
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing multifunctional collagen mimetic peptides to create bioinspired stimuli responsive hydrogels for controlled cell culture†
Eden M.
Ford‡
 a,
Amber M.
Hilderbrand‡
a and
April M.
Kloxin
*ab
a,
Amber M.
Hilderbrand‡
a and
April M.
Kloxin
*ab
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, DE 19716, USA. E-mail: akloxin@udel.edu
bDepartment of Materials Science and Engineering, University of Delaware, Newark, DE 19716, USA
First published on 12th August 2024
Abstract
The demand for synthetic soft materials with bioinspired structures continues to grow. Material applications range from in vitro and in vivo tissue mimics to therapeutic delivery systems, where well-defined synthetic building blocks offer precise and reproducible property control. This work examines a synthetic assembling peptide, specifically a multifunctional collagen mimetic peptide (mfCMP) either alone or with reactive macromers, for the creation of responsive hydrogels that capture aspects of soft collagen-rich tissues. We first explored how buffer choice impacts mfCMP hierarchical assembly, in particular, peptide melting temperature, fibril morphology, and ability to form physical hydrogels. Assembly in physiologically relevant buffer resulted in collagen-like fibrillar structures and physically assembled hydrogels with shear-thinning (as indicated through strain-yielding) and self-healing properties. Further, we aimed to create fully synthetic, composite peptide-polymer hydrogels with dynamic responses to various stimuli, inspired by the extracellular matrix (ECM). Specifically, we established mfCMP–poly(ethylene glycol) (PEG) hydrogel compositions that demonstrate increasing non-linear viscoelasticity in response to applied strain as the amount of assembled mfCMP content increases. Furthermore, the thermal responsiveness of mfCMP physical crosslinks was harnessed to manipulate the composite hydrogel mechanical properties in response to changes in temperature. Finally, cells relevant in wound healing, human lung fibroblasts, were encapsulated within these peptide–polymer hydrogels to explore the impact of increased mfCMP, and the resulting changes in viscoelasticity, on cell response. This work establishes mfCMP building blocks as versatile tools for creating hybrid and adaptable systems with applications ranging from injectable shear-thinning materials to responsive interfaces and synthetic ECMs for tissue engineering.
Introduction
Synthetic materials with bioinspired structures, including peptides and polymers, are of continued and growing importance for a range of applications, including in vitro and in vivo tissue mimics and therapeutic delivery. The synthetic origins of these molecules allow precise control of their chemical identity and complement naturally-derived, harvested extracellular matrix (ECM) materials, with less batch-to-batch variability in addition to tunable properties relevant for bottom-up design and hypothesis testing.1,2 The use of synthetic building blocks provides molecular-level control over a range of material properties, including crosslinking, stiffness, degradability, bioactive content, or dynamic capabilities.3,4 However, creating synthetic materials that capture the complexity of tissues of interest while simultaneously affording control over these properties remains a challenge.Tissue characteristics throughout the body can vary widely based on tissue type and function, ranging from the collagen-rich compliant loose connective tissues of skin and lung to the stiff dense connective tissues of ligament and bone.5–7 The physical properties of these systems arise, in part, from the assembly and hierarchical organization of the large structural proteins and glycosaminoglycans that comprise the ECM.8,9 For example, collagen-based tissues rely heavily on the self-assembly of smaller collagen units to form multiscale structures of fibrils and fibers, which are further organized and modified by cells in response to stimuli (e.g., applied mechanical force) within these tissues.10 This bottom-up (e.g., assembly) and top-down (e.g., cellular remodeling) organization of the ECM allows adaptation of tissue properties in response to stimuli throughout the body, whether persistent, chronic, or acute.
Properties and responses of tissues change over time, whether cyclically or irreversibly. For example, during breathing, lung tissue non-linearly and reversibly stiffens in response to forces of expansion during inhalation.11 Additionally, collagen-rich tissues become stiffer and less compliant with aging or disease, owing to increased deposition/accumulation and crosslinking of collagens, and are at an increased risk for injury due to elevated enzyme-mediated degradation of collagen.12–15 The properties of these complex tissues provide inspiration for the design of engineered materials with hierarchical structures and adaptable mechanical properties, particularly as the biomaterials community continues to expand the synthetic materials available for tissue engineering applications.
Toward capturing some of the structural complexity found in native tissues, assembling peptides have been established as an effective approach for the creation of soft materials with multiscale structures and tunable properties.16,17 Peptides using amino acid sequences of a minimal length for imparting desired structure and function inspired by Nature or through de novo design have been created. Sequences assemble into smaller subunits (e.g., α-helices, triple helices, β-sheets, β-hairpins) and higher ordered structures (e.g., flat or twisted fibrils, ribbons, supramolecular structures) through specific physical interactions, such as hydrogen bonding, π–π stacking, or ionic interactions, complementing protein-based systems.18–27 Beyond providing control over nanostructure, self-assembling peptide hydrogels have been designed using short peptide sequences functionalized with Fmoc groups and appended with integrin binding ligands inspired by structural ECM proteins (e.g., RGD, IKVAV, and GFOGER variants such as GFFGER) to impart biochemical functionality while spanning a range of moduli relevant for mimicking soft tissues.28 While these achievements have demonstrated new methods of creating hierarchical structures similar to those seen in native tissues, approaches that integrate strictly physical interactions can lack broad control over the mechanical properties of the resulting nanostructured hydrogels.
As an approach for imparting additional control over material properties with self-assembling peptides, hybrid materials have been formed to provide additional handles for tuning mechanical properties while retaining adaptability including through the reaction of assembling peptides with polymeric macromers.29 These hybrid hydrogels often exhibit adaptability to a variety of stimuli, including temperature, applied force, and other triggers.30,31 For example, thermoresponsive hydrogels have been formed by covalently linking assembled ‘bundlemers’ (reactive subunits made up of assembled coiled-coil peptides) with poly(ethylene glycol) (PEG) macromers, imparting cyclic changes in modulus with heating and cooling.32 Adaptable hydrogels also have been formed with assembling peptides (WW domain + proline-rich peptides) and a thermally responsive polymer (PNIPAM) attached to multiarm PEG, providing tunable mechanical properties and imparting responses to temperature and shear forces relevant for injectable applications.33 Advances such as those highlighted here demonstrate the utility of combining assembling peptides with polymer building blocks to create hybrid materials with hierarchical structure, controlled mechanical properties, and dynamic responsiveness to various stimuli. Such approaches provide opportunities for mimicking specific native tissues through bottom-up design paired with careful consideration of bioactivity, where bioactive moieties (e.g., integrin-binding sequences) and mechanisms for cellular remodeling processes need to be integrated for promoting specific cell–matrix interactions.
Informed by advances in synthetic hybrid materials, we have established an approach for the synthesis of assembling and reactive multifunctional collagen mimetic peptides (mfCMPs) that enable the creation of hybrid mfCMP–PEG hydrogels for three-dimensional (3D) stem cell culture.34 Toward designing synthetic 3D culture systems that better capture the biophysical and biochemical properties of collagen-rich human loose connective tissues, we now aim to use these building blocks to create new synthetic hybrid hydrogels with increasing concentrations of physically assembled mfCMPs integrated within a covalently crosslinked PEG–peptide network, imparting collagen-like structure and responsive properties. Specifically, we have designed these hydrogels to exhibit dynamic responses of various material properties (e.g., hierarchical structure, stiffness, viscoelasticity) to applied stimuli, integrating different combinations of physical and covalent crosslinks, and probe how those properties impact cellular responses.
We first examined how buffer or salt concentrations influence the assembly and stability of the mfCMPs from triple helices to fibrils, where physiologically relevant conditions were established for forming collagen-like fibrillar structures and physically assembled, shear-thinning and self-healing hydrogels. Robust hybrid peptide–polymer hydrogels then were formed with physically assembled mfCMP linkers in a photopolymerized PEG–peptide network, where incorporating increasing amounts of assembled mfCMPs imparted increasing non-linear viscoelasticity and responsiveness to applied strain. Further, we utilized the thermal responsiveness of the underlying mfCMP physical crosslinks to manipulate hybrid hydrogel mechanical properties with changes in temperature demonstrating collagen-like dynamic properties, where increased thermal responsiveness will have implications for the materials when subjected to physiological conditions. Human lung fibroblasts, cells relevant in wound healing and found within the collagen-rich interstitial loose connective tissue, were encapsulated within these hybrid materials. Utilizing the modularity of the system, we covalently incorporated an integrin-binding sequence for cell adhesion while achieving relevant structure and properties in 3D cultures with and without high concentrations of mfCMPs. The impact of increased thermoresponsiveness, non-linear viscoelasticity, and fibrillar structure on cell responses was probed. Specifically, cell viability, morphology, and motility were studied to improve our understanding of how these hydrogels influence cell behavior. Overall, this work demonstrates how mfCMP–polymer building blocks can be used for the creation of hybrid and adaptable systems for a range of applications, from shear-thinning materials relevant for injectables to responsive interfaces and synthetic ECMs for 3D fibroblast culture, and of relevance for future applications in tissue engineering and therapeutic delivery.
Experimental
Methods for multifunctional collagen mimetic peptide (mfCMP) synthesis and characterization
where θ represents the measured ellipticity (millidegrees), L the pathlength (mm), c the peptide concentration (mM), and N the number of amino acids in the peptide. To determine the melting temperature of the peptide (Tm, where approximately 50% of triple helices remain intact and 50% have thermally dissociated), a similar protocol was applied, this time looking specifically at the values of the 225 nm peak (indicating a polyproline type II helix) over a temperature range of 4 °C to 80 °C. Data points were collected at every degree over the temperature range (heating rate of 10 °C per hour) and the data conversion previously described was performed. The resulting data were graphed using Origin software (OriginLab Corporation, Northampton, MA) and fit to a Boltzmann curve. The temperature at which the second derivative of the curve crossed the x-axis was used to determine the inflection point, and therefore the Tm.
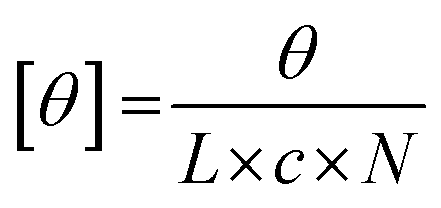
Methods for hydrogel formation
![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif)
![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif) ↓
↓![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif)
![[W with combining low line]](https://www.rsc.org/images/entities/char_0057_0332.gif) GK(alloc)K, identity confirmed using an Acquity UPLC H-class/SQD2 mass spectrometer; Waters Corporation, Milford, MA), and photoinitiator (lithium phenyl-2,4,6-trimethylbenzoyl-phosphinate; LAP). These components were synthesized and characterized using previously published protocols.34 PEG-SH macromer (∼60 mM SH), linker peptide (∼50 mM alloc), and photoinitiator (∼40 mM) stock solutions were prepared in DPBS and concentrations verified with Ellman's assay or UV-Vis spectroscopy as previously published.34 Component stock solutions were frozen for storage and thawed prior to hydrogel formation. Stock solutions of mfCMP at 5 mM in DPBS were melted at 85 °C for 15 minutes and allowed to cool to room temperature and assemble for 48 hours, as described in detail above.
GK(alloc)K, identity confirmed using an Acquity UPLC H-class/SQD2 mass spectrometer; Waters Corporation, Milford, MA), and photoinitiator (lithium phenyl-2,4,6-trimethylbenzoyl-phosphinate; LAP). These components were synthesized and characterized using previously published protocols.34 PEG-SH macromer (∼60 mM SH), linker peptide (∼50 mM alloc), and photoinitiator (∼40 mM) stock solutions were prepared in DPBS and concentrations verified with Ellman's assay or UV-Vis spectroscopy as previously published.34 Component stock solutions were frozen for storage and thawed prior to hydrogel formation. Stock solutions of mfCMP at 5 mM in DPBS were melted at 85 °C for 15 minutes and allowed to cool to room temperature and assemble for 48 hours, as described in detail above.
Hydrogel precursor solutions in DPBS were prepared at 10 wt% PEG-SH (20 mM thiol functional handles), 2.2 mM LAP, and different combinations of linker peptide and assembled mfCMP for a total of 20 mM alloc functional groups (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 thiol:alloc). Specifically, the assembled mfCMP concentration ranged from 0 mM to 20 mM (with 1 functional handle per individual peptide resulting in 0 mM to 20 mM alloc functional groups) and the linker peptide concentration inversely ranged from 10 mM to 0 mM (with 2 functional handles per peptide resulting in 20 mM to 0 mM alloc functional groups). Specific formulations and mixing protocols are noted below and concentrations are shown in Table S1 (ESI†). Composite mfCMP–PEG hydrogels then were formed through irradiation of the precursor solution in geometries of interest (in situ on a rheometer or in molds for hydrogel equilibrium swelling) using a low dose of long wavelength UV light (10 mW cm−2 at 365 nm for 4 minutes; Exfo Omnicure Series 2000 light source with 365 nm bandpass filter; Excelitas Technologies Corp., Waltham, MA) as noted below.
:1 thiol:alloc). Specifically, the assembled mfCMP concentration ranged from 0 mM to 20 mM (with 1 functional handle per individual peptide resulting in 0 mM to 20 mM alloc functional groups) and the linker peptide concentration inversely ranged from 10 mM to 0 mM (with 2 functional handles per peptide resulting in 20 mM to 0 mM alloc functional groups). Specific formulations and mixing protocols are noted below and concentrations are shown in Table S1 (ESI†). Composite mfCMP–PEG hydrogels then were formed through irradiation of the precursor solution in geometries of interest (in situ on a rheometer or in molds for hydrogel equilibrium swelling) using a low dose of long wavelength UV light (10 mW cm−2 at 365 nm for 4 minutes; Exfo Omnicure Series 2000 light source with 365 nm bandpass filter; Excelitas Technologies Corp., Waltham, MA) as noted below.
Hydrogel formulations were prepared with the goal of varying the percent of peptide linkages from assembled mfCMP compared to non-assembling linker peptides for imparting bioinspired structures and responsiveness. As noted above, the total amount of alloc functional handle (from mfCMP + linker peptide) in each hydrogel was kept consistent at 20 mM. To achieve this, when assembled mfCMP was added to the precursor solution, some amount of non-assembling peptide was subtracted to maintain a concentration of 20 mM alloc. For example, for 5 mM mfCMP in the hydrogel precursor solution, 5 mM of the alloc-functionalized peptide linkages (25%) were from mfCMP and 15 mM of alloc from the non-assembling linker and accordingly denoted as 25% mfCMP (Table S1, ESI†). For hydrogels that contained less than 15% of the alloc-functionalized peptide linkages from mfCMP (i.e., ≤3 mM alloc from mfCMP, ≥17 mM alloc from non-assembling peptide), solutions of 10 wt% PEG-SH, linker peptide, and assembled mfCMP were prepared with stoichiometric ratios of thiol groups to alloc groups (20 mM SH:20 mM alloc) and 2.2 mM LAP in the hydrogel precursor solution. For hydrogels that contained more than 15% of the alloc-functionalized peptide linkages from mfCMP (i.e., >3 mM alloc from mfCMP), the assembled mfCMP solution was frozen, lyophilized, and resuspended in diluted stock solutions of PEG-SH, linker peptide, and LAP to achieve a final hydrogel precursor solution of 10 wt% PEG-SH and balance between linker peptide and assembled mfCMP for achieving a stoichiometric ratio of thiol to alloc (20 mM SH:20 mM alloc). This procedure allowed assembly of the mfCMP at a consistent concentration (5 mM in DPBS following procedures delineated in mfCMP sample preparation) while still being able to incorporate high amounts of mfCMP within hydrogels, up to 100% peptide linkages from mfCMPs (all 20 mM alloc from mfCMP).
![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif)
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif)
![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif) (RGDS, identity confirmed with a Xevo G2-S QTof mass spectrometer), was synthesized on an automated Liberty Blue microwave peptide synthesizer (CEM Corporation, Matthews, NC) according to previously established protocols.34 This RGDS peptide was incorporated into the hydrogel system to promote cell–matrix interactions in cell culture applications, as it is derived from cell-binding proteins (e.g., denatured collagen I, fibronectin, vitronectin) and binds to a number of integrins.36,37 Note, for these hydrogel compositions, we increased the thiol concentration in the hydrogel precursor solution (from PEG-SH) to 22 mM, as 2 mM theoretically would be attached to the RGDS pendent peptide and thus not contributing to hydrogel crosslink density. Toward forming hydrogels with consistent moduli across different concentrations of mfCMP, we tuned all compositions to match the modulus of 10 mM mfCMP with 2 mM RGDS (specific component concentrations available in Table S2, ESI†).
(RGDS, identity confirmed with a Xevo G2-S QTof mass spectrometer), was synthesized on an automated Liberty Blue microwave peptide synthesizer (CEM Corporation, Matthews, NC) according to previously established protocols.34 This RGDS peptide was incorporated into the hydrogel system to promote cell–matrix interactions in cell culture applications, as it is derived from cell-binding proteins (e.g., denatured collagen I, fibronectin, vitronectin) and binds to a number of integrins.36,37 Note, for these hydrogel compositions, we increased the thiol concentration in the hydrogel precursor solution (from PEG-SH) to 22 mM, as 2 mM theoretically would be attached to the RGDS pendent peptide and thus not contributing to hydrogel crosslink density. Toward forming hydrogels with consistent moduli across different concentrations of mfCMP, we tuned all compositions to match the modulus of 10 mM mfCMP with 2 mM RGDS (specific component concentrations available in Table S2, ESI†).
Similar to the mfCMP–PEG hydrogels without RGDS, stock solutions of PEG-SH, linker peptide, RGDS, and LAP were prepared in DPBS, and the concentrations were verified with Ellman's assay or UV-Vis spectroscopy as previously published.34 mfCMP stock solutions (5 mM in DPBS) were melted at 85 °C for 15 minutes, cooled to room temperature, and assembled for 48 hours. The assembled mfCMP solutions were then aliquoted for the appropriate formulations, flash frozen in liquid nitrogen, and lyophilized. Hydrogel precursor solutions were prepared at 11 wt% PEG-SH (22 mM thiol functional handles), and contained 2.2 mM LAP, 2 mM RGDS peptide, linker peptide ranging from 10 mM to 11 mM, depending on the formulation (Table S2, ESI†), and DPBS. Lyophilized mfCMP was resuspended directly in the precursor solution for a final mfCMP concentration of 0 mM (0% peptide linkages from mfCMP), 5 mM (32% peptide linkages from mfCMP), or 10 mM (50% peptide linkages from mfCMP), where “percent mfCMP” indicates the percent of alloc coming from mfCMP out of the total alloc-functionalized peptide linkages (i.e., mfCMP and non-assembling linker peptide that contribute to crosslink density, not RGDS). Hydrogel precursor solutions were transferred to syringe molds (20 μL, 1 mL syringes with the tips removed; Thermo Fisher Scientific, Waltham, MA) followed by irradiation with the cytocompatible dose of long wavelength UV light described above.
Methods for hydrogel property measurements
where θ represents the measured ellipticity (millidegrees), L the pathlength (mm), c the peptide concentration (mM), and N the number of amino acids in the peptide. These measurements were repeated for a total of 3 heating–cooling cycles.
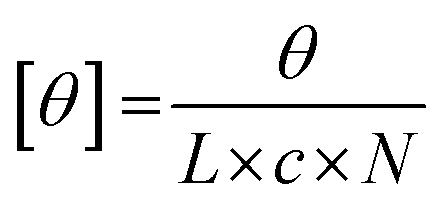
Methods for cell experiments and analysis
The Spot data were imported to MATLAB, and the data were sorted by track number then by frame number for each gel within each condition. To control for hydrogel drift during the timelapse, the Track output was compared with the original image file to identify a stationary point within the gel that was tracked for all 25 frames (for each timelapse file). For each file, the location (x,y) of the stationary point in each frame was subtracted from each individual cell/cell cluster location (x,y) in the corresponding frame. With the hydrogel drift accounted for, we were then able to measure the total track displacement (i.e., distance between the Spot in the first and last frames the Track was detected) and the total distance traveled by the cell/cluster (i.e., sum of displacement between each frame the Spot is detected along a Track). The data were then filtered to remove the stationary points used for subtraction of background movement as well as any cells that were detected for less than 6 frames (i.e., only cells/cell clusters that were tracked for at least 3 hours were analyzed to avoid bias from cells within the frame for only a short time). Finally, MATLAB was used to calculate moments of distribution and individual cell measurements, as well as to run statistical analysis. Specific individual cell measurements included cell speed (determined from the total distance traveled divided by the time over which the object was tracked) and directional persistence (determined by calculating the directionality ratio, i.e., the ratio of cell displacement, d, to total distance traveled, D), where values closer to 0 indicate low directional persistence, or a meandering movement, and values closer to 1 indicate high directional persistence, or direct movement.45
Methods for statistical analysis
All reported values are the mean ± standard error for each condition (excluding box plots), where three independent samples were measured unless otherwise stated. Statistical significance was determined using a one-way ANOVA with Tukey's post-hoc test. On plots, statistical significance is displayed as either not statistically different (n.s.) or as significantly different (*p < 0.05; **p < 0.01; ***p < 0.001).Results and discussion
Hybrid peptide–polymer materials offer the ability to incorporate properties of interest from both a multiarm polymer (e.g., mechanical properties) and assembling peptide (e.g., structural content) into a single material.29 We previously designed mfCMPs that integrate the ability to robustly assemble into fibrils through end-to-end interactions (ionic or aromatic) and include an alkene functional handle for incorporation into covalently crosslinked hydrogel materials.34,41 In our initial work with the two different mfCMP designs, we characterized the structure of the assembled peptides in dilute solution, ranging from triple helix formation to fibrils, and demonstrated the ability to covalently incorporate the preassembled peptides into a bulk PEG hydrogel (6 wt% PEG-4SH) at concentrations up to 2.5 mM mfCMP. The compatibility (cell viability and morphology) of these different mfCMP designs within the covalently crosslinked hydrogels was then examined with human mesenchymal stem cells.34 Focusing on the ionically charged designs, we have since rigorously improved upon our methods for mfCMP synthesis and purification, as well as mfCMP–PEG hydrogel characterization.41 Here, in this new contribution inspired by tissues, we aimed to expand upon the range of properties that can be created through the incorporation of increasing amounts of assembled peptides to leverage properties from both physically and covalently crosslinked hydrogels. We hypothesized that hydrogels with high concentrations of assembling peptides, reminiscent of systems rich in native collagen, would exhibit strain-yielding behavior and thermoresponsiveness and promote responses of wound healing cells (human fibroblasts) found within interstitial tissues.These mfCMPs contain (i) a middle block consisting of proline–hydroxyproline–glycine (POG) repeats to facilitate hydrogen bonding between mfCMP strands; (ii) a positively charged block of proline–lysine–glycine (PKG) repeats and a negatively charged block of aspartic acid–hydroxyproline–glycine (DOG) repeats on either end of the POG block to promote end-to-end interactions and the formation of elongated fibrils; and (iii) an alkene functional group through the incorporation of an allyloxycarbonyl (alloc)-protected lysine group that participates in photoinitiated thiol–ene click chemistry to create covalent peptide linkages (mass spectrometry results for mfCMP available in Fig. S1, ESI†). Notably, these mfCMPs do not contain a specific integrin-binding sequence. This design choice allows for independent control over hierarchical structure and biochemical content within the hydrogel, which we explore in this work. With this in mind, we hypothesized that, with increasing concentrations of mfCMP, hydrogels with physical crosslinks, covalent crosslinks, or a combination of the two could be formed in a physiologically relevant buffer and with adaptable properties in response to applied strain and changes in temperatures. Further, we explored how these responsive hydrogels could be applied to 3D cell culture applications toward better recapitulating aspects of responsive, viscoelastic tissues.
Buffer conditions impact mfCMP assembly from triple helices to physical hydrogels
We first examined multiple levels of mfCMP hierarchical assembly and physical hydrogel formation (Fig. 1(A)) in DPBS (as it has a physiological pH and concentration of salts). We compared this condition to controls of DI H2O (no salts; negative control) and 10 mM sodium phosphate buffer (10 mM salts; positive control previously shown to promote hydrogel formation with similar charged mfCMP sequences).19 Here, we will focus on results obtained in DPBS, which are relevant for use of these materials in a biological context; data collected for mfCMPs in DI H2O and 10 mM sodium phosphate can be found in the ESI,† Fig. S2. In all cases the mfCMPs were heated to 85 °C for 15 minutes to dissociate any previously formed triple helices and then slowly cooled to room temperature and assembled for ∼48 hours. Using CD, we observed that triple helices formed in all conditions. Specifically, these peptides showed a distinct peak at ∼225 nm, which is characteristic of collagen-like triple helices (Fig. 1(B) and Fig. S2A, B, ESI†). Further, when the samples were progressively heated and measured, there was a decrease in the 225 nm peak, indicating the mfCMPs were ‘melting’ (dissociating into individual peptide strands). To determine the melting temperature (Tm, point at which 50% of the mfCMPs are associated into triple helices and the remaining 50% are individual peptides), the value of the peak at 225 nm was measured as the samples were heated from 4 °C to 85 °C (Fig. 1(C) and Fig. S2C, D, ESI†). We observed that salt did influence the stability of the triple helices as shown by the decrease in melting temperature with increasing salt concentration, consistent with prior observations of similar sequences.19 mfCMPs in DI water had the highest Tm of ∼40 °C, and DPBS had the lowest Tm of ∼37 °C. We hypothesize that this is due to charges from the DPBS salts screening the ionic amino acids on the ends of the mfCMPs and therefore destabilizing the triple helices and lowering the melting temperature. | ||
| Fig. 1 Multiscale characterization of mfCMPs assembled in DPBS, from triple helices to hydrogels. (A) Schematic of mfCMP hierarchical assembly, starting from the left and moving clockwise (not to scale). Individual mfCMPs associate into triple helices (characterized with circular dichroism, (B) and (C)), which then further assemble into fibrils (characterized with transmission electron microscopy, (D)). With enough physical interfibrillar interactions, the mfCMP assembly will result in a hydrogel (characterized with rheometry, (E) and (F)). (B) Wavelength scans at various temperatures for mfCMP (0.3 mM) in DPBS demonstrates the presence of triple helices, identified through the characteristic peak at 225 nm. (C) Temperature scan for mfCMP (0.3 mM) at 225 nm and resulting melting temperature (Tm ∼ 37 °C). (D) TEM image of assembled mfCMP fibrils (assembled at 1 mM, diluted and imaged at 0.3 mM), counterstained with 2% Uranyl Acetate in water (scale bars = 500 nm). (E) Rheometry of mfCMP hydrogels assembled at 5 mM measured at 0.1% strain and 1 rad s−1 over 20 hours, demonstrating hydrogel formation. (F) mfCMP hydrogels exhibited strain-yielding behavior in response to applied strain (indicative of shear-thinning properties), to the point of reverse gelation (G′ < G′′). After reducing the strain, the mfCMP hydrogel underwent ‘self-healing’, returning to a hydrogel and recovering the initial mechanical properties. (Results shown are representative of multiple trials with similar results). | ||
Similarly, buffer influenced the fibrillar assembly of the mfCMPs, where different fibrillar morphologies were observed with TEM (Fig. 1(D) and Fig. S3, ESI†). mfCMP assembly in DPBS showed long fibrils with a range of diameters: both thicker and thinner fibrils were present, where thinner fibrils were integrated into the assembly of much larger fibrils (on the order of ∼250+ nm in diameter) that can span lengths beyond the field of view (>3.5 μm). In contrast, the negative control (assembly in DI H2O) showed thin fibrils (on a size scale similar to the thinnest, shortest fibrils in DPBS) sparsely throughout the sample grid, whereas the positive control (assembly in 10 mM sodium phosphate buffer) showed similarly thin fibrils but more prevalently throughout the grid and rigid in appearance. Importantly, hierarchical assembly into larger fibrils was only observed in DPBS. We speculate that the higher concentration of salt ions in DPBS are screening charged residues on the mfCMPs, potentially slowing the rate of assembly while allowing the formation of salt bridges to achieve larger sized fibrils.
With these end-to-end fibrillar assembly mechanisms, we were curious if the physical assembly was strong enough to produce a stable hydrogel that contained only these mfCMPs in DPBS. Concentrated solutions (5 mM) of mfCMPs in DPBS were heated to 85 °C to dissociate all formed triple helices and then placed on a temperature-controlled rheometer stage at 60 °C. A 20 mm 1° cone geometry was lowered onto the sample, and a solvent trap was filled to keep the sample hydrated throughout the measurement. The temperature was then ramped down to 25 °C over the course of 10 minutes to mimic similar cooling to when samples were heated and subsequently slowly cooled in a dry heating block. The samples were then measured at 0.1% strain and 1 rad s−1 over 20 hours (Fig. 1(E)). Specifically, gel formation was assessed with measurements of storage (G′) and loss (G′′) moduli: here, G′ > G′′ was defined as exhibition of gel-like properties, where G′ = G′′ is close to the gel point as defined by the Winters and Chambon criterion.46 After cooling was complete, a physically crosslinked hydrogel was observed for mfCMP in DPBS with a storage modulus of ∼4–5 Pa, which is similar to mechanical properties of a harvested collagen hydrogel.47,48 The positive control (mfCMPs assembled in 10 mM sodium phosphate buffer) exhibited a similar modulus of ∼3–5 Pa (Fig. S4A, ESI†), whereas the negative control (mfCMPs assembled in DI water) did not exhibit gel formation under the conditions probed.
Once a physical hydrogel was formed, a strain sweep was performed to determine the behavior of these mfCMP hydrogels outside the linear viscoelastic regime. The physically crosslinked mfCMP hydrogels were strained from 0.1% to 300% over 3 minutes. In both the DPBS samples and the positive control, the materials remained within a linear viscoelastic regime at low strains. As the strain was increased, the materials underwent strain-yielding and the loss modulus crossed over the storage modulus (G′ < G′′), indicating the materials have reverted to a liquid-like state and exhibit shear-thinning properties (Fig. 1(F) and Fig. S4B, ESI†) consistent with previous reports.19,49 Subsequently, the samples were allowed to recover unperturbed for ∼15 minutes after the high strain event, at which point the strain and frequency were restored to their initial parameters and the samples were remeasured. In both DPBS and the positive control, the mfCMP hydrogels returned to similar mechanical properties as those prior to the strain event, exhibiting self-healing behavior (Fig. 1(F) and Fig. S4C, ESI†).22,39,50 Excitingly, these observations demonstrate similarities between mfCMP materials and physically assembled harvested collagen hydrogels. Further, these mfCMP hydrogels exhibit similar properties in response to strain as natural polymer hydrogels modified for guest–host interactions (e.g., hyaluronic acid; alginate and gelatin),51,52 as well as other peptide-based hydrogels with viscoelastic properties of relevance for applications as cell culture scaffolds.19,49,53 Notably, with applications such as tissue engineering in mind, increased control over mechanical properties and hydrogel stability in cell culture conditions is vital.
Tuning assembled mfCMP content within a polymer-based hydrogel affords control over mechanical properties
Having control over the mechanical properties of hydrogels while also having control over the fibrillar content (provided by hierarchically assembled mfCMP) is desirable for the design of materials for specific biological applications. For independent control of the mechanical and biochemical properties of hydrogels, PEG, a biologically inert, hydrophilic polymer, often is used as a building block. One of the advantages of PEGs as building blocks is that they are amenable to modification with a variety of reactive handles, including click chemistries that are desirable for the formation of hydrogels and tuning of their mechanical properties in a biological context, as well as allow the incorporation of specific biological cues for controlled hypothesis testing.3 Toward controlling both mechanical properties and fibrillar content, hybrid collagen–PEG hydrogels have demonstrated improved mechanical properties compared to solely collagen hydrogels while maintaining the shear-thinning and self-healing properties of harvested collagen.54,55 We hypothesized that with increasing concentrations of mfCMP in an mfCMP–PEG hybrid hydrogel, we could similarly capture some of these collagen-like properties with a fully synthetic material.Here, we utilized photoinitiated thiol–ene click chemistry allowing for rapid covalent hydrogel formation with spatiotemporal control over the initiation of gelation. Briefly, we used a 4-arm PEG macromer functionalized with thiol groups (PEG-SH, Fig. S5, ESI†) that, in the presence of light-activated lithium phenyl-2,4,6-trimethylbenzoyl-phosphinate (LAP) photoinitiator (Fig. S6, ESI†), could be covalently linked with an alloc-functionalized non-assembling MMP-degradable linker peptide (Fig. S7, ESI†) and the assembled mfCMPs to form hydrogels. To enable integration of high concentrations of assembled mfCMPs, the assembled mfCMP in DPBS was frozen and lyophilized prior to resuspension directly into the hydrogel precursor solution (PEG-4SH, the appropriate balance of linker peptide, LAP, and DPBS); this approach allowed for consistent assembly of the mfCMP at 5 mM while incorporating higher concentrations of mfCMP (up to 20 mM) within the hydrogels. TEM characterization of assembled mfCMPs after lyophilization and resuspension is available in the ESI† (Fig. S8). We used different ratios of non-assembling peptide to assembling peptide while keeping the concentration of PEG-SH constant and maintaining a 1:1 alloc:thiol stoichiometry (Table S1, ESI†). Specifically, the ratio of functional groups within hydrogels is 20 mM thiol:20 mM alloc, and all hydrogel conditions are presented as “percent mfCMP”, indicating the percent of alloc linkages coming from mfCMP (where the total alloc linkages are from assembled mfCMP + non-assembling linker peptide); for example, 50% mfCMP indicates 10 mM mfCMP where there is 1 alloc per mfCMP peptide. This approach was taken to explore the impact of adjusting the peptide structure (i.e., ratio of non-assembling to assembling linker peptides) on the hydrogel mechanical properties and to determine the maximum amount of mfCMP linkers that could be used to consistently form these mfCMP–PEG hydrogels.
Hydrogels were formed in situ on a rheometer, and the mechanical properties were measured (Fig. 2(A)). The storage moduli of hydrogels made with 10 wt% PEG-SH (20 mM thiol functional groups) were measured with an increasing percentage of peptide linkages coming from mfCMPs. As assembled mfCMP content was increased from 0% mfCMP (0 mM mfCMP) to 15% mfCMP (3 mM mfCMP), there was no statistical change in the modulus; however, as the percentage of peptide linkages from assembled mfCMP was increased to 25%, a statistical decrease in the modulus was observed. This trend continues as more mfCMPs were added (Fig. 2(B), diagram in Fig. 2(C)). Encouragingly, robust hydrogels with 100% of the peptide linkages coming from the assembled mfCMPs (20 mM mfCMP) were still able to be formed in situ, where previously we had explored incorporation of mfCMP only up to 2.5 mM.34 These results indicate that the potential design space for high concentrations of mfCMP is large.
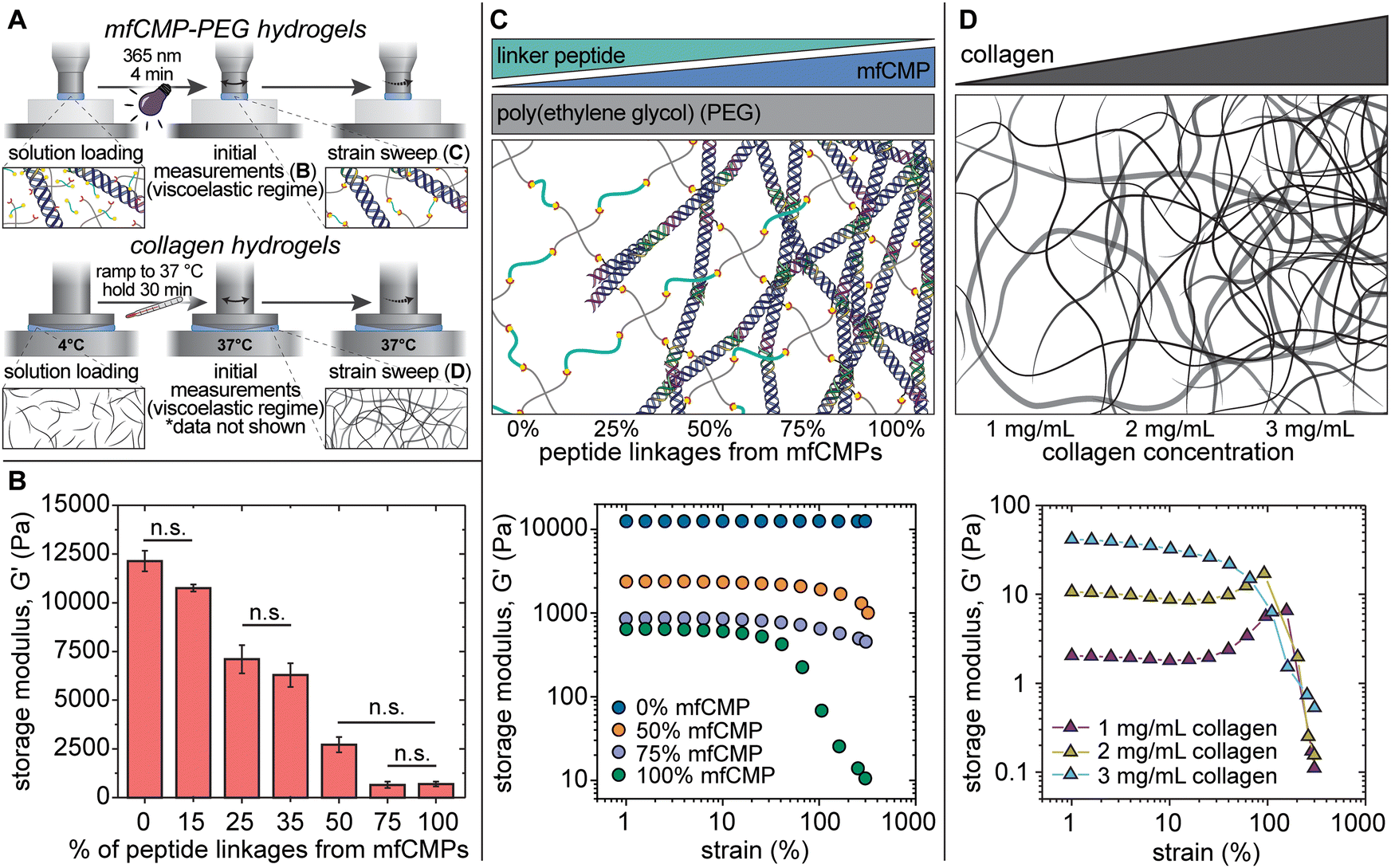 | ||
| Fig. 2 In situ characterization of mechanical properties of composite mfCMP–PEG hydrogels compared with harvested collagen hydrogels. (A) Hydrogels (either mfCMP–PEG hybrid or physically assembled collagen) were formed in situ on a rheometer, where initial modulus measurements were taken within the viscoelastic regime (B), followed by strain sweeps (C) and (D). (B) Average storage modulus of covalent hydrogels with increasing amounts of mfCMPs, where % mfCMP corresponds to the % of alloc from alloc-functionalized mfCMP peptide linker (n = 4). Conditions are statistically different (p < 0.05) unless otherwise noted as not significant (n.s.). (C) Top: Schematic representing the mfCMP–PEG hydrogel compositions for data shown in (B) and (C). Bottom: Strain sweeps of hydrogels with increasing amounts of peptide linkages from mfCMPs (n = 4). Loss modulus data are available in Fig. S9 (ESI†). (D) Top: Schematic representing the harvested collagen hydrogel compositions for data shown in (D). Bottom: Strain sweeps of collagen hydrogels formed at varying concentrations (n = 3). Graph of the initial storage moduli of collagen hydrogels within the viscoelastic regime are available in Fig. S10 (ESI†) and loss modulus data are available in Fig. S11 (ESI†). (Note: schematics are not to scale). | ||
After establishing the mechanical properties of these materials within the linear viscoelastic regime, we studied their response to high strain, similar to the study conducted with the physically assembled mfCMP-only hydrogels (Fig. 2(C)). When no mfCMPs were present, the PEG–peptide hydrogels exhibited a full linear viscoelastic response even at 300% strain. As the amount of mfCMP within the hydrogel increased, the deviations from linear viscoelasticity become both greater in magnitude and happen at increasingly lower strain (i.e., higher mfCMP hydrogels exhibit a lower yield strain). For example, in a mfCMP–PEG hydrogel with 100% mfCMP, deviations from linear viscoelasticity at ∼15% strain were observed, whereas hydrogels with 50% mfCMP showed deviations only at strains greater than 100%. As a comparison, physically assembled harvested collagen hydrogels were formed, and the same strain procedure was performed on these hydrogels (Fig. 2(D)). In all conditions a linear regime was observed at low strain, and then a significant deviation from linear viscoelasticity was observed at higher strain. This comparison shows some similarities in the behavior of mfCMP–PEG hydrogels and collagen hydrogels as shown by the strain-yielding results, which are indicative of shear-thinning behavior, while the mfCMP–PEG hydrogels provide tunability in storage modulus. The strain-yielding demonstrated by the mfCMP–PEG hydrogels with increasing fibrillar content further reflects the strain-yielding nature of various soft collagenous tissues, including lung.56 Such material properties are of particular interest toward increasing the complexity of 3D culture platforms and tissue model systems while maintaining the control afforded by a synthetic system. Ultimately, the approach of incorporating high concentrations of mfCMPs into a synthetic PEG–peptide hydrogel produces a hybrid material that exhibits properties of both covalently crosslinked PEG hydrogels (control over modulus) and physically assembled mfCMP hydrogels (shear-thinning behavior).
Composite mfCMP–PEG hydrogels exhibit dynamic properties in response to temperature cycling
As these mfCMPs have a distinct melting temperature (Fig. 1(C)), we aimed to determine if this property could be leveraged to dynamically change the modulus of these hybrid hydrogels using temperature after equilibrium swelling in DPBS. Rheometry was used to measure changes in the storage modulus of a hydrogel that contained 50% mfCMP (10 mM alloc from mfCMPs; 10 mM alloc from a non-assembling linker peptide) in response to programmed changes in temperature (Fig. 3(A)). This hydrogel composition was chosen as the maximum concentration of mfCMP moving forward, as it maximized mfCMP content while also maintaining a hydrogel that allowed easy handling after equilibrium swelling and could withstand cell culture conditions for at least one week. Temperature was cycled step-wise between 25 °C and 40 °C to create a state where the mfCMPs are intact (T = 25 °C < Tm = 37 °C) and another where they are partially melted (T = 40 °C > Tm = 37 °C). We hypothesized that this temperature cycling would allow the mfCMP within the network to assemble and disassemble according to the temperature, changing the effective crosslink density and thereby the modulus, serving as a demonstration that these hybrid hydrogels retain the thermoresponsive properties of natural collagen and collagen derivatives.48,57,58 Excluding the first 2 temperature steps (the first T1 and T2), where large amounts of variability were observed (perhaps due to hydrogel equilibration from seasonal ambient temperature variations from 25 °C), hydrogels with mfCMPs exhibited average modulus changes of ∼260 Pa between temperatures (a change of 20% based on initial modulus) (Fig. 3(A)). In hydrogels that did not contain mfCMPs, average changes of only ∼72 Pa (2% based on initial modulus) were observed.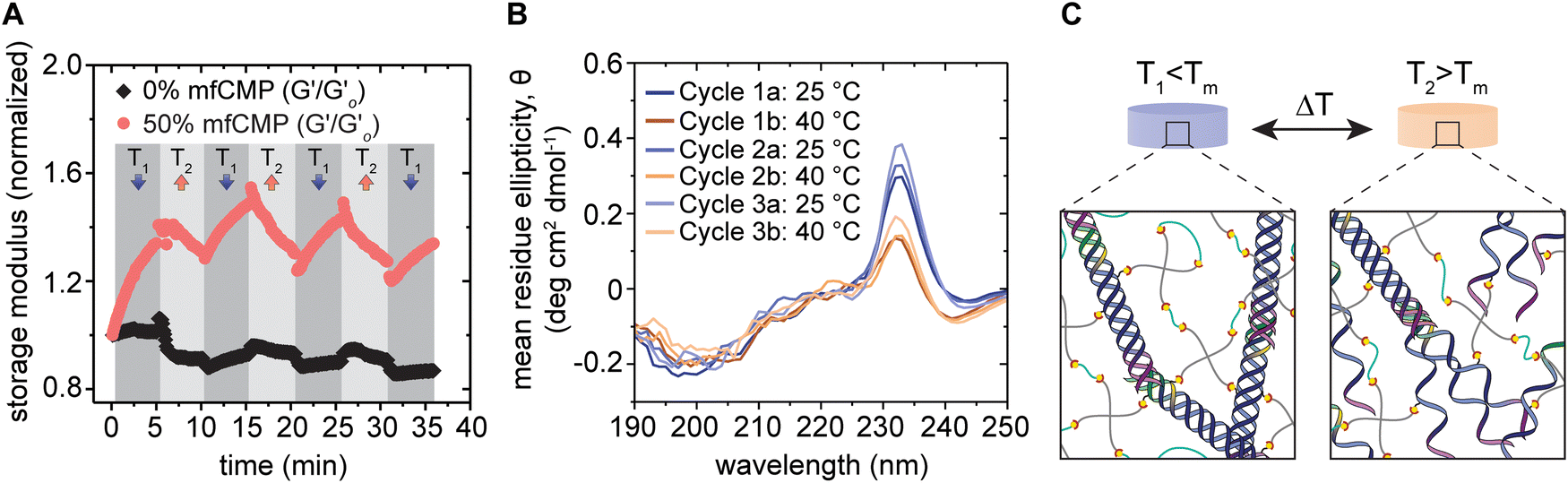 | ||
| Fig. 3 Thermoresponsive properties of mfCMP–PEG hybrid hydrogels. (A) Equilibrium swollen hydrogels with 0% mfCMP (control, 0 mM mfCMP) or 50% mfCMP (10 mM mfCMP) were cycled between 25 °C (T1) and 40 °C (T2) while measuring the storage modulus (normalized to the initial modulus for each condition). Loss modulus data are available in Fig. S12 (ESI†). (B) Circular dichroism was performed on in situ 50% mfCMP hydrogels after each ramp between 25 °C (T1) and 40 °C (T2), where spectra were measured between wavelengths of 190 nm and 250 nm and measurements from 0% mfCMP hydrogels were subtracted as background. (C) Schematic of potential network morphologies at T1 and T2, where relative to Tm, at T1 all triple helices should remain intact, and at T2 only about 25% of the triple helices should remain intact (schematic not to scale). (Results shown are representative of multiple trials with similar results). | ||
To confirm this change in modulus was due to the assembly and disassembly of the mfCMPs, CD was performed to observe the presence of triple helices within mfCMP–PEG hybrid hydrogels (Fig. 3(B)). Hydrogels were formed in situ within a demountable cuvette. Peaks at ∼232 nm were observed once the mfCMPs had been incorporated within the hydrogels, rather than at ∼225 nm, as seen in solution. This observed shift in peak wavelength is similar to other reports, where it is hypothesized that this red shift in signal is due to the PEG hydrogel providing a more locally hydrophilic environment for the mfCMPs.59 When the hydrogels were heated to 40 °C and held there for 5 minutes prior to the measurements being taken, a decrease in the mean residue ellipticity (MRE) was observed, indicating that the triple helices of the mfCMPs were dissociating. Upon changing the temperature back to 25 °C, the MRE increased again, demonstrating the recovery of the mfCMPs back into triple helices within the hydrogel. This behavior correlates with what was observed using rheometry. Taken together, these data support the hypothesis that (i) the decrease in modulus with increasing temperature is a result of the ‘melting’ of mfCMP triple helices that serve as physical crosslinks with the mfCMP–PEG–peptide hydrogels, and that (ii) the restoration of mechanical properties upon cooling results from the assembly of triple helical physical crosslinks. The triple helices formed upon cooling could be either the same mfCMP peptides that comprised the original triple helix or different individual mfCMP peptides, either resulting in a return to similar original crosslink density.
In short, the mfCMP–PEG hydrogel appears to exhibit reduced modulus and crosslink density upon melting of the mfCMP-based crosslinks, and subsequently increased modulus and crosslink density upon reassembly, similar to hydrogels utilizing partially-degraded collagen (gelatin) and temperature sensitive crosslinks.58,60 This heat cycling demonstration establishes that we retain the reversible unfolding functionality of collagen in response to changes in temperature. This property is important as collagen natively is unfolding and refolding under physiological conditions, which is hypothesized to give collagen fibers elasticity and strength.61 Aside from better capturing specific properties of collagenous tissues for 3D culture and model systems, these hybrid materials offer opportunities for a range of potential applications. For example, gold nanorods that convert near-infrared (NIR) light into heat have been embedded within thermoresponsive hydrogels to locally and reversibly alter the surface topography.62 With the thermoresponsiveness demonstrated here, such an approach could be used within these mfCMP materials in future studies to create a photo- and thermoresponsive system, where application of NIR light in the presence of nanorods would cause dissociation of mfCMP triple helices and lead to localized decreases in modulus. Manipulating hydrogel properties in this manner may permit one to locally direct cell behavior and fate or could be used as a delivery mechanism for therapeutics appended with peptides hybridized with mfCMP fibrils during assembly.63 Importantly, temperatures up to 40 °C remain below the threshold for heat shock protein production in human cells and associated irreversible negative cell responses; further, mild thermal elevations may even induce a positive cell response and lead to improved tissue regeneration.64,65 In this context, such thermal approaches for actuation of material properties would not be expected to inherently have detrimental effects on cell health, presenting opportunities for future exploration.
While thermoresponsiveness was examined in this demonstration, there are opportunities for future studies to probe the impacts of other stimuli on these hybrid materials. During the mfCMP assembly process, we have previously demonstrated the impact of temperature holds and cooling rates during mfCMP assembly on final fibril morphology,41 and here we noted that salt identity and concentration also impact fibril morphology. These observations point to opportunities for future exploration of how salt affects mfCMP stability after assembly in DPBS, covalent incorporation into the mfCMP–PEG hydrogel, and equilibrium swelling for both mechanistic understanding and actuation. Our thermoresponsive studies above, in addition to prior work, suggest the retention of assembled mfCMP building blocks within the hydrogel in physiologically relevant buffer.34,41 However, given the influence of salt on mfCMP assembly, opportunities for future responsive materials studies include examining the impact of changes in buffer identity, salt concentration, and pH on hydrogel mechanical properties, as well as determining whether ionic exchange after hydrogel formation impacts fibrillar structure. Such studies of the effects of ion concentration on mfCMP–PEG hydrogel properties would be of particular relevance toward translating these or similar materials into in vivo applications where salt concentration and pH locally vary.
Tuning composite mfCMP–PEG hydrogel formulations for cell culture applications
Fully synthetic hydrogels that allow for control over both modulus and fibrillar structure offer an opportunity to replicate aspects of the properties for a range of native tissues while minimizing batch-to-batch variability.66 Toward assessing the relevance of mfCMP–PEG hydrogels as platforms for applications in cell culture, wound healing models, or delivery, we sought to evaluate the impact of high concentrations of mfCMP on human lung fibroblast cell response. Lung fibroblasts natively exist within the collagen-rich loose connective tissue of the lung interstitium and are key players in all stages of wound healing.67 Fibroblasts are recruited to tissue injury sites and, when activated, produce various regulatory molecules, interact with immune cells, promote additional cell migration, and participate in ECM remodeling through MMP secretion and protein deposition, including collagens.68–70 Due to the prevalence of these cells in tissue and the active role they play, particularly during wound healing, we determined fibroblasts to be an appropriate cell type with which to perform an initial evaluation of these responsive mfCMP–PEG hybrid hydrogels with high mfCMP concentrations.To promote cell–matrix interactions in the mfCMP–PEG system, we introduced an alloc-functionalized pendent peptide containing an integrin-binding sequence for covalent incorporation into the hydrogel. As previously noted, these mfCMPs do not contain a specific integrin-binding motif, allowing us to take a modular approach in tuning the hydrogel properties. In this case we exploited this modularity to adjust the hierarchical structural content within the system while maintaining consistent biochemical functionality through the integrin-binding site present on the pendent peptide. Here, we incorporated an alloc-functionalized RGDS pendent peptide into the hydrogel network at a concentration of 2 mM, which has been shown to promote adequate cell–matrix interactions in both 2D and 3D culture applications of synthetic hydrogels and to be uniformly distributed throughout the hydrogel in similar materials (Fig. S13, ESI†).40,71–73 The peptide sequence RGDS is found in a number of ECM proteins, including fibronectin, vitronectin, and collagen type I where it typically is only accessible for cell interactions in thermally unfolded or otherwise damaged collagen type I.74–76 This peptide sequence binds to a number of integrins presented on the surface of human cells, making it a useful sequence for integration within synthetic ECMs for promoting cell adhesion.36,77 Notably, as RGDS is a pendent peptide, its incorporation into the hydrogel networks does not add to the crosslink density. To maintain consistency with the hydrogel compositions explored earlier in the work presented here, we increased the hydrogel thiol concentration from 20 mM to 22 mM (11 wt% PEG-SH) to keep the 1:1 thiol:alloc ratio while integrating 2 mM alloc from RGDS with the 20 mM alloc from linker peptides, the same concentration as the prior experiments without RGDS.
Importantly, under cell culture conditions, these mfCMP–RGDS–PEG hydrogels would also be exposed to different conditions than in the previous experiments, namely incubation in cell culture media at physiological temperature (37 °C), which is similar to the mfCMP Tm in DPBS where ∼50% of mfCMPs will be associated into triple helices and contribute to the hydrogel crosslink density. As previously mentioned, 50% mfCMP was determined to be the hydrogel composition with the highest concentration of mfCMP that would maintain robust hydrogel structure under these physiological conditions. Aiming to tune these hydrogel formulations to study the impact of different concentrations of mfCMP on cell response, the goal was to vary the number of peptide linkages coming from assembled mfCMP while maintaining a consistent bulk modulus between conditions to avoid confounding variables. As the 50% mfCMP (10 mM) was the highest concentration of mfCMP that consistently maintained a robust hydrogel structure under these conditions for at least one week, we took advantage of the modularity of this system to tune all compositions to match the modulus of these hydrogels after equilibrium swelling at 37 °C (10 mM mfCMP, or 50% of alloc-functionalized peptide linkage from mfCMP; 2 mM RGDS). To achieve modulus matching between hydrogel compositions at 37 °C, the concentration of the linker peptide was decreased as the mfCMP concentration was decreased (from 10 mM to 5 mM or 0 mM mfCMP, where the mfCMP concentrations were derived from the 50%, 25%, and 0% mfCMP–PEG hydrogel compositions, respectively). This resulted in an excess of thiols in the hybrid mfCMP–RGDS–PEG hydrogels (Table S2, ESI†), leading to lower crosslink densities and thereby similar moduli amongst the 0 mM to 10 mM mfCMP formulations at 37 °C.
Cell culture conditions impact mfCMP–RGDS–PEG hydrogel mechanical properties
Prior to studying cell response to mfCMP–RGDS–PEG hydrogels, we wanted to better understand the mechanical properties that encapsulated cells would experience in vitro. To do this, we prepared and maintained the hydrogels under the conditions that would be used for cell encapsulation and culture and conducted rheometric measurements at physiological temperatures. Specifically, 20 μL hydrogels with 2 mM RGDS and different amounts of mfCMP were prepared and allowed to equilibrium swell in DPBS overnight under mammalian cell culture conditions (37 °C, 5% CO2, under humidity, Fig. 4(A)). Rheometric measurements were conducted at 37 °C (ambient CO2, ambient humidity) on 3 hydrogel compositions: 0 mM, 5 mM, and 10 mM mfCMP with 2 mM RGDS (Fig. 4(B)). Initial storage modulus (G′) measurements were collected within the linear viscoelastic regime of these hydrogels, which established that the moduli between the 3 hydrogel compositions were statistically the same (Fig. 4(C)), demonstrating the modularity of this system for independently tuning assembled content, biochemical content, and bulk hydrogel modulus. | ||
| Fig. 4 Equilibrium swollen hydrogel mechanical properties under physiologically relevant conditions. (A) Timeline depicting hydrogel formation, equilibrium swelling, and rheometry, highlighting the environmental conditions at each step. (B) Schematic representing the mfCMP–RGDS–PEG hydrogel compositions for data shown in (C) and (D), where compositions for 0 mM and 5 mM mfCMP were adjusted to match the modulus of equilibrium swollen 10 mM mfCMP hydrogels (50% mfCMP peptide linkages) at 37 °C and concentrations refer to alloc concentration coming from each type of peptide linker (e.g., 5 mM mfCMP composition contains 5 mM alloc from mfCMP and 10.5 mM alloc from non-assembling peptide linker). (C) Storage moduli of mfCMP–RGDS–PEG hydrogels after equilibrium swelling under physiologically relevant conditions. (D) Strain sweeps of mfCMP–RGDS–PEG hydrogels with varying concentrations of mfCMP. Loss modulus data are available in Fig. S14 (ESI†). (All measurements were performed at 37 °C; n = 4; * indicates a significant difference from the control hydrogels (0 mM mfCMP) at the indicated strain (*p < 0.05, ***p < 0.001); n.s. = means are not significantly different; schematics are not to scale). | ||
After establishing the mechanical properties of these hydrogels within the linear viscoelastic regime, we conducted an experiment to study their response to high strain (Fig. 4(D)). Under physiologically relevant conditions, all 3 hydrogel formulations exhibited deviations from linear viscoelasticity at high strains, with an eventual yield strain indicating shear-thinning properties. Specifically, the 0 mM mfCMP and 5 mM mfCMP formulations initially underwent a slight increase in modulus beginning around 60% strain, followed by a significant drop in modulus (strain-yielding) between 200% and 300% strain. Notably, there were no significant differences in the modulus-strain profiles between these two conditions. In comparison, the 10 mM mfCMP formulation shows deviations from linear viscoelasticity that were both greater in magnitude and began at a lower strain than the hydrogels with 0 mM or 5 mM mfCMP. 10 mM mfCMP hydrogels appear to depart from linear viscoelasticity around 10% strain, where the strain-yielding begins around 100% strain, demonstrating significant differences in modulus from the 0 mM mfCMP control. Interestingly, the modulus profiles of these hydrogels in response to strain mirror those of harvested collagen shown in Fig. 2(D). Specifically, at low concentrations of collagen (1 mg mL−1 and 2 mg mL−1) as well as our hydrogels without mfCMP and with a lower concentration of mfCMP (0 mM mfCMP and 5 mM mfCMP), the moduli undergo a slight increase, or “strain overshoot”, prior to dropping off as the strain increases. In contrast, at high concentrations of collagen (3 mg mL−1) and high concentrations of mfCMP (10 mM mfCMP), the hydrogels do not exhibit this strain overshoot prior to strain-yielding. Interestingly, in rheological models, stain overshoots are a result of larger spring constants, which may indicate stronger elastic behavior in 0 mM and 5 mM mfCMP hydrogels and 1–2 mg mL−1 harvested collagen hydrogels compared to hydrogels with higher concentrations of fibrillar structure (10 mM mfCMP and 3 mg mL−1 harvested collagen).78,79 These results highlight similarities in mechanical property trends with increasing assembled content (e.g., mfCMP or collagen I, respectively) between the bioinspired synthetic mfCMP composite and the harvested collagen I hydrogels; however, the mfCMP–RGDS–PEG hydrogels allow for the formation of hydrogels at higher moduli and afford more control over hydrogel modulus, even with significant changes in fibrillar content. These mfCMP–RGDS–PEG hydrogels offer an approach to mimic structural and mechanical properties of a wide range of tissues while providing significant control over the materials with the potential for minimizing batch-to-batch variability.
Note, the earlier studies in this work indicate that the 0% mfCMP–PEG (0 mM mfCMP) condition did not deviate from linear viscoelasticity (Fig. 2(C)), whereas the 0 mM mfCMP–RGDS–PEG formulation in these later studies does deviate from linear viscoelasticity. There are a number of factors that may contribute to this observed difference. For example, the previous hydrogel properties were measured in situ and at 25 °C, whereas in this and the following studies, the hydrogels were equilibrium swelled at 37 °C prior to measurements at 37 °C. Additionally, the previous hydrogel had 1:1 stoichiometric ratios of thiol groups:alloc groups, whereas in this and the following studies, there are fewer non-assembling linker peptides (in order to modulus match to the 10 mM mfCMP–RGDS–PEG hydrogels), resulting in excess thiol groups and a lower crosslink density, which may allow for more chain flexibility within the hydrogel and the resulting response to strain. Ultimately, the strain-yielding behavior across all three hydrogel conditions reflects the strain-responsive properties of soft collagenous tissues, such as the lungs, making them of interest as fully synthetic 3D cell culture platforms.56 Nevertheless, by comparison, 10 mM mfCMP–RGDS–PEG hydrogels (50% mfCMP) exhibited interesting properties with significant differences in nonlinear viscoelasticity from the 0 mM mfCMP–RGDS–PEG control hydrogels (0% mfCMP) and was selected for further evaluation in 3D cell culture.
High concentrations of mfCMP content significantly impact cell response
Importantly, viscoelasticity and deviations from linear viscoelastic behavior play a large part in mechanotransduction and is important in mimicking normal healthy tissues.80,81 As viscoelastic and strain responsive hydrogels have allowed for cell migration not mediated by enzymatic degradation,82 and given the significant differences in structural content and viscoelastic response to strain between hydrogel compositions with 0% vs. 50% mfCMP, we were interested in how cells would respond differently to the two hydrogel compositions. Toward studying cell response to these materials, healthy human lung fibroblasts (CCL 151) were suspended in hydrogel precursor solution for encapsulation within each hydrogel formulation. The fibroblasts were encapsulated at 5 × 106 cells per mL, a cell density high enough to support cell viability (e.g., cell–cell interactions via paracrine signaling) but also low enough to promote predominantly cell–matrix interactions at the time of encapsulation (e.g., individual to small clusters of cells surrounded by synthetic ECM), even after equilibrium swelling in media.50 Specifically, these hydrogel formulations undergo swelling such that the equilibrium swollen volume is approximately 4.5× the volume immediately after preparation, resulting in a final fibroblast density of approximately 1.1 × 106 cells per mL. After encapsulation, cells were cultured for up to 6 days, and over that time we examined the fibroblasts for any differences in cell viability, morphology, and motility between the 0% and 50% mfCMP hydrogel formulations as an initial assessment of the relevance of these mfCMP–RGDS–PEG hydrogels with high mfCMP concentrations as a 3D cell culture platform.First, cells were stained for live/dead analysis 24 hours and 6 days after encapsulation, and then cytoskeletal and nuclear staining of fixed samples was performed at day 6. Following staining, z-stack images of the fibroblasts were taken using confocal microscopy (orthogonal projections available in Fig. 5(A) and Fig. S15, ESI†). Cell viability was quantified for each hydrogel condition at day 1 (24 hours) and day 6 (Fig. 5(B)), as hydrogels used for cell culture, as well as other applications including drug delivery, must support cell viability and function.2 There were no differences in viability between the 0% and 50% mfCMP hydrogels at either day 1 or day 6, indicating that the high concentration of mfCMPs within the 50% mfCMP condition did not impact fibroblast viability relative to the control (0% mfCMP).
 | ||
| Fig. 5 Human lung fibroblast viability and morphology in response to mfCMP–RGDS–PEG hydrogels. (A) Orthogonal projections of confocal z-stacks of fibroblasts in 3D culture within mfCMP–RGDS–PEG hydrogels with either 0% or 50% mfCMP (6 days after encapsulation; scale bars = 100 μm): left, stained for live/dead (green = live cells; red = dead cell nuclei); right, cytoskeletal staining (red = F-Actin; blue = nuclei). (B) Quantification of live/dead in (A) (see Fig. S15, ESI† for day 1 images). Viability results were normalized to the control condition (0% mfCMP) within each timepoint. (C) Half violin plots with individual data points indicating the number of individual cells within each object (defined as an individual cell or group of cells in contact). Associated histograms are available in Fig. S17 (ESI†). (***p < 0.001; n.s. = means are not significantly different). | ||
For characterizing fibroblast morphology, 3D renderings of the encapsulated cell bodies and nuclei were generated from the z-stacks. Qualitatively, the fibroblasts appeared to be elongated in both hydrogel formulations; however, larger clusters of cells were observed within the 50% mfCMP composition (Fig. 5(A), right). Qualitatively, the cell morphologies observed in response to the 50% mfCMP composition are consistent with published reports of human fibroblasts cultured within harvested collagen hydrogels, an encouraging result as we aim to capture aspects of collagen-rich tissues in these hydrogels.83–85 To understand cell morphology more within mfCMP–RGDS–PEG hydrogels, cell shape and the number of cells per cluster were quantified. Specifically, 3D shape factor (a measurement of sphericity) was calculated for each cell object identified within the images, where a cell object was defined as either a single cell or a cluster of cells in contact with each other. The shape factor for each object was sorted into 3 categories (elongated, spread, and rounded) for comparison between the two hydrogel conditions. While there is a trend toward more elongated cells in the 50% mfCMP condition, there was statistically no difference in the average shape factor between the 0% and 50% mfCMP conditions, and no significant differences emerged when comparing the fraction of objects within each shape factor category between the compositions (Fig. S16, ESI†). In previous work, we observed significant differences in human mesenchymal stem cell elongation between 6 wt% PEG hydrogels with 0 mM mfCMP (0% mfCMP peptide linkages) and 2.5 mM mfCMP (25% mfCMP peptide linkages).34 There are a few factors that may be contributing to the lack of statistical differences in elongation in this study with fibroblasts and the mfCMP–RGDS–PEG formulations. In particular, the hydrogels in the 3D culture studies have significantly lower modulus than the mfCMP hydrogels previously examined, as considerably higher concentrations of mfCMP were incorporated into these hydrogels, and the hydrogels without mfCMP were appropriately tuned to exhibit a matching modulus, in addition to differences between cell types (here, human lung fibroblast cells vs. previous studies of human mesenchymal stem cells).
In addition to analysis of cell elongation, cell clustering was quantified by calculating the number of nuclei contained within each cell object. Significantly more cells per cluster were observed in hydrogels with 50% mfCMP compared to hydrogels with 0% mfCMP (Fig. 5(C) and Fig. S17, ESI†). These results suggest that mfCMP hybrid hydrogels promote cell clustering and thereby a related increase in direct cell–cell contact.
The ECMs of native tissues direct cell responses, including migration, where migration becomes vital in the context of wound healing as various cell populations, including fibroblasts, are recruited to the injury site throughout the wound healing process.68,86 Because cell migration is a key part of healthy tissue function, it was important to assess whether the mfCMP–RGDS–PEG hydrogels allow for cell motility. To probe the impact of hydrogel composition on fibroblast motility, live cell imaging was conducted from 24 hours to 36 hours after encapsulation to monitor cell movement. This time range was chosen to allow time for hydrogel swelling and the potential for some initial cell-triggered pericellular degradation of the hydrogel and the initiation of general cell-mediated hydrogel remodeling based on literature reports.87,88 Each cell object (an individual cell or cluster of interacting cells) was identified and tracked over the course of the timelapse imaging. That information was used to generate track plots of cell motility and measure the displacement of each cell object from the first frame it was detected to the last, as well as the total distance traveled in those frames (Fig. 6(A)). Using the track plots, we can visualize the displacement and distance measurements of all the identified cells at once and compare those metrics between the two hydrogel compositions. Visually, we observed what appeared to be significantly larger displacements from the initial fibroblast location in 50% mfCMP hydrogels when compared to 0% mfCMP hydrogels (quantification in Fig. S18, ESI†).
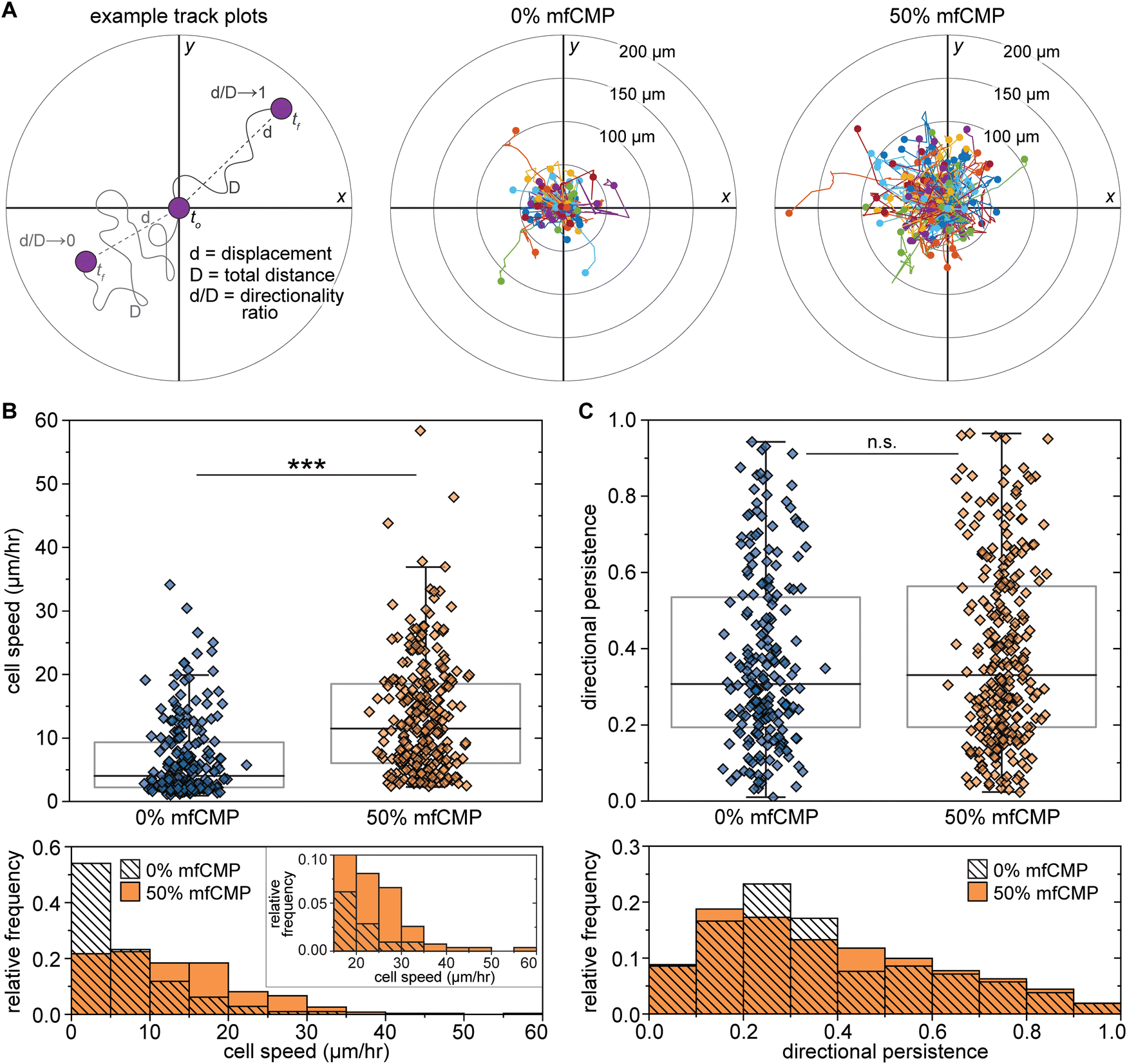 | ||
| Fig. 6 Impact of mfCMP–RGDS–PEG hydrogels on human lung fibroblast motility. Human lung fibroblasts were encapsulated within mfCMP–RGDS–PEG hydrogels (0% or 50% mfCMP compositions) and cell motility was monitored through timelapse imaging (24 hours to 36 hours after encapsulation). (A) Using timelapse imaging, each cell object (individual cells or clusters of cells) was tracked between frames to determine the total distance traveled (total distance, D, Fig. S18A, ESI†) and the displacement (d, Fig. S18B, ESI†), which were then used to determine the directionality ratio as a measurement of directional persistence (left). Track plots of fibroblast motility were generated for 0% mfCMP (center) and 50% mfCMP (right) conditions. Representative timelapse videos of cell motility are available in online ESI† (Videos S1 and S2). (B) Box plots (with individual data points) representing the speed of each cell object identified, with the associated histograms below. Individual cell object speed was determined from the total distance traveled divided by the time over which the object was tracked. (C) Box plots (with individual data points) representing the directionality ratio of each cell object identified, with the associated histograms below. Values closer to 0 indicate low directional persistence (meandering), where values closer to 1 indicate high directional persistence (direct movement). (***p < 0.001; n.s. = means are not significantly different; whiskers indicate outliers.) Data presentation for (B) and (C) as the mean ± SE is available in Fig. S19 (ESI†). | ||
To quantify fibroblast motility, we then evaluated 2 measurements: cell speed (total distance, D, traveled over time) and directional persistence (i.e., the directionality ratio, or the ratio of displacement, d, to total distance, D). For each cell object, the speed was determined by dividing the total distance traveled by the amount of time the object was tracked; for example, if a cell object traveled a total distance of 75.96 μm over the course of 7.5 hours (16 frames captured every 30 minutes), the speed would be 10.13 μm h−1. Similarly, the directional persistence was calculated for each object by dividing the displacement by the total distance traveled; for example, if a cell object traveled a total distance of 75.96 μm with a start-to-finish displacement of 49.22 μm, the directional persistence would be 0.6479, where values closer to 0 indicate low directional persistence (meandering movement) and values closer to 1 indicate high directional persistence (direct movement). With these measurements, we found that cells encapsulated within the 50% mfCMP composition moved significantly faster than the cells encapsulated within the 0% mfCMP composition (Fig. 1(B) and Fig. S19A, ESI†). While the bulk modulus between 0% and 50% mfCMP hydrogels were engineered to be statistically the same, the deviations from linear viscoelasticity with increasing strain were significantly different. This difference in non-linear viscoelastic response to strains may contribute in part to the difference in cell motility. Specifically, the increased sensitivity of 50% mfCMP hydrogels to imposed strains may allow cells to move through the hydrogel by applying forces to physically rearrange the matrix rather than relying solely, or primarily, on MMP-mediated degradation of the non-assembling linker peptide.82 Although we saw significant differences in cell speeds, there was no difference in directional persistence between the two conditions (Fig. 1(C) and Fig. S19B, ESI†). Taken together, these results suggest that the mfCMP hybrid hydrogels promote an increase in the rate of cell movement through the hydrogel, but not with any particular directionality. The lack of directional persistence in response to mfCMPs in these hydrogels is not surprising, as the alignment of fibrillar content has a significant impact on directional cell migration,89 and here there is no externally imposed directionality to the mfCMPs, synthetic ECM, or more broadly within microenvironments that are inspired by loose connective tissue. The non-directional, but significant, increase in cell speed within 50% mfCMP hydrogels compared to hydrogels without mfCMP demonstrates potential utility in further cell culture and delivery applications, particularly in the context of loose connective tissue. Looking forward, approaches to promote alignment of mfCMP fibrils prior to or during incorporation into the hydrogel matrix (e.g., applying electrical or magnetic fields or flow)90–93 provide opportunities for further enhancing motility or even directing migration in future studies with these materials.
Overall, the structural and mechanical properties of these mfCMP–PEG hydrogels, and their impacts on cell behavior, make them relevant for 3D cell culture applications. While these materials lack the full complexity found in native tissue ECMs, they provide a model system with high degrees of control for testing specific hypotheses and limit confounding variables that are inherent to more complex and in vivo models. Through the incorporation of high concentrations of mfCMP, we have aimed to capture some of the structural complexity and the viscoelastic behavior that structural proteins impart to the ECM within tissues, while maintaining the ability to modularly tune the hydrogel to mimic different collagen-rich microenvironments as desired. In particular, RGDS is one of many integrin-binding sequences found in the native ECM, and the hydrogel formulations discussed here do not fully capture the range of prominent integrin-binding sequences of a given tissue. However, the modular design of these hydrogels allows for incorporation of a variety of integrin binding peptides based on the application of interest, or a combination thereof; for example, peptides that present integrin-binding sequences found on collagen type-I (GFOGER), laminin (IKVAV, YIGSR, A5G81), and fibronectin (PHSRN + RGD) are well-defined and commonly used in synthetic ECMs.37,77 Note, these types of integrin-binding sequences, including the RGDS sequence utilized in these studies, may have non-specific interactions with assembling peptides, with the potential for formation of integrin-binding peptide clustering throughout the hydrogel that may impact cell morphology.94 Given that the PEG–peptide precursor solution is prepared and then briefly (∼10 seconds) and gently mixed with the assembled mfCMPs prior to rapid photopolymerization (gelation at ∼60 seconds),34 the time for non-specific interactions between RGDS and mfCMPs in solution is limited. However, the possibility for interactions between integrin-binding peptides and mfCMPs, whether non-specific or designed, and the control the approach affords over material formation presents an opportunity for future mechanistic studies around peptide sequestration, including ways to leverage it. Indeed, while in this work we separated assembling content from biochemical functionality to allow decoupling of their effects, another approach would be to incorporate a collagen-specific integrin-binding motif (e.g., GFOGER) within the hierarchically assembling mfCMP itself. Integration of integrin-binding sequences within the assembled peptide structure would be of particular interest for future work to inherently link hierarchical structure and biofunctionality together in a synthetic material and study the synergistic effects of structure and function. Further, this system complements other hydrogen bonding-driven and thermally or ionically responsive hydrogels, as the modular approach used for controlling mechanics and biochemical content with a combination of physical and chemical crosslinks could be applied with other assembling peptides.
Conclusions
In this publication, we have examined how mfCMPs [(PKG)4PK(alloc)G(POG)6(DOG)4] can be used for the creation of synthetic ECMs with a range of compositions and related responsive properties inspired by collagen-rich loose connective tissues like the lung interstitium. Specifically, we probed how assembly, concentration, and physical and covalent crosslinking conditions can be tuned to achieve responsive and bioinspired properties relevant for 3D culture. Here we have shown the influence of buffer and associated salt content on triple helix stability, fibrillar morphology, and physical hydrogel mechanical properties, where high salt concentrations result in collagen-like fibrillar assembly and physical hydrogel formation with mechanical properties, strain-response behaviors, and affinity for self-healing similar to that of harvested collagen. These results indicate that one could exploit the use of salt content even beyond what we have presented here, whether salt concentration or ion composition, to achieve specific melting temperatures, fibril morphologies, and hydrogel properties with these building blocks for a variety of applications.Further, we investigated how much alloc-functionalized, assembled mfCMP could be incorporated within composite mfCMP–PEG hydrogels and their resulting responsive properties. With this approach, we were able to form stable materials with a spectrum of assembled mfCMP content, creating hydrogels with a range of moduli, shear-thinning properties, and thermoresponsiveness. Harnessing the ability to independently tune mechanical properties (namely modulus), hierarchical structure, and biofunctional content, we developed hydrogel formulations with an integrin-binding peptide and varied mfCMP content (resulting in different viscoelastic behaviors) toward implementing these materials in cell culture applications. Hydrogel formulations with high concentrations of mfCMP supported cell spreading and elongation, promoted direct cell–cell interactions, and increased cell motility. These composite mfCMP–PEG hydrogels provide an opportunity to mimic the mechanical, structural, and biochemical properties of a wide range of collagenous tissues in a fully synthetic system for a variety of biomedical applications.
Ultimately, we have established a workflow to create adaptable hydrogel materials that exhibit properties associated with physically assembling systems and covalently crosslinked materials. These fully synthetic hydrogels allow control over fibrillar structure and hierarchical assembly on the nano- and micro-scale and the assembled content, biochemical functionality, and mechanical properties on the macro-scale. The extensive control over material properties and the wide range of properties that can be achieved sets up these adaptable materials to provide a platform for mimicking collagenous tissues in vitro while also allowing for adaptability to thermal stimuli.
Author contributions
E. M. F., A. M. H., and A. M. K. conceived the ideas and designed the experiments. E. M. F. and A. M. H. synthesized and characterized materials. A. M. H. performed mfCMP characterization, in situ rheometry experiments, and temperature cycling experiments. E. M. F. performed the experiments to determine hydrogel formulations for cell culture studies, rheometry experiments under physiological conditions, and all cell culture studies. E. M. F., A. M. H., and A. M. K. analyzed the data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
Data for this article are available at Harvard Dataverse Repository [https://dataverse.harvard.edu/dataverse/JMCB_FordHilderbrandKloxin_2024]. Data include: Cell morphology [https://doi.org/10.7910/DVN/P1OTRR, https://doi.org/10.7910/DVN/ZOZJ6I, https://doi.org/10.7910/DVN/OIQ9IC]. Cell motility [https://doi.org/10.7910/DVN/BFDOLH, https://doi.org/10.7910/DVN/2RZDVA, https://doi.org/10.7910/DVN/3W4TRK]. Cell viability [https://doi.org/10.7910/DVN/KRLLX7, https://doi.org/10.7910/DVN/UPX44R]. Circular dichroism [https://doi.org/10.7910/DVN/ROTFJ1]. Rheometry [https://doi.org/10.7910/DVN/EEUIDJ]Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the New Innovator Award funded by the National Institutes of Health (NIH) (DP2-HL152424), Pew Charitable Trusts (26178), and the University of Delaware Center for Hybrid, Active, and Responsive Materials (UD CHARM) Materials Research Science and Engineering Center (MRSEC) program supported by the National Science Foundation (NSF) (DMR-2011824). Support for instrumentation at the University of Delaware was provided in part by the Delaware Centers of Biomedical Research Excellence (COBRE) programs supported by grants funded by an Institutional Development Award from the National Institute of General Medical Sciences (NIGMS) of the NIH (P20GM104316, 5 P30 GM110758-02) and the UD CHARM MRSEC program supported by the NSF (DMR-2011824). This publication was also made possible by UD Core facilities including the Keck Center for Advanced Microscopy and Microanalysis and the University of Delaware NMR and Mass Spectrometry Core facilities. The authors would like to acknowledge the Millicent Sullivan group for their use of equipment and Christopher Kloxin for discussions regarding hydrogel properties.Notes and references
- X. Ding, H. M. Zhao, Y. Z. Li, A. L. Lee, Z. S. Li, M. J. Fu, C. N. Li, Y. Y. Yang and P. Y. Yuan, Synthetic peptide hydrogels as 3D scaffolds for tissue engineering, Adv. Drug Delivery Rev., 2020, 160, 78–104 CrossRef CAS PubMed.
- L. J. Lei, Y. J. Bai, X. Y. Qin, J. Liu, W. Huang and Q. Z. Lv, Current Understanding of Hydrogel for Drug Release and Tissue Engineering, Gels, 2022, 8(5), 31 Search PubMed.
- A. Z. Unal and J. L. West, Synthetic ECM: Bioactive Synthetic Hydrogels for 3D Tissue Engineering, Bioconjugate Chem., 2020, 31(10), 2253–2271 CrossRef CAS.
- U. Blache, E. M. Ford, B. Ha, L. Rijns, O. Chaudhuri, P. Y. W. Dankers, A. M. Kloxin, J. G. Snedeker and E. Gentleman, Engineered hydrogels for mechanobiology, Nat. Rev. Method. Prim, 2022, 2(1), 22 CrossRef.
- A. L. Kheng Lim Gohl, Daniel Béchet, Hierarchical mechanics of connective tissues: integrating insights from nano to macroscopic studies, J. Biomed. Nanotechnol., 2014, 10, 2464–2507 CrossRef.
- N. Reznikov, M. Bilton, L. Lari, M. M. Stevens and R. Kröger, Fractal-like hierarchical organization of bone begins at the nanoscale, Science, 2018, 360(6388), eaao2189 CrossRef PubMed.
- M. Sharabi, Structural Mechanisms in Soft Fibrous Tissues: A Review, Front. Mater., 2022, 8, 28 Search PubMed.
- L. D. Muiznieks and F. W. Keeley, Molecular assembly and mechanical properties of the extracellular matrix: A fibrous protein perspective, Biochim. Biophys. Acta, Mol. Basis Dis., 2013, 1832(7), 866–875 CrossRef CAS PubMed.
- Y. Tanino, Roles of extracellular matrix in lung diseases, Fukushima J. Med. Sci., 2024, 70(1), 1–9 CrossRef CAS PubMed.
- A. Sorushanova, L. M. Delgado, Z. Wu, N. Shologu, A. Kshirsagar, R. Raghunath, A. M. Mullen, Y. Bayon, A. Pandit, M. Raghunath and D. I. Zeugolis, The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development, Adv. Mater., 2019, 31(1), 1801651 CrossRef PubMed.
- I. Jorba, G. Beltrán, B. Falcones, B. Suki, R. Farré, J. M. García-Aznar and D. Navajas, Nonlinear elasticity of the lung extracellular microenvironment is regulated by macroscale tissue strain, Acta Biomater., 2019, 92, 265–276 CrossRef PubMed.
- J. G. Snedeker and A. Gautieri, The role of collagen crosslinks in ageing and diabetes – the good, the bad, and the ugly, Muscles Ligaments Tendons J., 2014, 4(3), 303–308 Search PubMed.
- A. J. Bailey, Molecular mechanisms of ageing in connective tissues, Mech. Ageing Dev., 2001, 122(7), 735–755 CrossRef CAS PubMed.
- I. Miler, M. D. Rabasovic, M. Aleksic, A. J. Krmpot, A. Kalezic, A. Jankovic, B. Korac and A. Korac, Polarization-resolved SHG imaging as a fast screening method for collagen alterations during aging: Comparison with light and electron microscopy, J. Biophotonics, 2021, 14(3), 11 CrossRef PubMed.
- L. E. Iannucci, C. S. Dranoff, M. A. David and S. P. Lake, Optical Imaging of Dynamic Collagen Processes in Health and Disease, Front. Mech. Eng., 2022, 8, 20 Search PubMed.
- M. C. Branco and J. P. Schneider, Self-assembling materials for therapeutic delivery, Acta Biomater., 2009, 5(3), 817–831 CrossRef CAS.
- M. E. Pitz, A. M. Nukovic, M. A. Elpers and A. A. Alexander-Bryant, Factors Affecting Secondary and Supramolecular Structures of Self-Assembling Peptide Nanocarriers, Macromol. Biosci., 2022, 22(2), 16 CrossRef PubMed.
- C. H. Liang, D. B. Zheng, F. Shi, T. Y. Xu, C. H. Yang, J. F. Liu, L. Wang and Z. M. Yang, Enzyme-assisted peptide folding, assembly and anti-cancer properties, Nanoscale, 2017, 9(33), 11987–11993 RSC.
- L. E. R. O’Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Multi-hierarchical self-assembly of a collagen mimetic peptide from triple helix to nanofibre and hydrogel, Nat. Chem., 2011, 3(10), 821–828 CrossRef PubMed.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Self-assembled peptide-based hydrogels as scaffolds for anchorage-dependent cells, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- M. Wang, J. Wang, P. Zhou, J. Deng, Y. Zhao, Y. Sun, W. Yang, D. Wang, Z. Li, X. Hu, S. M. King, S. E. Rogers, H. Cox, T. A. Waigh, J. Yang, J. R. Lu and H. Xu, Nanoribbons self-assembled from short peptides demonstrate the formation of polar zippers between β-sheets, Nat. Commun., 2018, 9(1), 5118 CrossRef PubMed.
- J. D. Tang, C. Mura and K. J. Lampe, Stimuli-Responsive, Pentapeptide, Nanofiber Hydrogel for Tissue Engineering, J. Am. Chem. Soc., 2019, 141(12), 4886–4899 CrossRef CAS PubMed.
- B. Ozbas, J. Kretsinger, K. Rajagopal, J. P. Schneider and D. J. Pochan, Salt-Triggered Peptide Folding and Consequent Self-Assembly into Hydrogels with Tunable Modulus, Macromolecules, 2004, 37(19), 7331–7337 CrossRef CAS.
- P. Worthington, K. M. Drake, Z. Q. Li, A. D. Napper, D. J. Pochan and S. A. Langhans, Beta-hairpin hydrogels as scaffolds for high-throughput drug discovery in three-dimensional cell culture, Anal. Biochem., 2017, 535, 25–34 CrossRef CAS PubMed.
- K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Peptide supramolecular materials for therapeutics, Chem. Soc. Rev., 2018, 47(20), 7539–7551 RSC.
- S. Chu, A. L. Wang, A. Bhattacharya and J. K. Montclare, Protein based biomaterials for therapeutic and diagnostic applications, Prog. Biomed. Eng., 2022, 4(1), 44 Search PubMed.
- S. R. Caliari and J. A. Burdick, A practical guide to hydrogels for cell culture, Nat. Methods, 2016, 13, 405 CrossRef CAS PubMed.
- E. Y. Du, F. Ziaee, L. Wang, R. E. Nordon and P. Thordarson, The correlations between structure, rheology, and cell growth in peptide-based multicomponent hydrogels, Polym. J., 2020, 52, 947–957 CrossRef CAS.
- E. Radvar and H. S. Azevedo, Supramolecular Peptide/Polymer Hybrid Hydrogels for Biomedical Applications, Macromol. Biosci., 2019, 19(1), 16 CrossRef CAS PubMed.
- H. Wang and S. C. Heilshorn, Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering, Adv. Mater., 2015, 27(25), 3717–3736 CrossRef CAS PubMed.
- C. Chen, D. Y. W. Ng and T. Weil, Polymer bioconjugates: Modern design concepts toward precision hybrid materials, Prog. Polym. Sci., 2020, 105, 101241 CrossRef CAS.
- D. Wu, N. Sinha, J. Lee, B. P. Sutherland, N. I. Halaszynski, Y. Tian, J. Caplan, H. V. Zhang, J. G. Saven, C. J. Kloxin and D. J. Pochan, Polymers with controlled assembly and rigidity made with click-functional peptide bundles, Nature, 2019, 574(7780), 658–662 CrossRef CAS PubMed.
- L. Cai, R. E. Dewi and S. C. Heilshorn, Injectable Hydrogels with In Situ Double Network Formation Enhance Retention of Transplanted Stem Cells, Adv. Funct. Mater., 2015, 25(9), 1344–1351 CrossRef CAS PubMed.
- A. M. Hilderbrand, E. M. Ford, C. Guo, J. D. Sloppy and A. M. Kloxin, Hierarchically structured hydrogels utilizing multifunctional assembling peptides for 3D cell culture, Biomater. Sci., 2020, 8(5), 1256–1269 RSC.
- A. M. Hilderbrand, P. A. Taylor, F. Stanzione, M. LaRue, C. Guo, A. Jayaraman and A. M. Kloxin, Combining simulations and experiments for the molecular engineering of multifunctional collagen mimetic peptide-based materials, Soft Matter, 2021, 17(7), 1985–1998 RSC.
- M. W. Tibbitt and K. S. Anseth, Hydrogels as extracellular matrix mimics for 3D cell culture, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- P. Dhavalikar, A. Robinson, Z. Y. Lan, D. Jenkins, M. Chwatko, K. Salhadar, A. Jose, R. Kar, E. Shoga, A. Kannapiran and E. Cosgriff-Hernandez, Review of Integrin-Targeting Biomaterials in Tissue Engineering, Adv. Healthcare Mater., 2020, 9(23), 26 Search PubMed.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nonlinear elasticity in biological gels, Nature, 2005, 435(7039), 191–194 CrossRef CAS PubMed.
- S. Uman, A. Dhand and J. A. Burdick, Recent advances in shear-thinning and self-healing hydrogels for biomedical applications, J. Appl. Polym. Sci., 2020, 137(25), 48668 CrossRef CAS.
- R. C. Locke, E. M. Ford, K. G. Silbernagel, A. M. Kloxin and M. L. Killian, Success Criteria and Preclinical Testing of Multifunctional Hydrogels for Tendon Regeneration, Tissue Eng., Part C, 2020, 26(10), 506–518 CrossRef CAS.
- E. M. Ford and A. M. Kloxin, Rapid Production of Multifunctional Self-Assembling Peptides for Incorporation and Visualization within Hydrogel Biomaterials, ACS Biomater. Sci. Eng., 2021, 7(9), 4175–4195 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676 CrossRef CAS PubMed.
- J. Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, TrackMate: An open and extensible platform for single-particle tracking, Methods, 2017, 115, 80–90 CrossRef CAS PubMed.
- D. Ershov, M. S. Phan, J. W. Pylvänäinen, S. U. Rigaud, L. Le Blanc, A. Charles-Orszag, J. R. W. Conway, R. F. Laine, N. H. Roy, D. Bonazzi, G. Duménil, G. Jacquemet and J. Y. Tinevez, TrackMate 7: integrating state-of-the-art segmentation algorithms into tracking pipelines, Nat. Methods, 2022, 19(7), 829–832 CrossRef CAS PubMed.
- R. Gorelik and A. Gautreau, Quantitative and unbiased analysis of directional persistence in cell migration, Nat. Protoc., 2014, 9(8), 1931–1943 CrossRef CAS PubMed.
- H. H. Winter and F. Chambon, Analysis of Linear Viscoelasticity of a Crosslinking Polymer at the Gel Point, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- L. Slyker, N. Diamantides, J. Kim and L. J. Bonassar, Mechanical performance of collagen gels is dependent on purity, α1/α2 ratio, and telopeptides, J. Biomed. Mater. Res., Part A, 2022, 110(1), 11–20 CrossRef CAS PubMed.
- Y. L. Yang, L. M. Leone and L. J. Kaufman, Elastic Moduli of Collagen Gels Can Be Predicted from Two-Dimensional Confocal Microscopy, Biophys. J., 2009, 97(7), 2051–2060 CrossRef CAS PubMed.
- I. C. Tanrikulu, L. N. Dang, L. Nelavelli, A. J. Ellison, B. D. Olsen, S. Jin and R. T. Raines, Synthetic Collagen Hydrogels through Symmetric Self-Assembly of Small Peptides, Adv. Sci., 2024, 11(3), 11 Search PubMed.
- M. E. Smithmyer, C. C. Deng, S. E. Cassel, P. J. LeValley, B. S. Sumerlin and A. M. Kloxin, Self-Healing Boronic Acid-Based Hydrogels for 3D Co-cultures, ACS Macro Lett., 2018, 7(9), 1105–1110 CrossRef CAS PubMed.
- M. H. Chen, L. L. Wang, J. J. Chung, Y. H. Kim, P. Atluri and J. A. Burdick, Methods To Assess Shear-Thinning Hydrogels for Application As Injectable Biomaterials, ACS Biomater. Sci. Eng., 2017, 3(12), 3146–3160 CrossRef CAS PubMed.
- E. Jalalvandi and A. Shavandi, Shear thinning/self-healing hydrogel based on natural polymers with secondary photocrosslinking for biomedical applications, J. Mech. Behav. Biomed. Mater., 2019, 90, 191–201 CrossRef CAS PubMed.
- H. Z. Huang, Y. Ding, X. Z. S. Sun and T. A. Nguyen, Peptide Hydrogelation and Cell Encapsulation for 3D Culture of MCF-7 Breast Cancer Cells, PLoS One, 2013, 8(3), 15 Search PubMed.
- J. Pupkaite, J. Rosenquist, J. Hilborn and A. Samanta, Injectable Shape-Holding Collagen Hydrogel for Cell Encapsulation and Delivery Cross-linked Using Thiol-Michael Addition Click Reaction, Biomacromolecules, 2019, 20(9), 3475–3484 CrossRef CAS PubMed.
- L. Zhang, Y. T. Zhou, D. D. Su, S. Y. Wu, J. Zhou and J. H. Chen, Injectable, self-healing and pH responsive stem cell factor loaded collagen hydrogel as a dynamic bioadhesive dressing for diabetic wound repair, J. Mater. Chem. B, 2021, 9(29), 5887–5897 RSC.
- S. R. Polio, A. N. Kundu, C. E. Dougan, N. P. Birch, D. E. Aurian-Blajeni, J. D. Schiffman, A. J. Crosby and S. R. Peyton, Cross-platform mechanical characterization of lung tissue, PLoS One, 2018, 13(10), 17 CrossRef PubMed.
- V. Kokol, Y. B. Pottathara, M. Mihelcic and L. S. Perse, Rheological properties of gelatine hydrogels affected by flow- and horizontally-induced cooling rates during 3D cryo-printing, Colloids Surf., A, 2021, 616, 10 CrossRef.
- Q. Mao, O. Hoffmann, K. Yu, F. Lu, G. Q. Lan, F. Y. Dai, S. M. Shang and R. Q. Xie, Self-contracting oxidized starch/gelatin hydrogel for noninvasive wound closure and wound healing, Mater. Des., 2020, 194, 12 Search PubMed.
- Y. Liang, M. V. Coffin, S. D. Manceva, J. A. Chichester, R. M. Jones and K. L. Kiick, Controlled release of an anthrax toxin-neutralizing antibody from hydrolytically degradable polyethylene glycol hydrogels, J. Biomed. Mater. Res., Part A, 2016, 104(1), 113–123 CrossRef PubMed.
- C. Tang, K. Zhou, Y. C. Zhu, W. D. Zhang, Y. Xie, Z. M. Wang, H. Zhou, T. T. Yang, Q. Zhang and B. C. Xu, Collagen and its derivatives: From structure and properties to their applications in food industry, Food Hydrocolloids, 2022, 131, 19 CrossRef.
- E. Leikina, M. V. Mertts, N. Kuznetsova and S. Leikin, Type I collagen is thermally unstable at body temperature, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(3), 1314–1318 CrossRef CAS PubMed.
- A. C. Nava, I. C. Doolaar, N. Labude-Weber, H. Malyaran, S. Babu, Y. Chandorkar, J. Di Russo, S. Neuss and L. De Laporte, Actuation of Soft Thermoresponsive Hydrogels Mechanically Stimulates Osteogenesis in Human Mesenchymal Stem Cells without Biochemical Factors, ACS Appl. Mater. Interfaces, 2023, 16(1), 30–43 CrossRef PubMed.
- L. C. Dunshee, M. O. Sullivan and K. L. Kiick, Therapeutic nanocarriers comprising extracellular matrix-inspired peptides and polysaccharides, Expert Opin. Drug Delivery, 2021, 18(11), 1723–1740 CrossRef CAS PubMed.
- K. Lauber, N. Brix, A. Ernst, R. Hennel, J. Krombach, H. Anders and C. Belka, Targeting the heat shock response in combination with radiotherapy: Sensitizing cancer cells to irradiation-induced cell death and heating up their immunogenicity, Cancer Lett., 2015, 368(2), 209–229 CrossRef CAS PubMed.
- E. B. Dolan, M. G. Haugh, D. Tallon, C. Casey and L. M. McNamara, Heat-shock-induced cellular responses to temperature elevations occurring during orthopaedic cutting, J. R. Soc., Interface, 2012, 9(77), 3503–3513 CrossRef CAS PubMed.
- E. Prince and E. Kumacheva, Design and applications of man-made biomimetic fibrillar hydrogels, Nat. Rev. Mater., 2019, 4(2), 99–115 CrossRef.
- G. Burgstaller, A. Sengupta, S. Vierkotten, G. Preissler, M. Lindner, J. Behr, M. Konigshoff and O. Eickelberg, Distinct niches within the extracellular matrix dictate fibroblast function in (cell free) 3D lung tissue cultures, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2018, 314(5), L708–L723 CrossRef CAS PubMed.
- F. Cialdai, C. Risaliti and M. Monici, Role of fibroblasts in wound healing and tissue remodeling on Earth and in space, Front. Bioeng. Biotechnol., 2022, 10, 18 Search PubMed.
- J. Roman, Fibroblasts-Warriors at the Intersection of Wound Healing and Disrepair, Biomolecules, 2023, 13(6), 19 CrossRef PubMed.
- S. S. Mathew-Steiner, S. Roy and C. K. Sen, Collagen in Wound Healing, Bioengineering, 2021, 8(5), 15 CrossRef PubMed.
- M. S. Rehmann, J. I. Luna, E. Maverakis and A. M. Kloxin, Tuning microenvironment modulus and biochemical composition promotes human mesenchymal stem cell tenogenic differentiation, J. Biomed. Mater. Res., Part A, 2016, 104(5), 1162–1174 CrossRef CAS PubMed.
- A. G. Karakecili, T. T. Demirtas, C. Satriano, M. Gümüsderelioglu and G. Marletta, Evaluation of L929 fibroblast attachment and proliferation on Arg-Gly-Asp-Ser (RGDS)-Immobilized chitosan in serum-containing/serum-free cultures, J. Biosci. Bioeng., 2007, 104(1), 69–77 CrossRef CAS PubMed.
- L. A. Sawicki and A. M. Kloxin, Design of thiol–ene photoclick hydrogels using facile techniques for cell culture applications, Biomater. Sci., 2014, 2, 1612–1626 RSC.
- A. P. Kishan, R. M. Nezarati, C. M. Radzicki, A. L. Renfro, J. L. Robinson, M. E. Whitely and E. M. Cosgriff-Hernandez, In situ crosslinking of electrospun gelatin for improved fiber morphology retention and tunable degradation, J. Mater. Chem. BJ. Mater. Chem. B, 2015, 3(40), 7930–7938 RSC.
- S. L. Bellis, Advantages of RGD peptides for directing cell association with biomaterials, Biomaterials, 2011, 32(18), 4205–4210 CrossRef CAS PubMed.
- A. V. Taubenberger, M. A. Woodruff, H. F. Bai, D. J. Muller and D. W. Hutmacher, The effect of unlocking RGD-motifs in collagen I on pre-osteoblast adhesion and differentiation, Biomaterials, 2010, 31(10), 2827–2835 CrossRef CAS PubMed.
- E. Ruoslahti, RGD and other recognition sequences for integrins, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS PubMed.
- A. K. Townsend and H. J. Wilson, Small- and large-amplitude oscillatory rheometry with bead-spring dumbbells in Stokesian Dynamics to mimic viscoelasticity, J. Non-Newtonian Fluid Mech., 2018, 261, 136–152 CrossRef CAS.
- G. Mattei, L. Cacopardo and A. Ahluwalia, Engineering Gels with Time-Evolving Viscoelasticity, Materials, 2020, 13(2), 14 CrossRef PubMed.
- I. Levental, P. C. Georges and P. A. Janmey, Soft biological materials and their impact on cell function, Soft Matter, 2007, 3(3), 299–306 RSC.
- E. Hui, L. Moretti, T. H. Barker and S. R. Caliari, The Combined Influence of Viscoelastic and Adhesive Cues on Fibroblast Spreading and Focal Adhesion Organization, Cell. Mol. Bioeng., 2021, 14(5), 427–440 CrossRef CAS PubMed.
- Y. Liu, B. Liu, J. J. Riesberg and W. Shen, In Situ Forming Physical Hydrogels for Three-dimensional Tissue Morphogenesis, Macromol. Biosci., 2011, 11(10), 1325–1330 CrossRef CAS PubMed.
- K. Zuniga, M. Gadde, J. Scheftel, K. Senecal, E. Cressman, M. Van Dyke and M. N. Rylander, Collagen/kerateine multi-protein hydrogels as a thermally stable extracellular matrix for 3D in vitro models, Int. J. Hyperthermia, 2021, 38(1), 830–845 CrossRef CAS PubMed.
- T. Jiao, Q. Lian, W. L. Lian, Y. H. Wang, D. C. Li, R. L. Reis and J. M. Oliveira, Properties of Collagen/Sodium Alginate Hydrogels for Bioprinting of Skin Models, J. Bionic Eng., 2023, 20(1), 105–118 CrossRef.
- M. Bacakova, J. Pajorova, A. Broz, D. Hadraba, F. Lopot, A. Zavadakova, L. Vistejnova, M. Beno, I. Kostic, V. Jencova and L. Bacakova, A two-layer skin construct consisting of a collagen hydrogel reinforced by a fibrin-coated polylactide nanofibrous membrane, Int. J. Nanomed., 2019, 14, 5033–5050 CrossRef CAS PubMed.
- S. Sasikumar, S. Chameettachal, B. Cromer, F. Pati and P. Kingshott, Decellularized extracellular matrix hydrogels-cell behavior as a function of matrix stiffness, Curr. Opin. Biomed. Eng., 2019, 10, 123–133 CrossRef.
- C. Loebel, R. L. Mauck and J. A. Burdick, Local nascent protein deposition and remodelling guide mesenchymal stromal cell mechanosensing and fate in three-dimensional hydrogels, Nat. Mater., 2019, 18(8), 883–891 CrossRef CAS PubMed.
- M. Daviran, J. Catalano and K. M. Schultz, Determining How Human Mesenchymal Stem Cells Change Their Degradation Strategy in Response to Microenvironmental Stiffness, Biomacromolecules, 2020, 21(8), 3056–3068 CrossRef CAS PubMed.
- W. Y. Wang, C. D. Davidson, D. Lin and B. M. Baker, Actomyosin contractility-dependent matrix stretch and recoil induces rapid cell migration, Nat. Commun., 2019, 10, 12 CrossRef PubMed.
- N. Debons, K. Matsumoto, N. Hirota, T. Coradin, T. Ikoma and C. Aimé, Magnetic Field Alignment, a Perspective in the Engineering of Collagen-Silica Composite Biomaterials, Biomolecules, 2021, 11(5), 16 CrossRef PubMed.
- L. J. Martin, B. Akhavan and M. M. M. Bilek, Electric fields control the orientation of peptides irreversibly immobilized on radical-functionalized surfaces, Nat. Commun., 2018, 9, 11 CrossRef PubMed.
- G. Pandey, J. Saikia, S. Sasidharan, D. C. Joshi, S. Thota, H. B. Nemade, N. Chaudhary and V. Ramakrishnan, Modulation of Peptide Based Nano-Assemblies with Electric and Magnetic Fields, Sci. Rep., 2017, 7, 9 CrossRef PubMed.
- A. Ahmed, M. Mansouri, I. M. Joshi, A. M. Byerley, S. W. Day, T. R. Gaborski and V. V. Abhyankar, Local extensional flows promote long-range fiber alignment in 3D collagen hydrogels, Biofabrication, 2022, 14(3), 15 CrossRef PubMed.
- J. Lam and T. Segura, The modulation of MSC integrin expression by RGD presentation, Biomaterials, 2013, 34(16), 3938–3947 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectrometry results (mfCMP1a, non-assembling peptide linker, RGDS); circular dichroism of mfCMP in water and sodium phosphate buffer; TEM images of mfCMP assembled in water, sodium phosphate buffer, and DPBS; TEM images of mfCMP assembled in DPBS, lyophilized, and resuspended; NMR results (PEG-SH, LAP); mfCMP physical hydrogel rheometry results (assembled in sodium phosphate buffer); full shear modulus data (in situ-formed mfCMP–PEG hydrogels, in situ-formed collagen hydrogels, temperature cycling for equilibrium swollen mfCMP-PEG hydrogels, equilibrium swollen mfCMP–RGDS–PEG hydrogels under physiologically relevant conditions); initial collagen storage modulus data within the linear viscoelastic regime; live/dead day 1 images; shape factor results and quantification; cells per cluster histograms; cell displacement and total distance results and quantification; cell motility results (mean ± standard error presentation); hydrogel formulation tables; and cell motility videos (PDF). Cell motility videos (MOV files). See DOI: https://doi.org/10.1039/d4tb00562g |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |