiimidazole, and 1,12-diaminododecane were bought from Sigma Aldrich. Norbornene-2-methylamine (a mixture of isomers) was bought from TCI Chemicals.
2.2 MSN synthesis and characterization
MSNs with thiol surface modifications (MSN-SH) were synthesized according to previously published protocol with minor modifications.26 First, a mixture of TEA (14.3 g, 95.6 mmol) and TEOS (1.466 g, 7.37 mmol) was heated at 90 °C for 20 min under static conditions (solution 1). A mixture of ammonium fluoride (100 mg, 2.7 mmol) and CTAC (2.41 mL, 1.83 mmol) in 21.7 mL Mili Q-water (solution 2) was heated at 60 °C for 20 min. Solution 2 was quickly added to solution 1 at room temperature (RT), and the mixture was left stirring at 700 rpm for 20 min to cool down. Then, TEOS (138.2 mg, 0.922 mmol) was added to the solution mixture in four equal increments every 3 min, and the mixture was left stirring for another 30 min at RT. To introduce free thiol groups on the MSN surface, TEOS (19.3 mg, 92.5 μmol) and MPTES (20.5 mg, 92.5 μmol) were added to the mixture and left stirring overnight at RT. The particles were collected by centrifugation at 7800 rpm for 20 min and washed with absolute ethanol. For organic template removal, the particle solution was heated for 45 min under reflux (90 °C) in an acidic ethanol solution (2 g NH4NO3 in 100 mL ethanol). MSNs were collected by centrifugation and washed with absolute ethanol before the second template extraction in 100 ml of a 3.7% hydrochloric acid solution in ethanol for 45 min at 90 °C. After two washing steps with absolute ethanol, the MSN suspension was stored at −20 °C in absolute ethanol.Dynamic light scattering (DLS) using a Malvern Zetasizer Nano (Sysmex, NL) was used to analyze the hydrodynamic diameter and zeta potential of synthesized MSNs (0.5 mg mL−1). Scanning electron microscopy (SEM; Teneo, FEI, US) and transmission electron microscopy (TEM; FEI Tecnai electron microscope) were used to visualize the shape and porosity of the nanoparticles. For SEM imaging, the samples were sputter-coated with 4 nm iridium. For TEM imaging, MSN suspension in ethanol was dropped on a TEM carbon grid and imaged after air drying at RT. The successful thiol functionalization on the MSN surface was confirmed with ATTO 647N-maleimide labeling, following the procedure described earlier.26
2.3 MSN-CaP synthesis and characterization
For MSN-CaP particles, we first synthesized amine surface-functionalized MSNs by following a previously reported protocol with minor changes.31 In our protocol, free amine groups were introduced by the addition of APTES (20.5 mg, 92.5 μmol) and TEOS (19.3 mg, 92.5 μmol) mixture followed by four incremental TEOS additions. Later, the amine groups on the MSN surface were converted to carboxylic acid groups. Briefly, 3 g of succinic anhydride was dissolved in 20 mL DMF solution and left stirring for 30 min at RT (mixture 1). Next, 0.2 g of MSNs were re-suspended in 20 mL DMF solution (mixture 2) and it was added to mixture 1 and left stirring at 60 °C for 48 h. The MSNs were collected via centrifugation and washed three times with absolute ethanol. To form calcium phosphate coating on carboxyl functionalized MSN surface, 100 mg of MSNs were re-suspended in first CaCl2 (5 mM in Mili-Q water, pH 9) solution and the mixture was stirred for 30 min. After the nanoparticles were collected by centrifugation, they were washed with Mili-Q water, pH 9. Later, MSNs were re-suspended in Na2PO4 solution (3 mM in Mili-Q water, pH 9) to introduce the phosphate layer to the MSN surface and the mixture was left to stir for 30 min. Alternating phosphate and calcium ion addition was repeated two more times. After the last phosphate coating step, MSNs were collected by centrifugation and washed with Mili-Q water. In the last step, calcium phosphate-coated MSNs were dispersed in a 12-mercaptodecylphosphonic acid solution (1 mM in Mili-Q water, pH 9) to introduce free thiol groups on the MSN surface.2.4 Synthesis of bis-norbornyl amide mono-pentafluoroester BTA precursor (perfluorophenyl 3,5-bis((bicyclo[2.2.1]hept-5-en-2 ylmethyl)carbamoyl)benzoate)
The tris(perfluorophenyl) benzene-1,3,5-tricarboxylat, the (perfluorophenyl 3,5-bis((bicyclo[2.2.1]hept-5-en-2-ylmethyl)carbamoyl)benzoate), was synthesized in our previously reported study14 and used in this study for synthesis of NBTA macromonomer. Prior to use for synthesis of the NBTA macromonomer, the 1H NMR spectrum was matched with the previously reported characterization of this molecule.162.5 Synthesis of NBTA macromonomers
A dry round-bottom flask was charged with the BTA precursor (perfluorophenyl 3,5-bis((bicyclo[2.2.1]hept-5-en-2-ylmethyl)carbamoyl)benzoate) (0.24 g, 0.40 mmol, 2.2 equiv.) and dissolved in 10 mL anhydrous DCM.14 Subsequently, dry DIPEA (96 μL, 0.55 mmol, 3 equiv.) was added into the reaction flask. Then, PEG bisaminododecane (3.75 g, 0.18 mmol, 1 equiv.) was dissolved in anhydrous DCM (10 ml) by vigorous stirring and was added dropwise to the reaction flask.14 The reaction mixture was stirred for 40 hours at room temperature (∼20 °C) under a nitrogen atmosphere. Excess solvent was removed in a vacuum and the crude reaction mixture was precipitated in excess cold diethyl ether twice and NBTA macromolecule was obtained as a white powder in 82% yield (3.2 g). 1NMR of the compound was matched to the previously synthesized compound to confirm that the molecule is pure.142.6 Molecular weight measurements
Gel permeation chromatography (GPC) was performed using N,N-dimethylformamide (DMF) containing 0.035 M LiCl as eluent. The Alliance Waters e2695 separation module was employed which comprised of a GRAM precolumn 10 μm (8 × 50 mm), followed by a GRAM column 10 μm 30 Å (8 × 300 mm), and a GRAM column 10 μm 1000 Å (8 × 300 mm). The module has a 2414 refractive index detector and a 2489 UV/Vis detector. The measurement was performed at a flow rate of 1 mL min−1. The sample was prepared in DMF at the final concentration of 2 mg ml−1. The samples were filtered through polytetrafluorethylene (PTFE) membranes with a pore size of 0.2 μm before injection.2.7 Formation of NBTA–MSN supramolecular nanocomposite hydrogels
The NBTA macromonomer solid powder was dispersed in a clear glass vial containing either DPBS or alpha MEM medium. NBTA (7% (w/v), initial concentration) was heated above the lower critical solution temperature (LCST ≈ 80 °C) of the PEG20K until the solution became opaque and then cooled to room temperature (RT) to form the hydrogel. After multiple cycles of heating and cooling and overnight aging at RT, the NBTA formed stable hydrogels. Next, an MSN suspension in LAP photoinitiator solution was added to the NBTA hydrogels and mixed using a spatula. The final NBTA polymer concentration was adjusted to 4% w/v and the final LAP concentration was 2 mM. Subsequently, the mixture was heated using a heat gun and vortexed for uniform distribution of MSN nanoparticles within the hydrogel. The same procedure was repeated twice and then the hydrogel solution was centrifuged at 2000 g for around 5 minutes at room temperature to remove the bubbles. Next, the hydrogel solution was aged for 3 hours for equilibration to form an NBTA–MSN hydrogel at room temperature. In situ covalent crosslinking was performed by exposing the hydrogel to UV light (at 365 nm). The final concentrations of MSNs in NBTA hydrogel networks were adjusted to 0.5 wt%, 1 wt%, and 2 wt%. NBTA–MSN-CaP-SH nanocomposites were prepared by mixing 1 wt% of MSN-CaP-SH with the same concentration of NBTA (4% w/v).2.8 In situ photorheometry measurements
The mechanical properties of the hydrogels were determined by performing time and strain sweeps using oscillatory rheology (TA instruments, Discovery HR-2). Gelation kinetics of the hydrogels was observed by oscillatory time sweep test at a constant angular frequency (ω) of 1 Hz and a 1% strain amplitude (γ). The strain sweep was performed from 0.1% to 1000% strain at a constant angular frequency (1 Hz). The self-healing behaviour was studied by a cyclic strain sweep. In these measurements, the strain amplitude was shifted between 500% and 1% for four cycles at a constant angular frequency of 1 Hz. All the measurements were done using a 20 mm (diameter), 2.002° cone geometry with a gap of 53 μm at 20 °C and analyzed using TA Instruments TRIOS software. The hydrogels were formed in situ on the rheometer stage by exposing them to UV light (200 mA, at 365 nm).2.9 Cell culture
Human osteoblasts-like cells (MG-63) were purchased (Gibco) and grown in an a-MEM growth medium supplemented with 10% fetal bovine serum (FBS) and 100 U mL−1 penicillin + 100 mg mL−1 streptomycin (Gibco) and 0.2 mM of ascorbic acid (ASAP). Cells were kept in 175 cm2 culture flasks at 37 °C with a controlled atmosphere of 5% CO2 and grown until 80% confluence. The cell medium was changed every 2–3 days.2.10 MG-63 cell encapsulation and cytocompatibility of NBTA–MSN hydrogels
The NBTA–MSN hydrogels were prepared at a final concentration of 4% (w/v) NBTA and with 0.5, 1, and 2% (w/v) of MSNs as described above in Section 2.7. In short, NBTA macromonomer powder was dissolved in alpha MEM medium with a 1% (v/v) addition of Penicillin–Streptomycin (P/S) instead of DPBS. MG-63 cells were encapsulated in NBTA–MSN hydrogels with the final cell density of 3 million cells per mL of hydrogel. Cells were encapsulated using the self-healing ability of the NBTA–MSN hydrogels. Briefly, the hydrogel was gently broken with a spatula which then turned into a highly viscous slurry. Next, the cell suspension was added to the mixture and the hydrogel was aged for 45 minutes to facilitate self-healing. Then, cell-encapsulated hydrogels were cross-linked by UV light at 365 nm for 2 minutes. Following this, cell culture medium was added on top of the cell-laden hydrogels, and they were placed in the incubator. Cell viability was assessed on both day 1 and day 4 using a live/dead assay and PrestoBlue®.For the live/dead assay, the hydrogel was washed twice gently with PBS and then a freshly prepared solution of calcein AM (1 μM) and ethidium homodimer (2 μM) in a cell culture medium were added to cover the hydrogel. The hydrogel was incubated in the dark incubator maintained at 37 °C with 5% CO2. After 50 minutes of waiting time, the hydrogel was washed with PBS solution and the fluorescent images were taken using a live-cell microscope (Nikon Eclipse Ti-e) under the conditions of 37 °C and 5% CO2. Live dead assay was performed in duplicate and multiple areas of the hydrogel were probed for investigation of cell viability.
PrestoBlue® assay was done to determine metabolic activity of cells on days 1 and 4. Cell culture media on top of the hydrogels was removed and PrestoBlue® solution (1:10 dilution in cell culture medium) was added to each well. After 2 hours of incubation at 37 °C (in the dark), 100 μL of the medium was removed and transferred to an opaque 96-well plate. The fluorescence was recorded using a spectrophotometer (Cary Eclipse Fluorescence Spectrometer, Agilent) with excitation at 535 nm and emission detection at 590 nm.
2.11 SBF immersion of nanocomposite hydrogels
The modified version of stimulated body fluid buffer (m-SBF) was prepared according to the previously published protocol.32 First, 9 g HEPES was dissolved in 50 mL of 0.2 M NaOH (buffer 1). Later, 2.7 g NaCl, 0.25 g NaHCO3, 0.21 g Na2CO3, 0.11 g KCl, 0.12 g K2HPO4·3H2O and 0.16 g MgCl2·6H2O were dissolved in 400 mL of Mili-Q water (buffer 2). Buffer 1 solution was added to buffer 2, followed by the addition of 0.15 g CaCl2 and 0.04 g Na2SO4. The final volume of the solution was 500 mL and the pH was adjusted to 7.4 by using NaOH (1 M).NBTA–MSN hydrogels were prepared as described in Section 2.7 and incubated in a m-SBF for 1 and 3 days at 37 °C. Later, hydrogels were washed with Mili-Q water and freeze-dried. The microstructure and Ca and P content of the lyophilized hydrogels were characterized by scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX, IT-200, Jeol). SEM-EDX measurements were performed at an acceleration voltage of 10 kV. Samples for SEM-EDX were fixed on carbon tape without any coating.
2.12 Statistical analysis
The experimental data were compared by two-way ANOVA followed by Turkey's multiple comparison test using GraphPad Prism software. Differences were considered statistically significant for values of p < 0.05.3. Results and discussion
3.1 Synthesis and characterization of MSNs and NBTA molecules
To fabricate photocrosslinkable NBTA–MSN nanocomposite hydrogels (Fig. 1), we first synthesized thiol-functionalized MSNs (MSN-SH) similar to what was reported previously.26 The synthesized MSN-SH had an average hydrodynamic radius of 313.1 ± 2.3 nm and their zeta potential was −16.6 ± 0.3 mV (Fig. 2(a) and (b)). Moreover, they showed uniform spherical morphology and porous structure with an average size of 81.8 ± 8.0 nm, as observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 2(c) and (d)).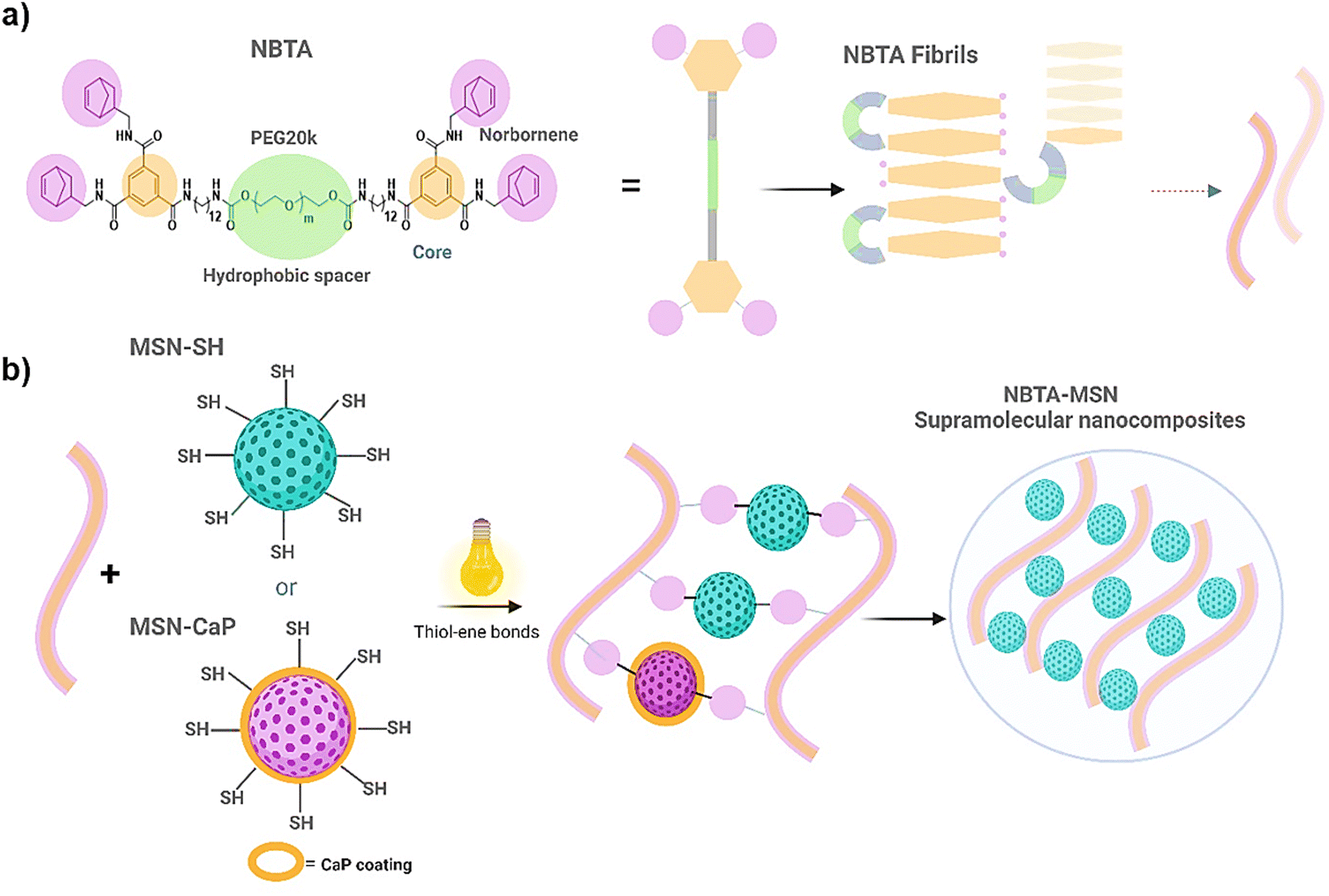 | ||
| Fig. 1 Molecular structure of NBTA macromonomer and MSN-SH crosslinkers employed in the study. (a) Chemical structure of NBTA macromonomer (left), cartoon representation (middle), and proposed stacking and fibre formation (right); (b) schematic representation of crosslinking of NBTA fibre with MSN-SH thiol crosslinkers using thiol–ene chemistry for formation of NBTA–MSN nanocomposites. | ||
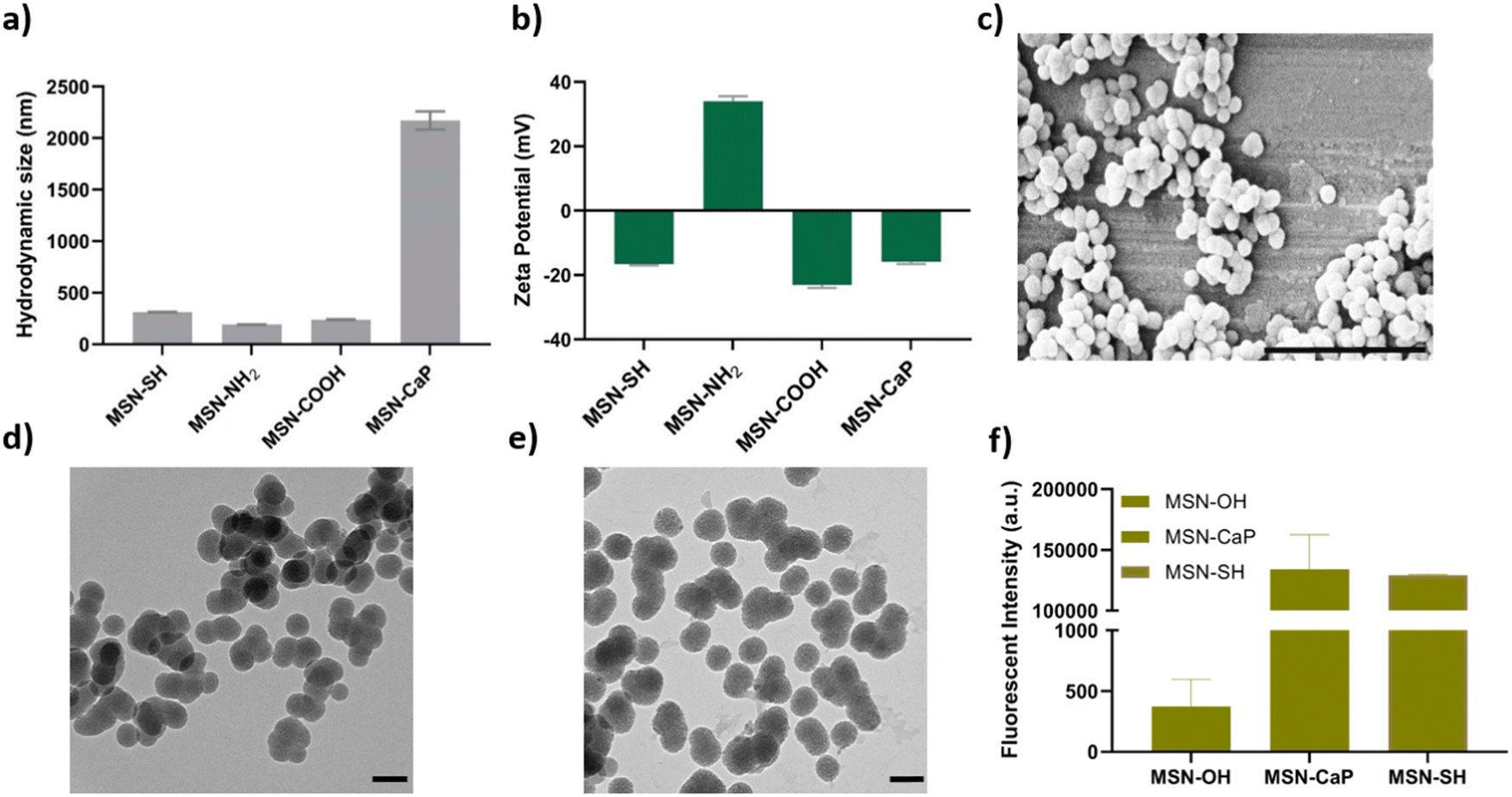 | ||
| Fig. 2 Characterization of the synthesized MSNs. (a) Hydrodynamic size of synthesized MSNs; (b) zeta potential of MSNs; (c) SEM imaging of MSN-SH (scale bar: 1 μM); (d) TEM imaging of MSN-SH; and (e) MSN-CaP (scale bar: 50 nm); (f) ATTO647N-maleimide labeling of control particles (MSN-OH), MSN-SH and MSN-CaP. | ||
Next, our aim was to use MSNs as ion reservoirs while also serving as covalent crosslinkers within supramolecular nanocomposites. To achieve this, we synthesized calcium (Ca) and phosphate (P) coated MSNs with free thiol groups on their surface (MSN-CaP, Fig. 1). To obtain MSN-CaP particles, we first synthesized amine functionalized MSNs (MSN-NH2) with a positive zeta potential of 34.3 ± 1.2 mV and an average hydrodynamic diameter of 192.3 ± 0.1 nm (Fig. 2(a) and (b)). Then, the free amines on the MSN-NH2 surface were converted to carboxylic acid groups (MSN-COOH). The surface charge of MSN-NH2 decreased significantly after carboxyl modification (i.e. from 34.3 ± 1.2 mV to −23.1 ± 0.7 mV for MSN-COOH, Fig. 2(b)). This significant decrease in the zeta potential indicated that the free amine groups on the nanoparticle surface were successfully converted into carboxylic acid groups. The MSN-COOH with a hydrodynamic size of 239.1 ± 2.5 nm were then coated with a calcium and phosphate layer as we reported previously.31 In the final step, a 12-mercaptodecylphosphonic linker was used to introduce free thiols on the CaP-coated MSN surface to create MSN-CaP. MSN-CaP exhibited a negative zeta potential of −15.9 ± 0.5 mV (Fig. 2(b)). The presence of a calcium phosphate layer on the MSN surface increased the hydrodynamic size to 2171.2 ± 71.1 nm, likely due to aggregation of the nanoparticles during the measurement (Fig. 2(a)). In addition, MSN-CaP were spherical in morphology and showed a porous structure with a slight change in size compared to MSN, with 90.0 ± 2.1 nm (Fig. 2(e)). All synthesized nanoparticles showed low polydispersity index (PDI) values (<0.15), indicating homogeneous size distribution. MSNs without additional surface modifications (MSN) were also synthesized as a control group (Fig. S1, ESI†). Thiol functionalization on the MSN surface was further confirmed by performing fluorescent labeling of MSN, MSN-SH, and MSN-CaP with a thiol-reactive dye (ATTO647N-Maleimide). Only thiol functionalized MSNs and MSN-CaPs showed high fluorescent intensity upon incubation with the dye and no significant fluorescent signal was observed for MSN-OH nanoparticles, confirming the presence of reactive thiol groups on the surface of these particles (Fig. 2(f)).
Norbornene functionalized BTA macromonomers (NBTA) were used as building blocks to create the fibrous supramolecular hydrogels. NBTA macromonomers were synthesized as previously described.15 NBTA were obtained pure (1H NMR, Fig. S2, ESI†) with an average molecular weight (Mw) of ≈47000 g mol−1 and Đ of 1.04 (gel permeation chromatography, Table S1 in ESI†). These building blocks were specifically synthesized to feature norbornene groups at both termini of a telechelic BTA polyethylene glycol, Consequently, thiol-functionalized MSNs can crosslink with the norbornene groups of NBTA fibres through UV-initiated thiol–ene interactions (Fig. 1).
3.2 Formation of NBTA–MSN supramolecular nanocomposite hydrogels and their structural and mechanical analysis
Supramolecular nanocomposite hydrogels were prepared using 0.5, 1, or 2 weight% (wt%) MSN-SHs mixed with NBTA (4 wt%) to create NBTA–MSN0.5, NBTA–MSN1, and NBTA–MSN2 hydrogels, respectively. The photocrosslinking was initiated using a LAP initiator under ultraviolet (UV) radiation at 365 nm. The color of the NBTA–MSN nanocomposites was milky white when higher concentrations of the MSNs were incorporated into the hydrogels, while NBTA hydrogels without thiol crosslinkers were transparent (Fig. 3(a)). Similar color changes based on nanoparticle incorporation have been observed in previous studies.28,33 Additionally, NBTA–MSN hydrogels exhibited improved structural stability compared to bare NBTA hydrogels (Fig. 3(a)). The morphology of the nanocomposite networks before and after photocrosslinking was analyzed using cryo-SEM imaging (Fig. 3(b)). Before UV exposure, both NBTA–MSN0.5 and NBTA–MSN1 supramolecular nanocomposites showed a highly fibrous microstructure. After covalent MSN crosslinking upon UV exposure, NBTA–MSN0.5 preserved its fibre morphology. In the case of NBTA–MSN1, we consistently observed a fracture pattern where it appeared that the fibre bundles were perpendicular to the surface. | ||
| Fig. 3 Structural analysis of NBTA–MSN nanocomposites. (a) Optical images of NBTA–MSN hydrogels with various concentrations of MSNs. (b) Cryo-SEM images of NBTA–MSN0.5 (scale bar: 5 μm) and NBTA–MSN1 (scale bar: 5 μm before UV exposure, and 1 μm for after UV exposure) before and after MSN crosslinking upon UV exposure. | ||
The rheological properties of NBTA–MSN nanocomposites were analyzed using dynamic in situ photorheometry at room temperature (Fig. 4). To monitor the gelation kinetics of the hydrogels, time sweep photorheometry was performed before and during the UV light exposure at a constant frequency (1 Hz) and strain value (1%) (Fig. 4(a)–(c) and Fig. S3, S4a, ESI†). Here it is important to note that MSNs were not crosslinked and thus served as nanofiller components within the supramolecular network before UV exposure (Fig. S3, ESI†). The storage modulus of NBTA–MSN nanocomposites was 125.2 Pa for NBTA–MSN0.5, 182.8 Pa for NBTA–MSN1, and 249.6 Pa for NBTA–MSN2. During UV exposure, MSNs could crosslink with the ancillary norbornene groups due to thiol–ene reactions forming inter fibrillary crosslinks (Fig. 4 and Fig. S3, ESI†). As a result, after UV light exposure, the storage modulus of NBTA–MSN nanocomposites reached 383.8 Pa (212% increase) for NBTA–MSN0.5, 371.1 Pa (103% increase) for NBTA–MSN1, 602.1 Pa (%141 increase) for NBTA–MSN2 (Fig. S3, ESI†). Additionally, similar to MSN-SH crosslinking, the addition of MSN-CaP (1 wt%) also notably enhanced the storage modulus of nanocomposites (NBTA–MSN-CaP1, 1100.2 Pa) upon UV exposure (Fig. S4, ESI†). Importantly, the CaP coating on the MSN surface did not hinder the nanoparticles’ ability to crosslink the NBTA network.
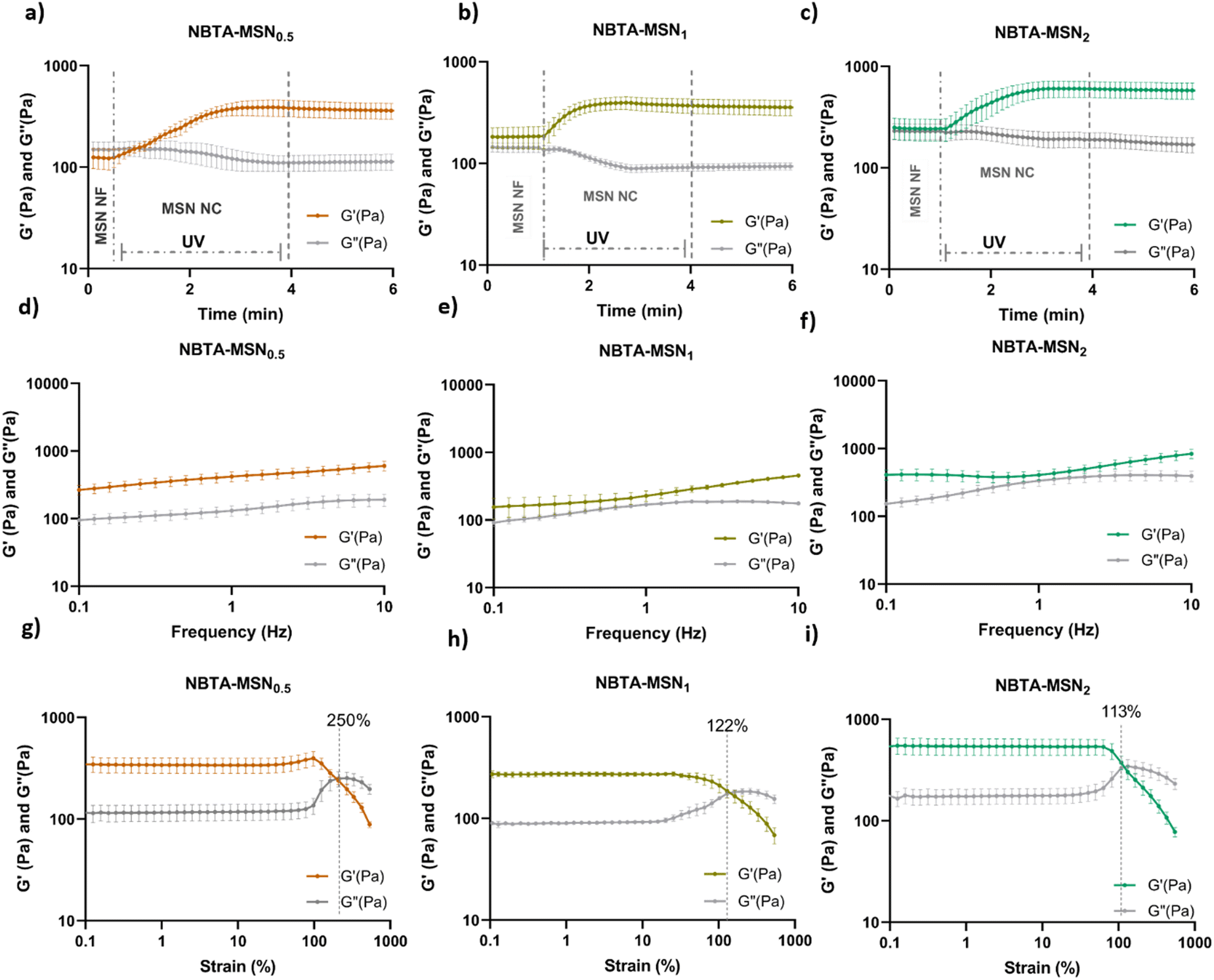 | ||
| Fig. 4 In situ photorheological characterization of NBTA–MSN0.5, NBTA–MSN1, and NBTA–MSN2 supramolecular nanocomposites. Time sweep test of (a) NBTA–MSN0.5, (b) NBTA–MSN1, and (c) NBTA–MSN2 hydrogels; frequency sweep test of (d) NBTA–MSN0.5, (e) NBTA–MSN1, and (f) NBTA–MSN2 hydrogels; strain sweep test of (g) NBTA–MSN0.5, (h) NBTA–MSN1, and (i) NBTA–MSN2 hydrogels. | ||
Frequency sweep tests demonstrated that NBTA–MSN exhibited viscoelastic properties before crosslinking, characterized by a crossover between G′ and G′′ (as shown for NBTA–MSN0.5 in Fig. S5, ESI†), which is similar to most supramolecular hydrogels. After crosslinking, both NBTA–MSN and NBTA–MSN-CaP nanocomposite hydrogels displayed a nearly constant storage modulus across the frequency range of 0.1 Hz to 10 Hz (Fig. 4 and Fig. S4b, ESI†), indicative of dominant elastic behaviour. This enhanced elasticity after crosslinking is likely attributed to the covalent bonding of supramolecular fibres facilitated by MSNs.
Next, the strain sweep tests were performed between strain ranges of 1% to 1000% at a constant angular frequency of 1 Hz (Fig. 4(d)–(f) and S4c, ESI†). At low strain values, the storage modulus (G′) is greater than the loss modulus (G′′), indicating solid-like behaviour. At high strain values, the loss modulus (G′′) was increased and exceeded the storage modulus which shows solid-to-liquid transformation and network disruption. NBTA–MSN0.5, NBTA–MSN1, and NBTA–MSN2 hydrogels were completely broken when the applied strain values were 250%, 122%, and 113%, respectively (Fig. 4(g)–(i)). Interestingly, NBTA–MSN-CaP1 nanocomposites were completely broken at a slightly higher strain (268%) compared to NBTA–MSNs.
Overall, both MSN-SHs and MSN-CaPs could successfully crosslink the supramolecular fibres through UV-initiated thiol–ene interactions. Although our recent study demonstrated the integration of MSNs as chemical crosslinkers into norbornene-modified PEG hydrogels using thiol–ene chemistry,27 this particular mode of nanoparticle incorporation within supramolecular hydrogels has not been reported in the literature.
A recent study by Hafeez et al. demonstrated that NBTA supramolecular assemblies could be crosslinked with small thiol functionalized PEG molecules.15 They observed a 2-fold increase in the storage modulus of NBTA hydrogels (containing 5 wt% NBTA macromonomer) upon crosslinking with short PEG dithiol crosslinkers. The higher increase in storage modulus observed in our system suggests that thiol-functionalized MSNs create a larger number of effective crosslink junctions compared to the PEG dithiol crosslinkers within these supramolecular hydrogels. Therefore, these findings emphasize that employing MSNs as crosslinkers within supramolecular hydrogels represents a powerful strategy for notably enhancing their mechanical properties compared to their use as nanofillers or to the use of small molecule crosslinkers.
Specifically before UV-crosslinking, we observed a nearly two-fold increase in the storage modulus as the MSN concentration increased from 0.5 to 2 wt%. Upon UV exposure, the covalent crosslinking of MSNs further enhanced the storage modulus of these nanocomposites. However, no clear correlation between nanoparticle concentration and storage modulus was observed after UV crosslinking. Specifically, 0.5 wt% MSN resulted in the highest percentage increase (212%) in the storage modulus of NBTA–MSN hydrogels after UV exposure, whereas 103% and 141% increase was observed when 1 and 2 wt% MSN were used, respectively. This observation suggests that MSN crosslinking of supramolecular fibres is more effective at lower nanoparticle concentrations. It is likely that MSN can serve as covalent crosslinkers up to a certain threshold concentration after which they incorporate in the hydrogels as nanofillers. This may interfere with the hydrogel network, eventually leading to diminishing of the mechanical properties. A similar observation was made in a related study, investigating the effect of covalently grafted silica nanoparticles versus silica nanoparticle fillers in poly(2-acrylamido-2-methylpropane sulfonic acid)/polyacrylamide double network hydrogels.34 A significantly higher compressive strength was observed for the silica-grafted hydrogels when the nanoparticle concentration was lower than 3 wt% within the network. However, increasing the nanoparticle concentration further resulted in similar compressive strength for both silica-grafted and silica-filled hydrogels. A negative impact of increasing nanoparticle concentration on the mechanical properties of hydrogels has also been reported in other studies.20–22 For example, the incorporation of hydroxyapatite nanoparticles (HA) at a concentration of 50 wt% into peptide amphiphile hydrogels led to a 2-fold enhancement in the viscoelastic properties of pristine hydrogels.20 However, increasing the nanoparticle concentration to 66.7% resulted in a decrease in their mechanical properties.
In summary, here we demonstrated that the incorporation of MSNs as crosslinkers into supramolecular fibrous hydrogels significantly enhanced their mechanical properties.
3.3 Self-healing behaviour and injectability of NBTA–MSN nanocomposites
Next, we investigated the self-healing properties and injectability of our supramolecular nanocomposites. The self-healing behaviour of these hydrogels was determined by a macroscopic self-healing test and rheological recovery tests. For the macroscopic self-healing test, disk-shaped hydrogels were cut into equal halves by a scalpel and put into direct contact with each other at the side of the cut. After 24 h of incubation at RT in a closed environment, the two different pieces appeared to reconnect and were able to hold their weight (Fig. 5(a)–(d)). This behaviour might be attributed to enhanced interactions between the polymers and MSN surface, as well as likely the reformation of hydrogen bonds between the NBTA units at the interface upon bringing together the two hydrogel pieces.35 Moreover, the supramolecular nanocomposites seemed to exhibit enhanced stretchability after healing compared to bare NBTA hydrogels (Fig. 5(a)–(d)). Similarly, the increased stretchability upon nanoparticle incorporation has also been observed in other types of hydrogels.36–38 Furthermore, a cyclic strain sweep test was performed to confirm the self-healing capability of the different formulations of NBTA–MSN hydrogels, shifting between 1% (non-deformative) to 500% (highly deformative) strain amplitude (Fig. 5(e)–(g)). When high strain (500%) was applied, the hydrogels exhibited a liquid-like behaviour (G′ < G′′). Upon removal of the destructive strain, NBTA–MSN hydrogels recovered spontaneously and showed solid-like behaviour (G′ > G′′). Importantly, rheological measurements confirmed an almost complete recovery of G′ within a few seconds after applying and removing the high strain amplitude (500%) and this process was reversible (Fig. 5(e)–(g)). We also observed that higher MSN concentrations (2 wt%) did not significantly influence the self-healing behaviour of supramolecular nanocomposite hydrogels. Moreover, the composite hydrogels were injectable through 20-gauge syringe needles (Fig. S6, ESI†).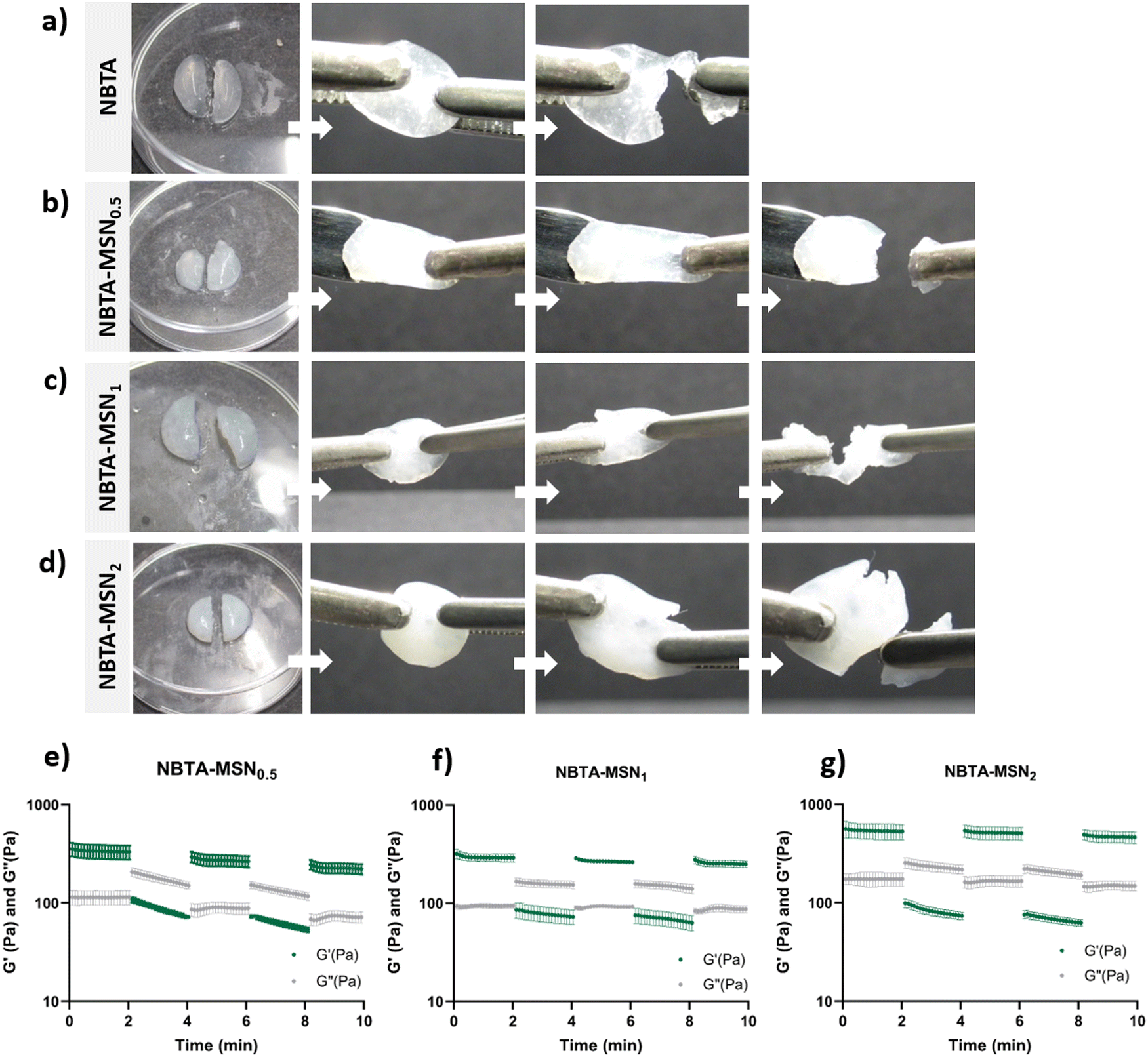 | ||
| Fig. 5 Self-healing behaviour of NBTA–MSN hydrogels. Macroscopic self-healing test of (a) NBTA hydrogels, (b) NBTA–MSN0.5, (c) NBTA–MSN1, (d) NBTA–MSN2. Rheological self-healing test of (e) NBTA–MSN0.5 gels, (f) NBTA–MSN1 gels, (g) NBTA–MSN2 gels. | ||
The presence of thiol–ene bonds in the supramolecular network did not significantly disrupt the self-healing behaviour of the hydrogels. This can be mainly attributed to the dominance of reversible, non-covalent interactions in these supramolecular nanocomposite hydrogels. Additionally, we anticipate that adhesive forces between nanoparticles and polymers can further contribute to the dynamic environment. It is known that silica surfaces exhibit high adhesion towards polymer chains and have been utilized as glue between hydrogel pieces.35 Our observation on the self-healing properties of NBTA–MSN hydrogels also aligns with a previously published study where norbornene-modified BTAs were crosslinked with thiol-functionalized PEG molecules.15 Their rheological self-healing tests demonstrated a rapid recovery of the covalent crosslinked NBTA hydrogels. However, in macroscopic self-healing studies, they observed that hydrogels with both the lowest (0.5 molar equivalent) and highest amount of thiol crosslinkers (2 molar equivalent) exhibited better self-healing properties. This was attributed to reduced covalent crosslinking, which provided more flexibility to the supramolecular macromonomers.
3.4 Cytocompatibility of NBTA–MSN nanocomposite hydrogels
After characterizing the mechanical properties and self-healing behaviour of NBTA–MSN hydrogels, we investigated the cytocompatibility of the NBTA–MSN nanocomposites (Fig. 6 and Fig. S7, S8, ESI†). Here, we selected MG-63 cells since they show the osteoblast-like phenotype and are widely used in in vitro studies for evaluating 3D scaffolds for bone tissue engineering.39–41 The viability of encapsulated MG-63 cells was assessed on both day 1 and 4 using a live/dead assay; cells were stained with calcein AM (green; live cell indicator) and ethidium homodimer-1 (red; dead cells indicator). Notably, the majority of the cells remained alive after 4 days of incubation in cell culture as fewer red-stained cells were observed. MG-63 cells were mostly visible as single cells on day 1 but formed multicellular aggregates on day 4, likely attributed to the dynamic properties of the supramolecular composite hydrogels as seen in the self-healing and injection tests. Similar cellular aggregation behaviour was also observed in a recently published study where chondrocytes (ATDC5) and human mesenchymal stem cells (hMSCs) formed multicellular aggregates upon incorporation within similar BTA hydrogels.14 Other studies also highlighted that the viscoelastic properties of self-healing hydrogels can tune the fusion behaviour of spheroids.42,43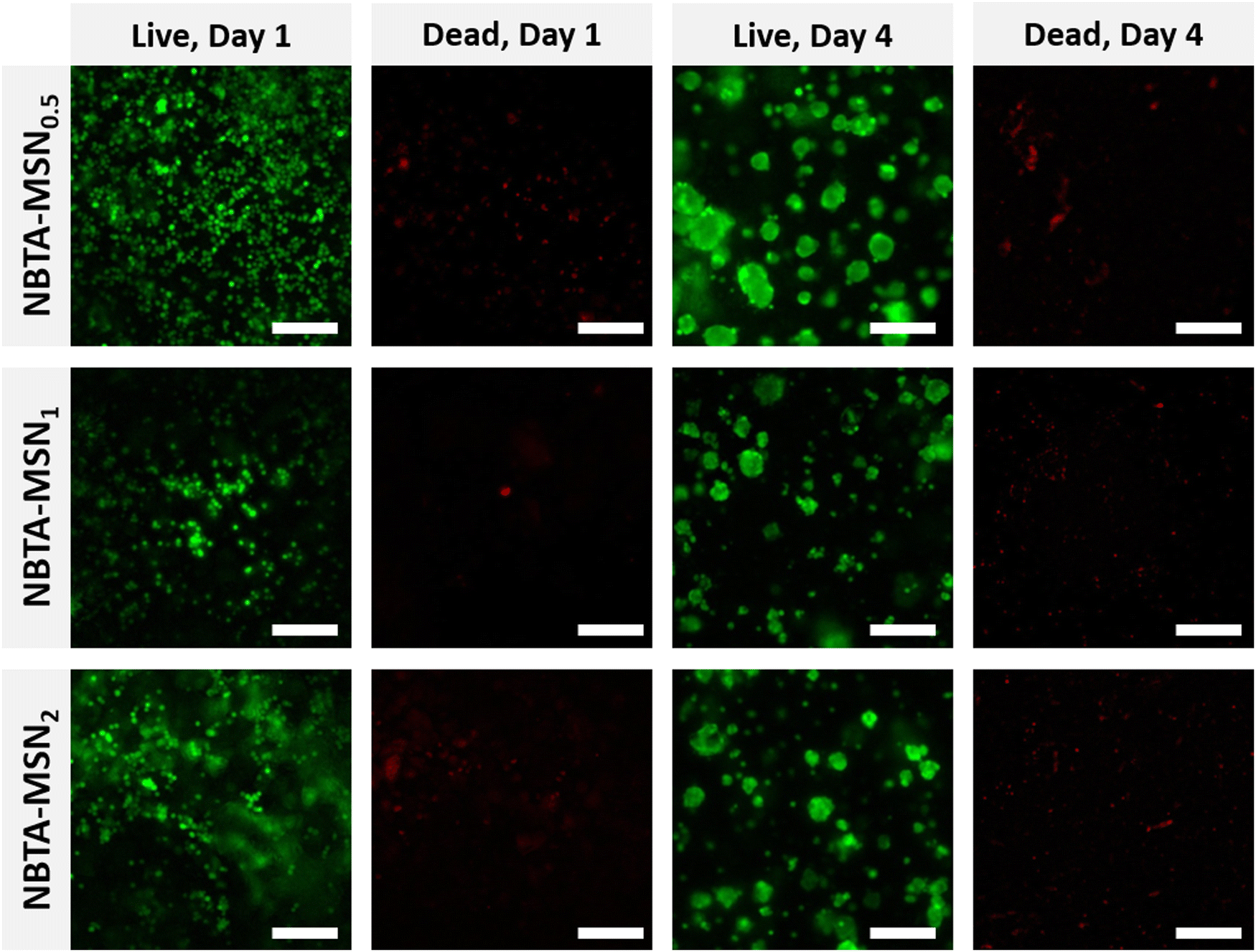 | ||
| Fig. 6 Viability of encapsulated MG63 cells inside NBTA–MSN0.5, NBTA–MSN1 and NBTA–MSN2 hydrogels. The representative images of encapsulated cells stained with calcein AM (green = live cells) and ethidium homodimer (EthD-1, red = dead cells) at day 1 and day 4. Scale bar: 200 μm. | ||
Furthermore, we studied the metabolic activity of encapsulated MG-63 cells on day 1 and day 4 using the Presto Blue assay (Fig. S9, ESI†). The results revealed a significant increase in cellular metabolic activity after 4 days of incubation, demonstrating the excellent cytocompatibility of the NBTA–MSN nanocomposites.
3.5 Stability and mineralization of NBTA–MSN nanocomposite hydrogels in m-SBF
Several studies have reported that mimicking the composition of natural bone tissue in synthetic bone graft hydrogels is a powerful strategy to accelerate bone tissue regeneration.44 The bone is a natural organic–inorganic composite network composed of mostly calcium phosphate (CaP) in the form of nanocrystalline hydroxyapatite (HA) embedded within collagen fibrils.45 To promote in situ mineralization of BTA fibres for bone tissue engineering applications, we synthesized MSN-CaPs to allow sustained and local calcium and phosphate release, as these ions are known to promote bone formation.46 We then evaluated the stability and bioactive nature of the synthesized hybrid hydrogels containing MSN-CaP and MSN-SHs in a modified simulated body fluid (m-SBF) buffer which closely mimics the ionic concentration of blood plasma during 3 days of incubation (Fig. S10, ESI†).SEM images showed that both freeze-dried NBTA–MSN-CaP1 and NBTA–MSN1 hydrogels had similar, randomly distributed pores (Fig. S10, ESI†). After 3 days of incubation in m-SBF solution, the pores became larger (Fig. S10a and b, ESI†). This might be attributed to the degradation of these hydrogels in the solution (Fig. S11, ESI†). Indeed, we observed complete degradation after 5 days of incubation in m-SBF. Most likely, the ionic strength of the SBF solution plays an important role in accelerating the degradation of the supramolecular matrix. In the literature, SBF solution has been widely used to evaluate the bioactivity of materials in terms of their ability to form apatite.18,47,48 Only a few studies reported degradation of materials when they are incubated in SBF buffer. For example, Tan et al. reported faster degradation of alginate hydrogels containing nano-hydroxyapatite/collagen particles in SBF solution than in deionized water at 37 °C. However, their in vivo results did not exhibit any significant degradation even after 8 weeks.49
The presence of Ca and P ions in the hydrogels was examined using EDX analysis of different areas on the samples (n = 7, Fig. S10c, ESI†). Our results showed that NBTA–MSN-CaP1 hydrogels exhibited higher Ca and P content for both day 1 and day 3 compared to NBTA–MSN1. This might be due to the very short incubation period (3 days) within the SBF solution compared to previously reported studies in the literature which were up to 2 weeks.47,50–52 Nevertheless, the proposed supramolecular nanocomposites might be beneficial for localized delivery of Ca and P ions within the hydrogels as bioactive bone substitutes. Our group also recently showed that similar CaP modification of MSN surface could induce osteogenic differentiation of human mesenchymal stem cells.31 Moreover, several studies reported that the incorporation of CaP nanoparticles into polymer networks could increase osteogenic differentiation of stem cells and in vivo new bone formation.53–55 Similar to our approach, Zhai et al. incorporated laponites into supramolecular poly (N-acryloyl glycinamide) (PNAGA) hydrogels to release essential ions such as Mg2+ and Si4+ to increase the bioactivity of PNAGA hydrogels for bone regeneration applications.24 Their results revealed that the controlled release of intrinsic Mg2+ and Si4+ from the PNAGA-LAPONITE® scaffolds effectively promoted the osteogenic differentiation of primary rat osteoblasts. These studies support that our strategy for localized ion delivery within the supramolecular hydrogels holds promise for bone tissue engineering applications. An additional benefit of our material is that it can be locally applied. This in combination with sustained ion release from MSNs may minimize the risk of ectopic calcification in other tissues.
4. Conclusions
In this study, we developed injectable, supramolecular nanocomposite hydrogels, combining non-covalent and covalent interactions. First, we created a supramolecular network based on hydrogen bonds between NBTA macromonomers. By the addition of thiol-functionalized MSNs into this supramolecular network, we were able to create secondary crosslinking based on thiol–ene reactions upon UV exposure. To the best of our knowledge, this is the first report that demonstrates the covalent crosslinking of supramolecular fibres with nanoparticles through UV light-initiated thiol–ene chemistry. We also demonstrated that the mechanical properties of the resulting supramolecular nanocomposites could be tailored by adjusting MSN concentrations. Furthermore, these hybrid hydrogels preserved their self-healing properties and autonomously restored their mechanical properties after deformation. In vitro studies further confirmed that NBTA–MSN hydrogels exhibit good cytocompatibility and effectively support MG-63 cell viability over the 4-day incubation period. As a proof of concept, we also showed that MSNs could function as ion reservoirs for calcium and phosphate within the nanocomposite network in addition to their role as covalent crosslinkers. Consequently, our strategy of integrating MSNs within a fibrillary supramolecular hydrogel is expected to be broadly applicable as an injectable, biomimetic filling material for bone tissue engineering, bioinks for 3D printing applications, and drug/ion delivery vehicles.Data availability
The data supporting this article have been included as part of the ESI.† Raw data is available upon request at the authors.Conflicts of interest
The authors declare no conflict of interest in this work.Acknowledgements
This research was financially supported by the Gravitation Program “Materials Driven Regeneration”, funded by the Netherlands Organization for Scientific Research (024.003.013). We also would like to thank to Hans Duimel for providing assistance with EDX analysis of hydrogels.References
- D. E. Discher, D. J. Mooney and P. W. Zandstra, Science, 2009, 324, 1673–1677 CrossRef CAS PubMed.
- F. Burla, Y. Mulla, B. E. Vos, A. Aufderhorst-Roberts and G. H. Koenderink, Nat. Rev. Phys., 2019, 1, 249–263 CrossRef.
- C. Frantz, K. M. Stewart and V. M. Weaver, J. Cell Sci., 2010, 123, 4195–4200 CrossRef CAS PubMed.
- F. M. Watt and W. T. Huck, Nat. Rev. Mol. Cell Biol., 2013, 14, 467–473 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. K. Hirschberg, R. F. Lange, J. K. Lowe and E. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- M. Fernandez-Castano Romera, R. P. Lafleur, C. Guibert, I. K. Voets, C. Storm and R. P. Sijbesma, Angew. Chem., Int. Ed., 2017, 129, 8897–8901 CrossRef.
- W. Y. Seow and C. A. Hauser, Mater. Today, 2014, 17, 381–388 CrossRef CAS.
- R. Binaymotlagh, L. Chronopoulou, F. H. Haghighi, I. Fratoddi and C. Palocci, Materials, 2022, 15, 5871 CrossRef CAS PubMed.
- B. G. Soliman, A. K. Nguyen, J. J. Gooding and K. A. Kilian, Adv. Mater., 2024, 2404235 CrossRef CAS PubMed.
- A. Chrisnandy, D. Blondel, S. Rezakhani, N. Broguiere and M. P. Lutolf, Nat. Mater., 2022, 21, 479–487 CrossRef CAS PubMed.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- S. Varela-Aramburu, G. Morgese, L. Su, S. M. C. Schoenmakers, M. Perrone, L. Leanza, C. Perego, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2020, 21, 4105–4115 CrossRef CAS PubMed.
- S. Hafeez, A. Aldana, H. Duimel, F. A. A. Ruiter, M. C. Decarli, V. Lapointe, C. van Blitterswijk, L. Moroni and M. B. Baker, Adv. Mater., 2023, e2207053, DOI:10.1002/adma.202207053.
- S. Hafeez, M. C. Decarli, A. Aldana, M. Ebrahimi, F. A. A. Ruiter, H. Duimel, C. van Blitterswijk, L. M. Pitet, L. Moroni and M. B. Baker, Adv. Mater., 2023, 35, e2301242 CrossRef PubMed.
- S. Hafeez, H. W. Ooi, D. Suylen, H. Duimel, T. M. Hackeng, C. van Blitterswijk and M. B. Baker, J. Am. Chem. Soc., 2022, 144, 4057–4070 CrossRef CAS PubMed.
- M. A. VandenBerg, S. Xian, Y. Xiang and M. J. Webber, Macromol. Biosci., 2023, e2300001, DOI:10.1002/mabi.202300001,.
- S. Varela-Aramburu, L. Su, J. Mosquera, G. Morgese, S. M. C. Schoenmakers, R. Cardinaels, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2021, 22, 4633–4641 CrossRef CAS PubMed.
- C. Li, A. Iscen, H. Sai, K. Sato, N. A. Sather, S. M. Chin, Z. Alvarez, L. C. Palmer, G. C. Schatz and S. I. Stupp, Nat. Mater., 2020, 19, 900–909 CrossRef CAS PubMed.
- J. M. Anderson, J. L. Patterson, J. B. Vines, A. Javed, S. R. Gilbert and H.-W. Jun, ACS Nano, 2011, 5, 9463–9479 CrossRef CAS PubMed.
- A. Carnicero, A. Gonzalez, S. D. Dalosto, M. C. G. Passeggi, Jr., R. J. Minari, C. I. Alvarez Igarzabal, M. Martinelli and M. L. Picchio, ACS Biomater. Sci. Eng., 2022, 8, 5027–5037 CrossRef CAS PubMed.
- R. Contreras-Montoya, A. B. Bonhome-Espinosa, A. Orte, D. Miguel, J. M. Delgado-López, J. D. G. Duran, J. M. Cuerva, M. T. Lopez-Lopez and L. Álvarez de Cienfuegos, Mater. Chem. Front., 2018, 2, 686–699 RSC.
- M. Mihajlovic, M. Mihajlovic, P. Y. W. Dankers, R. Masereeuw and R. P. Sijbesma, Macromol. Biosci., 2019, 19, e1800173 CrossRef PubMed.
- X. Zhai, Y. Ma, C. Hou, F. Gao, Y. Zhang, C. Ruan, H. Pan, W. W. Lu and W. Liu, ACS Biomater. Sci. Eng., 2017, 3, 1109–1118 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthcare Mater., 2018, 7, 1700831 CrossRef PubMed.
- A. Zengin, J. P. O. Castro, P. Habibovic and S. H. van Rijt, Nanoscale, 2021, 13, 1144–1154 RSC.
- A. Zengin, F. C. Teixeira, T. Feliciano, P. Habibovic, C. D. Mota, M. B. Baker and S. van Rijt, Biomater. Adv., 2023, 154, 213647 CrossRef CAS PubMed.
- S. Yang, J. Wang, H. Tan, F. Zeng and C. Liu, Soft Matter, 2012, 8, 8981–8989 RSC.
- M. Zhu, Y. Zhu, L. Zhang and J. Shi, Sci. Technol. Adv. Mater., 2013, 14, 045005 CrossRef CAS PubMed.
- N. Wang, M. Ma, Y. Luo, T. Liu, P. Zhou, S. Qi, Y. Xu and H. Chen, ChemNanoMat, 2018, 4, 631–641 CrossRef CAS.
- P. Sutthavas, M. Schumacher, K. Zheng, P. Habibovic, A. R. Boccaccini and S. van Rijt, Nanomaterials, 2022, 12, 2918 CrossRef CAS PubMed.
- A. Oyane, H. M. Kim, T. Furuya, T. Kokubo, T. Miyazaki and T. Nakamura, J. Biomed. Mater. Res., Part A, 2003, 65, 188–195 CrossRef PubMed.
- A. A. Mohammed, N. G. Merrild, S. Li, A. Pinna and J. R. Jones, ACS Omega, 2022, 7, 43904–43914 CrossRef CAS PubMed.
- Q. Wang, R. Hou, Y. Cheng and J. Fu, Soft Matter, 2012, 8, 6048–6056 RSC.
- S. Rose, A. Prevoteau, P. Elziere, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382–385 CrossRef CAS PubMed.
- F. K. Shi, X. P. Wang, R. H. Guo, M. Zhong and X. M. Xie, J. Mater. Chem. B, 2015, 3, 1187–1192 RSC.
- M. I. Sujan, S. D. Sarkar, S. Sultana, L. Bushra, R. Tareq, C. K. Roy and M. S. Azam, RSC Adv., 2020, 10, 6213–6222 RSC.
- M. Zhong, X. Y. Liu, F. K. Shi, L. Q. Zhang, X. P. Wang, A. G. Cheetham, H. Cui and X. M. Xie, Soft Matter, 2015, 11, 4235–4241 RSC.
- A. Grigore, B. Sarker, B. Fabry, A. R. Boccaccini and R. Detsch, Tissue Eng., Part A, 2014, 20, 2140–2150 CrossRef CAS PubMed.
- B. Sarker, J. Rompf, R. Silva, N. Lang, R. Detsch, J. Kaschta, B. Fabry and A. R. Boccaccini, Int. J. Biol. Macromol., 2015, 78, 72–78 CrossRef CAS PubMed.
- C. Vitale-Brovarone, E. Verne, L. Robiglio, P. Appendino, F. Bassi, G. Martinasso, G. Muzio and R. Canuto, Acta Biomater., 2007, 3, 199–208 CrossRef CAS PubMed.
- A. C. Daly, M. D. Davidson and J. A. Burdick, Nat. Commun., 2021, 12, 753 CrossRef CAS PubMed.
- D. T. Wu, M. Diba, S. Yang, B. R. Freedman, A. Elosegui-Artola and D. J. Mooney, Bioeng. Transl. Med., 2023, 8, e10464 CrossRef CAS PubMed.
- H. Wei, B. Zhang, M. Lei, Z. Lu, J. Liu, B. Guo and Y. Yu, ACS Nano, 2022, 16, 4734–4745 CrossRef CAS PubMed.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y.-Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- J. Jeong, J. H. Kim, J. H. Shim, N. S. Hwang and C. Y. Heo, Biomater. Res., 2019, 23, 4 CrossRef PubMed.
- X. Li, J. Lan, M. Ai, Y. Guo, Q. Cai and X. Yang, Colloids Surf., B, 2014, 123, 753–761 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- R. Tan, Q. Feng, Z. She, M. Wang, H. Jin, J. Li and X. Yu, Polym. Degrad. Stab., 2010, 95, 1736–1742 CrossRef CAS.
- A. Houaoui, I. Lyyra, R. Agniel, E. Pauthe and J. Massera, Mater. Sci. Eng., C, 2020, 107, 110340 CrossRef CAS PubMed.
- A. Fathi, F. Kermani, A. Behnamghader, S. Banijamali, M. Mozafari, F. Baino and S. Kargozar, Biomed. Glasses, 2020, 6, 57–69 Search PubMed.
- A. Houaoui, A. Szczodra, M. Lallukka, L. El-Guermah, R. Agniel, E. Pauthe, J. Massera and M. Boissiere, Biomolecules, 2021, 11, 444 CrossRef CAS PubMed.
- M. Bongio, J. J. van den Beucken, M. R. Nejadnik, Z. Tahmasebi Birgani, P. Habibovic, L. A. Kinard, F. K. Kasper, A. G. Mikos, S. C. Leeuwenburgh and J. A. Jansen, Acta Biomater., 2013, 9, 5464–5474 CrossRef CAS PubMed.
- L. Kuang, X. Ma, Y. Ma, Y. Yao, M. Tariq, Y. Yuan and C. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 17234–17246 CrossRef CAS PubMed.
- A. Bhattacharyya, G. Janarthanan, T. Kim, S. Taheri, J. Shin, J. Kim, H. C. Bae, H. S. Han and I. Noh, Biomater. Res., 2022, 26, 54 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb00499j |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |