
Diffusion doping of analgesics into UHMWPE for prophylactic pain management†
Nicoletta
Inverardi
 ab,
Sashank
Lekkala
a,
Maria F.
Serafim
a,
Amita
Sekar
ab,
Keith K.
Wannomae
a,
Brad
Micheli
a,
Hany
Bedair
ab,
Orhun K.
Muratoglu
ab and
Ebru
Oral
*ab
ab,
Sashank
Lekkala
a,
Maria F.
Serafim
a,
Amita
Sekar
ab,
Keith K.
Wannomae
a,
Brad
Micheli
a,
Hany
Bedair
ab,
Orhun K.
Muratoglu
ab and
Ebru
Oral
*ab
aHarris Orthopaedic Laboratory, Massachusetts General Hospital, Boston, Massachusetts 02114, USA
bDepartment of Orthopaedic Surgery, Harvard Medical School, Boston, Massachusetts 02114, USA. E-mail: eoral@mgh.harvard.edu; Tel: +1 (617) 726-0657
First published on 27th August 2024
Abstract
Pain management after total joint arthroplasty is often addressed by systemic delivery of opioids. Local delivery of non-opioid analgesic drugs directly in the joint space from the UHMWPE component of the prosthesis would be highly beneficial to increase the efficacy of the drugs, decreasing the overall side effects and the risk of opioid addiction. It has been shown that effective concentrations of local analgesics can be achieved by eluting from analgesic-blended UHMWPE; however, this approach is limited by the decrease in mechanical properties resulting from the extent of phase separation of the blended drugs from the polymeric matrix. Here we hypothesized that mechanical properties could be maintained by incorporating analgesics into solid form UHMWPE by diffusion as an alternative method. Lidocaine or bupivacaine were diffused in solid form UHMWPE with or without radiation crosslinking. The loaded drug content, the spatial distribution of the drugs and their chemical stability after doping were characterized by FTIR and NMR spectroscopy, respectively. Drug release kinetics, tensile mechanical properties and wear rates were assessed. The results showed that diffusion doping could be used as a promising method to obtain a therapeutic implant material without compromising its mechanical and structural integrity.
Introduction
Total joint arthroplasty is a widely used and effective surgery by which the bony and cartilaginous surfaces of the diseased joint are replaced by synthetic components to recover the articulating and load-bearing functions of the joint. The prostheses are most often comprised of an articular pair, one of which is made of metal or ceramic and the other of ultra-high molecular weight polyethylene (UHMWPE).1 This semicrystalline polymer has proven effective in orthopedics due to its relatively high strength, wear resistance, low friction, and biocompatibility.2 The wear performance of UHMWPE bearing surfaces was improved by crosslinking,3,4 which decreased the wear rate and the incidence of wear-related osteolysis.5–8 State-of-the-art UHMWPE bearing materials are processed by radiation and chemical crosslinking to obtain high wear resistance,8–14 and are stabilized by an antioxidant such as α-tocopherol (vitamin E) for long-term oxidation resistance.15–23 Crystallinity is maintained while increasing crosslink density to preserve high fatigue propagation resistance for load-bearing performance, especially under adverse conditions such as for tibial inserts under higher cyclic stresses.24–28More recently, the UHMWPE component of the joint implant was proposed as a local drug delivery device in addition to its primary mechanical function.29,30 Some potential benefits of using UHMWPE as an eluting device are an increase in bioavailability of drugs at the local site compared to drugs administered systemically, and a decrease in the systemic toxicity and side effects. A variety of non-opioid analgesic drugs (such as bupivacaine30), non-steroidal anti-inflammatory drugs (such as tolfenamic acid31,32) and antibiotic drugs (such as gentamicin, vancomycin and tobramycin29,33,34) were selected and incorporated into UHMWPE to address various challenges associated with joint replacement such as post-operative pain and infection. The incorporation method proposed was mechanical mixing or blending of the solid drug powders with the polymer resin powder before compression molding into solid forms to obtain a composite matrix made of UHMWPE embedded with drug domains. Such a method leads to a phase-separated material, where drug domain size and eccentricity have been shown to be affected by such factors as drug concentration, the polar surface area and the physicochemical properties of the drug, and compression during solid-form fabrication.29,31,32,35,36 The mechanical strength and ductility were decreased with increasing concentration of the incorporated drugs. There is a need to improve the mechanical strength and toughness of therapeutic-incorporated UHMWPE further for long-term, state-of-the-art performance in permanent implants.
Drug transport by diffusion through polymeric matrices is commonly studied in degradable and non-degradable polymeric matrices;37,38 however, it has not been studied in UHMWPE, which is non-porous and non-degradable and has not been extensively used for agent delivery. Diffusion in UHMWPE has been studied for vitamin E, which is a lipophilic antioxidant with high solubility in UHMWPE.39,40 In this compatible system without phase separation, the crystallinity, the mechanical strength, and ductility of the doped UHMWPE are unaffected by vitamin E. The diffusion coefficient was shown to decrease with increasing crosslinking in the network and to increase with increasing temperature. The impedance of the crystalline fraction to diffusion was reduced by conducting the diffusion process close to but below the peak melting point of radiation crosslinked UHMWPE. Doping temperature was maintained below the melting temperature because the mechanical strength of crosslinked UHMWPE is decreased if crosslinked material is melted due to the hindrance of efficient recrystallization in the presence of crosslinks. This process also minimized dimensional changes due to the incorporation of vitamin E. Based on these previous findings, we hypothesized that the mechanical strength and toughness of analgesic-eluting UHMWPE could be improved by doping it with the non-opioid analgesic drugs lidocaine and bupivacaine using diffusion.
Experimental section
Materials
UHMWPE powder was obtained by Celanese Corporation (GUR® 1020, average molecular weight: 4.7 × 106 g mol−1, average particle size: 140 μm, density: 930 kg m−3).41 The freebase form of the analgesic drugs lidocaine, LD (L7757, Sigma-Aldrich Co.) and bupivacaine, BP (16618, Cayman Chemical and J62742, Thermo Fisher Scientific) were used. Deionized water (751–628), o-xylene (X1040), DMSO-d6 (151874), and preserved bovine serum (12133C) were obtained by Sigma-Aldrich. Ethanol, 200 proof (04-355-223) and Phosphate Buffered Saline (1× Solution, pH 7.4, BP243820) were purchased by Thermo Fisher Scientific.Manufacturing of virgin and crosslinked UHMWPE
UHMWPE blocks were obtained by compression molding (Carver Press, Wabash, IN) of the polymer powder into a pre-heated custom mold (cavity: 50 × 85 mm2) over a range of thickness in between 3 mm to 16 mm. The molding cycle included a heating step at 180 °C under 20 MPa for 10 or 20 min, for 4 mm thick – and 16 mm thick blocks, respectively, and a subsequent cooling step down to 10 °C under the same applied pressure for at least 45 min.A set of molded blocks with varying thickness was e-beam irradiated (beam energy 2 MeV) at a total dose of 100 kGy at 12.5 kGy per pass (High Voltage Research Laboratory, Massachusetts Institute of Technology, Cambridge, MA). Thicker blocks (16 mm thick) underwent irradiation on both sides, due to the limited penetration of e-beam irradiation. Prior to irradiation, blocks were vacuum packed under inert atmosphere by fluxing with argon.
Both the non-irradiated and 100 kGy irradiated blocks were machined by CNC machining (ShopBot Desktop) into various geometries for further processing and later testing: (i) prismatic strips (3 × 5 × 20 mm3, n = 18); (ii) plates (3.2 × 24 × 65 mm3, n = 2); (iii) cylindrical pins (Φ = 9 mm, h = 13 mm, n = 9).
The materials analyzed in this study can be classified into 4 groups: (1) virgin UHMWPE (no additives); (2) 100 kGy irradiated UHMWPE; (3) analgesic-doped UHMWPE (3a) lidocaine-doped or (3b) bupivacaine-doped; (4) analgesic-doped 100 kGy irradiated UHMWPE (4a) lidocaine-doped 100 kGy irradiated UHMWPE or (4b) bupivacaine-doped 100 kGy irradiated UHMWPE.
Diffusion doping of analgesics into UHMWPE
The diffusion doping process was performed by soaking UHMWPE samples into the molten drug under controlled conditions, as outlined in Fig. 1.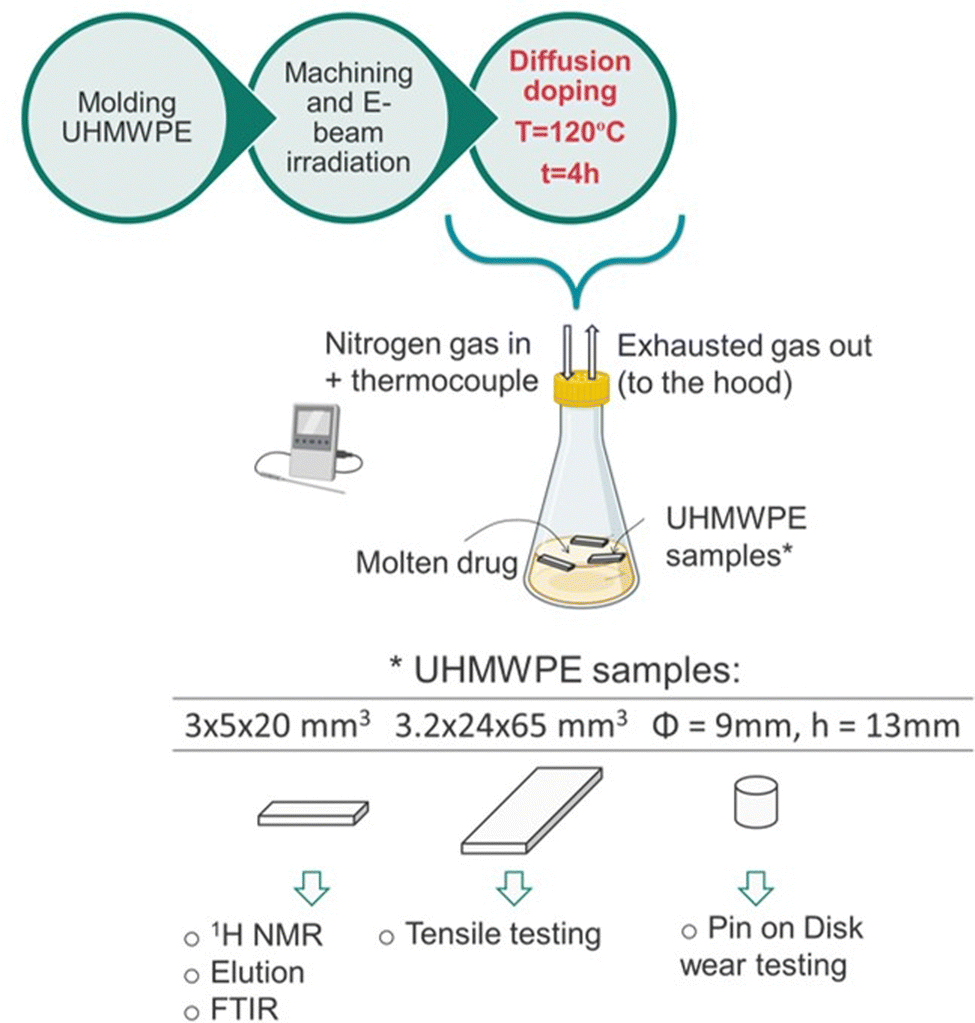 | ||
| Fig. 1 Schematics of the manufacturing process and the testing methodologies. The figure was partially created with BioRender.com. | ||
First, the freebase drug (about 50 g) lidocaine or bupivacaine was equilibrated at 120 °C, under nitrogen gas flow (8–10 cc min−1) in a chemical hood. The samples (the prismatic strips in (i), or the plates in (ii) or the cylindrical pins in (iii)) were introduced into the flask and the doping process was carried out at 120 °C for 4h. Every 30 min, the samples were flipped to expose all the surfaces to the molten drug and the temperature of the liquid was closely monitored with a thermocouple. At the end of the treatment, the samples were retrieved, gently blotted with a Kimwipes™ (Kimberly-Clark Professional™) and let cool down to room temperature. Additional diffusion doping experiments were performed to investigate the effect of the doping temperature on the drug uptake and its diffusion kinetics. Thin non-irradiated and 100 kGy irradiated UHMWPE samples (150 μm thin, section: 6.35 × 12.75 mm2) were doped with lidocaine or bupivacaine for increasing doping periods (from 0 to 10 min, n = 3 for each timepoint) at 120 °C. Their weight was measured before and after the doping treatment and the drug uptake was calculated as a percentage on the total mass of the sample. Thin non-irradiated UHMWPE samples were also doped with lidocaine for increasing doping periods (from 0 to 10 min) at various temperature (i.e., 100, 110 and 120 °C) and the drug uptake was measured likewise, by gravimetric measurements. For each timepoint and doping temperature, n = 3 samples were used. Thick non-irradiated UHMWPE samples (cylindrical pins, diameter: 9 mm; height: 13 mm) were also doped with lidocaine or bupivacaine for various periods (i.e., 1 h, 2 h and 4 h) at the doping temperature of 120 °C, and their weight was measured before and after the doping process.
Physicochemical characterization: gravimetric measurement
Each sample was weighed before and after the diffusion doping process on an analytical scale (Mettler Toledo XS205 DualRange Analytical Balance, 0.01 mg resolution; Mettler Toledo, Columbus OH) to evaluate the drug intake in terms of mass.Physicochemical characterization: 1H nuclear magnetic resonance (NMR) spectroscopy
Drug stock solutions (1 mg mL−1) were prepared in DMSO-d6 and the analgesic-doped prismatic strips were eluted into 1.7 mL DMSO-d6 for 24 h. 1H NMR spectra were recorded for both the unprocessed drug stock and the drug eluted from the doped strips with a 500 MHz Varian spectrometer (Palo Alto, CA). Each spectrum was an average of 64 scans.Physicochemical characterization: Fourier-transform infrared (FTIR) spectroscopy
FTIR was used to confirm the radiation dose profile and to investigate the diffusion profile of the doped drugs along the thickness of the prismatic samples. Samples were microtomed into thin sections (16 × 5 mm2 or 3 × 5 mm2 or circular sections with diameter of 9 mm, 150 μm thick) for FTIR analysis (Agilent, Varian 670-IR with a Varian 620-IR FTIR microscope attachment; Agilent Technologies Inc., Wilmington, DE). FTIR spectra were collected by scanning the thickness dimension (16, or 3 or 9 mm) every 0.20 mm. Thin samples (150 μm, section: 6.35 × 12.75 mm2) were also analyzed by FTIR after doping with lidocaine or bupivacaine. At least three FTIR measurements on three different samples of the same group were performed. Each spectrum was an average of 32 scans.The trans-vinylene index (TVI) was defined as the ratio of the area under the peak at 965 cm−1 (integration limit: 950–980 cm−1) and the polyethylene skeletal absorbance (i.e., 1895 cm−1, integration limit: 1850–1985 cm−1).42 TVI was measured on irradiated UHMWPE of different thickness (3 or 16 mm) before diffusion doping. The FTIR peaks used to calculate the amount of drug diffused in UHMWPE are shown on representative spectra (Fig. 2).
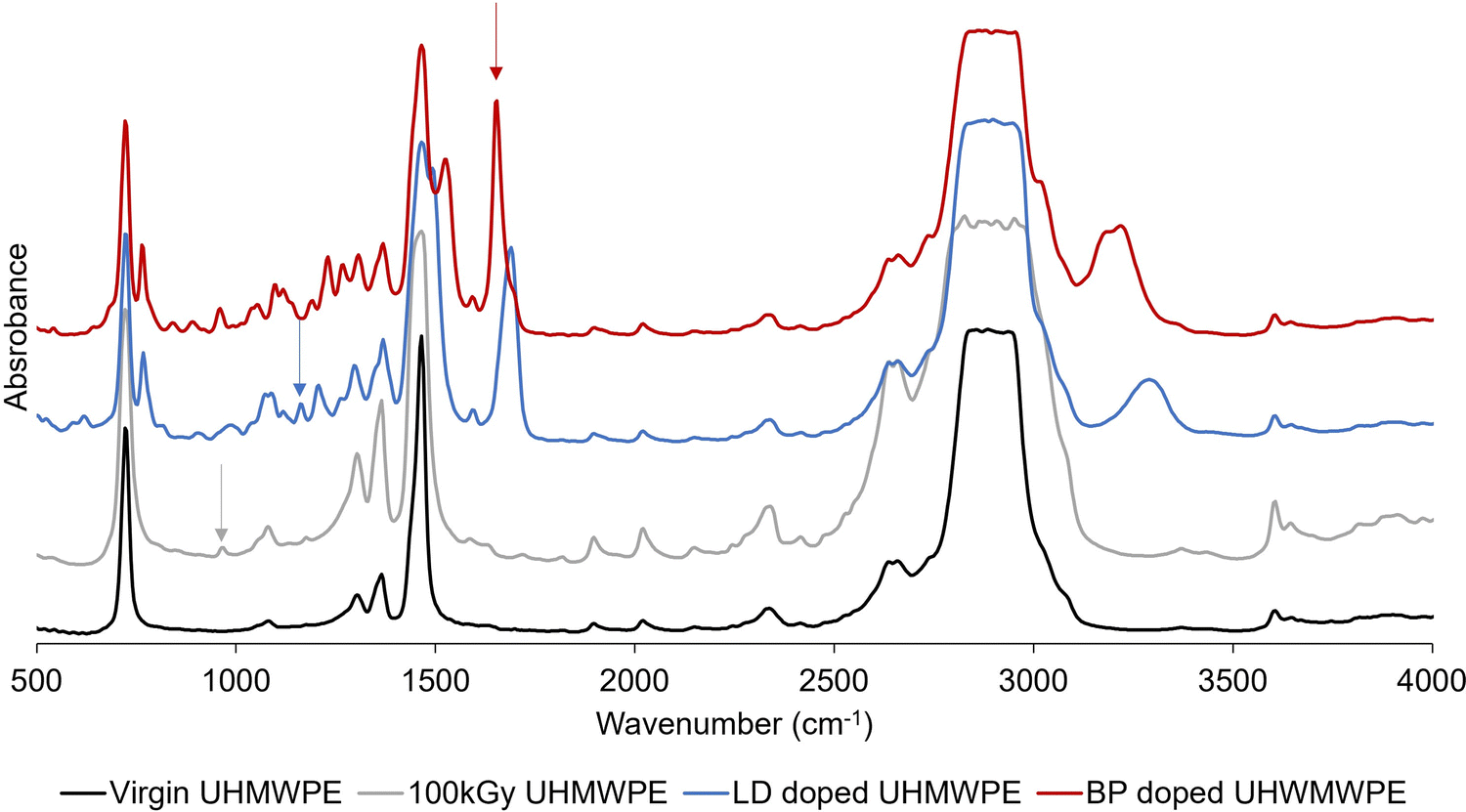 | ||
| Fig. 2 Fourier transform infrared spectroscopy (FTIR) peak absorbances for evaluating radiation exposure and the spatial distribution of incorporated lidocaine and bupivacaine. Representative FTIR spectra for non-irradiated and 100 kGy irradiated UHMWPE before and after doping. The arrows point to the peaks used for evaluating the trans-vinylene (TVI) index (grey arrow), the lidocaine index (blue arrow) and the bupivacaine index (red arrow). | ||
The lidocaine and bupivacaine indexes were defined as the ratio of the area under the selected drug peak (i.e., 1165 cm−1, integration limit: 1155–1175 cm−1; and 1674 cm−1, integration limit: 1610–1740 cm−1; for lidocaine and bupivacaine, respectively) and the polyethylene skeletal absorbance (i.e., 1895 cm−1, integration limit: 1850–1985 cm−1).
Physicochemical characterization: swelling measurement for crosslink density evaluation
Swelling experiments were performed in hot o-xylene to measure the crosslink density of the irradiated samples. Samples were obtained from the cylindrical pins by cutting approximately 3 × 3 × 3 mm3 cubes (approximately 15 mg, n = 3) and were weighed with an analytical scale (Mettler Toledo, resolution: 0.01 mg). The samples were immersed in pre-heated 25 mL o-xylene at 130 °C in an oil bath (Fisher Scientific, Isotemp 3016HD) for two hours to reach equilibrium swelling. The final weight of the swollen samples was recorded, the samples were dried at 45 °C in a vacuum oven for 16–18 h and their dried wight was recorded. The swell ratio ρ was calculated using eqn (1)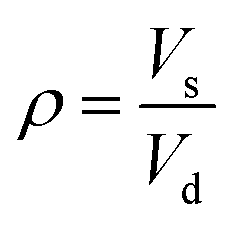 | (1) |
 | (2) |
Physicochemical characterization: thermogravimetric analysis
Thermogravimetric analysis was performed using a TGA Q50 (TA Instruments, New Castle, DE) on small samples (10 mg approximately) loaded on a platinum pan under nitrogen purging (60 mL min−1). The thermal program used involved thermal equilibration at 30 °C, followed by a 10 °C min−1 heating ramp up to 600 °C. TGA was performed on virgin UHMWPE, the free drug lidocaine, and the free drug bupivacaine (n = 3). Additionally, composition analysis by TGA was performed for the doped materials by testing a sample taken from the surface (depth position: 0 mm, average sample thickness: 0.40 ± 0.11 mm) and one from the core (depth position: 1.5 mm, average sample thickness: 0.48 ± 0.15 mm) of the doped prismatic strips (5 × 5 mm2 thin slices). TGA was also performed at the end of the elution study (day 28) on samples obtained from the surface and the core of the eluted strips, after gently drying the surface with a Kimwipes™ (Kimberly-Clark Professional™). TGA analysis was performed in duplicates for each material and each condition (i.e., before and after elution) at each depth position (i.e., surface and core).Physicochemical characterization: differential scanning calorimetry
Differential scanning calorimetry was performed using a Discovery DSC (TA Instruments, New Castle, DE) on small samples (10 mg approximately) under nitrogen purging (50 mL min−1). The thermal program involved a first heating scan from −20 °C to 180 °C, cooling down to −20 °C and a second heating scan up to 180 °C. Each scan was performed at 10 °C min−1 and in between each scan an isothermal step for 2 min was inserted. DSC tests were performed on UHMWPE before and after irradiation, on the free drugs and on the doped materials (n = 3). For the freebase lidocaine scan, the maximum temperature set was 130 °C. For each doped material, two samples were tested of which one was taken from the surface and on from the core of the doped prismatic strips (n = 3 for each condition).Tensile testing
Tensile samples (n = 3) were obtained by die-cutting 3.2 mm thick plates of the 4 study groups. Tensile testing was performed according to the ASTM D638-10 standard on an MTS Insight 2 (MTS Systems, Eden Prairie, MN) dynamometer equipped with a 2000 N load cell. The crosshead speed was set to 10 mm min−1. The displacement was obtained by a laser extensometer. Stress versus strain curves were plotted, and ultimate tensile strength (UTS) and elongation at break (EAB) were obtained. The parameter “work-to-failure” (in kJ m−2) was calculated as the area under the stress versus elongation curve until failure.Wear testing
Cylindrical pin-on-disk-samples were wear tested on a custom wear testing machine (OrthoPOD, Advanced Mechanical Technology Inc., Watertown, MA) with a rectangular pattern 5 × 10 mm2 against polished CoCr discs at 2 Hz in undiluted, preserved bovine serum as a lubricant. 100 kGy irradiated UHWPE pins were washed before testing. Bupivacaine-doped 100 kGy irradiated pins were washed and pre-eluted twice into DI water (for 30 min) in an ultrasonic cleaner (Branson), followed by ethanol (for 30 min) and 30 min of drying, before the start of the test. Lidocaine-doped 100 kGy pins underwent a similar washing followed by additional 5 cycles composed of washing into DI water (30 min) in the ultrasonic cleaner, followed by ethanol (2 h) and drying (30 min), before the test. The pins were cleaned and weighed before testing, after the first 0.5 million cycles (MC) and at each 0.157 MC until a total of 1.128 MC. The wear rate was determined by linear regression of the weight loss as a function of number of cycles from 0.5 to 1.128 MC.Elution experiments and pharmacokinetic modeling
Analgesics doped UHWMPE samples (3 × 5 × 20 mm3 prismatic strips, n = 3) were eluted in 29.7 mL de-ionized (DI) water or Phosphate Buffered Saline (PBS) under mild shaking (100 rpm) at room temperature or at 37 °C. The eluent was collected at given timepoints until day 28 and at each timepoint it was refreshed with new media. The drug concentration was measured by detecting the UV absorbance at 250 nm using a plate reader (Biotek, Agilent, Santa Clara, CA). The obtained data was used to calculate the cumulative drug mass release, the fractional release, and the release rate.Cumulative drug mass release profiles were fitted by using the semi-empirical Korsmeyer–Peppas equation, reported in eqn (3):43
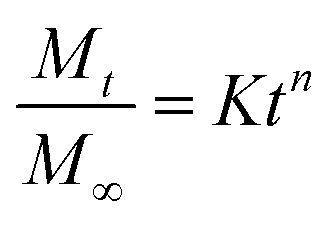 | (3) |
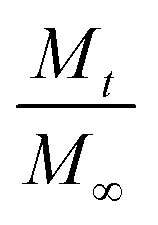 is a fraction of the drug released at time t, Mt is the released amount at time t, M∞ is the released amount at infinite time, K represents the release rate constant, and n is the release exponent. N ≤ 0.5 corresponds to Fickian diffusion, and 0.5 < n ≤ 1 corresponds to a non-Fickian diffusion (anomalous transport). The parameters K and n were estimated by log-weighted linear regression of the data points with
is a fraction of the drug released at time t, Mt is the released amount at time t, M∞ is the released amount at infinite time, K represents the release rate constant, and n is the release exponent. N ≤ 0.5 corresponds to Fickian diffusion, and 0.5 < n ≤ 1 corresponds to a non-Fickian diffusion (anomalous transport). The parameters K and n were estimated by log-weighted linear regression of the data points with 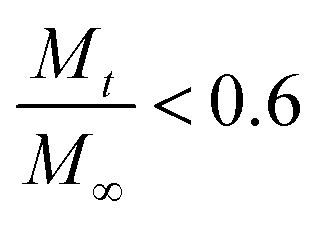 .
.
A pharmacokinetic model with a single compartment (the knee joint, nominal volume: 2 mL) and an infinite sink assumption was used to predict intraarticular drug concentration. The K and n fitting of the cumulative drug mass curves from the drug release study performed in DI water at 37 °C were used. The drug decay was assumed to follow first-order kinetics. The half-life of bupivacaine in the synovial fluid after intraarticular injection was 10 min.44 The half-life of lidocaine was reported to be 72 min, approximately, for the case of intraarticular administration in dogs.45 The drug eluting surface considered was that of a knee implant with 100 cm2 surface area. The predicted drug concentration versus time curves were plotted and compared to a bolus dose of 50 mg intraarticular injection of bupivacaine used as control.46 The peak concentration (Cmax), the peak time (Tmax) and the area under the curve (AUC) for different time intervals were calculated.
In vitro cell culture and biocompatibility
MG-63 human osteosarcoma cells (American Type Culture Collection, ATCC, VA) were cultured and maintained at 37 °C in 5% CO2 in 75 cm2 cell culture flasks (229341, CELLTREAT) using Eagle's Minimum Essential Medium with non-essential amino acids, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1500 mg L−1 sodium bicarbonate (EMEM, MT10009CV, Thermo Fisher Scientific) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS, A5670501, Thermo Fisher Scientific) and 1% antibiotic–antimycotic solution (100 units per mL penicillin, 100 μg mL−1 streptomycin, 0.25 μg mL−1 amphotericin B, MT30004CI, Thermo Fisher Scientific).The effect of the analgesic doped UHMWPEs on cell viability was investigated using the XTT colorimetric assay (Invitrogen™ CyQUANT™ XTT Cell Viability Assay, X12223, Thermo Fisher Scientific). Briefly, a 24-well plate (10062-896, VWR) was seeded with 1 × 105 cells for each well and incubated for 24 h at 37 °C in 5% CO2. Samples for each formulation (i.e., LD doped UHMWPE, LD doped 100 kGy UHMWPE, BP doped UHMWPE, BP doped 100 kGy UHMWPE, virgin UHMWPE, 100 kGy irradiated UHMWPE, n = 3 each, 3 × 5 × 8 mm3) were sterilized by ethylene oxide and were washed in media three times before placement in membrane inserts with a pore size of 0.4 μm (Millicell® 24 well hanging cell culture inserts, PTHT24H48, Thermo Fisher Scientific) and exposed to the pre-attached cells. After 24 h incubation, the samples were removed and the XTT reagent was added to each well and incubated for 4 h at 37 °C in 5% CO2 in a humid chamber. The specific UV absorbance was calculated according to the manufacturer's protocol after measuring the absorbances at 450 nm and 660 nm by using a plate reader (Biotek, Agilent, Santa Clara, CA). The viability of the cells exposed to the analgesic doped UHMWPE was compared to that of untreated cells, and cell culture media was used as blank.
Statistical analysis
The results are presented with average and standard deviation. A Student's t-test for two-tailed distribution of unequal variance was used to compare individual sets of data. The significance was assigned to p < 0.05.Safety statement
Bupivacaine (CAS: 38396-39-3) and lidocaine (CAS: 137-58-6) are classified under acute toxicity; their handling must be performed with extreme care, by using all the required PPEs and by working under a chemical food hood, according to the precautionary statements of the safety datasheet.Results
Diffusion doping
A preliminary thermal characterization of the materials (UHMWPE, lidocaine, and bupivacaine) was performed by DSC and TGA to determine the transition temperatures, melting enthalpy and peak degradation temperature (Fig. 3 and Table 1).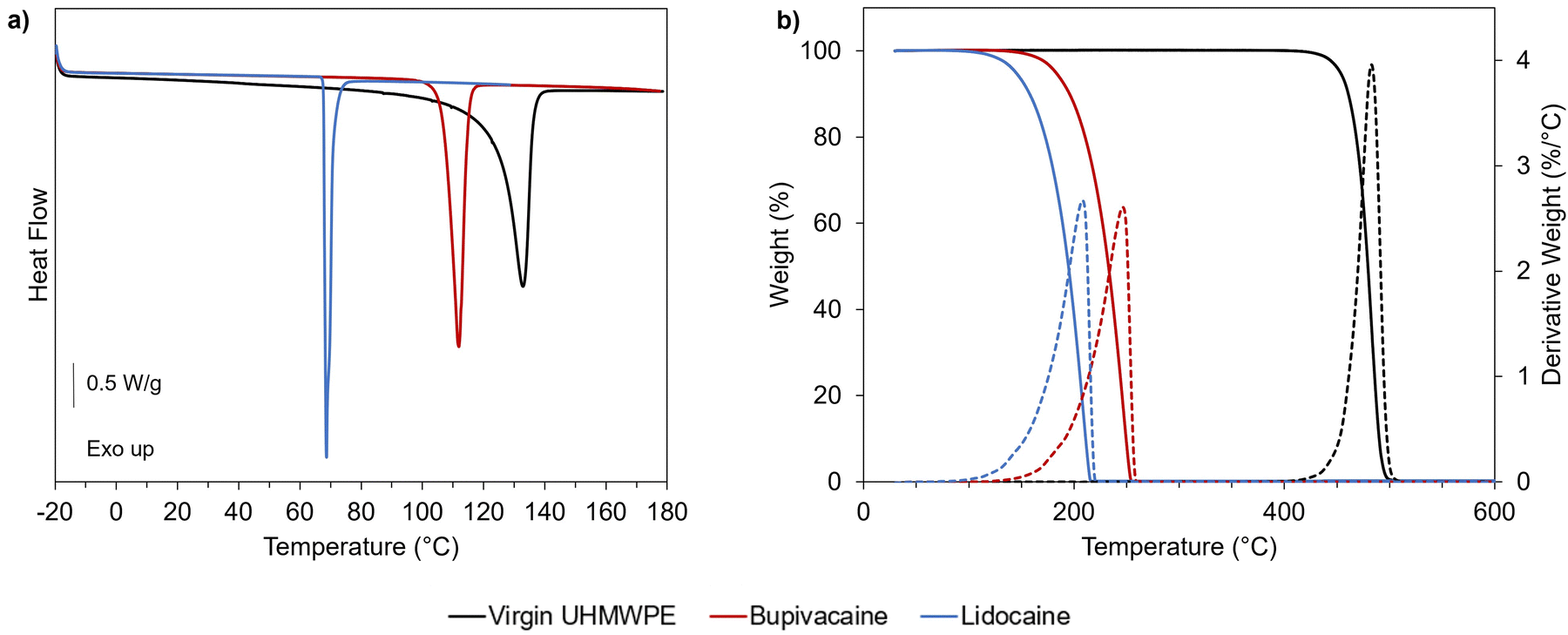 | ||
| Fig. 3 Thermal and thermogravimetric behavior of UHMWPE and selected analgesic drugs. (a) DSC first heating scan and (b) TGA heating ramp (10 °C min−1) for virgin UHMWPE (black line), bupivacaine (red line) and lidocaine (blue line). Dashed lines in (b) refer to the derivative weight curves. | ||
| T peak,melting (°C) | ΔHm (J g−1) | T peak,degradation (°C) | |
|---|---|---|---|
| Virgin UHMWPE | 134 ± 1 | 169 ± 4 | 483 ± 2 |
| Lidocaine | 69 ± 1 | 71 ± 2 | 208 ± 3 |
| Bupivacaine | 113 ± 1 | 95 ± 2 | 247 ± 2 |
A temperature of 120 °C was chosen for diffusion doping because it is above the melting point of the drugs and close but below the melting peak temperature for UHMWPE. Doping below the melting peak of UHMWPE was pursued as higher temperature resulted in significant dimensional and volumetric distortions in previous studies on diffusion of vitamin E inside UHMWPE.39 The chosen temperature also still ensures the thermal stability of the drugs under inert atmosphere, as supported by thermal analysis (Fig. 3 and Table 1). Preliminary diffusion experiments performed for lidocaine into non-irradiated UHMWPE at 100, 110 and 120 °C revealed lower drug uptake for lower temperatures, as expected based on the diffusion mechanism (Fig. S1, ESI†).
Assessment of the doped drug and diffusion profile
The amount of drug in non-irradiated UHMWPE prismatic samples after doping at 120 °C for 4 h was 20–27 mg, (corresponding to about 7–9 wt% of the total mass of the samples); whereas significantly less drug (corresponding to 5–8 wt% of the total mass of the samples) was diffused into irradiated UHMWPE (p < 0.05) (Table 2).| Sample geometry: 3 × 5 × 20 mm3 | Irradiation dose (kGy) | Total drug (mg) |
|---|---|---|
| Lidocaine-doped UHMWPE | 0 | 21.6 ± 0.9 |
| 100 | 16.1 ± 0.4 | |
| Bupivacaine-doped UHMWPE | 0 | 26.7 ± 4.1 |
| 100 | 22.7 ± 1.9 |
Diffused drug amount was higher in the case of bupivacaine-doped UHMWPE compared to lidocaine-doped UHMWPE (p < 0.05) for both irradiation conditions. Lidocaine and bupivacaine indices as a function of depth of drug-doped UHMWPEs confirmed the depth of the drug incorporation (Fig. 4).
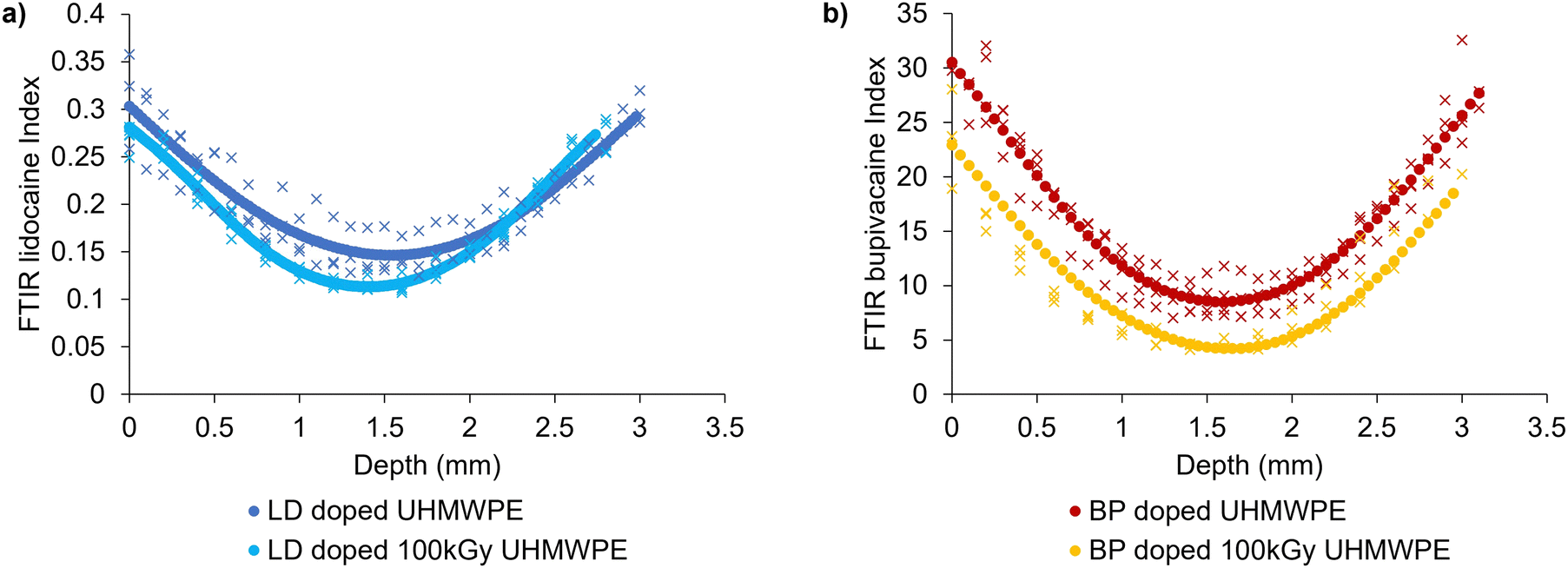 | ||
| Fig. 4 Spatial distribution of lidocaine and bupivacaine in UHMWPE. Diffusion profiles along the thickness of 3 × 5 × 20 mm3 prismatic strips for (a) lidocaine-doped materials and (b) bupivacaine-doped materials. Full circles represent spline interpolations of FTIR scans from multiple sections (n = 3). | ||
The drug diffused inside the polymer followed a parabolic profile which was symmetric with respect to the core of the sample: the maximum amount of drug was present at the sample surface and decayed as the distance from the surface increased. Drug diffused into irradiated UHMWPE was less than that diffused into non-irradiated UHMWPE throughout the whole thickness, as confirmed by FTIR.
Preliminary diffusion doping experiments performed on thick samples of non-irradiated UHMWPE (8.8 mm thick, approximately) demonstrated that diffusion kinetics followed Fick's second law of diffusion, where the concentration of the drug is function of the reciprocal of the square root of the depth and the doping time (Fig. S2, ESI†). The diffusion coefficient, D, was equal to 1.25 × 10−5 and 1.50 × 10−5 mm2 s−1 for lidocaine and bupivacaine, respectively, in non-irradiated, non-crosslinked UHMWPE. The drug content at the surface and the penetration depth increased with the doping time. Diffusion kinetics was also investigated by doping thin samples (150 μm thin) of non-irradiated UHMWPE and irradiated UHMWPE to reach the saturation concentration in a short period of time, supporting that higher drug uptake can be obtained for non-irradiated (i.e., non-crosslinked) UHMWPE compared to 100 kGy irradiated UHMWPE, for both lidocaine and bupivacaine (Fig. S3, ESI†). The drugs uptake as a function of time followed power-law trends (R2 value greater than 0.95) where the doped drug mass increased with time and its rate of increase decreased with time as the saturation concentration was approached. The exponents of the power law remained smaller than 0.5, corroborating Fickian diffusion.
The weight loss measured by TGA from 30 °C to 300 or 350 °C, for lidocaine and bupivacaine, respectively, was used to quantify the drug content (percentage) at the surface and in the core of the samples (Fig. S4 and S5, ESI†). The section at the surface had the highest drug loading across both the drugs and conditions tested. An estimation of the drug loading is provided in Table 3. Furthermore, drug uptake (percentage) measured by TGA was also confirmed by using the calibration curves prepared for both lidocaine and bupivacaine (Fig. S6, ESI†) to correlate the FTIR drug index to the weight percentage of the drug uptake.
| Drug loading weight estimation by TGA (%) | Non-irradiated UHMWPE | 100 kGy irradiated UHMWPE | |||||
|---|---|---|---|---|---|---|---|
| Pre-elution | Post 28 days elution | Average reduction | Pre-elution | Post 28 days elution | Average reduction | ||
| Lidocaine-doped UHMWPE | Surface | 8.6 ± 0.1 | 4.8 ± 0.2 | 45 | 5.2 ± 1.0 | 3.6 ± 0.1 | 30 |
| Core | 4.5 ± 0.3 | 4.5 ± 0.4 | 0 | 4.1 ± 1.0 | 3.6 ± 0.1 | 11 | |
| Bupivacaine-doped UHMWPE | Surface | 10.0 ± 2.1 | 7.4 ± 0.7 | 26 | 9.8 ± 3.9 | 5.7 ± 0.3 | 42 |
| Core | 4.5 ± 0.1 | 4.3 ± 0.5 | 4 | 3.0 ± 0.1 | 2.9 ± 0.3 | 2 | |
Crystallinity content and crosslink density
The TVI peak and its respective FTIR index along the thickness of the molded and irradiated UHMWPE before doping confirmed the achievement of a similar radiation dose profile regardless of the two different thicknesses tested and the relevant e-beam irradiation conditions (Fig. S7, ESI†). Crosslink density values from swelling experiments performed on the irradiated materials after doping and on irradiated UHMWPE confirmed the achievement of a highly crosslinked network due to the irradiation dose (Table 4). DSC confirmed the achievement of a high crystallinity content (∼60%) (Table 4). Representative DSC curves together with the methods of evaluation of the thermal properties and their evaluation are reported in Fig. S8 and S9 and Tables S1 and S2 (ESI†). No significant difference was found for any of the groups (p > 0.05).| χ c,UHMWPE (%) | Crosslink density (mol m−3) | ||
|---|---|---|---|
| n.t.: not tested. | |||
| Non-irradiated | Virgin | 58.0 ± 1.6 | n.t. |
| Lidocaine-doped | 60.3 ± 1.9 | n.t. | |
| Bupivacaine-doped | 60.2 ± 3.0 | n.t. | |
| 100 kGy irradiated | Virgin | 61.0 ± 2.5 | 184 ± 16 |
| Lidocaine-doped | 60.7 ± 0.8 | 212 ± 14 | |
| Bupivacaine-doped | 62.4 ± 2.1 | 203 ± 20 | |
Chemical stability of the drugs
1H NMR spectroscopy was used to compare the spectra of the unprocessed drug to that eluting out of UHMWPE after the doping process to assess the chemical stability of the drugs. No changes in the peaks and their respective chemical shifts were found suggesting the structural stability of the drug after the diffusion process at high temperature, according to 1H NMR spectroscopy (Fig. 5a and b).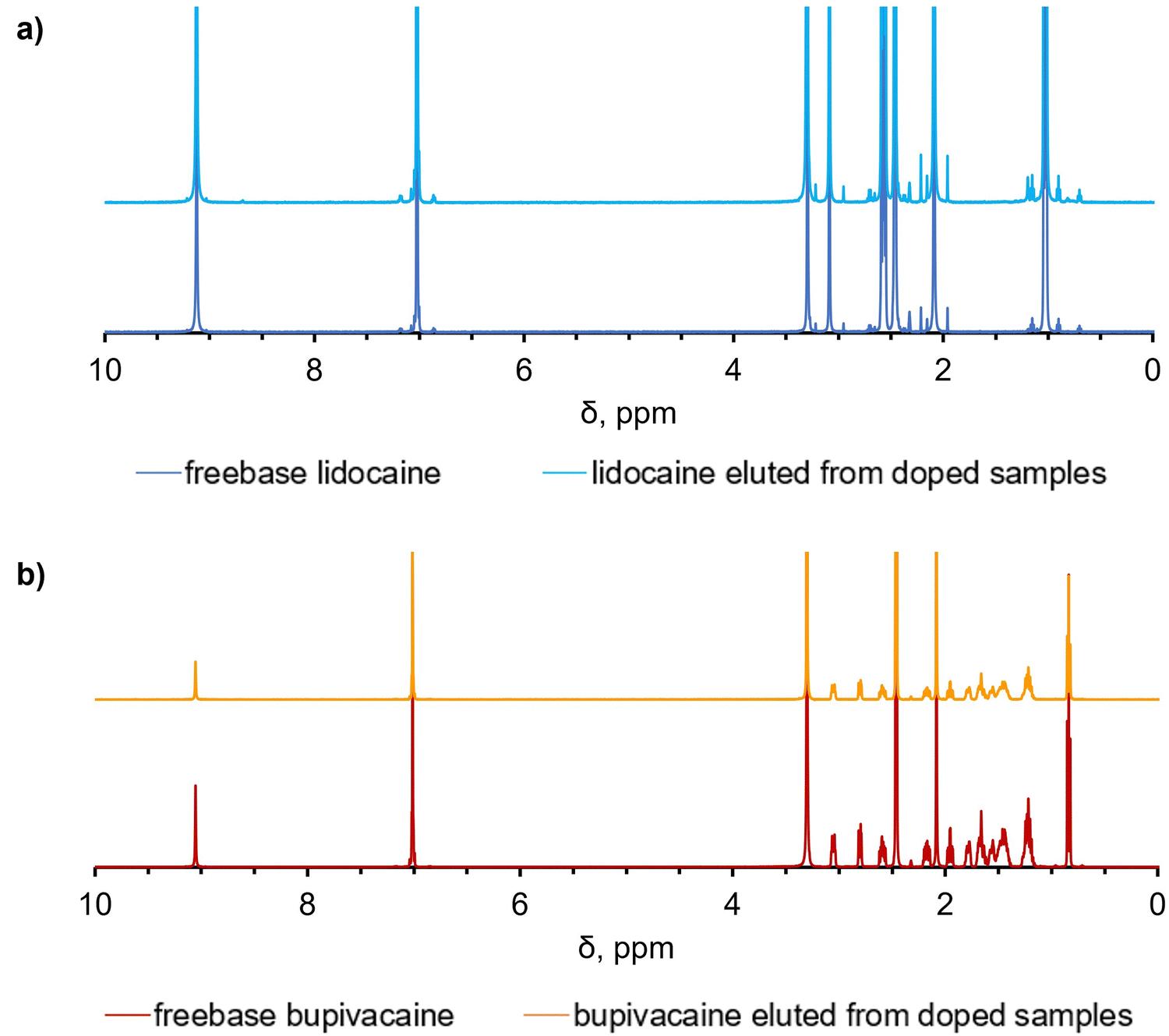 | ||
| Fig. 5 Chemical stability of analgesics incorporated into UHMWPE by diffusion doping. Comparison between the 1H NMR spectra of the freebase stock solution and the drug eluted from the doped UHMWPE samples for (a) lidocaine and (b) bupivacaine. | ||
The full list of the peaks is reported in the supplementary information, as well as the list of the multiplets and their assignments (Fig. S10–S13, ESI†).
Mechanical and wear properties
The UTS, EAB and work-to-failure of the doped materials were statistically similar to those of the controls (p > 0.05), namely, unirradiated and irradiated virgin UHMWPE (Table 5).| UTS (MPa) | EAB (%) | Work-to-failure (kJ m−2) | Wear rate (mg MC−1) | ||
|---|---|---|---|---|---|
| n.t.: not tested. | |||||
| Non-irradiated | Virgin | 50.8 ± 2.5 | 448 ± 34 | 713 ± 65 | n.t. |
| Lidocaine-doped | 49.2 ± 2.2 | 419 ± 14 | 661 ± 42 | n.t. | |
| Bupivacaine-doped | 53.8 ± 2.3 | 427 ± 14 | 650 ± 61 | n.t. | |
| 100-kGy irradiated | Virgin | 49.3 ± 3.5 | 300 ± 17 | 486 ± 33 | 1.3 ± 0.3 |
| Lidocaine-doped | 49.7 ± 1.7 | 324 ± 7 | 499 ± 49 | 1.5 ± 0.2 | |
| Bupivacaine-doped | 50.5 ± 1.4 | 299 ± 13 | 488 ± 10 | 1.7 ± 0.3 | |
All the compositions tested showed small wear rates; lidocaine- and bupivacaine-doped, irradiated UHMWPE had similar wear rates (p > 0.05), and the lidocaine-doped, irradiated UHMWPE had statistically similar wear rate to 100kGy irradiated UHMWPE (p > 0.05).
Drug elution properties
Lidocaine-doped samples showed a higher burst release than bupivacaine-doped samples, which was followed by similar rates of sustained release until day 28 for all the tested materials (Fig. 6a and c).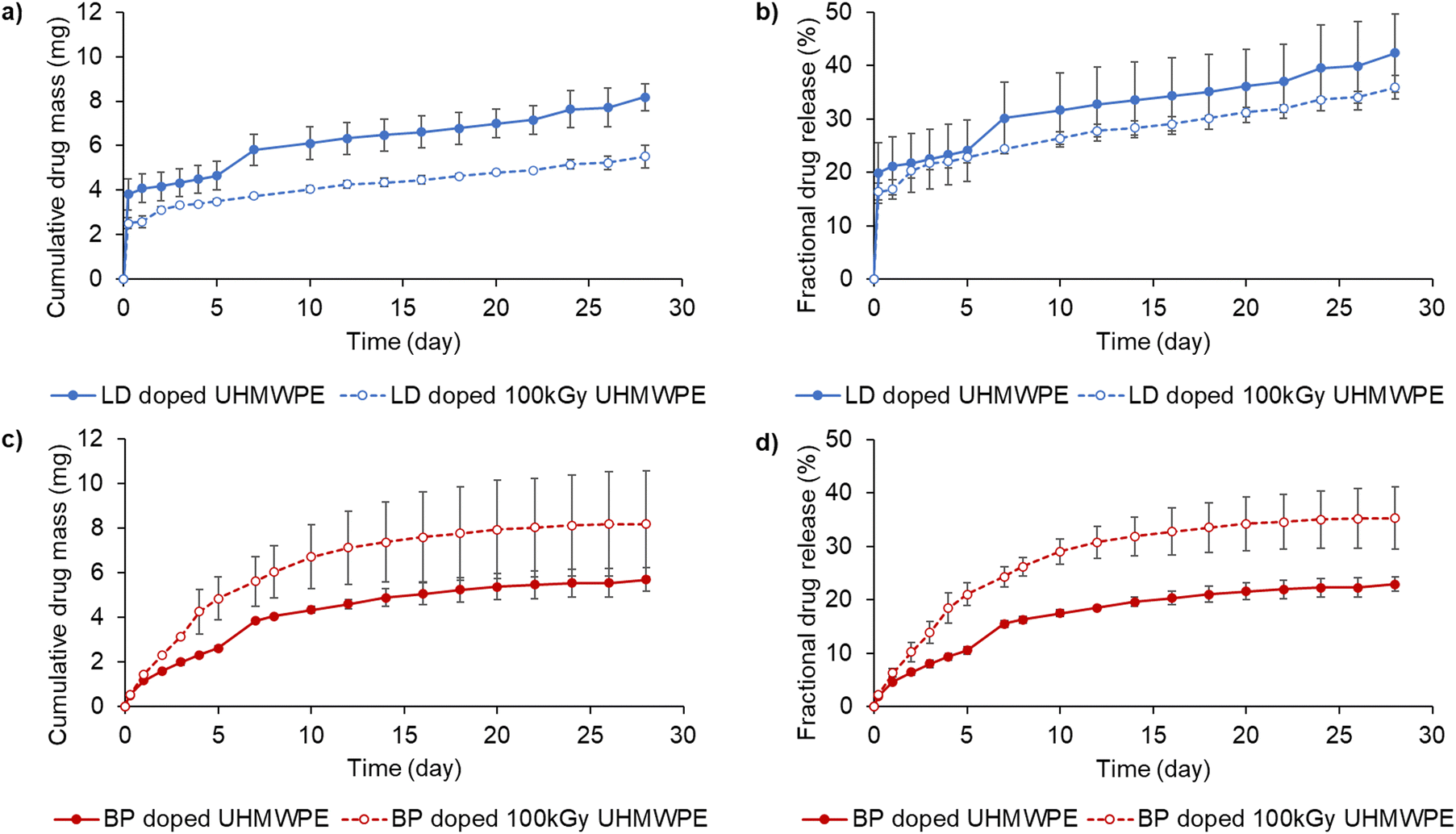 | ||
| Fig. 6 Drug elution from lidocaine and bupivacaine-doped UHMWPE. Cumulative and fractional drug release for lidocaine-doped UHMWPEs (a) and (b), and bupivacaine-doped UHMWPEs (c) and (d). Full circles represent the non-irradiated doped materials, while empty circles represent the doped materials after irradiation. Drug release study was performed at room temperature in de-ionized water. | ||
Lidocaine-doped, irradiated UHMWPE released less drug cumulatively than the lidocaine-doped, non-irradiated UHMWPE. The total fractional lidocaine release at day 28 was 36 and 42% for the irradiated and non-irradiated materials, respectively. Bupivacaine-doped, irradiated UHMWPE released more drug cumulatively than bupivacaine-doped, non-irradiated UHMWPE. Their fractional release at day 28 was 23 and 35% for the non-irradiated and the irradiated materials, respectively. Fractional release was also investigated by performing TGA composition analysis on the eluted samples. The surface of the eluted samples showed a more prominent decrease in the drug content compared to the core (Table 3). Fractional release increased at 37 °C as opposed to room temperature more prominently for lidocaine doped compositions compared to bupivacaine doped compositions, probably due to the lower melting point of lidocaine compared to bupivacaine. Using Phosphate Buffered saline instead of de-ionized water led to a further increase in fractional release for bupivacaine doped- and bupivacaine doped 100 kGy irradiated UHMWPE (Fig. S14, ESI†).
The elution of lidocaine was largely Fickian (release exponent n < 0.5); the elution of bupivacaine was characterized by higher release exponents than those of lidocaine (Table S3, ESI†). Pharmacokinetic modeling was used to predict the local intraarticular concentration of the drugs released from a 100 cm2 implant compared to that of a 50 mg intraarticular injection of bupivacaine (Fig. 7).
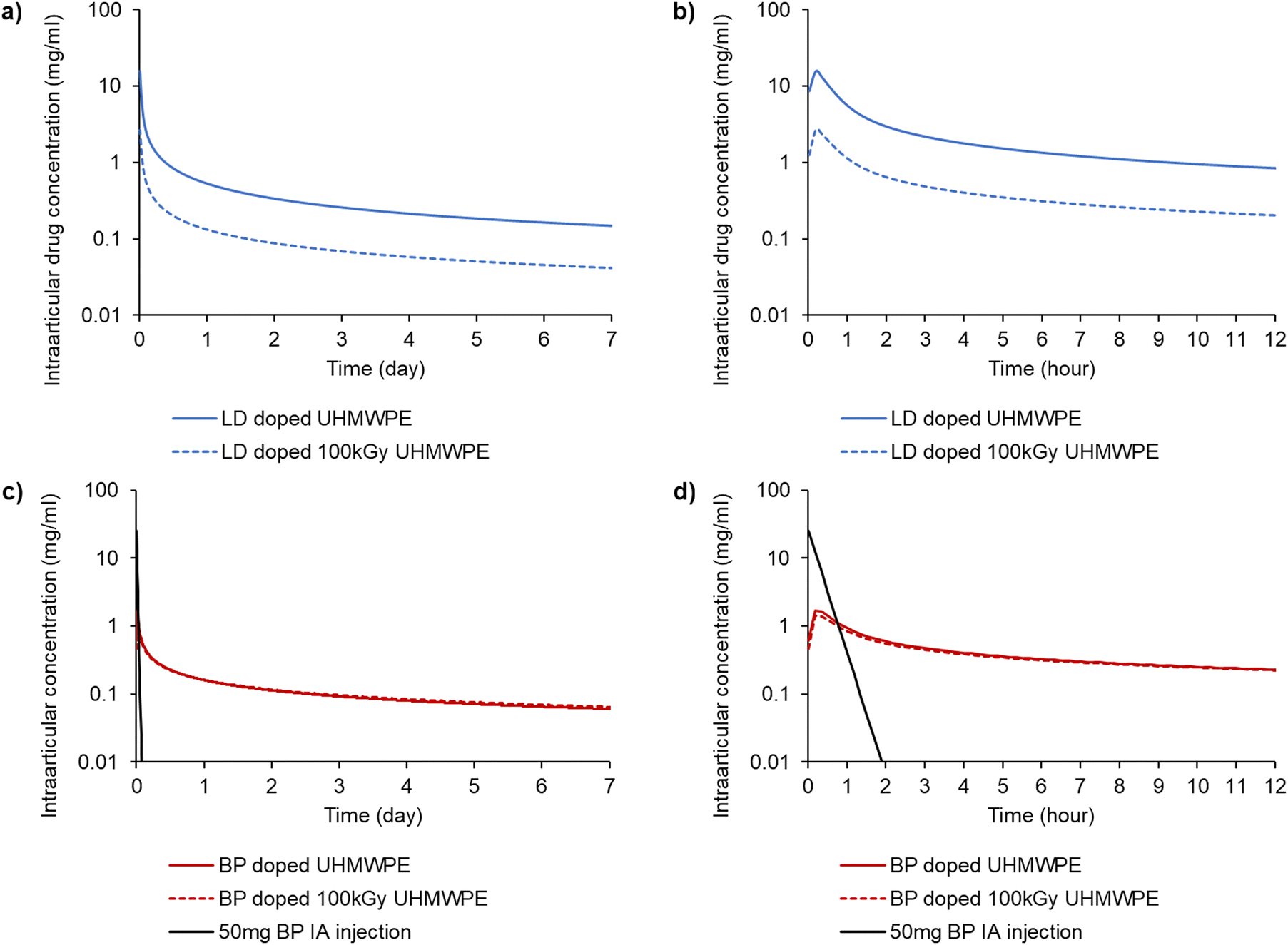 | ||
| Fig. 7 Predicted intraarticular concentration of the analgesic from the drug release experiments performed at 37 °C in de-ionized water. Drug concentration from a representative knee implant (surface area of 100 cm2) for (a) lidocaine-doped UHMWPEs over a period of 7 days and (b) over a period of 12 hours; and for (c) bupivacaine-doped UHMWPEs over a period of 7 days and (d) over a period of 12 hours. The black line in (c) and (d) represents the concentration resulting from 50 mg intraarticular (IA) injection of bupivacaine. | ||
The intraarticular concentration after the 50 mg bolus intraarticular injection of bupivacaine decayed much faster than that for the drugs released from the implant material. The AUC for the bupivacaine-doped implants reached similar values to the bolus administration within 24 h, and the value was approximately double within 72 hours (Table 6). Both lidocaine-doped compositions had a similar or higher Cmax and AUC compared to the bupivacaine-doped compositions for the 24 h and 72 h time periods.
| C max (mg mL−1) | T max (h) | AUC0–6 (mg h mL−1) | AUC0–12 (mg h mL−1) | AUC0–24 (mg h mL−1) | AUC0–72 (mg h mL−1) | |
|---|---|---|---|---|---|---|
| IA: intraarticular. | ||||||
| 50 mg BP IA injection | 25.0 | — | 6.5 | 6.5 | 6.5 | 6.5 |
| BP doped UHMWPE | 1.7 | 0.18 | 3.7 | 5.3 | 7.6 | 13.2 |
| BP doped 100 kGy UHMWPE | 1.4 | 0.18 | 3.4 | 4.9 | 7.2 | 12.9 |
| LD doped UHMWPE | 15.5 | 0.18 | 21.4 | 27.6 | 35.5 | 52.5 |
| LD doped 100 kGy UHMWPE | 2.6 | 0.18 | 4.3 | 5.8 | 7.7 | 12.1 |
In vitro cell culture and biocompatibility
Cell viability in the presence of the analgesic doped UHMWPEs was higher than 70% for all the formulations (Fig. 8).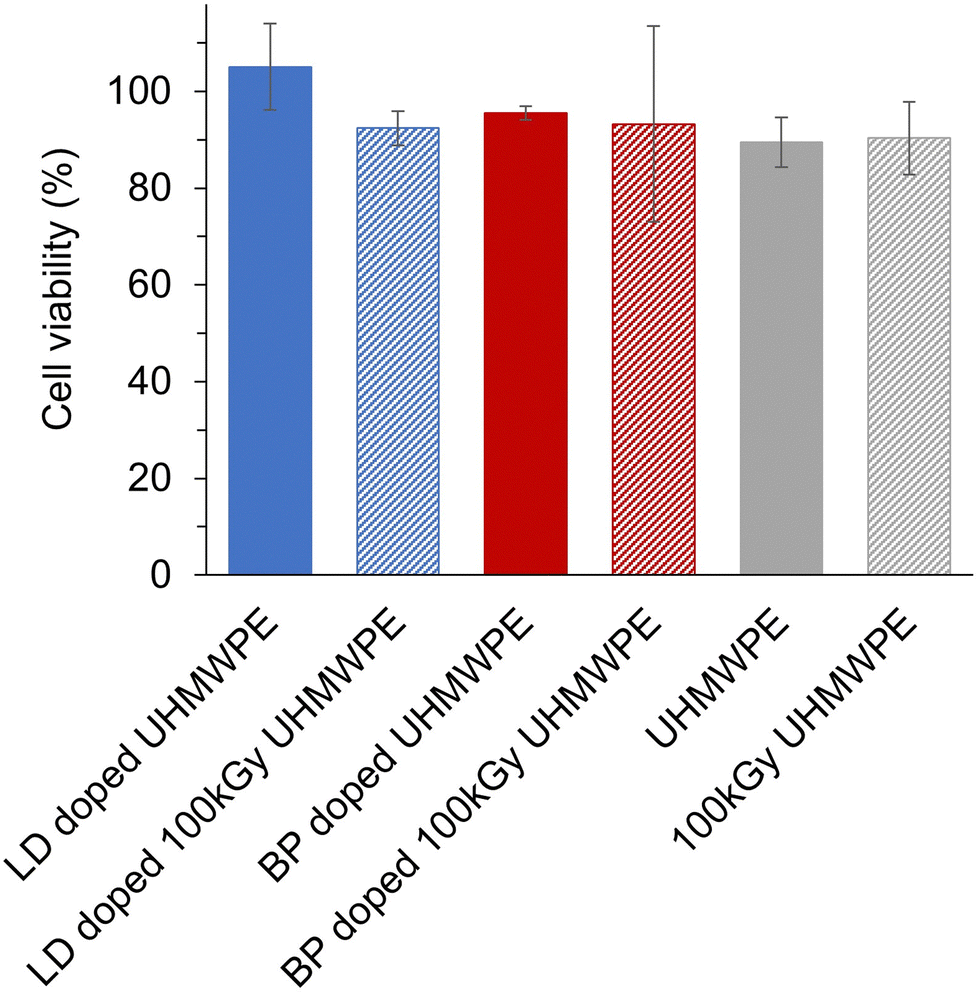 | ||
| Fig. 8 Cell viability in the presence of UHMWPE and of analgesic doped UHMWPEs. The cell line MG-63 (ATCC) was exposed to the analgesic doped UHMWPEs (3 × 5 × 8 mm3 samples, n = 3 for each group) for 24 h. Cell viability was measured with the XTT assay. | ||
Discussion
The mechanical and wear properties are crucial to meet the in vivo requirements for permanent joint implant materials with an expected lifetime of at least 20 years.47,48 The state-of-the-art bearing surfaces made of UHMWPE are highly crosslinked, and stabilized by antioxidants and display an ultimate tensile strength of about 45 MPa, elongation at break above 230–250%, IZOD impact strength around or above 55–65 kJ m−2, stress intensity factor at fatigue crack inception ranging in between 0.7 and 1.3 MPa·m1/2, and wear rates around 1–2 mg/MC.22 Drug-eluting UHMWPE implants are promising additional tools to address challenges associated with joint arthroplasty; the processes for manufacturing therapeutic formulations have to meet the mechanical and tribological properties of the state-of-the-art implants for permanent use.The morphology of drug-blended UHMWPE was described as phase-separated,29 where low affinity between the polymeric phase and the drug molecule resulted in drug domains, which can act as disrupters to the fusion/integration of the polymeric matrix during consolidation. Similar challenges have been observed while introducing a second phase of different chemical structure and properties in conductive polymer composites of UHMWPE.49,50 Fillers in polymer composites are known to act as reinforcements or as stress concentrators, depending on a variety of factors including the physical properties of the filler, its geometry, including factors such as shape, size, and aspect ratio,51,52 and its adhesion and interaction properties with the matrix.53,54 The introduction of drug molecules by blending and solid-state fabrication can adversely affect the properties that correlate well with the in vivo longevity for this biomedical polymer, limiting the use of drug blended UHMWPE on articulating and load-bearing regions of the implant.
Our central hypothesis of improving the mechanical strength and toughness of analgesic-doped UHMWPE by using the diffusion of undiluted hydrophobic forms of analgesics after molding and radiation crosslinking was strongly supported (Table 5). Doping of the drugs into UHMWPE followed a Fickian diffusion mechanism, where the driving force of the doping process is the concentration gradient between the sink of undiluted molten drug and the material to be doped (Fig. S2, ESI†). As time passes and driven by the temperature, the concentration of the drug into UHMWPE increased with a dependence on the depth position with respect to the doping surface up to reaching a saturation concentration (Fig. S1 and S3, ESI†). The ultimate tensile strength (UTS), which is used as a general indicator of strength in UHMWPE materials for intended use in joint replacement,55,56 was unchanged from that of virgin UHMWPE. The UTS can be affected by the crystallinity of the UHMWPE as well as the interaction of the crystalline and amorphous regions;57 its maintenance (Table 4) indicates maintenance of these structural regions.57–60 The elongation-at-break (EAB) is generally used as an indicator of ductility in UHMWPE materials for intended use in joint replacement. It is strongly affected (decreased) by radiation crosslinking, which occurs mainly in the amorphous regions of the material when irradiation is performed at room temperature.61 Radiation crosslinking decreased ductility, as expected, without an independent effect from drug diffusion. Toughness, which is a combination of strength and ductility, is correlated well with fatigue crack propagation resistance,28 a relevant property for the longevity of joint replacements working under cyclic fatigue conditions. The tensile toughness of drug-doped UHMWPEs being similar to their undoped counterparts corroborated that the mechanical properties of UHMWPEs doped using diffusion were unaffected by the drug diffusion under these conditions.
Increased wear resistance of UHMWPE materials by radiation crosslinking has, over the past few decades, decreased osteolysis caused by the accumulation of UHMWPE wear particles.62 Thus, for UHMWPE materials proposed for permanent implantation, having high wear resistance is crucial, especially in more demanding applications such as hip arthroplasty.63–67 For traditional radiation crosslinked UHMWPEs, crosslink density is the primary determinant of wear resistance.3,4 Diffusion of the analgesics into solid form UHMWPE is also advantageous because radiation crosslinking can be performed prior to the diffusion procedure, as opposed to the blending method.68 The uniformity of the radiation dose, which was confirmed by the TVI index for both the thin (∼3mm) and thick (∼16 mm) compression molded blocks (Fig. S7, ESI†)42 and the crosslink density (Table 4), confirmed that crosslinking was achieved as intended. The wear rates of the analgesic-doped UHMWPEs were similar to those before doping, demonstrating that drug diffusion did not have a significant effect on the wear behavior (Table 5).
There is currently no delivery device for analgesics equivalent to the one proposed here for peri-articular pain management in joint arthroplasty. The most common method of administration comprises a bolus injection of a cocktail of analgesics and vasoconstricting agents to increase the residence time of the agents relieving pain.69–73 There is no information on the local concentration of the delivered drugs; we based our comparison on the concentration profile for a bolus injection of 50 mg of bupivacaine, which is considered the recommended dose for relieving acute pain, as suggested from a meta-analysis in arthroscopic knee surgery.46 We predicted the concentration profiles of the locally administered drugs by using a pharmacokinetic model based on the half-lives of the drugs (10 and 72 min for bupivacaine and lidocaine, respectively) (Fig. 7). The exposure time of the knee compartment to the drug can be estimated by calculating the area under the curve (AUC) of the predicted intraarticular drug concentration versus time curves. For bupivacaine, the AUC over the first 24 hours was equivalent to that of the bolus injection while exceeding it at 72 hours (Table 6). The efficacy of pain relief is further affected by many physiological factors as well as treatment modalities such as the concomitant use of other drugs and patients’ factors such as age and comorbidities.74,75 Assuming that the main factor of interest is the concentration of the drugs, then the drug elution from the proposed analgesic-eluting UHMWPE should result in pain relief that is equivalent or better than that afforded by a bolus injection. The lidocaine concentrations were similar or higher than those for bupivacaine; however, it should be considered that the potency ratio of lidocaine to bupivacaine is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.76 Even considering this factor, the AUC of lidocaine over 24 and 72 hours is expected to result in pain relief equivalent to or better than that afforded by the 50 mg bupivacaine bolus injection.
:4.76 Even considering this factor, the AUC of lidocaine over 24 and 72 hours is expected to result in pain relief equivalent to or better than that afforded by the 50 mg bupivacaine bolus injection.
The maximum intraarticular concentration of bupivacaine and lidocaine resulting from elution from all four formulations was less than that of the bolus injection (Table 6), suggesting that any toxicity associated with these materials based on drug concentrations would be less for the proposed devices. The outcome of the diffusion process was a non-uniform concentration of drug-rich regions close to the surface (Fig. 4), which created a large early driving force for elution. The elution of the drugs, which were found chemically stable after the doping process as per NMR (Fig. 5), followed a largely Fickian behavior with an initial burst release followed by a slower rate (Fig. 6). The total fraction of released drugs was 30–45%, as characterized from elution media (Fig. 6) and confirmed by TGA (Table 3). The differences in the release profiles of the two drugs may be attributed to several factors, including the drug structure, polarity, solubility in the release medium and affinity to the polymeric matrix. For such materials where the incorporation of the drug is not homogeneous and it is dictated by diffusion through the thickness, the release of the drug may also display thickness-dependent phenomena. Because the driving force is higher from the surface, the drug release is higher at this location, as supported by the composition analysis after elution (Table 3). This profile is advantageous to limit the elution of the drug to the surface where it's needed and to lower elution concentration over the longer period of time.
The design of devices and formulations for sustained and prolonged release of analgesics is now receiving new attention due to the ongoing opioid epidemic.77 The sustained release of non-opioid analgesic drugs can be promising for longer-term pain relief, but it is of utmost importance to design the dosing and release profiles to avoid any side effects.78–80 Anesthetic drugs such as lidocaine and bupivacaine are associated with cardiac and central nervous system toxicity, and local chondrotoxicity.81–83 The lowering of maximum local drug concentrations from the eluting devices compared to that of the bolus injection (Table 6) suggests a lower probability of both local and systemic acute toxicity with the proposed drug-eluting devices. For a preliminary assessment of the biocompatibility of these novel analgesic eluting UHMWPEs, we performed a cytotoxicity study by exposing the osteoblast-like MG-63 cell line to the eluents from the polymer for an incubation period of 24 h. Cell viability remained higher than 70% for all the formulations (Fig. 8), thus, according to the ISO 10993, these materials are not expected to be cytotoxic, under the tested conditions. The probability of chronic effects due to the continued exposure to low amount of local anesthetics cannot be excluded and further in vitro and in vivo testing will be required.84,85 However, the proposed methodology of diffusion doping may be adapted to provide an optimized dosing and period of release. Key parameters that can be tuned for matching a desired release profile are the time and the temperature at which the diffusion process is performed. For achieving a shorter duration of release, both the time and temperature of the doping process can be decreased to limit the amount of diffused drug (Fig. S1, ESI†). A full leachable/extractable analysis may be required to analyze the extracts and further confirm the stability of the doped drugs. Terminal sterilization of the implant and incorporation of antioxidant for state-of-the-art properties are also required for long-term performance. Finally, further testing in vivo will be needed to test the efficacy of pain relief in addition to the evaluation of acute and chronic safety profiles.
Conclusions
A novel method for incorporating analgesics into UHMWPE implant materials was successfully developed by diffusion doping. Diffusion allowed to supplement non-opioid analgesic drugs into solid form UHMWPE, which could be irradiated a priori to achieve high wear resistance. Mechanical properties were not detrimentally affected by the incorporation of the drug: high strength and high ductility were maintained. The predicted intraarticular drug concentration obtained by elution from these therapeutic UHMWPEs can have a comparable, if not superior, analgesic effect to that of the clinically used intraarticular injections of bupivacaine. This study paves the way for the development of crosslinked UHMWPEs eluting various therapeutics for load-bearing implant materials, where high strength and wear resistance are required.Author contributions
Nicoletta Inverardi: conceptualization, data curation, formal analysis, investigation, methodology, supervision, writing – original draft, writing – review & editing. Sashank Lekkala: conceptualization, data curation, formal analysis, investigation, methodology, writing – review & editing. Maria F. Serafim: data curation, formal analysis, investigation, methodology, writing – review & editing. Amita Sekar: investigation, methodology, writing – review & editing. Keith K. Wannomae: conceptualization, investigation, methodology, writing – review & editing. Brad Micheli: investigation, methodology, writing – review & editing. Hany Bedair: conceptualization, supervision, writing – review & editing. Orhun K. Muratoglu: conceptualization, methodology, resources, supervision, writing – review & editing. Ebru Oral: conceptualization, data curation, methodology, funding acquisition, project administration, resources, supervision, writing – original draft, writing – review & editing. All authors have given approval to the final version of the manuscript.Data availability
Data are available upon request from the corresponding author and have been partially included as part of the ESI.†Conflicts of interest
The authors declare the following competing financial interest(s): H.B. is a paid consultant for Exactech, Smith & Nephew. O.K.M. declares royalties from Corin, Mako, Iconacy, Renovis, Arthrex, ConforMIS, Meril Healthcare, Exactech, and Cambridge Polymer Group and stake/equity from Cambridge Polymer Group, Orthopedic Technology Group, and Alchimist. E.O. declares royalties from Corin, Iconacy, Renovis, Arthrex, ConforMIS, Meril Healthcare, and Exactech, is a paid consultant for WL Gore & Associates, is on the Editorial Board of JBMR, and is an officer/committee for SFB and ISTA. None of these are in direct conflict with the study.Acknowledgements
This work was supported in part by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Medical Research Program under Award No. W81XWH-17-1-0614. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Support from the NIH shared instrument program, grant S10OD025234, is also acknowledged for the 11.7 T NMR spectrometer at the A.A. Martinos Center for Biomedical Imaging (MGH/HST). The authors would like to acknowledge Dr Chathan M. Cooke and the High Voltage Research Laboratory (Massachusetts Institute of Technology, Cambridge, MA) for the e-beam irradiation of the materials. The authors would like to thank the A.A. Martinos Center for Biomedical Imaging for providing access to the 11.7 T NMR spectrometer purchased with support of the NIH shared instrument program, grant S10OD025234. The table of contents was partially created with BioRender.com.References
- S. R. Knight, R. Aujla and S. P. Biswas, Orthop. Rev., 2011, 3, e16 Search PubMed.
- S. Kurtz, in UHMWPE Biomaterials Handbook, ed. S. Kurtz, Elsevier, London, 3rd edn, 2015, ch. 1, pp. 1–6 Search PubMed.
- H. McKellop, F.-w Shen, B. Lu, P. Campbell and R. Salovey, J. Orthop. Res., 1999, 17, 157–167 CrossRef CAS.
- O. K. Muratoglu, C. R. Bragdon, D. O. O’Connor, M. Jasty, W. H. Harris, R. Gul and F. McGarry, Biomaterials, 1999, 20, 1463–1470 CrossRef CAS PubMed.
- S. Kurtz, in UHMWPE Biomaterials Handbook, ed. S. Kurtz, Elsevier, London, 3rd edn, 2015, ch. 25, pp. 449–466 Search PubMed.
- S. Kurtz, H. Gawel and J. Patel, Clin. Orthop. Relat. Res., 2011, 469, 2262–2277 CrossRef PubMed.
- A. A. Edidin, L. Pruitt, C. W. Jewett, D. J. Crane, D. Roberts and S. M. Kurtz, J. Arthroplast., 1999, 14, 616–627 CrossRef CAS PubMed.
- O. Muratoglu, C. Bragdon, D. O'Connor, M. Jasty and W. Harris, in World Tribology Forum in Arthroplasty, ed. C. Rieker, S. Oberholzer and U. Wyss, Hans Huber, Bern, 2001, pp. 245–262 Search PubMed.
- J. R. Atkinson and R. Z. Cicek, Biomaterials, 1983, 4, 267–275 CrossRef CAS PubMed.
- J. R. Atkinson and R. Z. Cicek, Biomaterials, 1984, 5, 326–335 CrossRef CAS.
- H. McKellop, F.-W. Shen, W. DiMaio and J. G. Lancaster, Clin. Orthop. Relat. Res., 1998, 369, 73–82 CrossRef PubMed.
- F. Shen, H. McKellop and L. R. Salovey, J. Biomed. Mater. Res., 1998, 41, 71–78 CrossRef CAS PubMed.
- F. W. Shen, H. A. McKellop and R. Salovey, J. Polym. Sci.: Polym. Phys., 1996, 34, 1063–1077 CrossRef CAS.
- A. Wang, H. Zeng, S.-S. Yau, A. Essner, M. Manely and J. Dumbleton, J. Phys. D: Appl. Phys., 2006, 39, 3213–3219 CrossRef CAS.
- P. Bracco and E. Oral, Clin. Orthop. Relat. Res., 2011, 469, 2286–2293 CrossRef PubMed.
- E. Oral, K. K. Wannomae, N. E. Hawkins, W. H. Harris and O. K. Muratoglu, Biomaterials, 2004, 25, 5515–5522 CrossRef CAS.
- E. Oral, S. Christensen, A. Malhi, K. Wannomae and O. Muratoglu, J. Arthroplasty, 2006, 21, 580–591 CrossRef.
- E. Oral and O. Muratoglu, in UHMWPE Biomaterials Handbook, ed. S. Kurtz, Elsevier, London, 2nd edn, 2009, ch. 15, pp. 221–236 Search PubMed.
- E. Oral, A. Neils, S. Rowell, A. Lozynsky and O. Muratoglu, J. Biomed. Mater. Res., 2013, 101B, 436–440 CrossRef CAS.
- J. Green, N. Hallab, Y.-S. Liao, V. Narayan, E. Schwarz and C. Xie, Curr. Rheumatol. Rep., 2013, 15, 1–5 CAS.
- V. Narayan, Clin. Orthop. Relat. Res., 2015, 473, 952–959 CrossRef PubMed.
- E. Oral, B. Doshi, K. Fung, C. O'Brien, K. Wannomae and O. Muratoglu, J. Orthop. Res., 2019, 37, 2182–2188 CrossRef CAS PubMed.
- O. K. Muratoglu, M. D. Asik, C. M. Nepple, K. K. Wannomae, B. R. Micheli, R. L. Connolly and E. Oral, J. Orthop. Res., 2024, 42, 306–316 CrossRef CAS.
- E. Oral, A. Malhi and O. Muratoglu, Biomaterials, 2006, 27, 917–925 CrossRef CAS PubMed.
- L. A. Pruitt, Biomaterials, 2005, 26, 15 CrossRef PubMed.
- F. J. Medel, P. Pena, J. Cegonino, E. Gomez-Barrena and J. A. Puertolas, J. Biomed. Mater. Res., 2007, 83B, 380–390 CrossRef CAS PubMed.
- J. Furmanski and L. A. Pruitt, Polymer, 2007, 48, 3512–3519 CrossRef CAS.
- B. Doshi, J. Ward, E. Oral and O. Muratoglu, J. Orthop. Res., 2016, 34, 1514–1520 CrossRef CAS.
- V. Suhardi, D. Bichara, S. Kwok, A. Freiberg, H. Rubash, S. Yun, O. Muratoglu and E. Oral, Nat. Biomed. Eng., 2017, 1, 80 CrossRef CAS.
- S. Grindy, D. Gil, S. Suhardi, O. Muratoglu, H. Bedair and E. Oral, Acta Biomater., 2019, 93, 63–73 CrossRef CAS PubMed.
- D. Gil, S. Hugard, S. Grindy, N. Borodinov, O. Ovchinnikova, O. Muratoglu, H. Bedair and E. Oral, Materialia, 2020, 12, 100662 CrossRef CAS.
- D. Gil, S. Hugard, N. Borodinov, O. S. Ovchinnikova, O. K. Muratoglu, H. Bedair and E. Oral, J. Biomed. Mater. Res. Part B: Appl. Biomater., 2023, 111, 912–922 CrossRef CAS.
- D. Gil, A. Atici, R. Connolly, S. Hugard, S. Shuaev, K. Wannomae, E. Oral and O. Muratoglu, Bone Joint J., 2020, 102, 151–157 CrossRef PubMed.
- S. Lekkala, N. Inverardi, J. Yuh, K. K. Wannomae, P. Tierney, A. Sekar, O. K. Muratoglu and E. Oral, Macromol. Biosci., 2024, 24, 2300389 CrossRef CAS PubMed.
- Y. Ren, F.-Y. Wang, Z.-J. Chen, R.-T. Lan, R.-H. Huang, W.-Q. Fu, R. M. Gul, J. Wang, J.-Z. Xu and Z.-M. Li, J. Mater. Chem. B, 2020, 8, 10428–10438 RSC.
- F.-Y. Wang, Y. Ren, R.-T. Lan, W.-Q. Fu, Z.-J. Chen, S. Huang, R. M. Gul, J. Wang, J.-Z. Xu and Z.-M. Li, Mater. Sci. Eng., C, 2021, 124, 112040 CrossRef CAS PubMed.
- Y. Fu and W. J. Kao, Expert Opin. Drug Delivery, 2010, 7, 429–444 CrossRef CAS.
- G. G. Grassi, Curr. Drug Delivery, 2005, 2, 97–116 CrossRef PubMed.
- E. Oral, K. K. Wannomae, S. L. Rowell and O. K. Muratoglu, Biomaterials, 2007, 28, 5225–5237 CrossRef CAS PubMed.
- E. Oral and O. Muratoglu, Int. Orthop., 2011, 35, 215–223 CrossRef PubMed.
- GUR® 1020, Medical UHMW-PE powder grade; Celanese: Irving, Texas; Source: Celanese Materials Database, Printed: 2024-07-12, Revised: 2024-06-20.
- O. K. Muratoglu and W. H. Harris, J. Biomed. Mater. Res., 2001, 56, 584–592 CrossRef CAS.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- S. L. Barry, S. A. Martinez, N. M. Davies, C. M. Remsberg, C. L. Sayre and A. Bachelez, J. Vet. Pharmacol. Ther., 2015, 38, 97–100 CrossRef CAS.
- A. Di Salvo, A. Bufalari, V. De Monte, P. Cagnardi, M. L. Marenzoni, A. Catanzaro, V. Vigorito and G. della Rocca, J. Vet. Pharmacol. Ther., 2015, 38, 350–356 CrossRef CAS.
- Q.-B. Sun, S.-D. Liu, Q.-J. Meng, H.-Z. Qu and Z. Zhang, BMC Musculoskeletal Disord., 2015, 16, 21 CrossRef PubMed.
- A. Shah, D. Cieremans, J. Slover, R. Schwarzkopf and M. Meftah, J. Am. Acad. Orthop. Surg. Glob. Res. Rev., 2022, 6, e22.00116 Search PubMed.
- American Joint Replacement Registry (AJRR), 2022 Annual Report, American Academy of Orthopaedic Surgeons (AAOS), Rosemont, IL, 2022, https://www.aaos.org/registries/publications/ajrr-annual-report/ Search PubMed.
- P.-G. Ren, Y.-Y. Di, Q. Zhang, L. Li, H. Pang and Z.-M. Li, Macromol. Mater. Eng., 2012, 297, 437–443 CrossRef CAS.
- H. J. Favreau, K. I. Miroshnichenko, P. C. Solberg, I. I. Tsukrov and D. W. Van Citters, J. Appl. Polym. Sci., 2022, 139, 52175 CrossRef CAS.
- F. Ramsteiner and R. Theysohn, Composites, 1984, 15, 121–128 CrossRef CAS.
- J. Leidner and R. T. Woodhams, J. Appl. Polym. Sci., 1974, 18, 1639–1654 CrossRef CAS.
- Y. Bréchet, J.-Y. Cavaillé, E. Chabert, L. Chazeau, R. Dendievel, L. Flandin and C. Gauthier, Adv. Eng. Mater., 2001, 3, 571–577 CrossRef.
- S.-Y. Fu, X.-Q. Feng, B. Lauke and Y.-W. Mai, Composites, Part B, 2008, 39, 933–961 CrossRef.
- L. G. Malito, S. Arevalo, A. Kozak, S. Spiegelberg, A. Bellare and L. Pruitt, J. Mech. Behav. Biomed. Mater., 2018, 83, 9–19 CrossRef CAS PubMed.
- S. Spiegelberg, A. Kozak and G. Braithwaite, in UHMWPE Biomaterials Handbook (Third Edition), ed. S. M. Kurtz, William Andrew Publishing, Oxford, 2016, pp. 531–552 Search PubMed.
- E. Oral, C. A. Godleski Beckos, A. J. Lozynsky, A. S. Malhi and O. K. Muratoglu, Biomaterials, 2009, 30, 1870–1880 CrossRef CAS PubMed.
- E. M. Brach del Prever, A. Bistolfi, P. Bracco and L. Costa, J. Orthop. Traumatol., 2009, 10, 1–8 CrossRef PubMed.
- M. C. Sobieraj and C. M. Rimnac, J. Mech. Behav. Biomed. Mater., 2009, 2, 433–443 CrossRef CAS PubMed.
- E. Oral, A. S. Malhi and O. K. Muratoglu, Biomaterials, 2006, 27, 917–925 CrossRef CAS PubMed.
- S. M. Kurtz, M. L. Villarraga, M. P. Herr, J. S. Bergström, C. M. Rimnac and A. A. Edidin, Biomaterials, 2002, 23, 3681–3697 CrossRef CAS PubMed.
- S. M. Kurtz, H. A. Gawel and J. D. Patel, Clin. Orthop. Relat. Res., 2011, 469, 2262–2277 CrossRef PubMed.
- A. Bistolfi, F. Giustra, F. Bosco, L. Sabatini, A. Aprato, P. Bracco and A. Bellare, J. Orthop., 2021, 25, 98–106 CrossRef PubMed.
- P. R. T. Kuzyk, M. Saccone, S. Sprague, N. Simunovic, M. Bhandari and E. H. Schemitsch, J. Bone Joint Surg. Br. Vol., 2011, 93B, 593–600 CrossRef PubMed.
- T. C. J. Partridge, P. N. Baker, S. S. Jameson, J. Mason, M. R. Reed and D. J. Deehan, J. Bone Joint Surg. Am., 2020, 102, 119–127 CrossRef.
- T. S. Brown, D. W. V. Citters, D. J. Berry and M. P. Abdel, Bone Joint J., 2017, 99B, 996–1002 CrossRef PubMed.
- J. H. Currier, B. H. Currier, M. P. Abdel, D. J. Berry, A. J. Titus and D. W. V. Citters, Bone Joint J., 2021, 103B, 1695–1701 CrossRef PubMed.
- S. Lekkala, N. Inverardi, S. C. Grindy, S. Hugard, O. K. Muratoglu and E. Oral, Biomacromolecules, 2024, 25, 2312–2322 CrossRef CAS PubMed.
- A. W. Amundson, R. L. Johnson, M. P. Abdel, C. B. Mantilla, J. K. Panchamia, M. J. Taunton, M. E. Kralovec, J. R. Hebl, D. R. Schroeder, M. W. Pagnano and S. L. Kopp, Anesthesiology, 2017, 126, 1139–1150 CrossRef CAS PubMed.
- S. Summers, N. Mohile, C. McNamara, B. Osman, R. Gebhard and V. H. Hernandez, J. Bone Joint Surg. Am., 2020, 102, 719–727 CrossRef PubMed.
- J. A. Karam, E. S. Schwenk and J. Parvizi, J. Bone Joint Surg. Am., 2021, 103, 1652–1662 CrossRef PubMed.
- V. Sreedharan Nair, N. Ganeshan Radhamony, R. Rajendra and R. Mishra, Arthroplast. Today, 2019, 5, 320–324 CrossRef PubMed.
- D. Y. Kong, J. H. Oh, W. R. Choi, Y.-I. Ko and C. H. Choi, J. Arthroplasty, 2020, 35, 2439–2443 CrossRef PubMed.
- M. E. Hilton, T. Gioe, S. Noorbaloochi and J. A. Singh, BMC Musculoskeletal Disord., 2016, 17, 421 CrossRef PubMed.
- S. Mullins, F. Hosseini, W. Gibson and M. Thake, Clin. Med., 2022, 22, 307–310 CrossRef PubMed.
- P. J. MacMahon, S. J. Eustace and E. C. Kavanagh, Radiology, 2009, 252, 647–661 CrossRef PubMed.
- Y. Li, G. E. Owens and D. S. Kohane, ChemMedChem, 2023, 18, e202300009 CrossRef CAS PubMed.
- C. M. Santamaria, A. Woodruff, R. Yang and D. S. Kohane, Mater. Today, 2017, 20, 22–31 CrossRef CAS PubMed.
- N. C. Brigham, R.-R. Ji and M. L. Becker, Nat. Commun., 2021, 12, 1367 CrossRef CAS PubMed.
- S. Grindy, D. Gil, J. Suhardi, Y. Fan, K. Moore, S. Hugard, C. Leape, M. Randolph, M. D. Asik, O. Muratoglu and E. Oral, J. Controlled Release, 2023, 361, 20–28 CrossRef CAS PubMed.
- P. C. Kreuz, M. Steinwachs and P. Angele, Knee Surg., Sports Traumatol., Arthrosc., 2018, 26, 819–830 CrossRef PubMed.
- A. J. Rao, T. R. Johnston, A. H. S. Harris, R. L. Smith and J. G. Costouros, Am. J. Sports Med., 2014, 42, 50–58 CrossRef PubMed.
- S. T. Webb and S. Ghosh, BJA: Br. J. Anaesth., 2009, 102, 439–441 CrossRef CAS PubMed.
- N. Hussain, C. J. L. McCartney, J. M. Neal, J. Chippor, L. Banfield and F. W. Abdallah, Br. J. Anaesth., 2018, 121, 822–841 CrossRef CAS PubMed.
- R. Padera, E. Bellas, J. Y. Tse, D. Hao and D. S. Kohane, Anesthesiology, 2008, 108, 921–928 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Effect of the doping temperature on the drug uptake; diffusion kinetics; quantification of the drug uptake after diffusion doping; FTIR evaluation of radiation dose; DSC results; 1H NMR spectra with lists of peaks and multiplets; effect of the elution medium on the drug release; Peppas–Korsmeyer fitting of the cumulative drug mass release profiles. See DOI: https://doi.org/10.1039/d4tb01050g |
| This journal is © The Royal Society of Chemistry 2024 |