
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multimodal layer-by-layer nanoparticles: a breakthrough in gene and drug delivery for osteosarcoma†
Eugenia
Crisafulli‡
a,
Annachiara
Scalzone‡
b,
Chiara
Tonda-Turo
 c,
Joel
Girón-Hernández§
*d and
Piergiorgio
Gentile§
*ae
c,
Joel
Girón-Hernández§
*d and
Piergiorgio
Gentile§
*ae
aSchool of Engineering, Newcastle University, NE1 7RU Newcastle Upon Tyne, UK
bCenter for Advanced Biomaterials for Health Care, Istituto Italiano di Tecnologia, Largo Barsanti e Matteucci 53, 80125 Naples, Italy
cDepartment of Mechanical and Aerospace Engineering, Politecnico di Torino, 10129 Turin, Italy
dDepartment of Applied Sciences, Faculty of Health and Life Sciences, Northumbria University, NE1 8ST Newcastle Upon Tyne, UK. E-mail: joel.l.g.hernandez@northumbria.ac.uk
eCentre for Biomaterials and Tissue Engineering (CBIT), Universitat Politècnica de València, 46022 València, Spain. E-mail: pgentil@upvnet.upv.es
First published on 29th October 2024
Abstract
Osteosarcoma is one of the most common primary malignant bone tumours in children and adolescents, frequently arising from mesenchymal tissue in the distal femur. It is highly aggressive, often metastasising to the lungs. Current treatments, which include surgery combined with neoadjuvant chemotherapy and radiotherapy, are often unsatisfactory due to the inability of surgery to control metastasis and the side effects and drug resistance associated with chemotherapy. Thus, there is an urgent need for new treatment technologies. This study explored the use of nanoparticles for gene and drug delivery in osteosarcoma treatment. The nanoparticles were composed of biodegradable and biocompatible polymers, chitosan and PLGA, and were loaded with miRNA-34a, a short RNA molecule that functions as a tumour suppressor by inducing cell cycle arrest and apoptosis in osteosarcoma cells. Recognising that the co-delivery of multiple drugs can enhance treatment efficacy while reducing systemic toxicity and drug resistance, three additional classes of nanoparticles were developed by adding doxorubicin and resveratrol to the chitosan-PLGA-miRNA-34a core. A layer-by-layer technique was employed to create a bilayer nanocoating using pectin and chitosan as polyelectrolytes, for encapsulating the therapeutic payloads. The manufactured nanoparticles were tested on U2OS and Saos-2 cells to assess cell viability, metabolic activity, and morphology before and after treatment. Cells were treated in both two-dimensional cultures and three-dimensional osteosarcoma spheroids, creating a biomimetic cellular model. Increased apoptotic activity and disruption of cellular functions were primarily observed with nanoparticles co-delivering miRNA-34a and drugs, particularly those functionalised with the LbL nanocoating, as confirmed by PCR analysis.
1. Introduction
A bone tumour represents an alteration in bone tissue and is classified, based on histological findings, architecture, and the type of matrix produced, as either benign or malignant. While benign tumours are noncancerous, they have the potential to become malignant if left untreated.1 Osteosarcoma (OS) stands as the most common primary malignant bone tumour, predominantly affecting individuals in their first and second decades of life. Its annual incidence is reported at 3.4 per million individuals worldwide.2 This aggressive tumour typically metastasises to the lungs and primarily develops in the metaphyseal regions of the distal femur, proximal tibia, and proximal humerus. Current treatments primarily involve surgery, complemented by neoadjuvant chemotherapy and radiotherapy. As a result, the five-year survival rate for OS patients has seen a significant increase, reaching approximately 60–70% over recent decades. Nevertheless, the efficacy of these conventional therapeutic approaches seems to have reached a plateau. In addition, chemotherapy can lead to drug resistance and potentially fatal adverse effects, such as cardiotoxicity and nephrotoxicity.3 One of the main challenges contributing to these limitations is the lack of selective drug delivery, leading to systemic toxicity. Consequently, there is an urgent need to develop innovative treatment methods that can enhance the effectiveness of OS treatments while mitigating the adverse effects associated with chemotherapy.4Recent advancements in nanomedicine offer promising solutions to existing challenges. A primary focus within this field is the development of nanoscale delivery systems,5 specifically nanoparticles (NPs), which are solid colloidal particles with a nano-sized diameter.6 These nanoparticle drug delivery systems have garnered significant attention due to their biocompatibility, enhanced drug penetration, and prolonged in vivo circulation time, making them viable alternatives for tumour treatment.7 Notably, these systems facilitate high drug loading and controlled drug release, thereby increasing drug bioavailability, reducing the frequency of administration, and necessitating lower dosages.
However, the manufacturing of nanoparticles for genetic and drug delivery presents challenges, particularly regarding biodegradation rates. Nanoparticles resistant to nuclease action may accumulate in the body to problematic concentrations, while those lacking sufficient resistance may degrade prematurely, compromising their efficacy before reaching the target site. To address these challenges, various synthetic polymers, such as poly lactide-co-glycolic acid (PLGA) and PEG, along with natural polymers like hyaluronan and chitosan, have been extensively explored due to their biocompatibility and biodegradability. Clinical trials have investigated a range of biodegradable polymeric drug delivery systems for localised or systemic administration of therapeutic drugs for OS treatment.8,9 Both single and dual chemotherapeutic drug delivery systems using polymeric nanoparticles have demonstrated promising results in numerous studies.10,11 For instance, in therapeutically relevant animal models of osteosarcoma, chitosan nanoparticles loaded with DNA enzymes exhibited favourable outcomes, inhibiting tumour growth without damaging surrounding bone tissue.12 In addressing bone cancer metastases, Salerno et al.13 developed doxorubicin (Dx)-loaded bioconjugate nanoparticles comprising PLGA–alendronate (ALN), demonstrating superior efficacy compared to free Dx in preventing osteolytic bone metastases.
Over the past years, there has been a significant surge in interest in the role of microRNAs (miRNAs), small endogenously expressed noncoding RNAs.3 These molecules play a crucial role in the post-transcriptional regulation of genes,14 exerting their influence by inhibiting the expression of target mRNAs. This regulatory function can impact various biological processes, including development and behaviour. Notably, each miRNA targets a specific mRNA, and alterations in mRNA expression can have profound implications for the development of malignant tumours.
Dysregulation of miRNAs has been implicated in numerous cancers, including OS.15 Several studies have highlighted significant alterations in the miRNA expression profiles of OS patients, suggesting a pivotal role for miRNAs in the onset, progression, and invasion of OS through various mechanisms.16 Specifically, miR-34a is known to act as a tumour suppressor in OS, often in conjunction with the tumour suppressor protein p53. Ectopic expression of miR-34a has been shown to induce cell cycle arrest in both primary and established OS cell lines.17,18
In this work, we proposed the co-delivery of therapeutic payload leading to significantly better results than a single-drug delivery. Indeed, the co-delivery of multiple chemotherapeutic drugs is the most popular combination therapeutic modality for successful cancer treatment in clinical practice,19 aiming at maximising the cancer treatment effectiveness while reducing systemic toxicity and drug resistance.20 Specifically, we propose the simultaneous delivery of doxorubicin hydrochloride (Dx), resveratrol (Rs) and miR-34. Dx is a chemotherapeutic agent that stimulate cell apoptosis, thus it is used as a drug to treat different cancers and it is provided intravenously in the course of OS treatment together with other chemotherapeutic agents like methotrexate and cisplatinum.21 Studies conducted in vitro to determine the impact of Dx on human OS cell lines revealed that endocytosis, which occurs 24 hours after Dx treatment, reduces U2OS osteosarcoma cells metabolic activity and proliferation.22 However, when exposed to high amount of Dx, U2OS cells can develop resistance to it.23 The use of NPs has shown promise in overcoming the drawbacks of free intravenous Dx in the treatment of cancers like breast cancer24 and hepatic cancers.25 This suggests that Dx-loaded NPs could be a practical way of promoting leftover osteosarcoma cell apoptosis. As further therapeutic agent, trans-3,4′,5 trihydroxystilbene, known as resveratrol, is a naturally occurring polyphenolic molecule that is extracted from a variety of plant species such as grapes, mulberries, and peanuts. Rs was found to be an efficient inhibitor for OS stem cells in vitro and in vivo,26 and this gives the basis for its clinical application as agent for OS treatment.27
This study aimed to explore the potential application of the layer-by-layer assembly technique-an environmentally friendly approach that involves the sequential exposure of a charged substrate to solutions containing positively and negatively charged polyelectrolytes – as an effective method for surface functionalising nanoparticles at the nanoscale.28 This process enables the incorporation of multiple therapeutic agents. Specifically, our goal was to evaluate the synergistic effects of conventional drugs commonly used to treat OS alongside emerging advanced techniques with promising prospects, including miRNA-34a. Beginning with the optimisation of the NP core, which consisted of chitosan and PLGA, we synthesised four distinct types of nanoparticles using the layer-by-layer assembly technique to incorporate the therapeutic payload. Remarkably, miRNA-34a was incorporated into the core due to its negative charge, facilitating electrostatic interactions with the positively charged chitosan. On the other side, both Dx and Rs were incorporated into both the core and the bilayered nanocoating, utilising chitosan as the polycation and pectin as the polyanion. The synthesised nanoparticles underwent comprehensive physicochemical characterisation to confirm their successful preparation. Subsequently, their biological efficacy was evaluated using U2OS and Saos-2 osteosarcoma cell lines, encompassing 2D cell culture as well as 3D spheroid cultures, with the aim of replicating the tumour microenvironment more faithfully.
2. Results and discussion
2.1. Optimisation of the manufacturing of the NPs before and after LbL assembly functionalisation
In this study, various manufacturing parameters were evaluated to establish the ideal process for the manufacturing of homogenous polyplexes with a size and surface charge suitable for the obtainment of multilayered nanocoatings. However, the precise size of the nanoparticles required to circumvent physiological biological barriers (such as glomerular filtration or extravasation through blood fenestration) remains unclear and poorly understood, hindering the clinical translation of NP.29 For example, NPs with a hydrodynamic diameter of 100–400 nm have traditionally been considered optimal for passive tumour targeting due to the enhanced permeability and retention (EPR) effect.29 Since the typical thickness of the resulting LbL nanocoating formed by 3 nanolayers ranges from a few to 30–50 nm, we determined the ideal size range for the polyplexes to be between 200–300 nm, which serves as the response variable in the design of experiment (DoE). Furthermore, ζ-potential values exceeding 20–25 mV (in absolute terms) are crucial for initiating the electrostatic interactions between the polyelectrolytes, and thus were considered as the second response variable in the DoE.DLS analysis revealed the resulting size, ζ-potential and PDI of polyplexes, which are summarised in Table S1 (ESI†). The polyplexes showed a mean diameter of 257 nm, PDI of 0.29, and a zeta potential of +29.2 mV. Furthermore, Fig. 1A–C display the factors (EO concentration, homogenization speed, and sonication time) and their interactions mostly influencing the evaluated processes. It was observed that the homogenization speed (indicated with S) and the homogenization time (indicated with t) influenced significantly both polyplexes size and ζ-potential value.
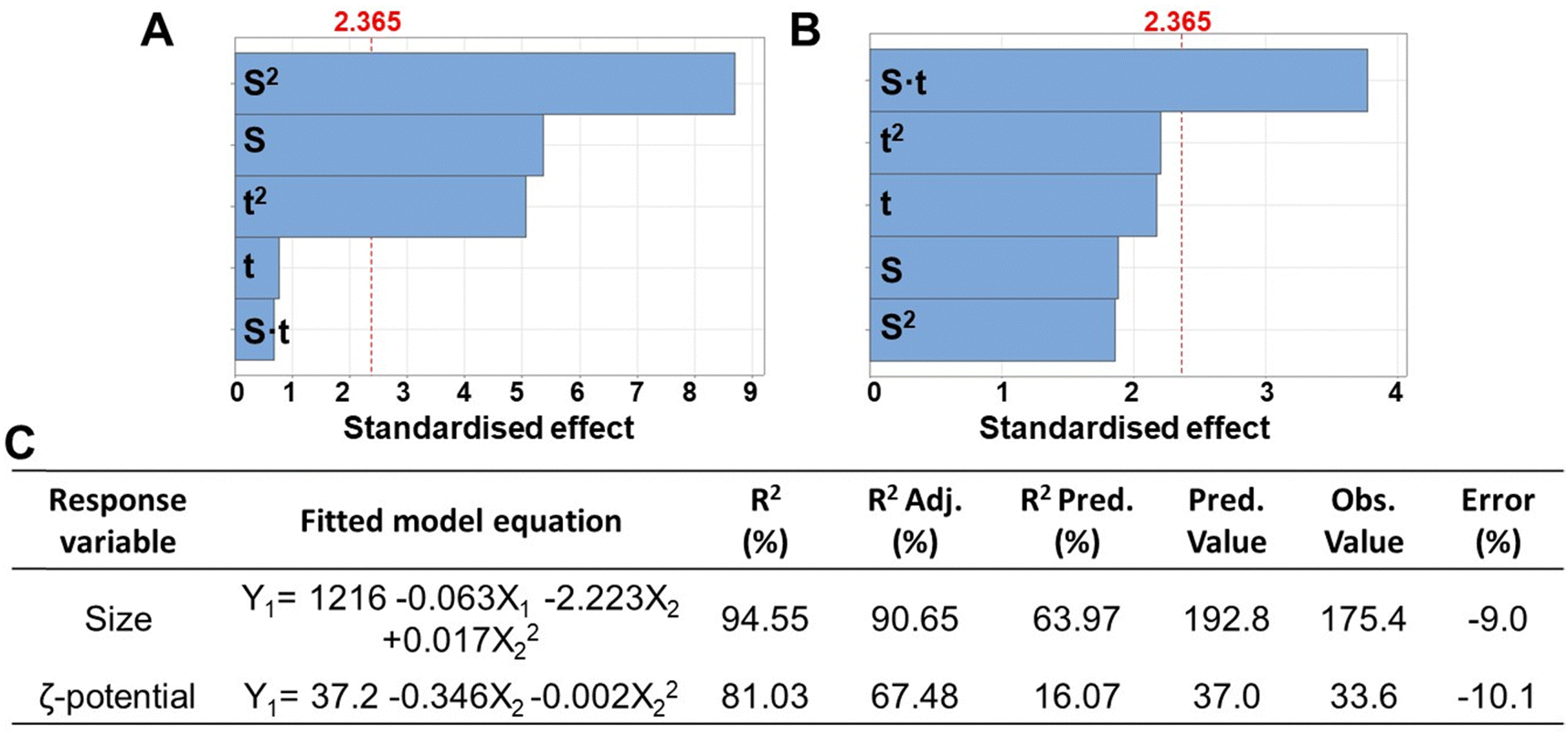 | ||
| Fig. 1 Pareto charts of the standardised effects of the factors (S: homogenization speed, t: homogenization time) and their interactions on the response variable (α = 0.05) of the resulting size (A) and ζ-potential (B). The bars crossing the red dashed line are considered significant (α = 0.05). Fitted model equations and metrics for each procedure calculated using Minitab software (C). Metrics include coefficient of determination (R2), adjusted coefficient of determination (R2 Adj.), predictive coefficient of determination (R2 Pred.), predicted value of size and ζ-potential from the fitted model (Pred. value), experimental value of size and ζ-potential (Obs. value), and experimental percentage error (error), calculated by dividing the absolute difference between the observed and predicted values by the observed value (%). | ||
Notably, the optimised conditions for manufacturing polyplexes with the minimum size (192.8 nm as predicted value) were found as 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750 rpm of homogenization speed and a homogenization time of 79.5 s, while for obtaining the maximum ζ-potential value a homogenization speed of 38107 rpm and a homogenization time of 138.6 s was requested (Table S1, ESI†). However, at the optimised conditions for the minimum size, a ζ-potential value of +31.9 was measured, showing a slight decrease compared the predicted maximum surface charge (+37.0). This suggests the potential of pre-charging the polyplexes with a positive core, followed by immersion in a polyanionic solution, such as one containing pectin for starting the LbL multilayered coating.
750 rpm of homogenization speed and a homogenization time of 79.5 s, while for obtaining the maximum ζ-potential value a homogenization speed of 38107 rpm and a homogenization time of 138.6 s was requested (Table S1, ESI†). However, at the optimised conditions for the minimum size, a ζ-potential value of +31.9 was measured, showing a slight decrease compared the predicted maximum surface charge (+37.0). This suggests the potential of pre-charging the polyplexes with a positive core, followed by immersion in a polyanionic solution, such as one containing pectin for starting the LbL multilayered coating.
For both size and ζ-potential, the R2 coefficients were above 80% and the prediction coefficients (R2 Pred) for the size was over 60%, whereas for the ζ-potential it was around 16%. This lower value could be related to the higher variability in the measurement of the surface charge using DLS.30 The percent error, calculated based on the predicted and observed values, was around 10%.
The PDI was not utilised as a response variable in the DoE. Instead, it served to assess the uniformity of polyplexes size distribution within each sample. PDI values range from 0 to 1, with values below 0.3 generally considered indicative of a homogeneous sample.31 In our work, the PDI remained mostly below 0.3, confirming that the samples were monodispersed, and each portion was representative of the entire sample (Table S1, ESI†). This is in accordance with literature that correlate surface charge with value >20 mV increases the repulsive forces between particles outweigh attractive forces, thereby ensuring particle dispersion.32
TEM analysis (Fig. 2A) revealed that the morphology of the manufactured polyplexes was predominantly spherical, consistent with the source material utilised in the method.33 Specifically, miR-NPs showed an average hydrodynamic diameter of 159.8 ± 20.9 nm (Fig. 2B). While the incorporation of Dx (miRDx-NPs) increased the resulting polyplex diameter, reaching a value of 242.0 ± 38.6 nm, no substantial differences were observed with the addition of Rs in the miRDxRs-NPs (242.4 ± 33.2 nm). After the deposition of two nanolayers onto the miRDxRs-NPs core, the size increased approximately 2-fold compared to the initial size of the miR-NPs (****p < 0.0001) and 1.5-fold compared to the size of the loaded polyplexes (miRDx-NPs and miRDxRs-NPs, **p < 0.01), reaching a final value of 343.5 ± 51.7 nm. Interestingly, as shown in Fig. 2C, the deposition of the second layer significantly increased the final size of the LbL-NPs (*p < 0.05 compared with miRDxRs-NPs) probably due to the higher amount of chitosan absorbed electrostatically for the formation of the bilayer nanocoating. On this regard, two main types of coating growth have been reported in the literature, where linear growth is observed with strongly charged synthetic polyelectrolytes, while exponential growth is seen with weak natural polyelectrolytes such as polypeptides or polysaccharides.34 By using pectin and chitosan as polyelectrolytes in this work, the observed exponential growth can be attributed to various phenomena, including increased film surface roughness with fractal-like growth,35 complexation of polyelectrolytes above the film surface,36 or the diffusion of at least one of the two polyelectrolytes within the film.37
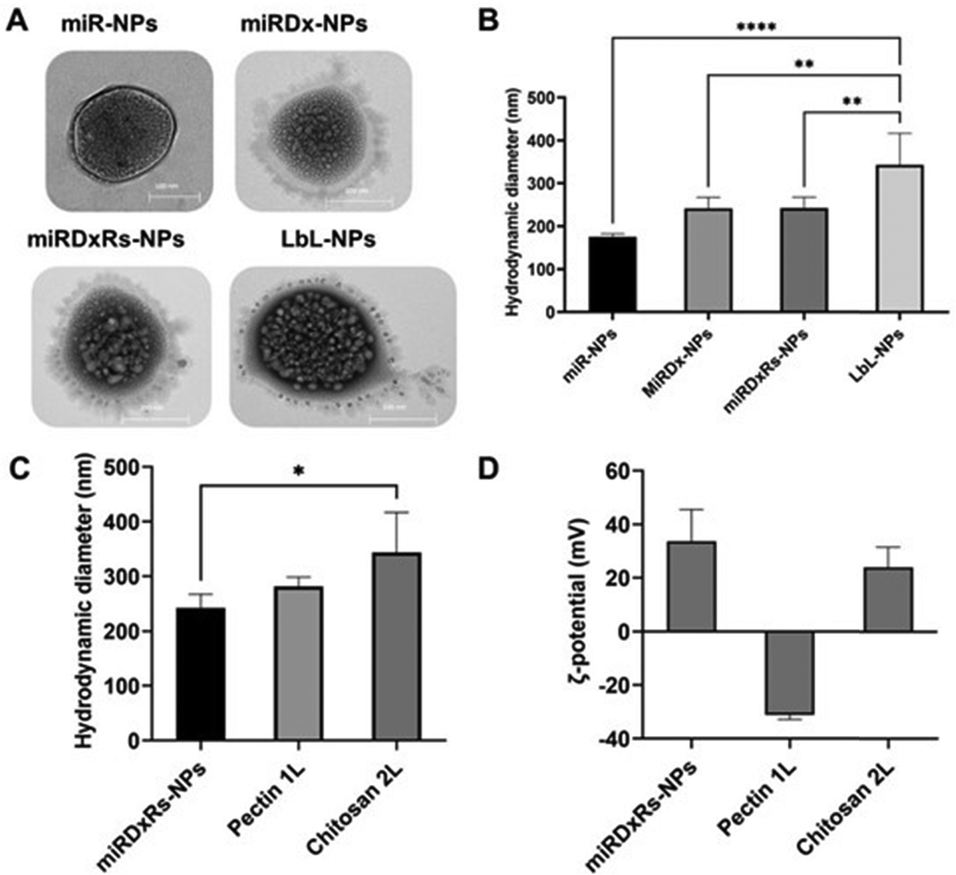 | ||
| Fig. 2 TEM images (A) and the hydrodynamic diameter (B) of the different manufactured nanoparticles; hydrodynamic diameter (C) and ζ-potential values (D) of miRDxRs-NPs before and during the build-up of the bilayered coating by LbL assembly. The values represent the average ± standard deviation (n = 3). Statistics: *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001. | ||
Furthermore, layer by layer assembly is confirmed by measuring the particle ζ-potential, which demonstrates charge reversal due to overcompensation (Fig. 2D).38 Adsorption of pectin layer results in negative ζ-potential (−31.1 ± 1.7 mV) over the positively charged surface of the miRDxRs-NPs (33.6 ± 11.9 mV), whereas adsorption of chitosan results in positive ζ-potential (24.0 ± 7.5 mV). The observed charge reversal, indicated by the particle ζ-potential, confirms that the adsorption of alternating charged polyelectrolytes modifies the particle's surface chemistry.
The encapsulation values of miRNA, Dx, and Rs for the manufactured NPs are shown in Fig. 2, reported as both entrapped amounts (μg) (Fig. 2A) and entrapment efficiency (%) (Fig. 2B). Dx and miRNA-34 exhibited efficient encapsulation within all types of manufactured NPs, with the highest entrapment efficiency observed in miR-NPs, where miRNA-34 was the sole drug, reporting an entrapment efficiency of ∼80%. The encapsulation of miRNA-34 was also higher than 60% in the other three types of NPs, demonstrating the efficient electrostatic interactions between the anionic miRNA and the cationic chitosan. Indeed, the entrapment efficiency is a crucial parameter, especially for delivering biologically active macromolecules such as the miRNA-34. A successful entrapment can positively influence the in vivo therapeutic response and enhance the potential for clinical translation.39 The entrapment efficiency found in our work is in accordance with the work published by Cosco et al.,40 where the authors have used various amounts of miRNA-34 (ranging from 100 to 500 μg) to the aqueous phase during nanoplex preparation. Specifically, using 300 and 400 μg of miRNA-34 achieved a drug load of ∼85% and ∼62 respectively, satisfactory values for potential in vivo applications (Fig. 3).
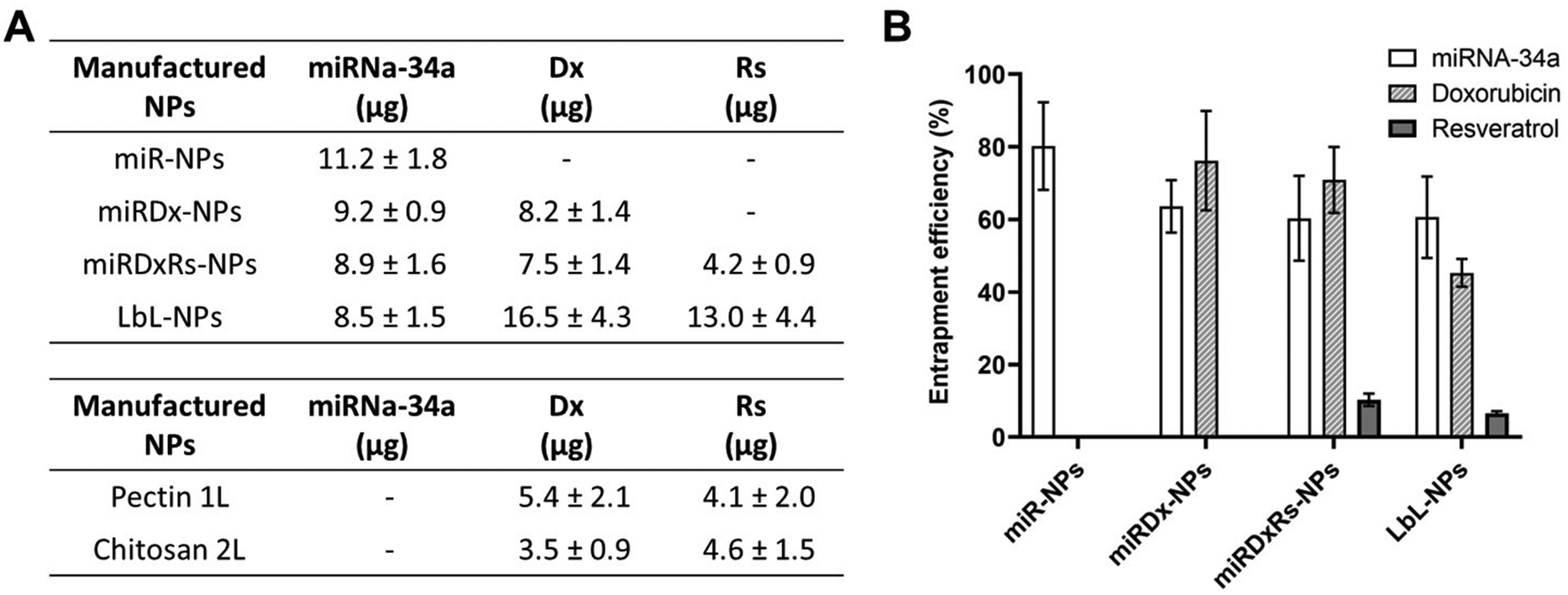 | ||
| Fig. 3 (A) Amounts (μg) of miRNA-34a, Dx and Rs entrapped within all the manufactured NPs with the specific values in the two-forming layers of LbL-NP. (B) Bar graph showing the entrapment efficacy (%) of miRNA-34a in miR-NPs, miRNA-34 and Dx in miRDx-NPs, miRNA-34a, Dx and Rs in miRDxRs-NPs, and all the payload in LbL-NPs (n = 3). | ||
Moreover, for the Dx, the highest entrapment efficiency was observed in miRDx-NPs with a value of ∼77%, while after the LbL assembly the entrapment efficiency decreased at ∼46%. Interestingly, in the LbL-NPs a greater quantity of Dx was detected in the first pectin layer, likely because this chemotherapeutic agent, slightly positively charged,41 could be able to create complexes with the negatively charged pectin, enhancing its amount entrapped in this nanolayer. However, the difference in encapsulation percentages between the layers was not significant: ∼20% in the first layer and ∼13% in the second layer, with higher variability in the pectin layer. Overall, Rs encapsulation was very low in both the core of miRDxRs-NPs and the layers of LbL-NPs. Compared to the original amounts of Rs dissolved in the solutions to prepare the core (40.1 μg) and the layers (100.4 μg each), the resulting entrapped amounts were much smaller, with 4.16 μg in the core and 12.96 μg in the overall LbL-NPs. This can be due to low solubility of Rs in aqueous solution (∼50 μg mL−1 in pH 7.4 buffer)42 and micro- and nano-encapsulation approaches or chemical modifications can be used to improve its bioavailability.41,43
To evaluate the release of miRNAa-34, DxDx, and Rs from the manufactured NPs, the nanoparticles were incubated at 37 °C in PBS at pH 7.4, mimicking storage and blood plasma conditions, and their cumulative release over a period of six days was measured (Fig. 4). In Fig. 4A, the cumulative release of miRNA-34a from miR-NPs indicated that ∼70% of the initially entrapped amount was released within two days. This release pattern was like that of miR-34a from both miRDx-NPs and miRDxRs-NPs, demonstrating that the presence of Dx and did not affect the miRNA-34a release trend. Looking at the release of Dx from miRDx-NPs and miRDxRs-NPs, it was observed that ∼70% of the chemotherapeutic agent was released within two days, with almost 45% released within the first five hours of incubation (Fig. 4B).
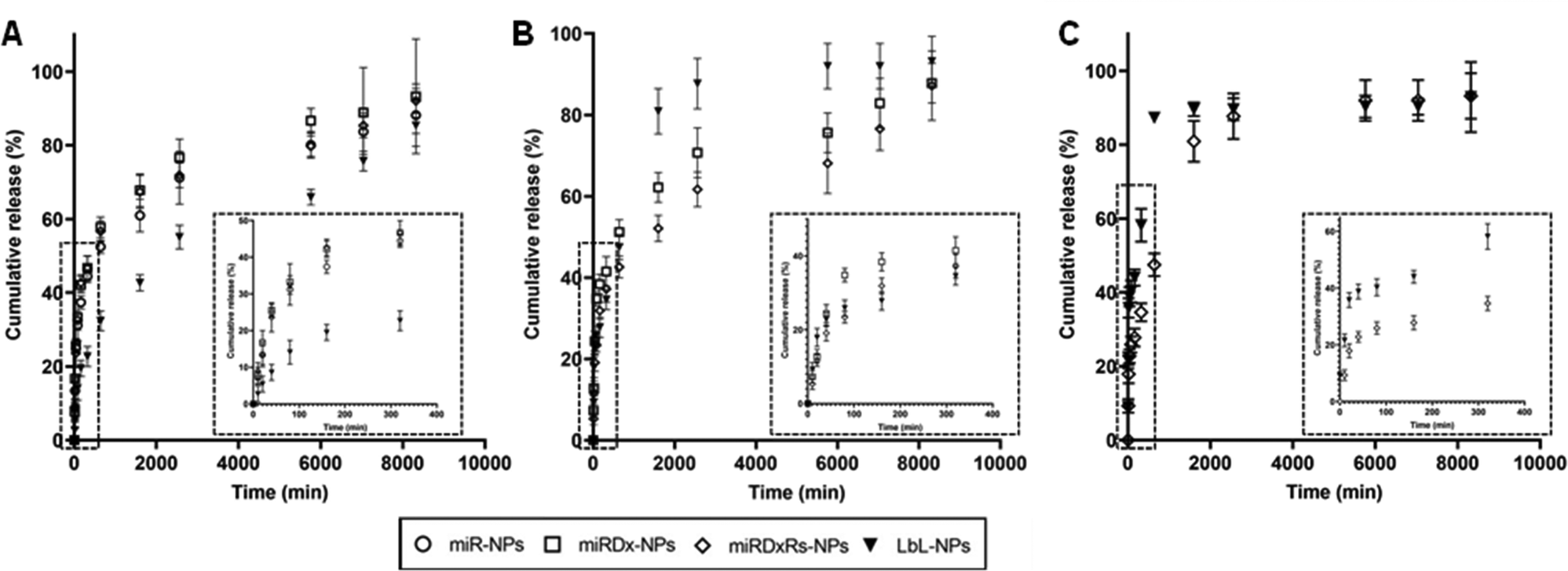 | ||
| Fig. 4 Cumulative release (%) of miRNA-34a (A), Dx (B) and Rs (C) from the different manufactured NPs after immersion in PBS (n = 3). | ||
For LbL-NPs, the release of miR-34a was slower compared to the other manufactured NPs with a very reduced burst release (10% within the 1 hour of immersion in PBS) followed by a gradual and liner release up to the 6 days of incubation. On the other hand, both Dx and Rs were released quickly compared to the miRDx-NPs and miRDxRS-NPs. Indeed, approximately 20% and 40% of Dx and Rs amounts respectively were released within the first 20 minutes of incubation, followed by a significant increase after 24 hours, which then plateaued until the end of the evaluation period (Fig. 4B and C). The observed irregular release pattern could be attributed to the either the high diffusion rate of these two therapeutics within the two nanolayers or to the quick degradation of the bilayered coating. Importantly, it is recognised that the incubation conditions in PBS did not perfectly mimic physiological or intracellular environments (such as pH 5.5 for endolysosomal conditions), and future studies should investigate miR-34a and drug release kinetics in serum and under conditions representing in vivo mixing.44 However, the results obtained provided valuable insights regarding the release profiles of miR-34a, Dx, and Rs from the manufactured NPs under extracellular pH conditions.
2.2. In vitro biological characterisation of the LbL-functionalised NPs
Live/dead images and PrestoBlue assays were used to evaluate cell viability and metabolic activity after incubation with the different manufactured nanoparticles at various time points (1, 2, and 3 days). Fig. 5 shows the results for treated cells, while Fig. S3 (ESI†) displays live/dead images of control cells incubated with NPs lacking therapeutic payloads (negative controls), where both cell types proliferated as the culture period increased and notably, Saos-2 cells exhibited their typical more elongated, spindle-shaped morphology compared to the rounded morphology of U2OS cells with Saos-2 assumed a more elongated spindle-shaped morphology compared to U2OS, which display a more rounded morphology.45 However, the incubation of U2OS cells with miRDxRs-NPs (Fig. 5A) affected the cell viability with an increase in the number of dead cells from 24 to 72 hours. This observation aligns with the PrestoBlue assay data (Fig. 5C), which shows a reduction in U2OS metabolic activity to one-third at 72 hours compared to 24 hours after treatment with miRDxRs-NPs. However, the highest decrease in cell viability was observed in U2OS cells when treated with LbL-NPs, indicating high apoptotic activity from 24 hours onwards (Fig. 5A). These findings suggest that the natural nanocoating materials achieved by the LbL technique might be more efficient in carrying the therapeutic payload (miRNA-34a, Dx, and Rs) to the core with minimal leakage and potentially increased drug capacity due to the additional layers. Furthermore, LbL-NPs displayed a decrease in fluorescence intensity after 48 hours (35802 ± 1508 RFU), like other NP-treated samples after 72 hours (31516 ± 3106 RFU and 34649 ± 3932 RFU for miRDx-NPs and miRDxRs-NPs, respectively). In contrast, miR-NPs exhibited higher fluorescence values (84415 ± 5514 RFU) at 72 hours, suggesting a lower impact on U2OS metabolic activity (Fig. 5C).
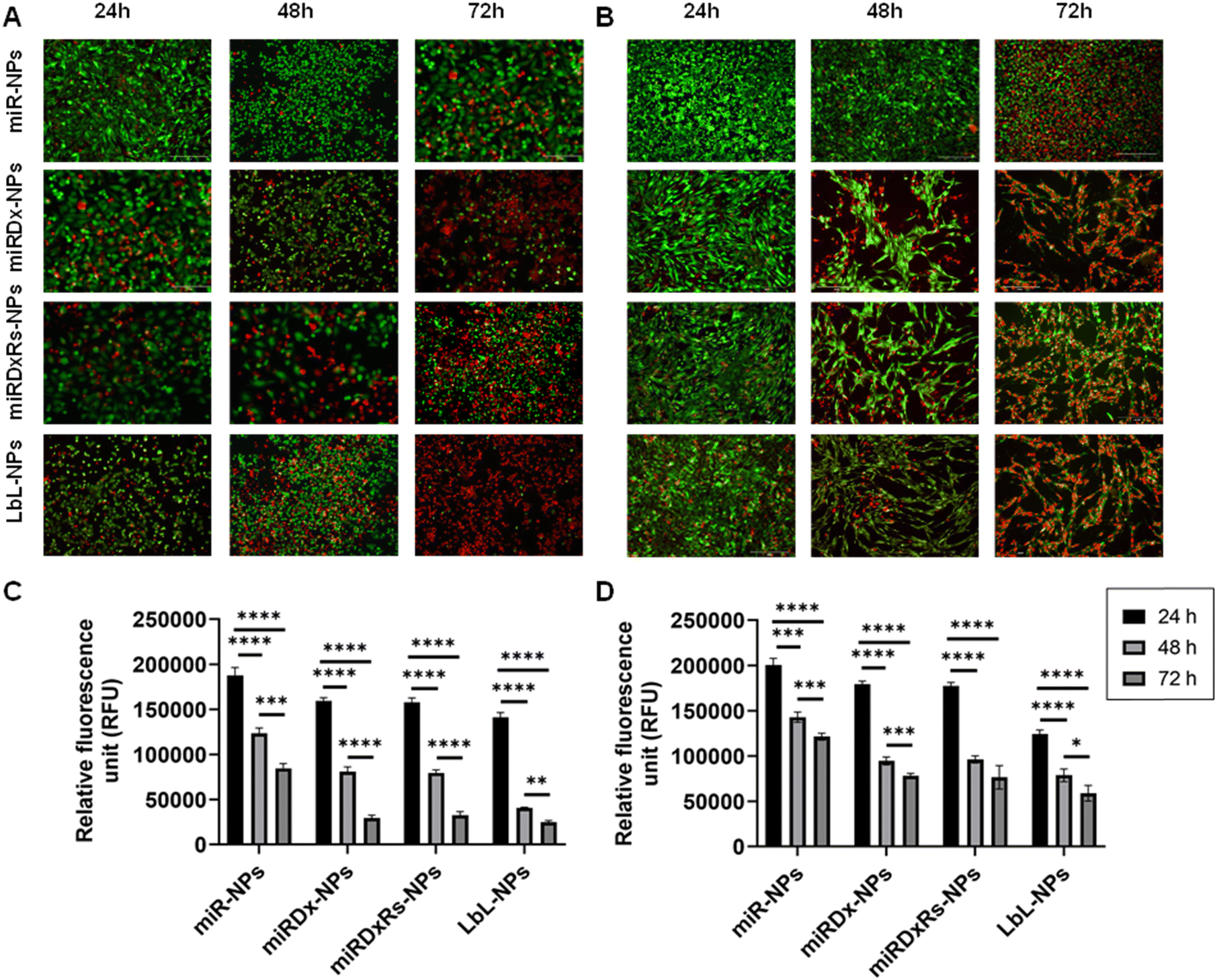 | ||
| Fig. 5 Live/dead images of U2OS (A) and Saos-2 cells (B) incubated with all the manufactured NPs for 24, 48 and 72 h. Bar = 300 μm; metabolic activity of U2OS (C) and Saos-2 (D) cells incubated with all the manufactured NPs for 24, 48 and 72 h. The results are shown as average ± SD. | ||
Regarding Saos-2 cells, it was observed a significant difference between the miR-NPs and all the other treatments (Fig. 5B) after 48, and 72 hours of incubation of the manufactured NPs. Indeed, miR-NPs showed less influence on cell viability after 24 and 48 hours of incubation with a larger amount of green (live) cells compared to red (dead) cells. However, after 72 hours, the number of live cells notably decreased. On the other hand, the addition of miRDx-NPs, miRDxRs-NPs and LbLNPs presented similar effects, with a low number of dead cells after 24 hours but almost all cells dying after 72 hours, suggesting efficient synergistic antitumor effects of the drug combinations. Cells treated with miRDx-NPs showed higher number of dead cells, suggesting that combined delivery of miRNA-34a and Dx led to apoptosis. The quantity of dead cells increased with longer incubation times (70% of red dead cells observed after 72 hours), aligning with previous studies that miRNAs have synergistic antitumor effects with conventional chemotherapy like Dx. Saos-2 cells showed similar fluorescence values to U2OS cells at the same time points however, after 72 hours, Saos-2 cells' metabolic activity was less affected than U2OS cells, suggesting greater resistance to NPs treatment (i.e. p > 0.05 for 72612 ± 6953 RFU and 63776 ± 8568 RFU for LbL-NPs). Saos-2 osteosarcoma cells exhibited greater resistance to treatments compared to U2OS cells. This resistance might be due to several factors. Firstly, Saos-2 cells have a higher potential for differentiation towards mature bone cells, which may be linked to enhanced survival mechanisms. Secondly, Saos-2 cells might express more drug efflux pumps that actively remove chemotherapeutic drugs from the cell, reducing their effectiveness. Finally, Saos-2 cells might have more efficient DNA repair mechanisms, allowing them to repair treatment-induced damage and survive longer. Understanding these resistance mechanisms is crucial for developing more effective osteosarcoma therapies.46,47
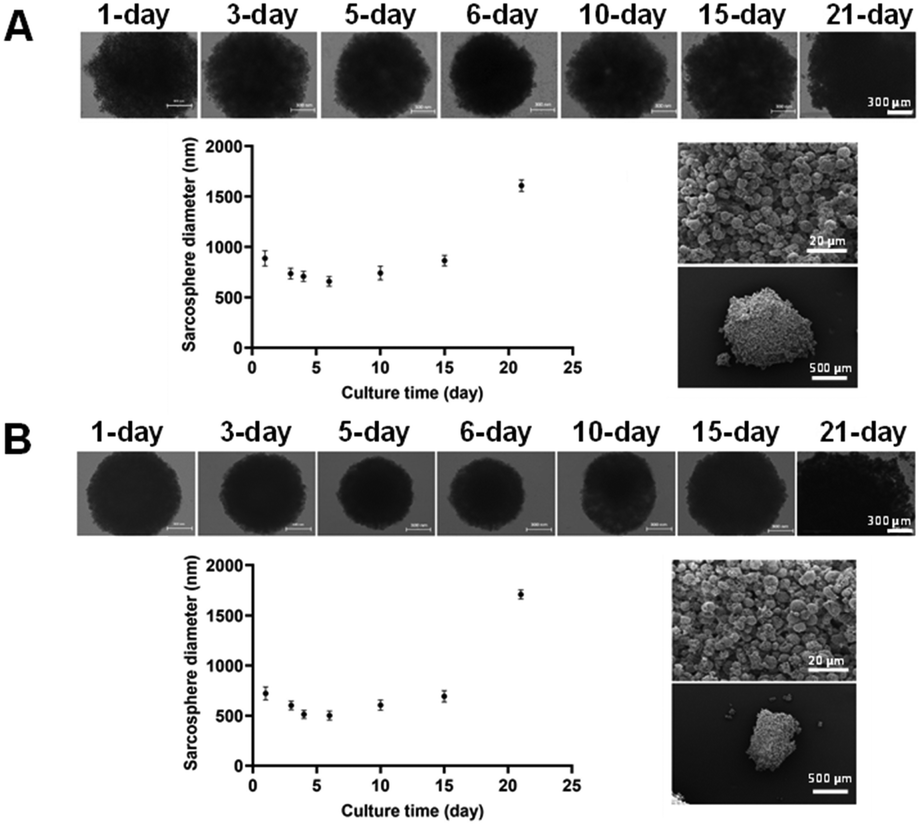 | ||
| Fig. 6 Sarcosphere diameter of U2OS (A) and Saos-2 (B) over 21 days analysing values with ImageJ software. Sarcosphere images (on the top) taken with EVOS M5000 microscope over 21 days and SEM images (on the right) of the sarcosphere at 21 days of culture time at two different magnifications. | ||
In both cases, the diameter gradually decreased until day 6 (501 ± 45 μm and 658 ± 47 μm for U2OS and Saos-2) and then increased again to almost twice the initial size at 21 days (1709 ± 45 μm and 1609 ± 57 μm for U2OS and Saos-2). However, after evaluation at the SEM, U2OS spheroids were characterised by a more compact configuration with greater roundness, making them slightly smaller than those with Saos-2 cells, which were less compact and homogeneous. The size decrease during the first 6 days occurred because the cells initially aggregated to form the spheroid. As they proliferated, their number increased, resulting in an increase in diameter over time, due to the high proliferation rate of these tumoral cell lines.49 The compact structure of the initial spheroids was disturbed by cell outward movement from the spheroid, which is typical of tumour outgrowth.50
Fig. 7A and B illustrate the viability of U2OS and Saos-2 cells in Sarcosphere models (100000 cells per well) after 24, 48, and 72 hours of incubation with the manufactured NPs. The upper images display both live and dead cells, whereas the lower images show only the dead cells. Both U2OS (Fig. 7A) and Saos-2 (Fig. 7B) sarcospheres exhibited similar responses. Indeed, as the incubation time increased, the green circular structures representing live cells decreased in size, while the necrotic cores representing dead cells enlarged, indicating increased cell death over time. The presence of the necrotic core confirmed the model's characterisation by hypoxic regions and necrotic centres, validating it as a suitable model for mimicking tumour micro-regions or micro-metastases, as reported in previous studies.51 No prior studies have investigated the effect of miR-34a on U2OS and Saos-2 spheroid models of osteosarcoma. However, our study found that the addition of Dx and Rs to the NPs enhanced their apoptotic and cytotoxic effects in the spheroids, as evidenced by the larger necrotic core compared to spheroids treated with NPs containing only miRNA-34a (miR-NPs). This observation aligns with other studies demonstrating the significant cytotoxic effect of Dx on osteosarcoma spheroids,22,52 and the synergistic antitumor effects of miRNAs combined with conventional chemotherapy drugs like Dx.52–54 Then, Fig. 7C and D presents the PrestoBlue assay results on both sarcosphere models, assessing metabolic activity before and after incubation with various manufactured nanoparticles, where a common trend was observed: the highest fluorescence values occur after 24 hours of incubation, with values decreasing over time. This trend is especially pronounced in U2OS cells, where fluorescence significantly decreases from 24 h to 48 h and 72 h, stabilising around a value of 25000 RFU. An exception is seen with miR-NPs treatment, where fluorescence values gradually decline over the incubation period. After 24 hours, spheroids did not exhibit significant changes in metabolic activity when incubated with any of the manufactured NPs. This initial stability might be attributed to spheroid densification and reduced drug permeability.22 However, by 72 hours, both U2OS and Saos-2 spheroids had largely disintegrated, except for those treated with miR-NPs, which displayed less pronounced cytotoxic effects.
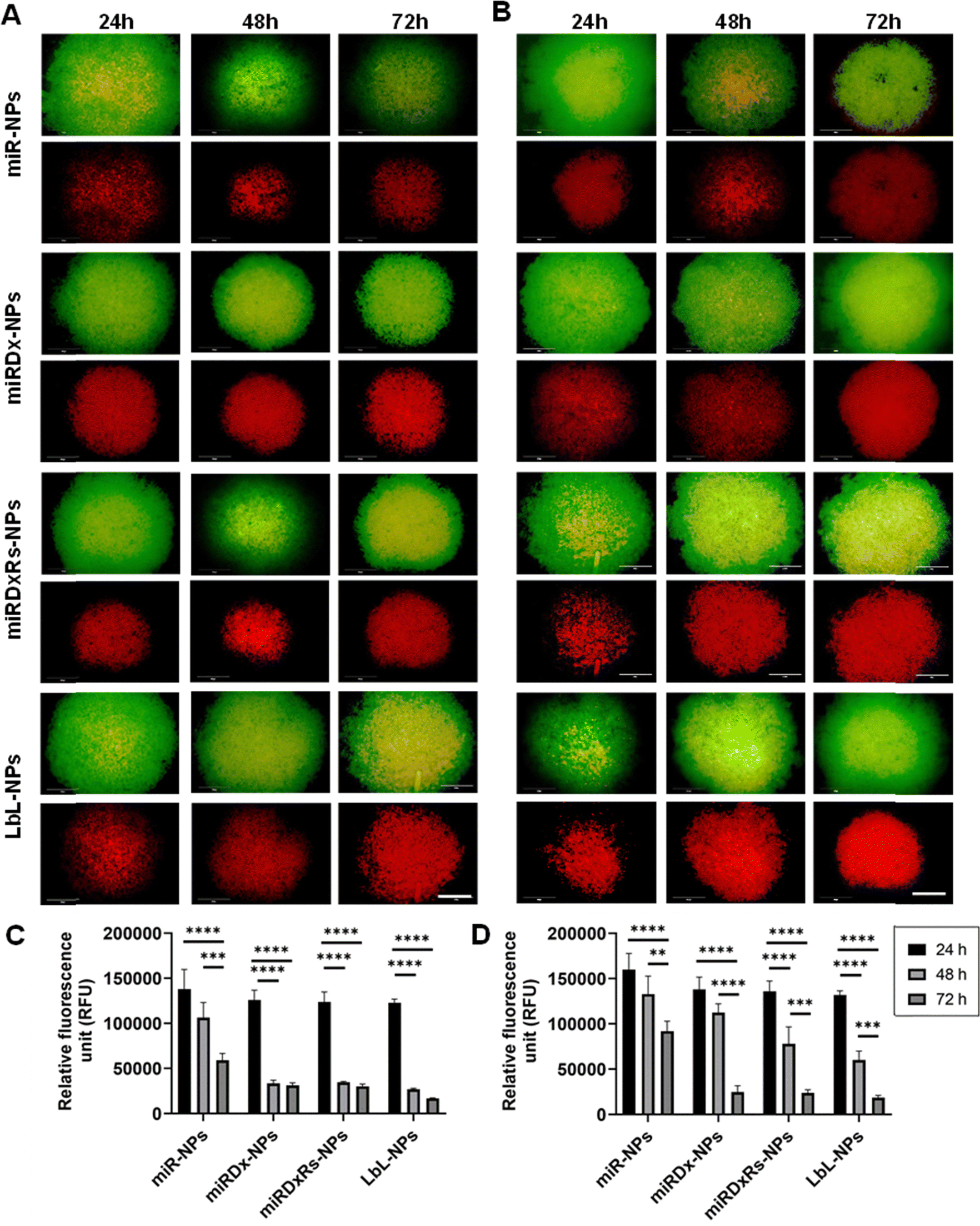 | ||
| Fig. 7 Live/dead images of U2OS (A) and Saos-2 (B) sarcospheres incubated with all the manufactured NPs for 24, 48 and 72 h. Bar = 300 μm; metabolic activity of U2OS (C) and Saos-2 (D) sarcospheres incubated with all the manufactured NPs for 24, 48 and 72 h. The results are shown as average ± SD. | ||
Finally, the expression of apoptosis-related genes Bcl-2, BAX, caspase-3, and caspase-9 in osteosarcoma spheroids was investigated using quantitative real-time PCR. As shown in Fig. 8, the expression of the anti-apoptotic gene Bcl-2 significantly decreased in all the manufactured NPs, while the expression of the pro-apoptotic genes BAX, caspase-3, and caspase-9 significantly increased in both osteosarcoma types. Moreover, compared to miR-NPs, a clear synergistic effect of miRNA-34a and Dx on the expression of Bcl-2, BAX, caspase-3, and caspase-9 was observed, with a more evident difference in the LbL-NPs. Particularly, in both osteosarcoma spheroids, a threefold down-regulation in Bcl-2 expression and at least a twofold up-regulation in BAX, caspase-3, and caspase-9 expression were measured compared to the sample containing only miRNA-34a.
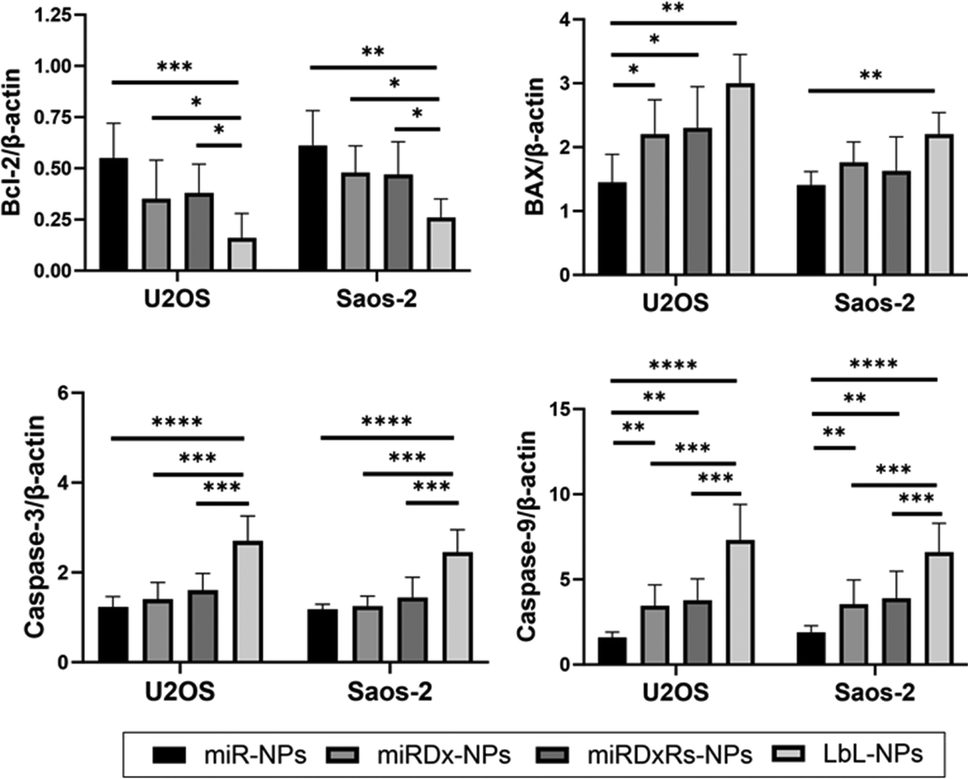 | ||
| Fig. 8 Gene expression analysis via RT-qPCR of U2OS and Saos-2 sarcospheres incubated with all the manufactured NPs for 24, 48 and 72 (n = 5). Data for Bcl-2, BAX, caspase-3, caspase-9 were normalised to β-actin housekeeping gene. Statistics: ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05. | ||
3. Experimental
3.1. Materials and chemicals
For NPs preparation poly(D,L-lactide-co-glycolide) (lactide:glycolide (75:25), MW = 66000–107000 Da), chitosan (low molecular weight, MW = 50000–190000 Da), pluronic F-127 (powder, BioReagent, suitable for cell culture), Dx hydrochloride (98–102% HPLC, powder), resveratrol (powder, ≥99% HPLC), sodium acetate, dimethyl sulfoxide (DMSO) (ACS reagent, ≥99.9%), acetic acid (glacial, ReagentPlus®, ≥99%), acetone (ACS reagent, ≥99.5%) were purchased from Merck, UK. MiRNA-34a (miRIDIAN microRNA Human has-miR-34a-5p) was supplied by Horizon Discovery Biosciences Ltd, UK; RNase-free water was purchased from ThermoFisher, UK. All solvents were of analytical grade and used with no further purification. They were all purchased from Merck, UK. Distilled water (dH2O) was obtained throughout PureA-Q+ System (SLS-LabPro, UK).
3.2. Manufacturing methods
| Factor | Level | |||||
|---|---|---|---|---|---|---|
| −α | −1 | 0 | 1 | α | ||
| X 1 | Homogenization speed (rpm) | 16893 |
20000 |
27500 |
35000 |
38106 |
| X 2 | Homogenization time (s) | 11.4 | 30 | 75 | 120 | 138.6 |
After the ultra-homogenization, the CH solution was combined with 200 μl of the solution containing the PLGA and miRNAs-34, and stirred at 600 rpm for 3 hours to allow acetone evaporation. Finally, the samples were centrifuged at 13000 rpm (Pico 17 Microcentrifuge, Thermo Scientific, UK) for 1 hour to separate the cores from the supernatant. Once the preparation of the NPs core with miR-34a was optimised, different types of cores were developed by incorporating different therapeutic payloads, that included anticancer and antioxidants agents (Scheme 1). Particularly, for the manufacturing of miRNA-Doxorubicin NP cores (coded as miRDx-NPs), 2 μL of 10 mM of Dx solution (previously dissolved in DMSO:PBS (1:1 v/v) as solvents) was added to the CH solution together with the miRNAs before the homogenization. For the miRNA-DX-resveratrol (Rs) cores (coded as miRDxRs-NPs), 2 μL of 88 mM of Rs solution (previously dissolved in DMSO) was added to the CH solution together with the miRNAs and Dx before the homogenization.
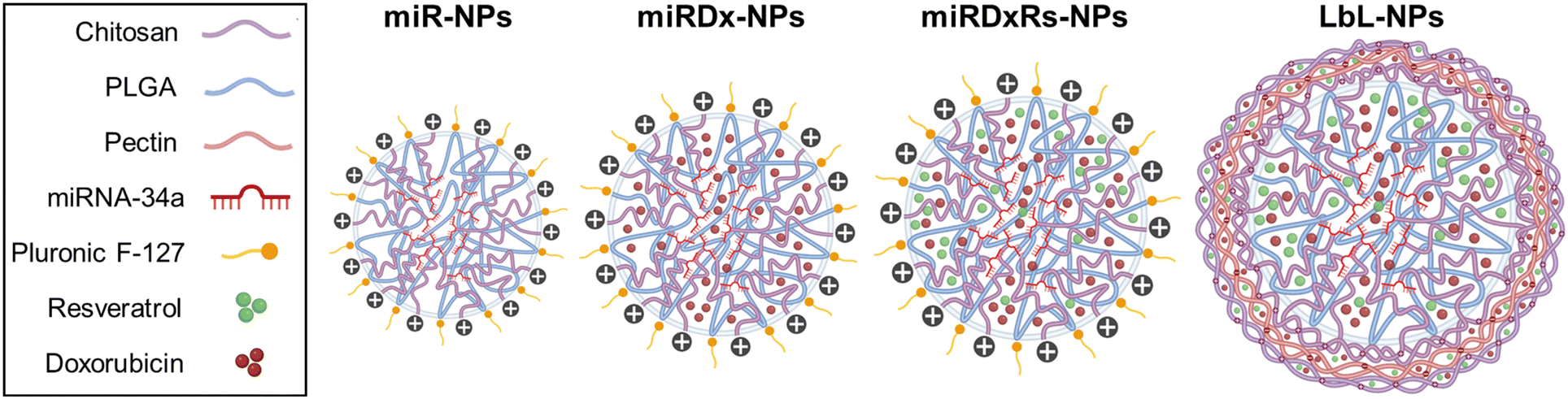 | ||
| Scheme 1 Illustration of the components and the sequence of formation of the different manufactured nanoparticles. | ||
300 rpm to separate the formed coated NPs from the PE, and, then, by two washing steps (for 5 minutes at 13300 rpm each) with the replacement of the supernatant with 1 mL of sodium acetate buffer. After the second washing steps, 100 μL of functionalised NPs were collected to measure the corresponding ζ-potential by DLS (see Section 3.3.1), and then, the remaining supernatant was replaced with the CH polyanionic solution for the deposition of the second nanolayer following the same procedure.
3.3. Physico-chemical characterisation of the manufactured NPs
:10 dilution in RNA-free water before being added to the zetasizer cuvette.
000×. Afterwards, the images were analysed with ImageJ software to measure their diameter size.
3.3.3.1 Encapsulation of miRNA-34a. The value of entrapment efficiency was assessed via QuantiFluor® RNA assay (Promega, UK), that contains a fluorescent RNA-binding dye (492 nm Ex/540 nm Em) to enable sensitive quantification of small amounts of RNA in purified samples. Briefly, according to the manufacturer procedure, a QuantiFluor® RNA Dye working solution was prepared by diluting the QuantiFluor® RNA Dye 1
:2000 in 1× TE buffer. Then, RNA standards were prepared by serially diluting QuantiFluor® RNA Standard (100 ng μL−1) to prepare RNA Standard Curve. Finally, 200 μL of QuantiFluor® RNA Dye working solution was pipetted into a 96-multiwell and 20 μL of the standards and unknown samples were added to their corresponding well, incubating for 5 minutes at room temperature protected from light before measuring the reading. The measures were taken using a multiplate reader (FLUOstar Omega, BMG Labtech, Germany). Each sample was read in triplicate and referred to the standard curve generated using the standard RNA concentration. The encapsulation efficiency was calculated as follow: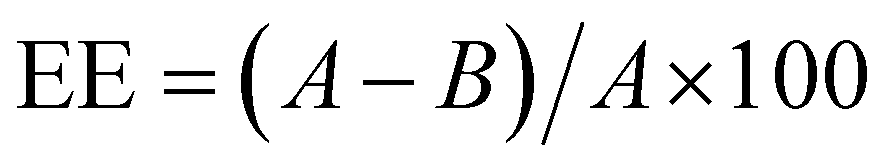 | (1) |
3.3.3.2 Dx and Rs encapsulation. The encapsulation efficiency of Dx and Rs in the manufactured NPs was evaluated by indirect measurements from the un-encapsulated Dx and Rs recovered from the supernatants. The encapsulation efficiency was calculated as previously, following eqn (1) where A and B were the weights of total amount of initial drugs and the drugs fraction recovered from the supernatant after the centrifugation, respectively. Specifically, supernatants collected were analysed at 480 nm to detect Dx and 285 nm to detect Rs. Each sample, for both the drugs, was read in triplicate and, referred to a standard curve generated using free Dx and Rs at known concentrations (Fig. S3, ESI†). The supernatant recovered from NPs without Dx and Rs was used for baseline correction.
000 rpm for 30 min. The supernatant was collected for the analysis of released miRNA, Dx and Rs following the protocols described before in par. 3.3.3.1 and 3.3.3.2 respectively. For each type of NP, the cumulative release was calculated adding the release of each time point to the previous one. This process was repeated by suspending the NPs in fresh PBS, vigorously shaking for 5 min, and incubated at 37 °C for the next time point.
3.4. In vitro biological evaluation
Two types of cell models were established for both cell types, utilising the four types of manufactured nanoparticles. The 2D cell model comprised bidimensional cell cultures in CELLSTAR® multiwell 96 plates (Greiner, UK). In this model, cells were seeded at a density of 8000 cells per well and treated with the nanoparticles (at concentration of 1 mg mL−1) prepared in an Eppendorf tube, following the manufacturing and functionalisation protocols described in Sections 3.2.1 and 3.2.2. Additionally, 3D Sarcospheres were created using Costar UltraLow Attachment 96-well plates (Greiner, UK). Both cell lines were cultured at a density of 100000 cells per well in DMEM culture medium supplemented with a specific factors cocktail, which included 0.25% w/v methylcellulose56 (Merck, UK). The cultures were maintained at 37 °C and in a 5% CO2 atmosphere. The cells were cultured for 21 days, with 50% of the culture media replaced every 48 hours, to observe the formation of sarcospheres.
:10 ratio, ensuring protection from light. The solution was then vortexed to ensure homogeneity. Subsequently, 500 and 200 μL of this solution was added for the 2D cell and 3D model respectively in each well to the previously washed samples using PBS. The fluorescence (Ex 544 nm/Em 590 nm) was measured after 1.5 hours of incubation using a Filter-based FLUOstar® Omega multimode plate reader.
Furthermore, to assess the impact of the manufactured nanoparticles on cell viability, a live/dead assay (live/dead Cell Imaging Kit, Life Technologies, UK) was conducted before and after incubating the cells with NPs (in both 2D and Sarcosphere models). This fluorescence-based assay utilises calcein AM and ethidium bromide to differentiate between live (green) and dead (red) cells.
For both cell types and models, cell samples were treated with the NPs for 24, 48, and 72 hours. At each time point, cells were rinsed twice with PBS and subsequently incubated with 100 μL of a staining solution, which was prepared by combining 1 μL of calcein and 4 μL of ethidium in 2 mL of DPBS. Following a 30-minute incubation at 37 °C, the samples were imaged using an EVOS M5000 fluorescence microscope from Thermo Fisher Scientific.
3.5. Statistical analysis
The results were presented as means ± standard deviations. Statistical analysis was carried out using GraphPad Prism Software (version 8.4.1). Initially, a one-way ANOVA with repeated measurements was employed. Subsequently, a Tukey's post hoc test was conducted to identify the main factors contributing to data variability. The level of statistical significance was set as follows: *p < 0.05, **p < 0.01, ***p < 0.0001 ****p < 0.00001.4. Conclusions
This study demonstrated the effective application of nanocarrier systems for delivering miRNA-34a, Dx, and Rs in a pre-clinical OS model. Chitosan and PLGA were identified as ideal materials due to their excellent mechanical properties, biodegradability, biocompatibility, and regulatory approval. Both miRDx and miRDxRs-NPs, showed slightly larger sizes compared to miR-NPs with distinct morphological changes due to drug inclusion. Both NP types efficiently encapsulated miRNA and Dx, though Rs encapsulation by miRDxRs-NPs was only 10%. The LbL-NPs, featuring a nanocoating of polyanionic pectin from cocoa biowaste and a polycationic chitosan layer, had larger sizes but were suitable in terms of size and charge. TEM images confirmed the presence of two distinct layers, and zeta potential results verified expected charge variations, and all four NPs exhibited suitable sustained release profiles, with gradual release over three days. Finally, the manufactured nanoparticles were tested on 2D in vitro cell models using U2OS and Saos-2 cells to evaluate their efficiency and then, incubated with sarcospheres mimicking the 3D tumour tissue. Initially, spheroids decreased in size due to cell clustering, then grew as tumour cells proliferated. Functional evaluation revealed potent cytotoxic activity of Dx and miRNA-loaded NPs against both U2OS and Saos-2 osteosarcoma cells, highlighting their synergistic effects. In spheroids, necrotic cores increased with incubation time, indicating higher cell death rates. Notably, LbL-NPs demonstrated superior cytotoxicity and metabolic activity interference in both 2D and 3D models, suggesting enhanced efficiency in delivering the complete therapeutic payload. In contrast, miRDx and miRDxRs-NPs showed similar but less cytotoxic behaviour compared to miR-NPs, as confirmed by PCR analysis. These findings warrant further investigation of LbL-NPs as a promising drug delivery platform for osteosarcoma therapy. Future studies could explore in vivo efficacy and optimize targeting strategies for improved therapeutic outcomes.Author contributions
Joel Girón-Hernández, Piergiorgio Gentile: conceived and designed the experiments; performed the experiments; analysed and interpreted the data; contributed reagents, materials, analysis tools or data; wrote and revised the paper. Eugenia Crisafulli and Annachiara Scalzone: performed the experiments; analysed and interpreted the data; contributed reagents, materials, analysis tools or data; wrote the paper. Chiara Tonda-Turo: performed the experiments; analysed and interpreted the data; revised the paper.Data availability
The datasets supporting this article are available under request to the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NERC Cross-disciplinary Research for Discovery Sciences (NE/X018229/1) and by Ministerio de Ciencia Tecnología e Innovación Colombia (project BPIN 2021000100065). The authors would like also to knowledge the Electron Microscopy Research Services (EMRS) at Newcastle university for the TEM analysis.References
- A. Misaghi, A. Goldin, M. Awad and A. A. Kulidjian, SICOT-J., 2018, 4, 1–8 CrossRef.
- I. G. Freedman, H. N. Dowd, M. M. Dhodapkar, S. J. Halperin and J. N. Grauer, JAAOS Global Res. Rev., 2023, 7, e22 CrossRef.
- J. Zhang, Y.-G. Yan, C. Wang, S.-J. Zhang, X.-H. Yu and W.-J. Wang, Clin. Chim. Acta, 2015, 444, 9–17 CrossRef CAS.
- P. Sharma, V. Jhawat, P. Mathur and R. Dutt, Med. Oncol., 2022, 39, 76 CrossRef.
- C. M. Dawidczyk, L. M. Russell and P. C. Searson, Front. Chem., 2014, 2, 69 Search PubMed.
- K. Wu, B. Yu, D. Li, Y. Tian, Y. Liu and J. Jiang, Front. Oncol., 2022, 12, 805978 CrossRef CAS PubMed.
- P. Huang, X. Wang, X. Liang, J. Yang, C. Zhang, D. Kong and W. Wang, Acta Biomater., 2019, 85, 1–26 CrossRef CAS PubMed.
- S. Doppalapudi, A. Jain, A. J. Domb and W. Khan, Expert Opin. Drug Delivery, 2016, 13, 891–909 CrossRef CAS.
- J. Suksiriworapong, V. Taresco, D. P. Ivanov, I. D. Styliari, K. Sakchaisri, V. B. Junyaprasert and M. C. Garnett, Colloids Surf., B, 2018, 167, 115–125 CrossRef CAS PubMed.
- S. Li, Y. Xiong and X. Zhang, Biomed. Pharmacother., 2017, 90, 872–879 CrossRef CAS PubMed.
- K. Sun, J. Wang, J. Zhang, M. Hua, C. Liu and T. Chen, Int. J. Biol. Macromol., 2011, 49, 173–180 CrossRef CAS.
- M. L. Tan, D. E. Dunstan, A. M. Friedhuber, P. F. Choong and C. R. Dass, J. Controlled Release, 2010, 144, 196–202 CrossRef CAS.
- M. Salerno, E. Cenni, C. Fotia, S. Avnet, D. Granchi, F. Castelli, D. Micieli, R. Pignatello, M. Capulli and N. Rucci, Curr. Cancer Drug Targets, 2010, 10, 649–659 CrossRef CAS.
- G. Zhang, Y. Li, J. Xu and Z. Xiong, Open Med., 2020, 15, 1003–1011 CrossRef CAS.
- G. Bottai, B. Pasculli, G. A. Calin and L. Santarpia, Expert Opin. Biol. Ther., 2014, 14, 1667–1683 CrossRef CAS.
- M. Nugent, Cancer Manage. Res., 2014, 15–25 CrossRef.
- L. He, X. He, L. P. Lim, E. De Stanchina, Z. Xuan, Y. Liang, W. Xue, L. Zender, J. Magnus and D. Ridzon, Nature, 2007, 447, 1130–1134 CrossRef CAS PubMed.
- N. Raver-Shapira, E. Marciano, E. Meiri, Y. Spector, N. Rosenfeld, N. Moskovits, Z. Bentwich and M. Oren, Mol. Cell, 2007, 26, 731–743 CrossRef CAS.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS.
- J. A. Kemp, M. S. Shim, C. Y. Heo and Y. J. Kwon, Adv. Drug Delivery Rev., 2016, 98, 3–18 CrossRef CAS PubMed.
- D. Yu, S. Zhang, A. Feng, D. Xu, Q. Zhu, Y. Mao, Y. Zhao, Y. Lv, C. Han and R. Liu, Medicine, 2019, 98, e15582 CrossRef CAS.
- N. Baek, O. W. Seo, M. Kim, J. Hulme and S. S. A. An, OncoTargets Ther., 2016, 7207–7218 CrossRef CAS PubMed.
- I. Buondonno, E. Gazzano, E. Tavanti, K. Chegaev, J. Kopecka, M. Fanelli, B. Rolando, R. Fruttero, A. Gasco and C. Hattinger, Cell. Mol. Life Sci., 2019, 76, 609–625 CrossRef CAS.
- J. Prados, C. Melguizo, R. Ortiz, C. Vélez, P. J. Alvarez, J. L. Arias, M. A. Ruiz, V. Gallardo and A. Aranega, Anti-Cancer Agents Med. Chem., 2012, 12, 1058–1070 CrossRef CAS.
- P. Krishnamurthy, M. Brown, S. Agrawal and R. F. Short, J. Gastrointest. Oncol., 2017, 8, E26 CrossRef.
- T. Taniguchi, Y. Iizumi, M. Watanabe, M. Masuda, M. Morita, Y. Aono, S. Toriyama, M. Oishi, W. Goi and T. Sakai, Cell Death Dis., 2016, 7, e2211 CrossRef CAS.
- H. Tian and Z. Yu, Int. J. Clin. Exp. Pathol., 2015, 8, 2755 Search PubMed.
- P. Gentile, I. Carmagnola, T. Nardo and V. Chiono, Nanotechnology, 2015, 26, 422001 CrossRef CAS.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed.
- G. Roebben, S. Ramirez-Garcia, V. A. Hackley, M. Roesslein, F. Klaessig, V. Kestens, I. Lynch, C. Garner, A. Rawle and A. Elder, J. Nanopart. Res., 2011, 13, 2675–2687 CrossRef.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- H. H. Liu, J. Lanphere, S. Walker and Y. Cohen, Nanotechnology, 2015, 26, 045708 CrossRef CAS PubMed.
- H. Kang, S. Rho, W. R. Stiles, S. Hu, Y. Baek, D. W. Hwang, S. Kashiwagi, M. S. Kim and H. S. Choi, Adv. Healthcare Mater., 2020, 9, 1901223 CrossRef CAS.
- M. Lundin, E. Blomberg and R. D. Tilton, Langmuir, 2010, 26, 3242–3251 CrossRef CAS.
- J. Ruths, F. Essler, G. Decher and H. Riegler, Langmuir, 2000, 16, 8871–8878 CrossRef CAS.
- D. L. Elbert, C. B. Herbert and J. A. Hubbell, Langmuir, 1999, 15, 5355–5362 CrossRef CAS.
- C. Picart, P. Lavalle, P. Hubert, F. Cuisinier, G. Decher, P. Schaaf and J.-C. Voegel, Langmuir, 2001, 17, 7414–7424 CrossRef CAS.
- A. Lee, N. Gosnell, D. Milinkovic, P. Taladriz-Blanco, B. Rothen-Rutishauser and A. Petri-Fink, ACS Appl. Bio Mater., 2023, 6, 83–92 CrossRef CAS.
- N. Arghiani and K. Shah, Cancer Biol. Med., 2022, 19, 289 CAS.
- D. Cosco, F. Cilurzo, J. Maiuolo, C. Federico, M. T. Di Martino, M. C. Cristiano, P. Tassone, M. Fresta and D. Paolino, Sci. Rep., 2015, 5, 17579 CrossRef CAS PubMed.
- C. Anandharamakrishnan, Techniques for nanoencapsulation of food ingredients, Springer, New York, 2014, vol. 8, pp. 65–67 Search PubMed.
- J. Zhu, J. Yang, L. Zhao, P. Zhao, J. Yang, J. Zhao and W. Miao, Int. J. Nanomed., 2021, 5167–5183 CrossRef PubMed.
- Š. Zupančič, Z. Lavrič and J. Kristl, Eur. J. Pharm. Biopharm., 2015, 93, 196–204 CrossRef.
- H. Jayan, M. M. Leena, S. S. Sundari, J. Moses and C. Anandharamakrishnan, J. Funct. Foods, 2019, 57, 417–424 CrossRef CAS.
- M. Ghezzi, S. Pescina, C. Padula, P. Santi, E. Del Favero, L. Cantù and S. Nicoli, J. Controlled Release, 2021, 332, 312–336 CrossRef CAS.
- N. Ayobian-Markazi, T. Fourootan and M. Kharazifar, Dent. Res. J., 2012, 9, 86 CrossRef CAS.
- C. Pautke, M. Schieker, T. Tischer, A. Kolk, P. Neth, W. Mutschler and S. Milz, Anticancer Res., 2004, 24, 3743–3748 CAS.
- C. Novello, L. Pazzaglia, A. Conti, I. Quattrini, S. Pollino, P. Perego, P. Picci and M. S. Benassi, PLoS One, 2014, 9, e114757 CrossRef PubMed.
- G. Bassi, S. Panseri, S. M. Dozio, M. Sandri, E. Campodoni, M. Dapporto, S. Sprio, A. Tampieri and M. Montesi, Sci. Rep., 2020, 10, 22294 CrossRef CAS PubMed.
- F. E. Freeman, R. Burdis, O. R. Mahon, D. J. Kelly and N. Artzi, Adv. Healthcare Mater., 2022, 11, 2101296 CrossRef CAS.
- C. F. Monteiro, C. A. Custódio and J. F. Mano, Acta Biomater., 2021, 134, 204–214 CrossRef CAS.
- A. De Luca, L. Raimondi, F. Salamanna, V. Carina, V. Costa, D. Bellavia, R. Alessandro, M. Fini and G. Giavaresi, J. Exp. Clin. Cancer Res., 2018, 37, 1–15 CrossRef PubMed.
- X. Deng, M. Cao, J. Zhang, K. Hu, Z. Yin, Z. Zhou, X. Xiao, Y. Yang, W. Sheng and Y. Wu, Biomaterials, 2014, 35, 4333–4344 CrossRef CAS PubMed.
- Y. Zhao, M.-J. Tu, Y.-F. Yu, W.-P. Wang, Q.-X. Chen, J.-X. Qiu, A.-X. Yu and A.-M. Yu, Biochem. Pharm., 2015, 98, 602–613 CrossRef CAS PubMed.
- J. Girón-Hernández, A. Tombe, M. C. Koyilot, K. T. Salas-Calderón, A. Charlton, C. Wills and P. Gentile, Eur. Polym. J., 2024, 210, 112967 CrossRef.
- A. Scalzone, X. N. Wang, K. Dalgarno, A. M. Ferreira and P. Gentile, Tissue Eng., Part A, 2022, 28, 84–93 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01541j |
| ‡ These authors contributed equally to this work. |
| § These authors supervised equally this work. |
| This journal is © The Royal Society of Chemistry 2024 |