
Highly crystalline polycyclic aromatic lactam-based regioregular wide-band gap polymer donors for organic solar cells†
Jong-Woon
Ha
 ab,
Byoungwook
Park
b,
Seo-Jin
Ko
b and
Do-Hoon
Hwang
*a
ab,
Byoungwook
Park
b,
Seo-Jin
Ko
b and
Do-Hoon
Hwang
*a
aDepartment of Chemistry, and Chemistry Institute for Functional Materials, Pusan National University, Busan 609-735, Korea. E-mail: dohoonhwang@pusan.ac.kr; Fax: +82-51-516-7421; Tel: +82-51-510-2232
bAdvanced Energy Materials Research Center, Korea Research Institute of Chemical Technology (KRICT), Daejeon 34114, Republic of Korea
First published on 27th November 2023
Abstract
The design of wide-band gap polymer donors with complementary absorption to non-fullerene acceptors (NFAs) is essential to increase the photovoltaic efficiency of organic solar cells (OSCs). However, some effective wide-band gap polymer donors comprising asymmetric units suffer from a lack of regioregularity, which negatively impacts the intermolecular packing among polymer backbones and decreases their degree of crystallinity and charge carrier mobility. In this study, asymmetrical electron-accepting building blocks of thienoquinolinone (TQO) or thienoisoquinolinone (TIQO) are modified to achieve dimeric structures for improving their symmetry. The different monomers are synthesized by connecting TQO and TIQO in a symmetrical dimeric structure and four kinds of wide-band gap polymer donors, namely PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, are synthesized by Stille cross-coupling polymerization with a benzodithiophene derivative. Grazing incidence wide-angle X-ray scattering (GIWAXS) results demonstrate that the synthesized polymer donors have a high degree of crystallinity. Among them, PBDT-PiQ exhibits both compact intermolecular π–π packing and a high degree of crystalline properties, which promotes effective charge transfer in the active layers. Consequently, PBDT-PiQ:Y6-based OSCs exhibit the maximum power conversion efficiency of 11.28%. The synthetic strategy proposed in this study strengthens the intermolecular interactions between polymer backbones and improves the degree of crystallinity, thereby enhancing the photovoltaic performance of OSCs.
Introduction
Recently, organic solar cells (OSCs) have attracted considerable attention as renewable energy sources owing to their potential for manufacturing large-area flexible devices using low-cost solution processes.1–5 Toward OSC development, various state-of-the-art organic semiconducting materials based on small molecules or polymers have been reported.6–12 In particular, the use of non-fullerene acceptors (NFAs) comprising malononitrile derivatives as a terminal group significantly increases the power conversion efficiency (PCE) (>18%) of OSCs in relation to that of conventional fullerene-based acceptors.7,13–17From the viewpoint of designing a polymer donor, the complementary absorption regions and appropriate energy levels between a polymer donor and NFAs should be carefully considered. Poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl]] (PTB7), which contains asymmetric thieno[3,4-b]thiophene (TT), is a well-known efficient polymer donor that is commonly used in fullerene- and NFA-based OSCs because its absorption region matches well with that of electron acceptors.18–24 In particular, when PTB7-Th, which is a derivative of PTB7, is blended with IEICO-4F, a high PCE of 12.8% is achieved.25 However, PTB7-Th exhibits a lack of regioregularity, which has an unfavorable effect on the intermolecular packing among polymer backbones and decreases its degree of crystallinity and charge carrier mobility. To address these issues, Lee et al. designed and synthesized a regioregular polymer donor comprising TT (regioregular PBDTTT-C-T) that has perfectly controlled TT orienation.26 Compared with an irregular polymer donor (random PBDTTT-C-T), the regioregular PBDTTT-C-T exhibited a significantly lower optical band gap (Eoptg) and higher degree of crystallinity, thereby improving the photovoltaic performance of the corresponding OSC.
Recently, there has been a lot of research on new wide band gap polymer donors, namely PM6 and D18, which are reported to achieve high-performance OSCs when blended with NFAs, which can absorb a longer wavelength.8,27–31 To achieve high performance, the development of wide band gap polymer donors with complementary absorption to NFAs and high crystallinity is essential.32–36 Polycyclic aromatic lactams are promising and weak-electron-accepting building blocks to make wide-band gaps.37–40 Recently, we reported two new semiconducting polymer donors that contained polycyclic aromatic lactam-based electron accepting building blocks, namely, thienoquinolinone (TQO) and thienoisoquinolinone (TIQO) (respective polymers denoted as PBDT-TQO and PBDT-TIQO) as polymer electron donors for OSCs.41 PBDT-TQO and PBDT-TIQO exhibited absorption complementary to that of non-fullerene acceptor IT-4F owing to their significantly wide band gaps (>2.0 eV). Moreover, the fabricated OSCs exhibited a high open circuit voltage (VOC) owing to the deep-lying highest occupied molecular orbital (HOMO) energy level of the polymer donors. However, TQO and TIQO have an asymmetric structure. Thus, it is predicted that if their symmetry can be improved, their crystallinity and mobility can be increased, allowing the enhancement of the efficiency of the corresponding OSCs.
In this study, the asymmetrical TQO or TIQO electron-accepting building blocks are modified to achieve dimeric structures in order to improve their symmetry. This strategy was proposed because dimeric electron-accepting building blocks are expected to a have lower-lying HOMO energy level owing to the enhanced electron-withdrawing strength.42 Four different accepter monomers were synthesized by connecting TQO and TIQO in a symmetrical dimeric structure, as shown in Scheme S1 (ESI†). 7,7′-Bithieno[3,2-c]quinoline-4,4′(5H,5′H)-dione (PQ) and 7,7′-bithieno[3,2-c]isoquinoline-5,5′(4H,4′H)-dione (PiQ) monomers were synthesized by connecting their phenyl to the phenyl of TQO and TIQO, respectively, and 2,2′-bithieno[3,2-c]quinoline-4,4′(5H,5′H)-dione (TQ) and 2,2′-bithieno[3,2-c]isoquinoline-5,5′(4H,4′H)-dione (TiQ) monomers were synthesized by connecting their thienyl to the thienyl of TQO and TIQO, respectively. Four kinds of polymer donors were synthesized by reacting the four acceptor monomers with a benzodithiophene derivative (BDT) via Stille cross-coupling polymerization. The synthesized polymer donors exhibited significantly wide-band gaps (>2.05 eV) and deep HOMO energy levels. Among them, PBDT-PiQ exhibited the highest PCE exhibiting 11.28% when blended with Y6, a well-known NFA.
Results and discussion
The symmetric electron-accepting building blocks constructed from TQO or TIQO were synthesized with high yields (Schemes S1 and S2, ESI†). For the PQ- and PiQ-comprising frameworks with a “phenyl-to-phenyl” segment in the center, TQO or TIQO reacted with bis(tributyltin) via typical Stille coupling to afford 1 and 2, respectively, which were further reacted with bromine to produce the final brominated monomer. TQ and TiQ were also easily synthesized through copper-catalyzed oxidative coupling between TQO and TIQO. The synthesized electron-accepting building blocks were polymerized with typical stannylated-BDT to obtain PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ. These polymers are soluble in common chlorinated organic solvents, such as chloroform, chlorobenzene, and o-dichlorobenzene. The molecular weights of the synthesized polymers were determined by high-temperature gel permeation chromatography using 1,2,4-trichlorobenzene as an eluent at 150 °C to avoid the aggregation of molecules. The number average molecular weights (Mn)/polydispersity indices were measured to be 26.0 kDa/3.05 for PBDT-PQ, 11.0 kDa/2.54 for PBDT-TQ, 15.0 kDa/2.51 for PBDT-PiQ, and 8.6 kDa/2.12 for PBDT-TiQ. Thermogravimetric analyses and differential scanning calorimetry were conducted to investigate the thermal properties of the synthesized polymers; the results are shown in Fig. S5 (ESI†). The synthesized polymers exhibit high thermal stability (5% decomposition temperature, Td > 350 °C) for manufacturing OSC devices.Fig. 1(a) shows the detailed molecular structures of the synthesized polymers and Y6. The optical properties of the synthesized polymers were investigated by UV-Vis absorption spectroscopy in the solution and thin film states. As shown in Fig. 1(b) and (c), the absorption edges of PBDT-PQ and PBDT-TQ are below 550 nm, whereas those of PBDT-PiQ and PBDT-TiQ exhibit relatively broad absorption profiles that extend to 600 nm. These results are attributed to the different intramolecular charge transfer strengths induced by the BDT and different accepting blocks.41 In the thin film state, the absorption profiles of the synthesized polymers shift to longer wavelengths than those in the solution state. The estimated Eoptg values of the synthesized polymers are 2.19, 2.27, 2.07, and 2.12 eV for PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, respectively, which are calculated from the corresponding absorption edges (λedge) in the thin film state. These optical features correspond well with those of Y6. Cyclic voltammetry was conducted to estimate the electrochemical properties of the synthesized polymers. The voltammograms are shown in Fig. S6 (ESI†). The HOMO energy levels estimated by the first onsets of the oxidation potentials were −5.53, −5.54, −5.40, and −5.39 eV for PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, respectively. The lowest unoccupied molecular (LUMO) energy level was estimated by the first onset of the reduction potential. The estimated LUMO energy levels were −3.44, −3.32, −3.34, and −3.37 eV for PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, respectively. The relatively low HOMO energy levels of the synthesized polymer donors were related to the strong electron-withdrawing ability of the symmetric accepting building blocks, which could effectively adjust the difference between the HOMO and LUMO energy levels of the electron donor and acceptor, respectively, resulting in an improved VOC. However, a too-close HOMO–HOMO energy level offset between the polymer donor and electron acceptor might reduce JSC because of the charge recombination behavior between the well-photogenerated charge carriers, which may dominate prior to their extraction to the corresponding electrodes. The energy diagram of the synthesized polymers and Y6 is shown in Fig. 1(d); this indicates suitable cascade stages for efficient exciton dissociation and charge carrier transport and collection, respectively. The optical and electrochemical properties of the synthesized polymers and Y6 are summarized in Table 1.
 | ||
| Fig. 1 (a) Molecular structures of the synthesized polymers and Y6. Absorption spectra of the synthesized polymers and Y6 in the (b) chloroform solution and (c) thin film, and (d) the corresponding energy level diagram. | ||
| M n [kDa] | PDIsa | λ max [nm] | λ edge [nm] |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b [eV] b [eV] |
E HOMO [eV] | E LUMO [eV] | |
|---|---|---|---|---|---|---|---|
|
a The Mn and PDIs of the polymers were determined by gel permeation chromatography using PS standards in trichlorobenzene at 150 °C.
b
E
g = 1240/λedge.
c
E
HOMOorLUMO = −e(4.80 + Eoxorred − Eferrocene).
|
|||||||
| PBDT-PQ | 26.0 | 3.05 | 520 | 566 | 2.19 | −5.53 | −3.44 |
| PBDT-TQ | 11.0 | 2.54 | 522 | 547 | 2.27 | −5.54 | −3.32 |
| PBDT-PiQ | 15.0 | 2.51 | 551 | 599 | 2.07 | −5.40 | −3.34 |
| PBDT-TiQ | 8.6 | 2.12 | 555 | 586 | 2.12 | −5.39 | −3.37 |
| Y6 | — | — | 832 | 916 | 1.35 | −5.67 | −3.93 |
Density functional theory (DFT) calculations at the B3LYP/6-31G(d) level were conducted to investigate the optimum geometric structure, electrical distribution, and frontier molecular orbital energy levels. The monomeric architecture with short side-alkyl chains was established to simplify the calculation. From the optimum geometric structures (Fig. 2(a) and (c)), it is obvious that the torsional angels (37.16° and 32.19°) between the phenyl groups in PQ and PiQ are considerably larger than those between the thienyl groups in TQ and TiQ (5.33° and 18.46°) because “phenyl-to-phenyl” segments have a larger steric hindrance than “thienyl-to-thienyl”. The results show that BDT-PQ and BDT-PiQ have more twisted conformations than BDT-TQ and BDT-TiQ. Fig. S7 (ESI†) shows the electrical charge distribution of the HOMO and LUMO. BDT-PQ and BDT-TQ had deeper-lying HOMO energy levels than BDT-PiQ and BDT-TiQ, which is consistent with the results of the electrochemical experiments. Furthermore, grazing incidence wide-angle X-ray scattering (GIWAXS) was conducted to analyze the relationship between the calculated DFT results and degree of crystallinity of neat polymer films. The GIWAXS images of the neat polymer films are shown in Fig. S8 (ESI†), and the corresponding line-cut out-of-plane (OOP) and in-plane (IP) profiles are shown in Fig. 2(b) and (d), respectively. The GIWAXS results of the previous polymers, namely, PBDT-TQO and PBDT-TIQO, are summarized in Table S1 (ESI†) for comparison. The new synthesized polymers displayed π–π stacking diffraction along the OOP direction and lamellar stacking diffraction along the IP direction, similar to the previous polymer donors, suggesting a predominant face-on orientation. From the peak locations in the corresponding line-cut profiles, obvious strong π–π stacking diffractions were located at 1.66, 1.70, 1.73, and 1.74 Å−1, which correspond to distances (d(010)) of 3.79, 3.70, 3.63, and 3.61 Å for PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, respectively. For PBDT-TQ and PBDT-TiQ, compact intermolecular interactions between the polymer backbones were observed in relation to those of polymer donors constructed from phenyl-to-phenyl segments, which is consistent with the DFT results. According to the Scherrer equation,43 the crystalline coherence length (LC) of the π–π stacking was measured to investigate the degree of crystallinity. The measured LC(010) values were 20.20, 23.56, 35.34, and 35.34 Å for PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ, respectively. The measured LC(010) values of the synthesized polymers were higher than those of the irregular polymers (PBDT-TQO and PBDT-TIQO), indicating that symmetric building blocks can significantly enhance the degree of crystallinity and inhibit the disturbance of intermolecular packing due to irregular repeating units.26 In particular, PBDT-PiQ and PBDT-TiQ exhibited compact intermolecular packing and a higher degree of crystallinity than PBDT-PQ and PBDT-TQ, which would facilitate charge carrier mobility. The relevant GIWAXS results, including the d-spacing and LC values of the synthesized and referenced polymers, are summarized in Table S1 (ESI†).
 | ||
| Fig. 2 (a) and (c) Density-functional-theory-simulated geometric structures of the monomeric structures. (b) and (d) GIWAXS scattering line-cut profiles of the neat polymer films. | ||
The OSCs with an inverted architecture consisting of indium–tin-oxide/ZnO/photoactive layer/MoOx/Ag were manufactured to evaluate their photovoltaic performance. The systematic procedure and corresponding photovoltaic parameters are summarized in Tables S2 and S3 (ESI†), respectively. Current density–voltage (J–V) and external quantum efficiency (EQE) curves of the optimum OSCs are shown in Fig. 3, and the detailed photovoltaic parameters are summarized in Table 2. PBDT-PiQ:Y6 exhibited the highest PCE of 11.28%, with VOC, JSC, and FF values of 0.81 V, 20.30 mA cm−2, and 63.51%, respectively, whereas PBDT-TiQ:Y6 had a lower PCE of 8.50% owing to its slightly reduced VOC and FF. In contrast to the OSCs based on PBDT-PiQ:Y6 and PBDT-TiQ:Y6, those based on PBDT-PQ:Y6 and PBDT-TQ:Y6 exhibited low PCEs (<2.5%). Despite the improvement in VOC owing to the deep-lying EHOMO of PBDT-PQ and PBDT-TQ, the decrease in JSC and FF was mainly attributable to the low PCE. This decrease was related to the relatively close HOMO–HOMO energy level offset between the synthesized polymer donor (PBDT-PQ or PBDT-TQ) and Y6, resulting in an accumulating charge recombination behavior between the photo-generated charge carriers.44 The EQE spectra of both polymer-based devices exhibited responses in the 300–900 nm range. The EQE values of the PBDT-PiQ:Y6- and PBDT-TiQ:Y6-based devices reached 60% at 400–900 nm, whereas PBDT-PQ:Y6- and PBDT-TQ:Y6-based devices exhibited low EQE responses (<20%). The calculated JSC values were well-matched within a 5% error range with the JSC values determined from the J–V curves.
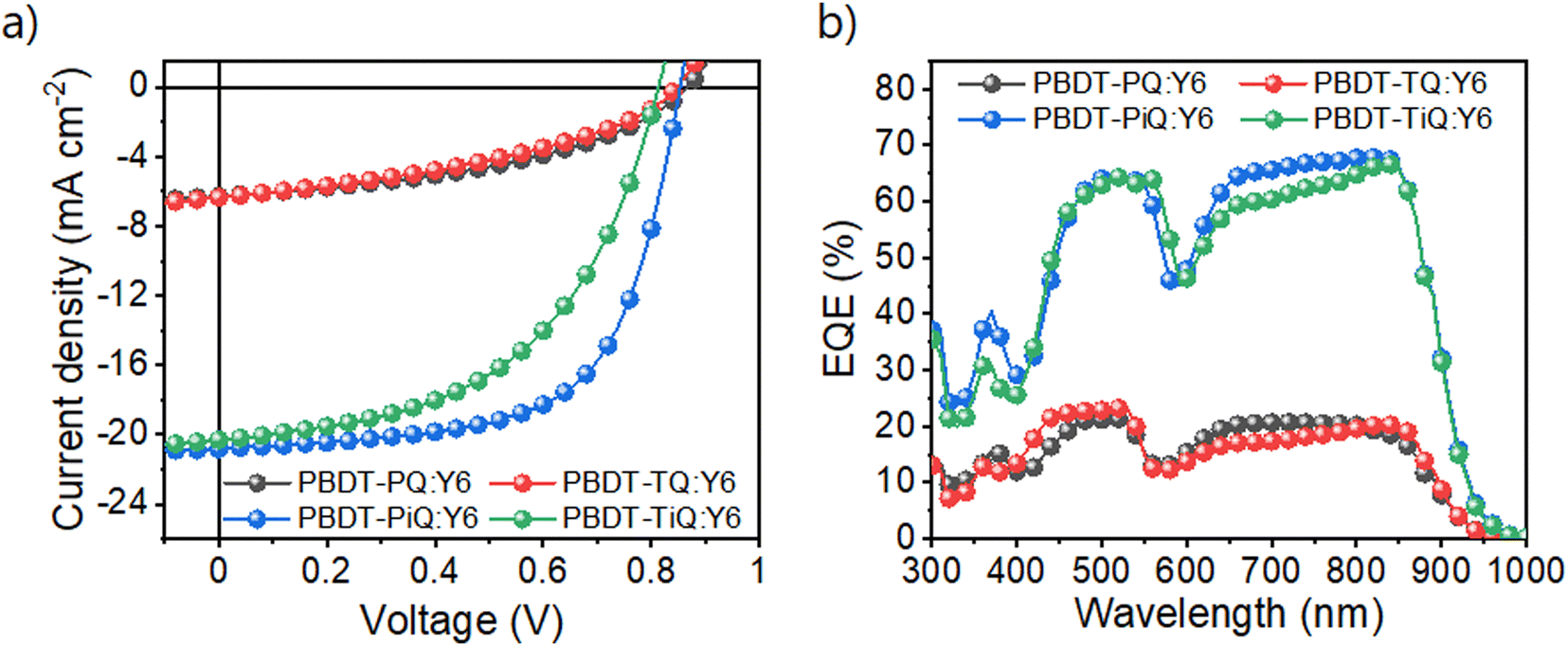 | ||
| Fig. 3 (a) Current density–potential curves and (b) external quantum efficiency spectra of the fabricated OSC devices under AM 1.5G-illumination (100 mW cm−2). | ||
| V OC [V] | J SC [mA cm−2] | Calc. JSCa [mA cm−2] | FF [%] | PCE [%] | G max [m−3 s−1] | μ [cm2 V−1 s−1] | P(E, T) | |
|---|---|---|---|---|---|---|---|---|
| a J SC values are calculated from the EQE spectra. b Mobilities are measured by the CELIV method. c Average parameters are calculated from 8 independent cells. | ||||||||
| PBDT-PQ:Y6 | 0.87 | 6.27 | 6.16 | 43.18 | 2.35 (2.34 ± 0.10)c | 3.97 × 1027 | 1.93 × 105 | 70.8 |
| PBDT-TQ:Y6 | 0.85 | 6.35 | 6.06 | 39.54 | 2.13 (2.13 ± 0.04)c | 7.23 × 1027 | 1.22 × 105 | 54.5 |
| PBDT-PiQ:Y6 | 0.85 | 20.82 | 20.15 | 63.51 | 11.28 (10.95 ± 0.26)c | 1.29 × 1027 | 4.04 × 105 | 88.4 |
| PBDT-TiQ:Y6 | 0.81 | 20.30 | 19.54 | 51.49 | 8.50 (8.26 ± 0.19)c | 1.61 × 1027 | 2.67 × 105 | 82.8 |
The effective charge carrier mobility was determined by the photocharge carrier extraction method with a linearly increasing voltage (CELIV), which was used to evaluate the charge carrier mobility of the photo-induced charge carriers of optimized devices for practical applications in terms of the device structure and film thickness of the photoactive layer.45–47 The effective charge carrier mobilities were estimated using the following equation:
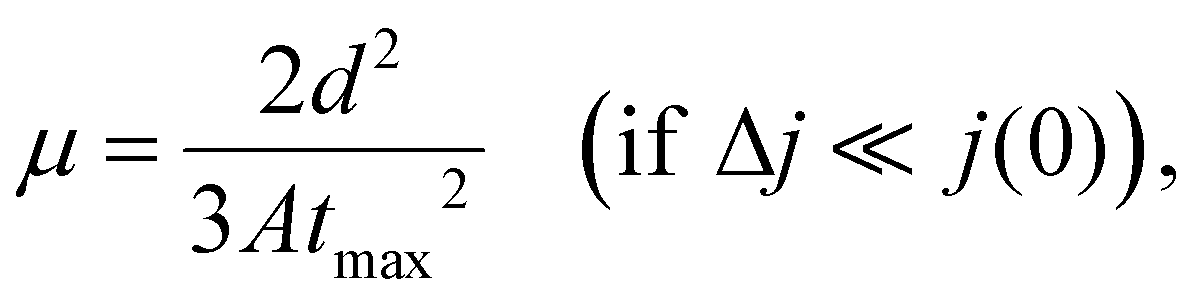
| Gmax = Jsat/qL, |
| P(E, T) = Jph/Jsat, |
 | ||
| Fig. 4 (a)–(d) Photo-carrier extraction method with linearly increasing voltage transients, (e) a saturated photocurrent density (JSC) versus light intensity plot, and (f) current density–voltage curves representing the saturation current of their respective OSCs. | ||
The crystalline properties and molecular orientation of the blend films comprising polymer donor:Y6 were investigated through GIWAXS analysis. Fig. 5(a) shows the GIWAXS images of the blend films and the corresponding line-cut profiles are shown in Fig. S9 (ESI†). In the OOP direction, the (010) peaks belonging to the synthesized polymers and Y6 overlapped in the blend films, which implies that the neat polymer had a face-on orientation and that the Y6 film was well maintained, and their corresponding diffraction locations/π–π stacking distances were located at 1.71 Å−1/3.67 Å for PBDT-PQ:Y6, 1.74 Å−1/3.61 Å for PBDT-TQ:Y6, 1.74 Å−1/3.61 Å for PBDT-PiQ:Y6, and 1.75 Å−1/3.59 Å for PBDT-TiQ:Y6. The estimated π–π stacking distances of the blend films were slightly closer than those of the neat polymer films. Notably, the PBDT-PiQ:Y6 and PBDT-TiQ:Y6 blend films exhibited significantly more compact π–π stacking distances, indicating a synergistic effect on the vertical charge carrier transport. The LC(010) values of the blend films were measured to investigate their crystallinity. The measured LC(010) values of PBDT-PiQ:Y6 and PBDT-TiQ:Y6 were 33.26 and 33.26 Å, respectively, whereas PBDT-PQ:Y6 and PBDT-TQ:Y6 were less crystalline in nature. The results demonstrated that the blend films of PBDT-PiQ:Y6 and PBDT-TiQ:Y6 showed closer π–π stacking and a higher degree of crystallinity, resulting in high JSC, FF, and effective charge carrier mobility values. The detailed GIWAXS results are summarized in Table S5 (ESI†). Atomic force microscopy was performed to investigate the correlation between the nanoscale morphology and photovoltaic performance (Fig. 5(b)). All films possessed well-mixed phases between the polymer donor and Y6, resulting in smooth surfaces with low root-mean-square (RMS) roughness values (<3.0 nm). Nevertheless, the PBDT-PiQ:Y6 and PBDT-TiQ:Y6 blend films presented higher RMS roughness values of 2.86 and 2.66 nm than the PBDT-PQ:Y6 (∼2.31 nm) and PBDT-TQ:Y6 (∼1.73 nm) blend films. In general, increasing the crystallinity of the blended molecules could increase the surface roughness of the blend films.56,57 Thus, the results of the RMS roughness of the blend films were consistent with the GIWAXS results.
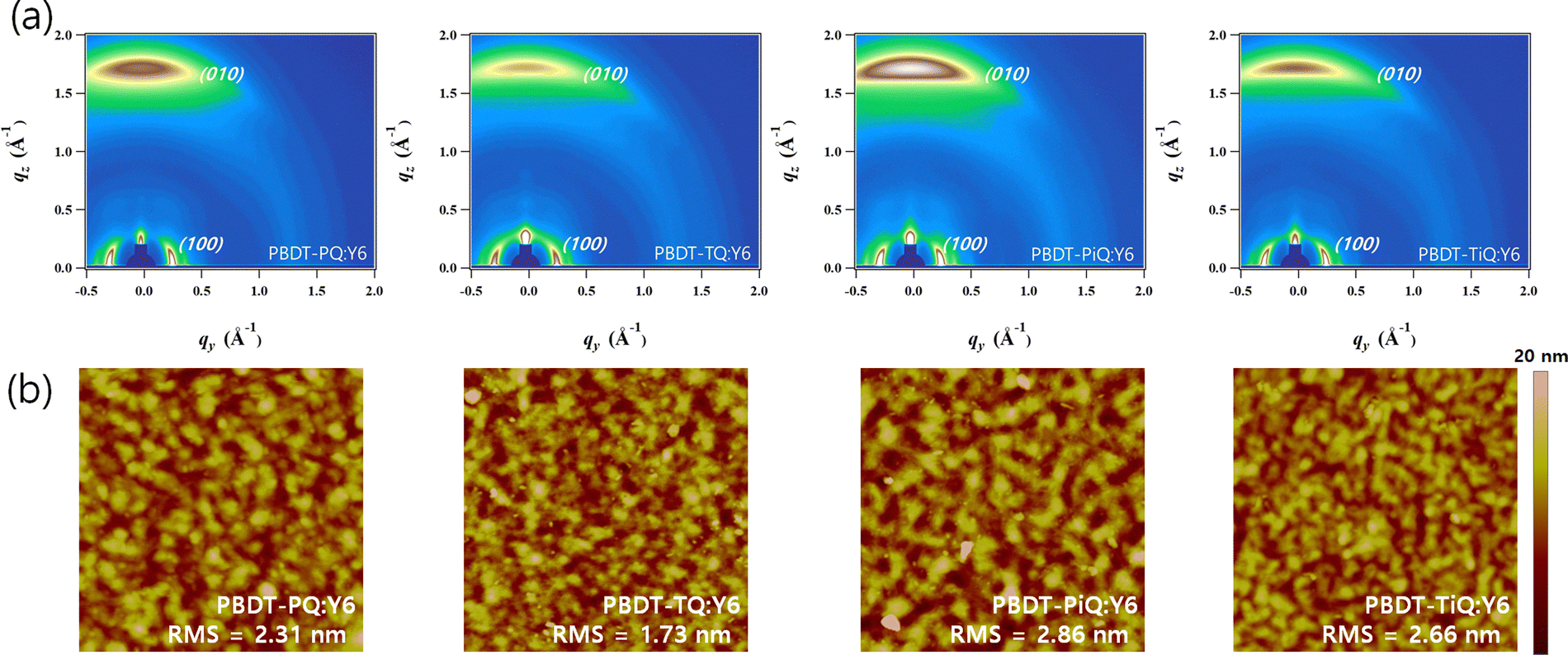 | ||
| Fig. 5 (a) 2D-GiWAXS and (b) atomic force microscopy images of the blend films. | ||
Conclusions
New symmetric electron-accepting building blocks based on TQO or TIQO were easily synthesized and used to construct polymer donors, namely, PBDT-PQ, PBDT-TQ, PBDT-PiQ, and PBDT-TiQ. PBDT-PiQ and PBDT-TiQ exhibited compact intermolecular packing between the polymer backbones and a high degree of crystallinity according to the 2D-GIWAXS results. PBDT-PiQ:Y6 showed the highest PCE (11.28%) owing to its high effective charge carrier mobility and effectively suppressed bimolecular charge recombination. On the other hand, PBDT-PQ:Y6 and PBDT-TQ:Y6 exhibited low PCEs (<2.50%) despite their high VOC values owing to the deep-lying HOMO energy levels of the polymers. This resulted in a significant narrowing of the HOMO–HOMO energy level offset between the synthesized polymer donors (PBDT-PQ and PBDT-TQ), which accelerated charge recombination between the generated charge carriers, leading to a decrease in JSC and FF. The synthetic strategy proposed in this study, which involves the conversion of an irregular polymer to a regioregular polymer, could improve both the intermolecular interactions between the polymer backbones and the degree of crystallinity, thereby improving the photovoltaic performance of OSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support granted by the Korea Research Institute of Chemical Technology (KRICT) of the Republic of Korea (No. KS2322-20) and the National Research Foundation (NRF) (NRF-2021R1C1C2007445 and RS-2023-00280495) of Republic of Korea.References
- G. P. Kini, S. J. Jeon and D. K. Moon, Adv. Funct. Mater., 2021, 31, 2007931 CrossRef CAS
.
- Y. Li, C. He, L. Zuo, F. Zhao, L. Zhan, X. Li, R. Xia, H.-L. Yip, C.-Z. Li, X. Liu and H. Chen, Adv. Energy Mater., 2021, 11, 2003408 CrossRef CAS
- Y. Zhang, D. Luo, C. Shan, Q. Liu, X. Gu, W. Li, W. C. H. Choy and A. K. K. Kyaw, Sol. RRL, 2022, 6, 2100785 CrossRef CAS
- J. Qin, L. Lan, S. Chen, F. Huang, H. Shi, W. Chen, H. Xia, K. Sun and C. Yang, Adv. Funct. Mater., 2020, 30, 2002529 CrossRef CAS
- K. Fukuda, K. Yu and T. Someya, Adv. Energy Mater., 2020, 10, 2000765 CrossRef CAS
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng, P. A. Johnson, M. Leclerc, Y. Cao, J. Ulanski, Y. Li and Y. Zou, Joule, 2019, 3, 1140–1151 CrossRef CAS
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170–1174 CrossRef CAS PubMed
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS
- H. Xia, Y. Zhang, W. Deng, K. Liu, X. Xia, C.-J. Su, U. S. Jeng, M. Zhang, J. Huang, J. Huang, C. Yan, W.-Y. Wong, X. Lu, W. Zhu and G. Li, Adv. Mater., 2022, 34, 2107659 CrossRef CAS
- M. Jeong, J. Oh, Y. Cho, B. Lee, S. Jeong, S. M. Lee, S.-H. Kang and C. Yang, Adv. Funct. Mater., 2021, 31, 2102371 CrossRef CAS
- A. Zeng, X. Ma, M. Pan, Y. Chen, R. Ma, H. Zhao, J. Zhang, H. K. Kim, A. Shang, S. Luo, I. C. Angunawela, Y. Chang, Z. Qi, H. Sun, J. Y. L. Lai, H. Ade, W. Ma, F. Zhang and H. Yan, Adv. Funct. Mater., 2021, 31, 2102413 CrossRef CAS
- R. Sun, W. Wang, H. Yu, Z. Chen, X. Xia, H. Shen, J. Guo, M. Shi, Y. Zheng, Y. Wu, W. Yang, T. Wang, Q. Wu, Y. Yang, X. Lu, J. Xia, C. J. Brabec, H. Yan, Y. Li and J. Min, Joule, 2021, 5, 1548–1565 CrossRef CAS
- T. Zhang, C. An, P. Bi, Q. Lv, J. Qin, L. Hong, Y. Cui, S. Zhang and J. Hou, Adv. Energy Mater., 2021, 11, 2101705 CrossRef CAS
- Y. Cai, Y. Li, R. Wang, H. Wu, Z. Chen, J. Zhang, Z. Ma, X. Hao, Y. Zhao, C. Zhang, F. Huang and Y. Sun, Adv. Mater., 2021, 33, 2101733 CrossRef CAS PubMed
- D. Wang, G. Zhou, Y. Li, K. Yan, L. Zhan, H. Zhu, X. Lu, H. Chen and C.-Z. Li, Adv. Funct. Mater., 2022, 32, 2107827 CrossRef CAS
- F. Liu, L. Zhou, W. Liu, Z. Zhou, Q. Yue, W. Zheng, R. Sun, W. Liu, S. Xu, H. Fan, L. Feng, Y. Yi, W. Zhang and X. Zhu, Adv. Mater., 2021, 33, 2100830 CrossRef CAS
- S. Bao, H. Yang, H. Fan, J. Zhang, Z. Wei, C. Cui and Y. Li, Adv. Mater., 2021, 33, 2105301 CrossRef CAS
- J. Lee, S.-J. Ko, M. Seifrid, H. Lee, B. R. Luginbuhl, A. Karki, M. Ford, K. Rosenthal, K. Cho, T.-Q. Nguyen and G. C. Bazan, Adv. Energy Mater., 2018, 8, 1801212 CrossRef
- Z. Xiao, X. Jia, D. Li, S. Wang, X. Geng, F. Liu, J. Chen, S. Yang, T. P. Russell and L. Ding, Sci. Bull., 2017, 62, 1494–1496 CrossRef CAS
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766–4771 CrossRef CAS PubMed
- C. Liu, C. Yi, K. Wang, Y. Yang, R. S. Bhatta, M. Tsige, S. Xiao and X. Gong, ACS Appl. Mater. Interfaces, 2015, 7, 4928–4935 CrossRef CAS PubMed
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef CAS
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS
- X. Song, N. Gasparini, L. Ye, H. Yao, J. Hou, H. Ade and D. Baran, ACS Energy Lett., 2018, 3, 669–676 CrossRef CAS
- H. Kim, H. Lee, D. Seo, Y. Jeong, K. Cho, J. Lee and Y. Lee, Chem. Mater., 2015, 27, 3102–3107 CrossRef CAS
- J.-W. Ha, C. E. Song, H. S. Kim, D. H. Ryu, W. S. Shin and D.-H. Hwang, ACS Appl. Mater. Interfaces, 2020, 12, 51699–51708 CrossRef CAS
- J. S. Park, C. Sun, Y. Han, G.-U. Kim, T. N.-L. Phan, Y.-H. Kim and B. J. Kim, Adv. Energy
Sustainability Res., 2022, 3, 2200070 CrossRef CAS
- C. Yang, Q. An, H.-R. Bai, H.-F. Zhi, H. S. Ryu, A. Mahmood, X. Zhao, S. Zhang, H. Y. Woo and J.-L. Wang, Angew. Chem., Int. Ed., 2021, 60, 19241–19252 CrossRef CAS
- Z. Abbas, S. U. Ryu, M. Haris, C. E. Song, H. K. Lee, S. K. Lee, W. S. Shin, T. Park and J.-C. Lee, Nano Energy, 2022, 101, 107574 CrossRef CAS
- J. Qin, L. Zhang, Z. Xiao, S. Chen, K. Sun, Z. Zang, C. Yi, Y. Yuan, Z. Jin, F. Hao, Y. Cheng, Q. Bao and L. Ding, Sci. Bull., 2020, 65, 1979–1982 CrossRef CAS PubMed
- S. Chen, Y. Liu, L. Zhang, P. C. Y. Chow, Z. Wang, G. Zhang, W. Ma and H. Yan, J. Am. Chem. Soc., 2017, 139, 6298–6301 CrossRef CAS
- Y. Tang, H. Sun, Z. Wu, Y. Zhang, G. Zhang, M. Su, X. Zhou, X. Wu, W. Sun, X. Zhang, B. Liu, W. Chen, Q. Liao, H. Y. Woo and X. Guo, Adv. Sci., 2019, 6, 1901773 CrossRef CAS PubMed
- B. Pang, C. Liao, X. Xu, L. Yu, R. Li and Q. Peng, Adv. Mater., 2023, 35, 2300631 CrossRef CAS PubMed
- K. He, P. Kumar, Y. Yuan, Z. Zhang, X. Li, H. Liu, J. Wang and Y. Li, ACS Appl. Mater. Interfaces, 2021, 13, 26441–26450 CrossRef CAS
- J. Kim, M. Kyeong, J.-W. Ha, H. Ahn, J. Jung, S. Seo, T. N.-L. Phan, C. Lee, S. C. Yoon, B. J. Kim and S.-J. Ko, J. Mater. Chem. A, 2021, 9, 27551–27559 RSC
- W. S. Yoon, D. W. Kim, J.-M. Park, I. Cho, O. K. Kwon, D. R. Whang, J. H. Kim, J.-H. Park and S. Y. Park, Macromolecules, 2016, 49, 8489–8497 CrossRef CAS
- A. M. Schneider, L. Lu, E. F. Manley, T. Zheng, V. Sharapov, T. Xu, T. J. Marks, L. X. Chen and L. Yu, Chem. Sci., 2015, 6, 4860–4866 RSC
- Y. Luo, X. Wang, C. Xiao, Z. Li, S. Lei, Y. Yu, B. Xiao, Z. Liu and R. Yang, ACS Appl. Energy Mater., 2021, 4, 13110–13119 CrossRef CAS
- D. Li, Z. Xiao, S. Wang, X. Geng, S. Yang, J. Fang, H. Yang and L. Ding, Adv. Energy Mater., 2018, 8, 1800397 CrossRef
- J.-W. Ha, H. S. Kim, C. E. Song, H. J. Park and D.-H. Hwang, J. Mater. Chem. C, 2020, 8, 12265–12271 RSC
- J.-W. Ha, H. J. Park, I.-N. Kang and D.-H. Hwang, Dyes Pigm., 2022, 200, 110176 CrossRef CAS
- J. Rivnay, S. C. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS
- Z. Yao, X. Liao, K. Gao, F. Lin, X. Xu, X. Shi, L. Zuo, F. Liu, Y. Chen and A. K. Y. Jen, J. Am. Chem. Soc., 2018, 140, 2054–2057 CrossRef CAS
- G. Juška, K. Arlauskas, M. Viliūnas and J. Kočka, Phys. Rev. Lett., 2000, 84, 4946–4949 CrossRef
- B. Park, J. Jung, D.-H. Lim, H. Lee, S. Park, M. Kyeong, S.-J. Ko, S. H. Eom, S.-H. Lee, C. Lee and S. C. Yoon, Adv. Funct. Mater., 2022, 32, 2108026 CrossRef CAS
- A. Pivrikas, N. S. Sariciftci, G. Juška and R. Österbacka, Prog. Photovolt., 2007, 15, 677–696 CrossRef CAS
- Y. Liu, M. Li, J. Yang, W. Xue, S. Feng, J. Song, Z. Tang, W. Ma and Z. Bo, Adv. Energy Mater., 2019, 9, 1901280 CrossRef
- V. D. Mihailetchi, H. X. Xie, B. deBoer, L. J. A. Koster and P. W. M. Blom, Adv. Funct. Mater., 2006, 16, 699–708 CrossRef CAS
- V. D. Mihailetchi, L. J. A. Koster, J. C. Hummelen and P. W. M. Blom, Phys. Rev. Lett., 2004, 93, 216601 CrossRef CAS
- S. R. Cowan, R. A. Street, S. Cho and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 035205 CrossRef
- Y. Duan, X. Xu, H. Yan, W. Wu, Z. Li and Q. Peng, Adv. Mater., 2017, 29, 1605115 CrossRef
- J. Zhao, Y. Li, H. Lin, Y. Liu, K. Jiang, C. Mu, T. Ma, J. Y. Lin Lai, H. Hu, D. Yu and H. Yan, Energy Environ. Sci., 2015, 8, 520–525 RSC
- A. J. Heeger, Adv. Mater., 2014, 26, 10–28 CrossRef CAS
- C. M. Proctor, C. Kim, D. Neher and T.-Q. Nguyen, Adv. Funct. Mater., 2013, 23, 3584–3594 CrossRef CAS
- L. Hong, H. Yao, Z. Wu, Y. Cui, T. Zhang, Y. Xu, R. Yu, Q. Liao, B. Gao, K. Xian, H. Y. Woo, Z. Ge and J. Hou, Adv. Mater., 2019, 31, 1903441 CrossRef
- L. Nian, Y. Kan, K. Gao, M. Zhang, N. Li, G. Zhou, S. B. Jo, X. Shi, F. Lin, Q. Rong, F. Liu, G. Zhou and A. K. Y. Jen, Joule, 2020, 4, 2223–2236 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02994h |
| This journal is © The Royal Society of Chemistry 2024 |