
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thieno[3,2-b]phosphole-based AIEgens: facile preparation and dual modulation of solid-state luminescence†
Nils
König
 a,
Justin
Mahnke
a,
Yokari
Godínez-Loyola
b,
Hendrik
Weiske
c,
Julian
Appel
a,
Peter
Lönnecke
a,
Cristian A.
Strassert
*b and
Evamarie
Hey-Hawkins
*a
a,
Justin
Mahnke
a,
Yokari
Godínez-Loyola
b,
Hendrik
Weiske
c,
Julian
Appel
a,
Peter
Lönnecke
a,
Cristian A.
Strassert
*b and
Evamarie
Hey-Hawkins
*a
aFakultät für Chemie und Mineralogie, Universität Leipzig, Institut für Anorganische Chemie, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
bInstitut für Anorganische und Analytische Chemie, CiMiC, SoN and CeNTech, Universität Münster, Heisenbergstraße 11, 48149 Münster, Germany. E-mail: ca.s@uni-muenster.de
cWilhelm-Ostwald-Institut für Physikalische- und Theoretische Chemie, Fakultät für Chemie und Mineralogie, Universität Leipzig, Linnéstraße 2, 04103 Leipzig, Germany
First published on 7th May 2024
Abstract
Thieno[3,2-b]phosphole-based AIEgens (Aggregation-Induced Emission Luminogens) have gained significant attention due to their unique solid-state emission properties. Here, we present a facile and efficient method for the preparation of thieno[3,2-b]phosphole-based AIEgens with dual tunability of solid-state emission. The synthesis involves the modification of the thiophene core with different substituents and changing the environment of the phosphorus atom, leading to a library of AIEgens with diverse photophysical properties. By controlling the molecular packing, intermolecular interactions and electronic structures, we successfully tuned the emission color and efficiency of the molecules in the solid state. The results demonstrate the remarkable dual tunability of the thieno[3,2-b]phosphole-based scaffold and the importance of a three-dimensional fluorophore design, making them promising candidates for advanced luminescent materials and devices.
Introduction
Luminescent materials have revolutionized numerous fields, from optoelectronics1 to bioimaging,2,3 by providing efficient and controllable emission of light. Among the diverse mechanisms that govern luminescence, aggregation-induced emission (AIE) has emerged as a captivating phenomenon that offers unique opportunities for manipulating solid-state emission.4–6 Traditionally, materials that emit light in solution tend to exhibit diminished or quenched emission upon aggregation in the solid state, a phenomenon known as aggregation-caused quenching (ACQ).7 However, the discovery of AIE materials reshaped this understanding, revealing a remarkable opposite effect: enhanced light emission upon aggregation.8 This paradoxical behavior can be observed in rotation-rich systems and arises from the suppression of non-radiative processes, such as molecular motion by the formation of intermolecular interactions, unlocking a plethora of intriguing optical properties and opportunities.9,10 The exploration of AIE in combination with tuneable solid-state emission has led to the discovery and design of a diverse range of ground-breaking materials.11–14 Through careful engineering of molecular structures and intermolecular interactions, researchers have achieved unprecedented control over emission wavelengths, intensity, and polarization in solid-state environments.15–21 Numerous stator frameworks have been effectively employed to achieve solid-state fluorescence tunability in aggregation-induced emission luminogens (AIEgens).23–31 In a study conducted by Zhang et al., these effects were demonstrated on the tetraphenylethene (TPE) scaffold through the strategic incorporation of donor and acceptor groups in the para position of the phenyl moieties, as well as the construction of push–pull systems (Fig. 1).22 This rational design strategy facilitated the synthesis of molecules exhibiting emission wavelengths spanning from 450 to 645 nm, accompanied by excellent quantum yields ranging from 48% to 61%.22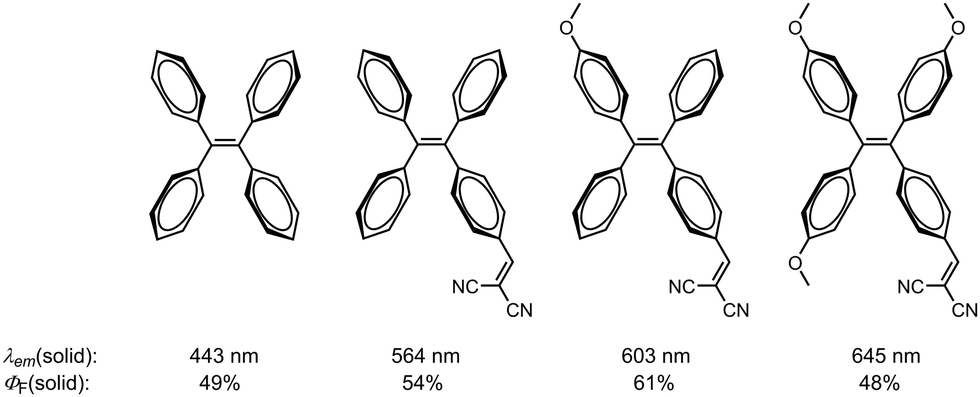 | ||
| Fig. 1 Tunable luminescence of TPE-based AIEgens in the solid state.22 | ||
Phospholes are a class of phospha-cyclopentadienes32 that exhibit unique electronic and structural properties, making them highly suitable for modulating the emission characteristics of fluorophores.33–45 Phosphole-based fluorophores have demonstrated efficacy in diverse applications, including organic light-emitting diodes (OLEDs)46–48 and biomolecular imaging,49–51 thus underscoring their utility and versatility. Benzo- and thieno-annellated phosphole oxides emerge as highly advantageous candidates for achieving color tunability in fluorophores due to their favorable electron-acceptor properties (Fig. 2).52–62 In a recent study, we have described the successful synthesis of propeller-like thieno[3,2-b]phosphole oxides that demonstrate strong fluorescence in the solid state, coupled with significant dependence on substituents for their optical properties.63
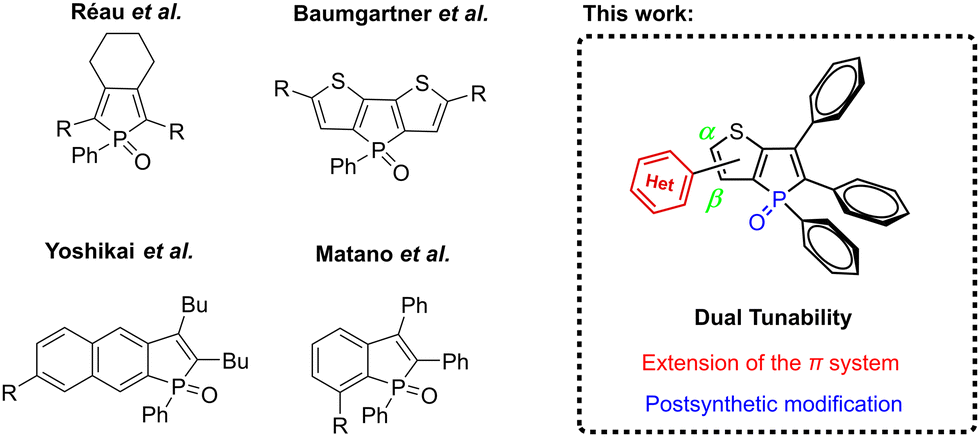 | ||
| Fig. 2 Known phosphole-based fluorophores with tuneable luminescence properties and the thieno[3,2-b]phosphole scaffold reported herein. | ||
Building upon these findings, our current investigation capitalizes on this molecular framework to achieve emission tunability spanning into the red region of the visible light spectrum. This is accomplished through the strategic introduction of distinct substituents at the α- and β-positions of the thiophene moiety. Moreover, we employed this system to explore the efficacy of our recently published post-synthetic derivatization approach for phosphole oxides, involving their reaction with tosyl isocyanate (TSI).64 This transformation led to a noteworthy enhancement in fluorescence efficiency and a significant bathochromic shift in emission wavelength. To investigate this further, selected examples were subjected to the conversion into the corresponding tosylimino phospholes, and the resulting impact on the solid-state structure and optical properties was thoroughly examined.
Results and discussion
Experimental section
In our previous study, we reported the successful synthesis of phosphole-based AIE emitters containing a thieno[3,2-b]phosphole moiety, showcasing their exceptional optical properties in the solid state.63 These emitters were synthesized through a straightforward oxidative annulation reaction of secondary thienyl phosphine oxides, employing a convenient one-pot synthesis method that yielded moderate product yields. Herein, we demonstrate the use of previously reported synthon 263 as a building block to construct novel AIE-based molecules. Starting from 2,5-dibromothiophene, the mono-Grignard compound can be selectively generated by temperature-controlled reaction with Mg in the presence of LiCl (Scheme 1, I). Reaction with PhPCl2 and subsequent hydrolysis gives the previously reported63 secondary phosphine oxide 1.65 Oxidative annulation with diphenylacetylene and Mn(OAc)3·2H2O leads to the formation of 2.63,66–68 To demonstrate the potential of compound 2, we have previously conducted experiments involving NiII-catalyzed cross-coupling69 phosphination.63 This methodology enabled the synthesis of highly efficient fluorophores, exhibiting solid-state quantum yields of up to 100%. Using cross-coupling reactions, different aromatic moieties can be easily linked to the thieno[3,2-b]phosphole scaffold (Scheme 1, II). Introducing substituents in the α-position of the thiophene segment results in an extended π system, leading to enhanced conjugation and a substantial red shift in fluorescence. To achieve this, phenyl (3a), thienyl (3b), biphenyl (3c), p-anisolyl (3d), triphenylamine (3e), and bis(methoxyphenyl) amine (3f) were introduced via a Suzuki–Miyaura coupling, yielding the corresponding fluorescent solids in yields ranging from 79% to 94% (Scheme 1, II). Compound 3a was previously prepared via an MnIII-mediated oxidative annulation reaction between diphenylacetylene and phenyl(5-bromothiophen-2-yl)phosphine oxide,63 albeit in a lower yield (49% vs. 85% via Suzuki coupling reported here) and serves as a reference within the present study for the purpose of comparative analysis with alternative α-substituted derivatives. Furthermore, an ethynyltriphenylamine unit was incorporated into the π system through a Sonogashira coupling, affording the product 3g as a yellow solid with a yield of 87%. Moreover, in order to examine the impact and adjustability of the β-position, several β-substituted derivatives (5, 6a–c) were synthesized. Following the synthesis of compound 1,63 the corresponding secondary phosphine oxide 4 was prepared through a Grignard reaction using 2,4-dibromothiophene as the starting material (Scheme 1, III). Subsequent oxidative cyclization, followed by a Suzuki–Miyaura coupling, yielded the desired β-substituted derivatives 6a–c in good yields (72–94%). To explore the dual modulation of fluorescence, a series of phosphole oxides was reacted with TSI, leading to the formation of their corresponding tosylimines via elimination of CO2.64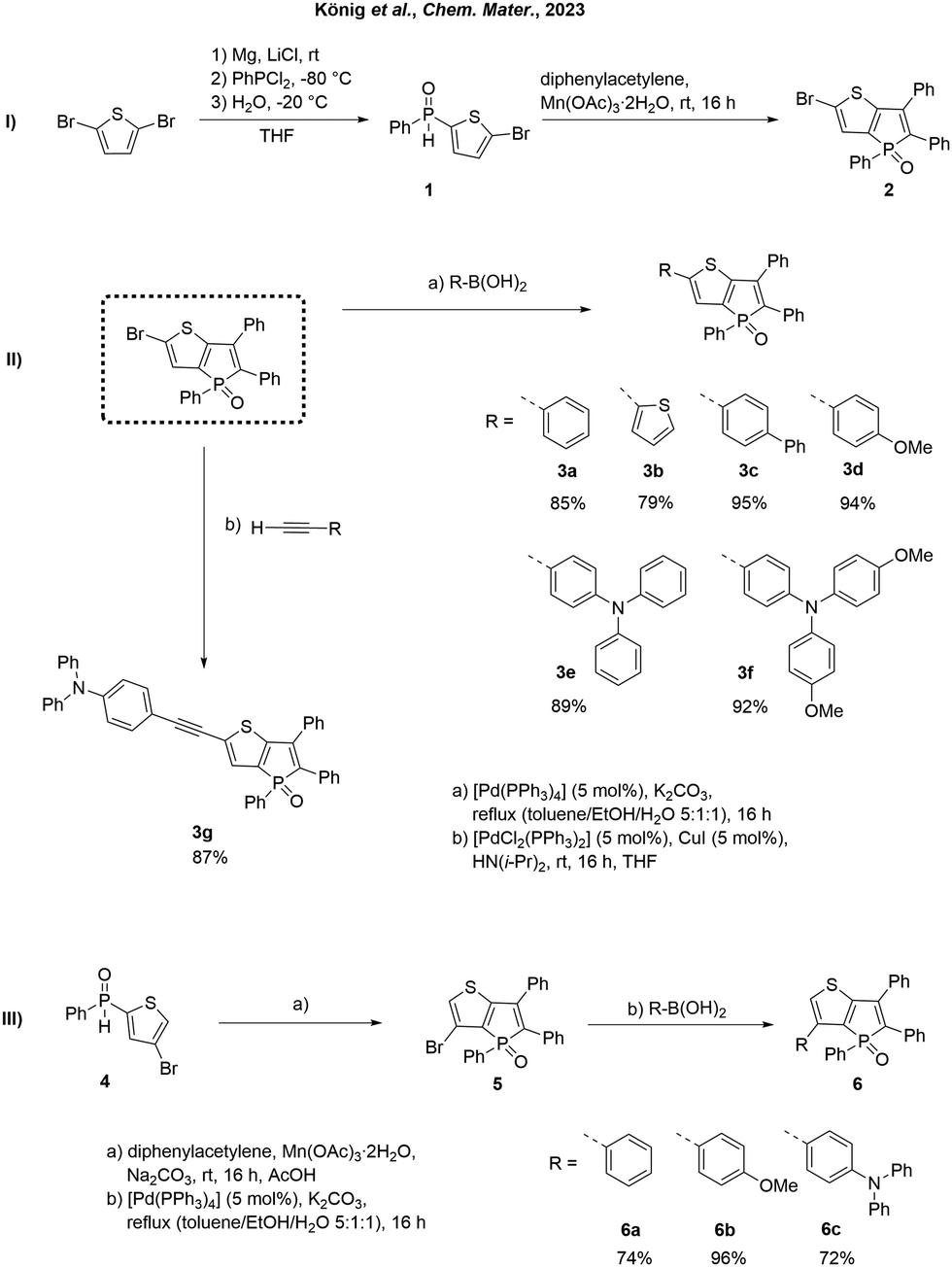 | ||
| Scheme 1 Synthesis of synthon 2 (I), extension of the π system in α-position via Pd-catalyzed cross-coupling reactions (II) and synthesis of β-substituted phospholes (III). | ||
In order to compare the results, the model compound triphenylthieno[3,2-b]phosphole oxide (TPTPO)63 as well as the phenyl-substituted derivatives pentaphenylthieno[3,2-b]- phosphole oxide (PPTPO),633a63 and 6a, were selected for the modification (Scheme 2). At 85 °C for 16 hours, all phosphole oxides underwent facile phosphole imination, resulting in the substitution with the tosyl group. The conversion process was quantitative, devoid of any observed side products. Consequently, a simple washing step with Et2O after the successful reaction, followed by recrystallization, proved to be sufficient. This procedure generated tosylimino phospholes 7a–d in very good yields ranging from 74% to 92%.
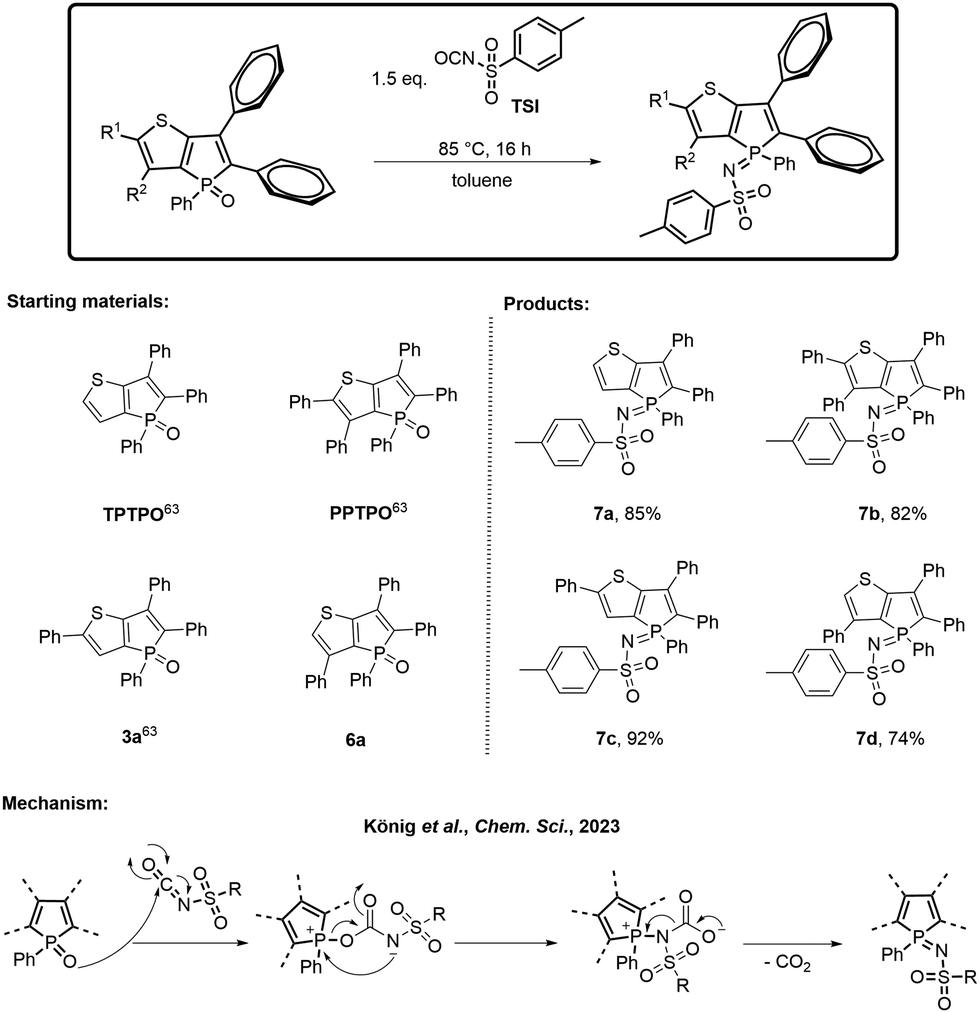 | ||
| Scheme 2 Transformation into the tosylimino phospholes 7a–d, their isolated yields and the proposed mechanism.64 | ||
Thieno[3,2-b]phosphole oxides
The thieno[3,2-b]phosphole unit is a powerful scaffold and exhibits remarkable photophysical properties, encouraging comprehensive investigations about the fluorescent characteristics of phosphole oxides 3a–g, 5 and 6a–c. The goal was to understand the influence of the substituents in the α- and β-position, and their capability to produce a strong bathochromic shift of luminescence into the deep red region. To investigate this, in-depth photoluminescence studies were conducted, comprising photoluminescence spectra, excited state lifetimes (τ), and photoluminescence quantum yields (PLQY, ΦF) in various environments, namely liquid solutions at room temperature (in CH2Cl2), in frozen glassy matrices at 77 K (in CH2Cl2/MeOH 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and solid state (Table 1). As a result of the complexity arising from the coexistence of different conformers under the studied conditions, the time-resolved photoluminescence measurements displayed multiexponential decays. To address this, amplitude-weighted average lifetimes (τav_amp) were employed for discussion and to estimate in a robust manner average radiative rate constants (kr = 1/τav_amp = ΦF/τav_amp) and non-radiative rates (knr = 1 − ΦF/τav_amp), as suggested by Engelborghs et al.64 These parameters provide valuable insights into the excited state dynamics of the fluorescence processes and their efficiency in the studied materials, allowing a deeper understanding of their photophysical properties.
:1) glassy matrices at 77 K
:1) and solid state (Table 1). As a result of the complexity arising from the coexistence of different conformers under the studied conditions, the time-resolved photoluminescence measurements displayed multiexponential decays. To address this, amplitude-weighted average lifetimes (τav_amp) were employed for discussion and to estimate in a robust manner average radiative rate constants (kr = 1/τav_amp = ΦF/τav_amp) and non-radiative rates (knr = 1 − ΦF/τav_amp), as suggested by Engelborghs et al.64 These parameters provide valuable insights into the excited state dynamics of the fluorescence processes and their efficiency in the studied materials, allowing a deeper understanding of their photophysical properties.
:1) glassy matrices at 77 K
| Compound | λ abs [nm] | λ em [nm] | τ av_amp(sol, rt) [ns] | Φ F(liq, rt) (±2) [%]ad | Glassy matrix at 77 K | |
|---|---|---|---|---|---|---|
| λ em [nm] | τ av_amp(77 K) [ns] | |||||
|
a Measured in liquid CH2Cl2 solutions at rt.
b Amplitude-weighted average lifetimes (τav_amp).70
c Frozen glassy matrices of CH2Cl2/MeOH (1:1) at 77 K.
d Absolute ΦF values were obtained using a calibrated integrating sphere.
e The photoluminescence properties of 3a have been previously reported in liquid MeCN solution at rt and in a frozen glassy matrix (butyronitrile) at 77 K.63
|
||||||
| 3a | 248, 308, 408 | 567 | 0.23 ± 0.01 | 2 | 552 | 8.44 ± 0.01 |
| 3b | 256, 420 | 584 | 0.42 ± 0.01 | 3 | 568 | 8.46 ± 0.01 |
| 3c | 265, 311, 416 | 572 | 0.45 ± 0.01 | 4 | 557 | 6.82 ± 0.02 |
| 3d | 256, 419 | 593 | 0.25 ± 0.01 | 2 | 579 | 7.55 ± 0.01 |
| 3e | 273, 323, 444 | 649 | 2.36 ± 0.01 | 25 | 425, 615 | 1.89 ± 0.03, 6.10 ± 0.02 |
| 3f | 288, 343, 458 | 703 | 0.89 ± 0.02 | 7 | 412, 645 | 0.87 ± 0.02, 5.89 ± 0.02 |
| 3g | 297, 348, 432 | 634 | 4.62 ± 0.01 | 60 | 590 | 4.81 ± 0.01 |
| 5 | 248, 379 | 522 | 0.51 ± 0.01 | 2 | 515 | 11.33 ± 0.02 |
| 6a | 250, 386 | 525 | 0.50 ± 0.02 | 2 | 510 | 10.67 ± 0.02 |
| 6b | 258, 392 | 530 | 0.21 ± 0.01 | 2 | 515 | 10.35 ± 0.02 |
| 6c | 242, 337, 400 | 640 | 0.29 ± 0.01 | 3 | 570 | 3.22 ± 0.06 |
In liquid CH2Cl2 solutions, the UV/Vis spectra of the phosphole oxides exhibit 2–3 maxima. Specifically, within the wavelength range of 242–292 nm, the transitions into n–π*-configured excited states of the thiophene units are observed. Moreover, for the push–pull systems 3e–g and 6c, bands are observed in the 323–343 nm range, which can be attributed to transitions into the n–π* states of the phenylamine units. The transition maxima corresponding to π–π* excitations of the α-substituted derivatives 3a–g are found in the 408–458 nm range.
Notably, the position of these absorption and emission maxima strongly relies on the nature of the substituent and its electronic structure. The β-substituted derivatives are clearly blue-shifted and show bands in the range of 379–400 nm. The synthesized thieno[3,2-b]phosphole oxides exhibit varying fluorescence intensities in liquid CH2Cl2 solutions, and are assigned to fluorescence processes. These compounds demonstrate a wide spectral coverage, spanning from 522 to 703 nm (Fig. 3). Notably, derivatives 3a–d display yellow-orange fluorescence with emission peaks falling within the range of 567 to 593 nm. These emission wavelengths are significantly red-shifted compared to the unsubstituted model compound TPTPO, for which the emission peak is located at 502 nm.63 The observed red shift in the emission of derivatives 3a–d is attributed to the extension of the π-conjugated system. The ΦF values for the synthesized compounds range from 2–4%, while the τ values fall within the range of 0.2–0.4 ns. The presence of a flexible, propeller-like structure facilitates rapid relaxation of the excited fluorophores to their ground state through vibrational relaxation. This is evident in the large values of knr (see Section S5.1, ESI†). Such characteristics are consistent with those commonly observed in AIE-based materials71 and are in agreement with our previously published work.63 To achieve red-emitting fluorophores, we previously designed and reported a push–pull system with strong amine donor groups (D)72–74 and utilized the phosphole oxide scaffold as the acceptor moiety (A). The derivatives 3e and 3g, which are substituted with diphenylamine, exhibit intense red luminescence with emission wavelengths of 649 and 634 nm, respectively. Despite their propeller-like structures, these compounds show notably high ΦF values, with 3e reaching 25% and 3g up to 60%. This enhancement in fluorescence efficiency can be attributed to the D–A system. The extended conjugation and subsequent intramolecular charge transfer (ICT) in the molecular framework induce increased rigidity, thereby reducing the occurrence of non-radiative relaxation channels, thus leading to higher fluorescence efficiency.75 Moreover, the increased polarization of the molecules and the resulting dipole moment are evident through pronounced solvatochromism (see Section S5.5, ESI†). Similar findings have been reported by Tang et al.46 and Hissler et al.48 in their studies on amine-substituted phosphindole derivatives. Due to the conjugation-induced rigidity, the investigated compounds lose their AIE characteristics and can be classified as dual-state emitters, exhibiting intense fluorescence both in the solid state and in solution.76–79 Remarkably, the introduction of a dimethoxydiphenylamine moiety results in an even more significant red shift. Specifically, phosphole oxide 3f demonstrates a high red emission with an emission maximum at 703 nm (Fig. 3) and a ΦF value of 7% even in liquid CH2Cl2 solution. Similar red-shifted phosphole derivatives have been previously reported by Baumgartner and Yamaguchi et al.80 for boryl- and amino-substituted dithienophospholes with distinct D–A–A π systems, but observed only in polar MeCN solutions. In contrast, the β-substituted derivatives 5, 6a and 6b do not exhibit a clear red shift compared to their α-substituted counterparts but display weak green luminescence with emission wavelengths ranging from 522 to 530 nm and low ΦF values of only 2%. The limited influence of the substituents in the β-position can be attributed to their inductive nature, as the conjugation is not extended through this position.81 However, the diphenylamine derivative 6c shows a pronounced red shift and weak emission at 640 nm, which can be attributed to an ICT process. Except for the push–pull systems, the thienophospholes generally exhibit low luminescence efficiencies in liquid CH2Cl2 solutions at rt. This can be ascribed to the high density of rotational degrees of freedom at the exocyclic substituents, leading to higher knr values. At low temperatures, these AIEgens experience reduced rotovibrational degrees of freedom, which has a significant impact on their optical characteristics. Consequently, an investigation into the performance of these compounds in frozen CH2Cl2/MeOH (1:1) glassy matrices at 77 K was conducted.
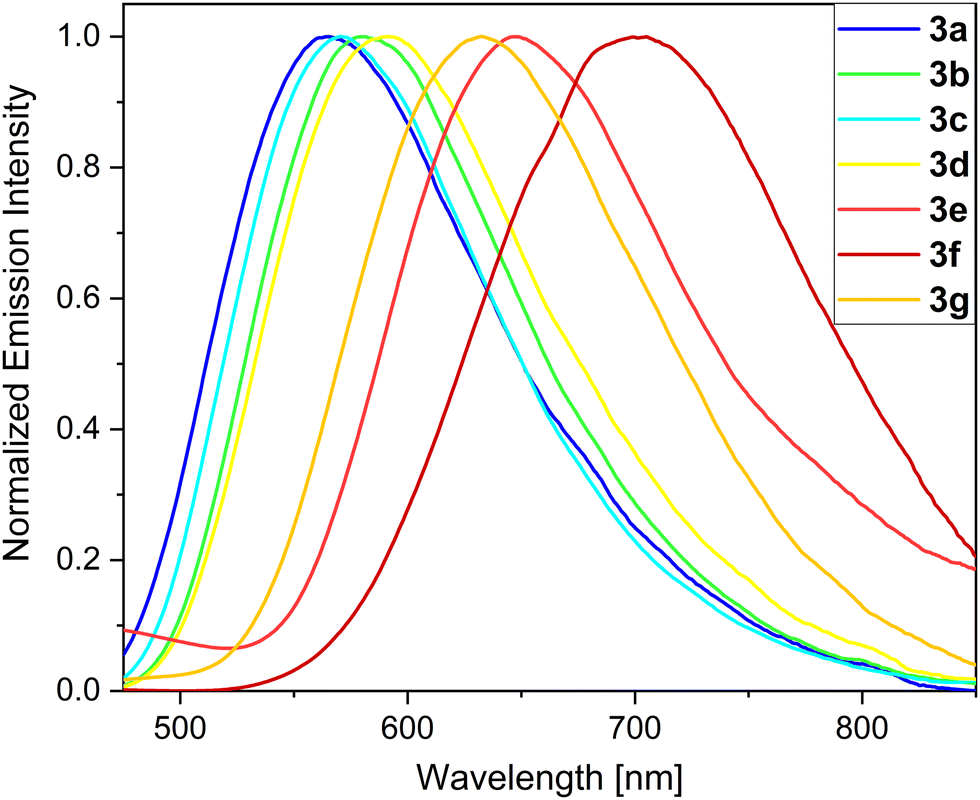 | ||
| Fig. 3 Photoluminescence spectra of α-substituted phospholes 3a–g in liquid CH2Cl2 solutions at rt. | ||
Under these specific conditions, all derivatives exhibited an enhanced fluorescence due to rotovibrational constraints of the exocyclic phenyl rings at low temperature. It is worth noting that, in frozen matrices without solvent reorganization, the emission bands of all phosphole oxides exhibited a blue shift compared to measurements conducted in liquid CH2Cl2 solutions. Additionally, a notable rise in the lifetime values up to 49 times (6b) was observed. The AIE-based thienophospholes presented in this study demonstrate a characteristically low-intensity fluorescence in solution, primarily attributed to the rotovibrational relaxation process occurring upon photoexcitation. However, in the solid state, this non-radiative decay pathway is effectively suppressed due to enhanced intermolecular interactions and increased molecular rigidity. Consequently, these thienophospholes exhibit intense fluorescence in the solid state (Table 2). In particular, the phenyl derivative 3a63 displays a strong yellow fluorescence emission at 548 nm, boasting a notable ΦF of 75% and an amplitude-weighted average lifetime of 6.2 ns. On the other hand, the thienyl species 3b exhibits a significant red shift in its fluorescence emission to 596 nm compared to its solution behavior, resulting in an orange emission. However, this luminescence is accompanied by a reduction in fluorescence efficiency, with a ΦF of 20%. It is worth mentioning that previous investigations on phosphinated thienophosphole oxides have demonstrated a similar fluorescence behavior, which aligns well with the findings reported by Tang et al.82 These observations suggest that AIE-active fluorophores containing thiophene residues generally experience elevated knr values due to the sulfur atoms in their molecular structure, which favour intersystem crossing. Substitution with a biphenyl moiety (3c) induces a red shift of 18 nm compared to the reference compound 3a.63 This red shift can be attributed to the extension of the delocalized π system, accompanied by a small torsion angle of 4.2° (Fig. 4), which enhances the molecular conjugation. However, the ΦF value decreases significantly to 51% due to the twisted structure and consequent molecular flexibility. In the case of compound 3d, which incorporates an anisole group with methoxy functionality, a yellow-orange solid-state fluorescence is observed. The ΦF value for 3d is 61%, and it displays a relatively long fluorescence lifetime of 8.7 ns. The D–A system 3e exhibits red emission at 646 nm, consistent with its behavior in solution. However, the fluorescence efficiency drops to 16%. This reduced efficiency arises from poor molecular packing and limited intermolecular interactions which consequently lead to high knr values (44 × 107 s−1). Efficient red emitters based on organic scaffolds pose a challenging task due to the energy gap law.83,84
| Compound | λ em [nm] | τ av_amp [ns] | Φ F(solid) (±2) [%]c |
|---|---|---|---|
| a Solid state at rt. b Amplitude-weighted average lifetimes at rt. c Absolute ΦF values of solids at rt were obtained by using a calibrated integrating sphere with a suitable sample holder. d The photoluminescence properties of 3a have been previously reported in solid state at rt.63 | |||
| 3a | 548 | 6.17 ± 0.02 | 75 |
| 3b | 596 | 7.16 ± 0.04 | 20 |
| 3c | 566 | 3.81 ± 0.03 | 51 |
| 3d | 585 | 8.72 ± 0.02 | 61 |
| 3e | 646 | 1.91 ± 0.06 | 16 |
| 3f | 649 | 4.43 ± 0.02 | 52 |
| 3g | 591 | 1.54 ± 0.04 | 27 |
| 5 | 492 | 5.09 ± 0.02 | 45 |
| 6a | 508 | 5.67 ± 0.02 | 40 |
| 6b | 499 | 6.46 ± 0.02 | 65 |
| 6c | 547 | 3.64 ± 0.02 | 16 |
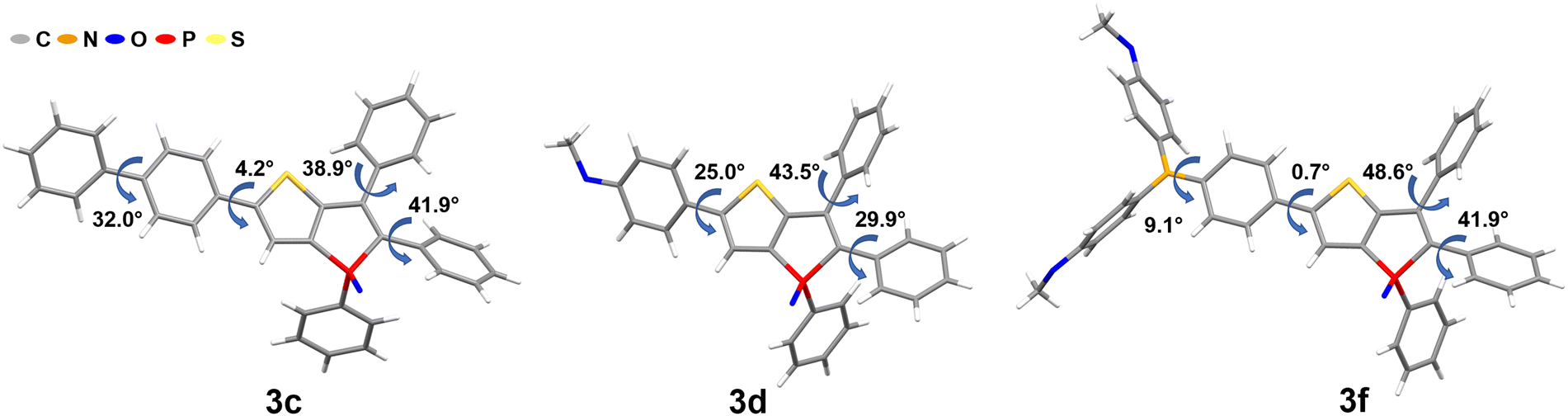 | ||
| Fig. 4 Molecular structures and torsion angles of phospholes 3c, 3d and 3f (molecular structures and details of compounds 5, 6a and 6c are given in Section S4, ESI†). | ||
Decreasing the HOMO–LUMO energy gap results in an exponential increase in knr values while hindering an efficient red fluorescence. Nevertheless, and improved molecular design may offer promising avenues to mitigate non-radiative decay and facilitate the development of efficient red emitters.85 The replacement of the diphenylamine moiety with a dimethoxydiphenylamine unit demonstrated a remarkable positive effect in the phosphole 3f. This structural modification resulted in the manifestation of intense deep red luminescence, characterized by a ΦF of 52% and a fluorescence lifetime of 4.4 ns. As a consequence, this compound stands out as one of the few highly efficient red-emitting solid-state fluorophores,86,87 and to the best of our knowledge, it represents the most potent red-emitting phosphole derivative identified to date. The heightened efficiency of this red emitter can be ascribed to its well-defined molecular structure. Both the torsion angles between the thiophene and phenyl units, as well as between the phenyl and amine moieties, exhibit very small values of only 0.7° and 9.1°, respectively. This observation implies a significant conjugation effect and a rigid molecular framework, both of which contribute to the resulting red-shift phenomenon. Additionally, the presence of methoxy groups in compound 3f facilitates intermolecular interactions, specifically involving Calkyl–H⋯π and Caryl–H⋯O interactions, thereby effectively decreasing the probability of non-radiative decay processes. In a broader context, it is recognized that the introduction of anisole substituents, beyond inducing a red shift and augmenting quantum yield, also correlates with extended τ values. The β-substituted derivatives 5 and 6a–c follow a similar trend as observed in liquid CH2Cl2 solutions. Among these, compound 6b, characterized by an emission maximum of 499 nm and a ΦF of 65%, attains the most superior performance characteristics.
Fig. 5 demonstrates the expansive potential for modulating the characteristics of the thieno[3,2-b]phosphole scaffold. The model compound TPTPO63 exhibits a blue emission profile (λem = 485 nm) accompanied by good quantum efficiencies. Through strategic incorporation of substituents in the α-position that exhibit either inductive or weak mesomeric effects (–Me, –Br, –P(O)Ph2), it becomes feasible to generate AIEgens with tunable cyan to green luminescence.63 The extension of the conjugated π system through the introduction of aromatic substituents leads to the development of compounds displaying luminescence spanning from yellow to orange. To engineer red-emitting species, the implementation of a push–pull configuration alongside the introduction of potent donor groups, such as diphenylamines, proves to be effective. In contrast, the β-position does not serve as a straightforward avenue for manipulating the color of the luminescence due to the inherent limitations imposed by inadequate conjugation phenomena.56
 | ||
| Fig. 5 Overview of the optical properties in the solid state of different substituted thieno[3,2-b]phosphole oxides and selected crystals under irradiation of UV light (395 nm). | ||
Tosylimino phospholes
The conversion of phosphole oxides (TPTPO,63PPTPO,633a,636a) to their respective tosylimino phospholes 7a–d induces substantial alterations in the photophysical characteristics.64 These changes arise due to modifications in the chemical environment of the phosphorus atom, alongside additional intermolecular interactions and variations in molecular packing. Across all instances, a notable red shift of the solid-state fluorescence spectra, ranging from 13 to 58 nm (Fig. 6), is observed when compared to their corresponding phosphole oxides. Two plausible explanations account for these observations. Firstly, an intramolecular π–π interaction between the tosyl group and the annulated thiophene moiety occurs within the conjugated π system (Fig. 6).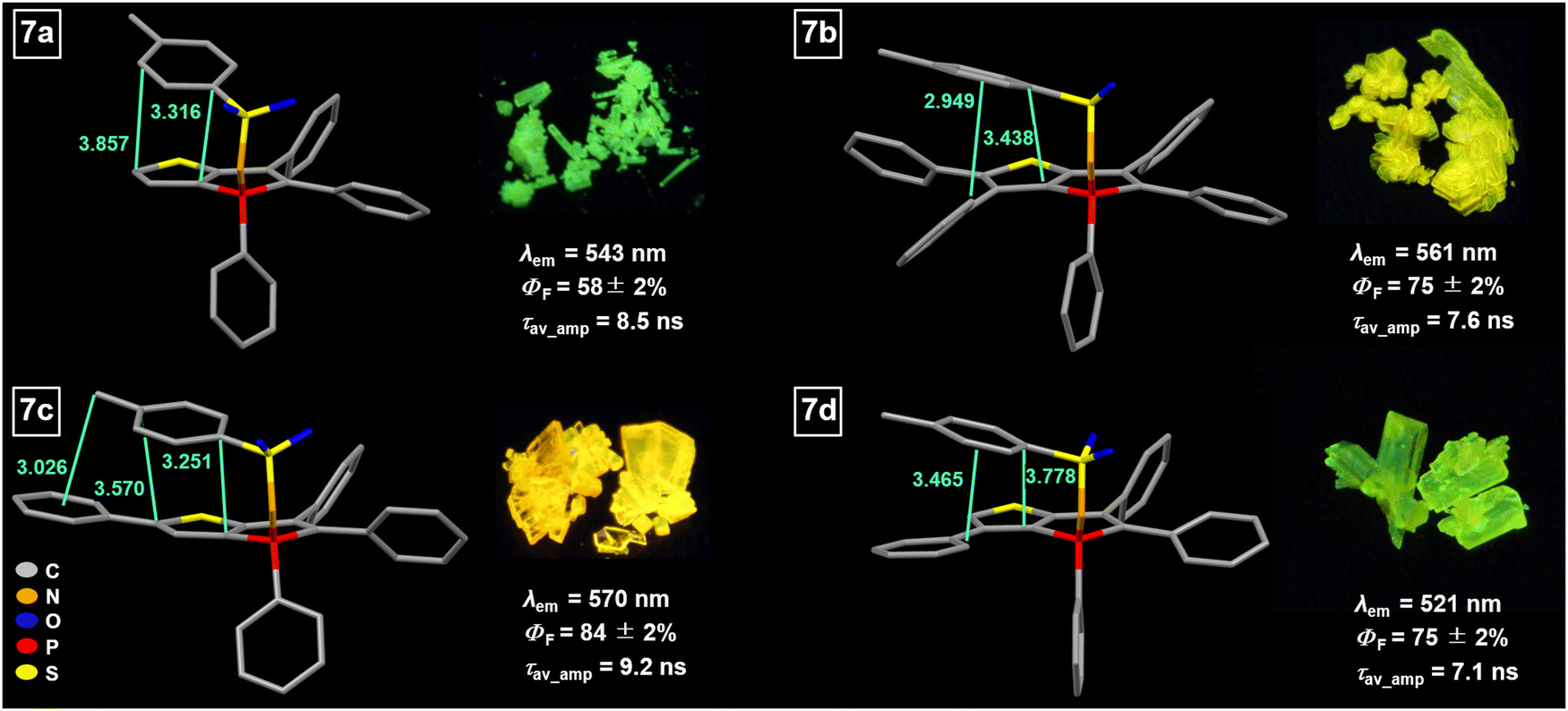 | ||
| Fig. 6 Molecular structures of 7a–d (hydrogen atoms have been omitted for clarity) with intramolecular interactions (distances in Å) and their photophysical properties in the solid state. Pictures of selected crystals under irradiation of UV light (395 nm). | ||
This through-space conjugation exerts a stabilizing effect on the excited state, leading to a reduction in the energy difference between the frontier orbitals, particularly the lowering of the LUMO level.88–90 Consequently, the decrease in the HOMO–LUMO energy gap is responsible for the observed bathochromic shift in the emission wavelength. The distances involved in these intramolecular π–π interactions range from 3.3 to 3.8 Å, consistent with previously reported compounds.64 To provide further insights, exemplary non-covalent interaction (NCI) plots were calculated for compounds 7a and 7c (Fig. 7).91,92 The non-covalent interactions predominantly arise from π–π interactions between the tosyl group and the thiophene unit. In the case of compound 7c, an additional Calkyl–H⋯π interaction occurs between the methyl group and the exocyclic phenyl ring. The stabilization associated with the π–π interaction was estimated by performing theoretical calculations involving the rotation of the tosyl moiety (see Section S6, ESI†).
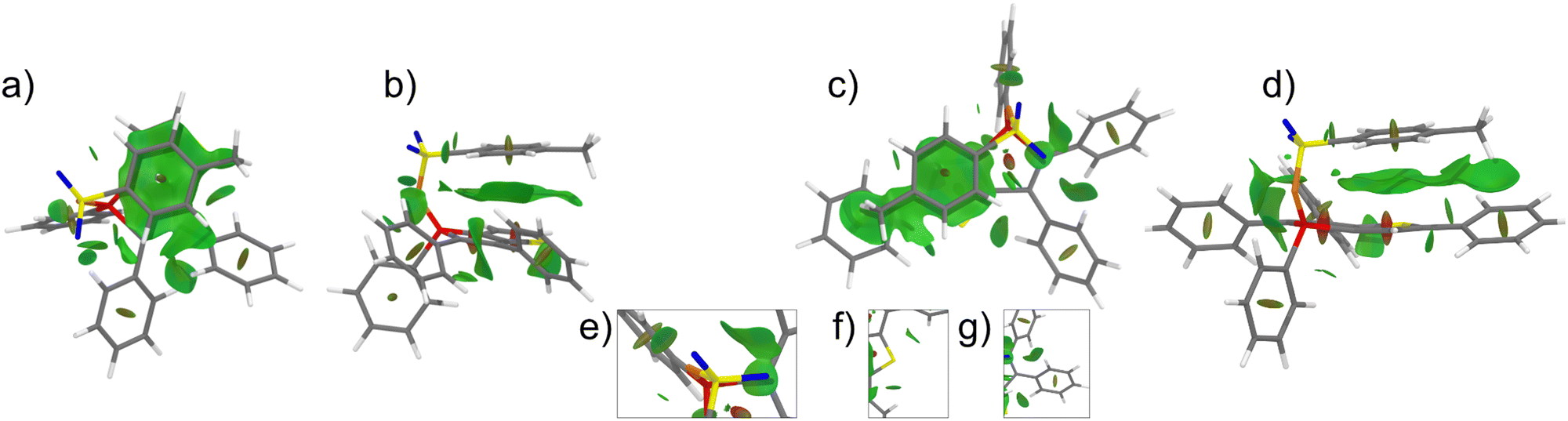 | ||
| Fig. 7 NCI plot of the phospholes 7a and 7c with top (a) and (c) and side view (b) and (d) of the tosylate group. The insets (e)–(g) show various N⋯H–C as well as C–H⋯π interactions. | ||
Compound 7a exhibits a stabilization energy of approximately −6.6 kJ mol−1, whereas compound 7c shows an energy of −14.4 kJ mol−1, attributed to the additional Calkyl–H⋯π interaction. Moreover, a secondary explanation for the observed changes in the photophysical properties arises from the alteration in the chemical nature of the phosphorus atom, which exerts a substantial influence on the emission wavelength within phosphole derivatives. In the case of tosylimino phospholes, a delocalized P–N–S moiety is present, resulting in an additional red shift in the emission spectrum.93 This is evidenced by P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond lengths within the expected range (1.58–1.59 Å), while the S–N bonds are significantly shorter (1.58–1.59 Å) than expected for a single bond, matching the lengths of the PN double bonds. This unique electronic structure is also manifested in the remarkable chemical and thermal stability exhibited by the tosylimino phospholes. In addition to influencing the emission maxima, certain effects on the efficiency and lifetime of the emitted light were observed. It is noteworthy that in the solid state, multi-exponential decays were observed due to the presence of various conformers and different proportions of mono-dispersed or aggregated states; in addition, the polycrystalline nature of the solids provides different microenvironments that also contribute to a complex photoluminescence decay. Therefore, the calculation of radiative and non-radiative constants using amplitude-weighted average lifetimes should be regarded as an approximation of the dynamics of the excited states. Notably, all tosylimines 7a–d demonstrated strong fluorescence in the solid state, with ΦF ranging from 58% to 84%, and fluorescence lifetimes ranging from 7.1 to 9.2 ns. Particularly noteworthy is the significant increase in ΦF values observed in 7b–d as compared to the starting materials. This enhancement is evident when comparing the average deactivation rate constants, as 7b–d exhibit significantly lower non-radiative deactivation rates. For example, in 7b and 7d, the modification led to at least a one-third reduction in the average non-radiative decay rates, resulting in values of 3.3 × 107 s−1 and 3.5 × 107 s−1, respectively. Consequently, the increase in fluorescence efficiency in these cases was 24% for 7b and 35% for 7d. Furthermore, the conversion of the strong yellow emitter 3a63 to 7c led to a noteworthy increase of ΦF by 9%. This enhancement can be attributed to two main factors. Firstly, the formation of intramolecular π–π interactions significantly increased the rigidity of the molecular framework, thereby enhancing the fluorescence properties. Secondly, the introduction of the relatively large tosyl group caused significant alterations in the molecular packing in the solid state, leading to additional C–H⋯heteroatom interactions (mainly SO⋯H–Caryl or PN⋯H–Caryl). These interactions further constrained the flexibility of the fluorophores, resulting in a notable reduction in the non-radiative relaxation channels. Among the tested compounds, only 7a exhibited no change in the average knr values (Fig. 8). In contrast, for the other compounds, variations were observed in the average kr values, which had a notable impact on the fluorescence efficiency. Specifically, when the kr was reduced to 6.9 × 107 s−1, a discernible 11% decrease in fluorescence efficiency was observed. In 7a–d, we observed a significant increase in fluorescence lifetime by 1.4–3.6 ns. In particular, compound 7b stands out with a lifetime of 7.6 ns, which is almost double the lifetime of the starting compound PPTPO63 (4.0 ns). The increase in the τ values can be attributed to the formation of the through-space conjugation and the resulting stabilization of the excited state.94
N bond lengths within the expected range (1.58–1.59 Å), while the S–N bonds are significantly shorter (1.58–1.59 Å) than expected for a single bond, matching the lengths of the PN double bonds. This unique electronic structure is also manifested in the remarkable chemical and thermal stability exhibited by the tosylimino phospholes. In addition to influencing the emission maxima, certain effects on the efficiency and lifetime of the emitted light were observed. It is noteworthy that in the solid state, multi-exponential decays were observed due to the presence of various conformers and different proportions of mono-dispersed or aggregated states; in addition, the polycrystalline nature of the solids provides different microenvironments that also contribute to a complex photoluminescence decay. Therefore, the calculation of radiative and non-radiative constants using amplitude-weighted average lifetimes should be regarded as an approximation of the dynamics of the excited states. Notably, all tosylimines 7a–d demonstrated strong fluorescence in the solid state, with ΦF ranging from 58% to 84%, and fluorescence lifetimes ranging from 7.1 to 9.2 ns. Particularly noteworthy is the significant increase in ΦF values observed in 7b–d as compared to the starting materials. This enhancement is evident when comparing the average deactivation rate constants, as 7b–d exhibit significantly lower non-radiative deactivation rates. For example, in 7b and 7d, the modification led to at least a one-third reduction in the average non-radiative decay rates, resulting in values of 3.3 × 107 s−1 and 3.5 × 107 s−1, respectively. Consequently, the increase in fluorescence efficiency in these cases was 24% for 7b and 35% for 7d. Furthermore, the conversion of the strong yellow emitter 3a63 to 7c led to a noteworthy increase of ΦF by 9%. This enhancement can be attributed to two main factors. Firstly, the formation of intramolecular π–π interactions significantly increased the rigidity of the molecular framework, thereby enhancing the fluorescence properties. Secondly, the introduction of the relatively large tosyl group caused significant alterations in the molecular packing in the solid state, leading to additional C–H⋯heteroatom interactions (mainly SO⋯H–Caryl or PN⋯H–Caryl). These interactions further constrained the flexibility of the fluorophores, resulting in a notable reduction in the non-radiative relaxation channels. Among the tested compounds, only 7a exhibited no change in the average knr values (Fig. 8). In contrast, for the other compounds, variations were observed in the average kr values, which had a notable impact on the fluorescence efficiency. Specifically, when the kr was reduced to 6.9 × 107 s−1, a discernible 11% decrease in fluorescence efficiency was observed. In 7a–d, we observed a significant increase in fluorescence lifetime by 1.4–3.6 ns. In particular, compound 7b stands out with a lifetime of 7.6 ns, which is almost double the lifetime of the starting compound PPTPO63 (4.0 ns). The increase in the τ values can be attributed to the formation of the through-space conjugation and the resulting stabilization of the excited state.94
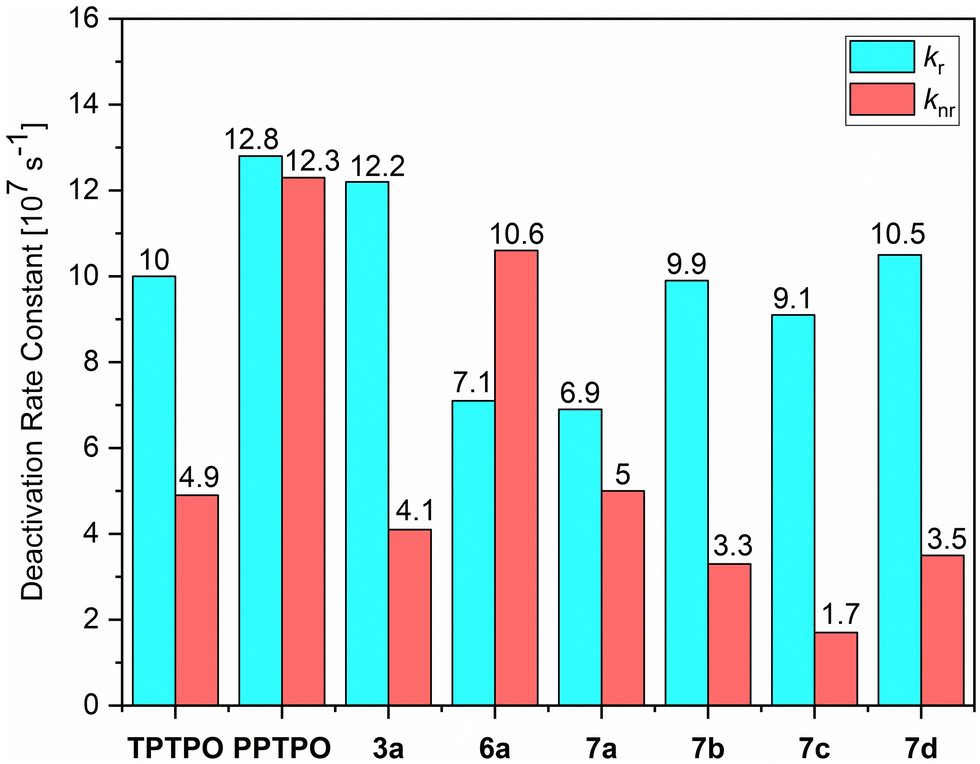 | ||
| Fig. 8 Average radiative (kr) and radiationless (knr) deactivation rate constants of phosphole oxides TPTPO,63PPTPO,633a,636a and tosylimino phospholes 7a–d in the solid state. The average rate constants are estimated according to kr = 1/τav_amp = ΦF/τav_amp and knr = 1 − ΦF/τav_amp by using amplitude-weighted average lifetimes, as suggested by Engelborghs et al.70 The uncertainties are not presented in this diagram for clarity (for kr and knr in liquid CH2Cl2 solutions at rt and frozen glassy matrix in CH2Cl2/MeOH (1:1) at 77 K, see Section S5, ESI†). | ||
Overall, the observations suggest that the strategic incorporation of tosyl groups in the molecular structure of the fluorophores can yield substantial improvements in fluorescence efficiency and lifetime, making these tosylimines promising candidates for various optoelectronic applications.
Conclusions
In conclusion, the study presented herein demonstrates the remarkable versatility and potential of thieno[3,2-b]phosphole-based fluorophores for achieving tuneable solid-state luminescence. The strategic manipulation of molecular structures and intermolecular interactions has enabled the design and synthesis of a diverse range of innovative materials with interesting optical properties. Through systematic exploration of different substituents in α- and β-positions on the thieno[3,2-b]phosphole scaffold, a comprehensive understanding of the factors influencing emission characteristics has been established. The introduction of various electron-donating groups and the extension of the conjugated π system has enabled the tuning of emission wavelengths across the visible spectrum, from cyan to deep red. Additionally, the incorporation of push–pull systems and potent donor moieties has led to the development of a highly efficient red-emitting fluorophore with a maximum at 649 nm and an efficiency of 52%, a challenging endeavor in the field of organic luminescent materials. The investigation also delved into the conversion of phosphole oxides to their corresponding tosylimino derivatives, revealing that these modifications can further enhance the fluorescence efficiency and prolong the excited state lifetimes. The synergistic effects of intramolecular interactions and altered molecular packing resulting from the introduction of tosyl groups contribute to these improvements and emphasize the significance of the 3-dimensional fluorophore design. This work showcases the power of rational molecular design in achieving precise control over solid-state emission properties by dual modulation of the thieno[3,2-b]phosphole framework, offering new avenues for the development of luminescent materials with tailored optical characteristics. The insights gained from this study provide a foundation for future research in the design and engineering of advanced materials for a wide range of applications.Author contributions
N. K. performed the experimental work and wrote the original draft. J. M. and J. A. supported in the synthesis of several derivatives. Y. G.-L. and N. K. analyzed the photophysical properties. P. L. performed single crystal diffractometric X-ray analysis and interpreted the structural data. H. W. performed the DFT calculations. E. H.-H. and C. A. S. supervised the project. All authors have corrected the final manuscript. All authors have read and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. A. S. gratefully acknowledges the generous financial support for the acquisition of an “Integrated Confocal Luminescence Spectrometer with Spatiotemporal Resolution and Multiphoton Excitation” (DFG/Land NRW: INST 211/915-1 FUGG; DFG EXC1003: “Berufungsmittel”).References
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- C. J. Weijer, Science, 2003, 300, 96–100 CrossRef CAS PubMed.
- R. N. Germain, E. A. Robey and M. D. Cahalan, Science, 2012, 336, 1676–1681 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CrossRef CAS PubMed.
- P. Ramasamy, N. Kim, Y.-S. Kang, O. Ramirez and J.-S. Lee, Chem. Mater., 2017, 29, 6893–6899 CrossRef CAS.
- R. Zhan, Y. Pan, P. N. Manghnani and B. Liu, Macromol. Biosci., 2017, 17, 1600433 CrossRef PubMed.
- R. Hu, X. Yang, A. Qin and B. Z. Tang, Mater. Chem. Front., 2021, 5, 4073–4088 RSC.
- P. Shen, Z. Zhuang, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 11835–11852 RSC.
- C. Kirst, F. Knechtel, M. Gensler, D. Fischermeier, J. Petersen, N. A. Danaf, J. Tietze, A. Wedel, D. C. Lamb, R. Mitrić and K. Karaghiosoff, Adv. Funct. Mater., 2023, 33, 2212436 CrossRef CAS.
- Y. Yu, Z. Xu, Z. Zhao, H. Zhang, D. Ma, J. W. Y. Lam, A. Qin and B. Z. Tang, ACS Omega, 2018, 3, 16347–16356 CrossRef CAS PubMed.
- S. Zhao, J. Sun, Z. Qin, Y. Li, H. Yu, G. Wang, X. Gu and K. Pan, Small, 2022, 18, 2201117 CrossRef CAS PubMed.
- S. Suzuki, S. Sasaki, A. S. Sairi, R. Iwai, B. Z. Tang and G. Konishi, Angew. Chem., Int. Ed., 2020, 59, 9856–9867 CrossRef CAS PubMed.
- M. Yu, R. Huang, J. Guo, Z. Zhao and B. Z. Tang, PhotoniX, 2020, 1, 11 CrossRef.
- J. Yang, Y. Zhang, X. Wu, W. Dai, D. Chen, J. Shi, B. Tong, Q. Peng, H. Xie, Z. Cai, Y. Dong and X. Zhang, Nat. Commun., 2021, 12, 4883 CrossRef CAS PubMed.
- J. Kuno, N. Ledos, P.-A. Bouit, T. Kawai, M. Hissler and T. Nakashima, Chem. Mater., 2022, 34, 9111–9118 CrossRef CAS.
- S.-H. Chen, X.-Y. Cao, P.-T. Hu, K. Jiang, Y.-T. Liang, B.-J. Xu, Z.-H. Li and Z.-Y. Wang, Mater. Adv., 2023, 4, 6612–6620 RSC.
- X. Gu, J. Yao, G. Zhang, C. Zhang, Y. Yan, Y. Zhao and D. Zhang, Chem. – Asian J., 2013, 8, 2362–2369 CrossRef CAS PubMed.
- N. Zhao, Z. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie, S. Chen, H. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637–8639 RSC.
- Y. Chen, Y. Fang, H. Gu, J. Qiang, H. Li, J. Fan, J. Cao, F. Wang, S. Lu and X. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 55094–55106 CrossRef CAS PubMed.
- Z. Zhang, B. Xu, J. Su, L. Shen, Y. Xie and H. Tian, Angew. Chem., Int. Ed., 2011, 50, 11654–11657 CrossRef CAS PubMed.
- C. Maeda, T. Todaka, T. Ueda and T. Ema, Chem. – Eur. J., 2016, 22, 7508–7513 CrossRef CAS PubMed.
- K. Pauk, S. Luňák, A. Růžička, A. Marková, K. Teichmanová, A. Mausová, M. Kratochvíl, R. Smolka, T. Mikysek, M. Weiter, A. Imramovský and M. Vala, RSC Adv., 2022, 12, 34797–34807 RSC.
- Q. Chen, D. Zhang, G. Zhang, X. Yang, Y. Feng, Q. Fan and D. Zhu, Adv. Funct. Mater., 2010, 20, 3244–3251 CrossRef CAS.
- L. Biesen, N. Nirmalananthan-Budau, K. Hoffmann, U. Resch-Genger and T. J. J. Müller, Angew. Chem., Int. Ed., 2020, 59, 10037–10041 CrossRef CAS PubMed.
- Y.-F. Sun, Z.-Y. Chen, L. Zhu, S.-H. Xu, R.-T. Wu and Y.-P. Cui, Color. Technol., 2013, 129, 165–172 CAS.
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef PubMed.
- F. Mathey, Chem. Rev., 1988, 88, 429–453 CrossRef CAS.
- F. Riobé, R. Szűcs, C. Lescop, R. Réau, L. Nyulászi, P.-A. Bouit and M. Hissler, Organometallics, 2017, 36, 2502–2511 CrossRef.
- Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258–1271 RSC.
- X. He, A. Y. Y. Woo, J. Borau-Garcia and T. Baumgartner, Chem. – Eur. J., 2013, 19, 7620–7630 CrossRef CAS PubMed.
- C. Fave, T.-Y. Cho, M. Hissler, C.-W. Chen, T.-Y. Luh, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2003, 125, 9254–9255 CrossRef CAS PubMed.
- D. Joly, D. Tondelier, V. Deborde, B. Geffroy, M. Hissler and R. Réau, New J. Chem., 2010, 34, 1603–1611 RSC.
- C. Hay, M. Hissler, C. Fischmeister, J. Rault-Berthelot, L. Toupet, L. Nyulászi and R. Réau, Chem. – Eur. J., 2001, 7, 4222–4236 CrossRef CAS PubMed.
- T. Higashino, K. Ishida, T. Satoh, Y. Matano and H. Imahori, J. Org. Chem., 2018, 83, 3397–3402 CrossRef CAS PubMed.
- Y. Dienes, M. Eggenstein, T. Kárpáti, T. C. Sutherland, L. Nyulászi and T. Baumgartner, Chem. – Eur. J., 2008, 14, 9878–9889 CrossRef CAS PubMed.
- M. Stolar and T. Baumgartner, Chem. – Asian J., 2014, 9, 1212–1225 CrossRef CAS PubMed.
- F. Riobé, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sánchez, R. Réau, L. Nyulászi and M. Hissler, Chem. – Eur. J., 2015, 21, 6547–6556 CrossRef PubMed.
- H.-C. Su, O. Fadhel, C.-J. Yang, T.-Y. Cho, C. Fave, M. Hissler, C.-C. Wu and R. Réau, J. Am. Chem. Soc., 2006, 128, 983–995 CrossRef CAS PubMed.
- J. Crassous and R. Réau, Dalton Trans., 2008, 6865–6876 RSC.
- M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- Z. Zhuang, F. Bu, W. Luo, H. Peng, S. Chen, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 1836–1842 RSC.
- M. P. Duffy, P.-A. Bouit, B. Geffroy, D. Tondelier and M. Hissler, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 845–853 CrossRef CAS.
- N. Ledos, D. Tondelier, B. Geffroy, D. Jacquemin, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2023, 11, 3826–3831 RSC.
- M. Schenk, N. König, E. Hey-Hawkins and A. G. Beck-Sickinger, ChemBioChem, 2024, 25, e202300857 CrossRef CAS PubMed.
- E. Rémond, J.-A. Fehrentz, L. Liénart, S. Clément, J.-L. Banères and F. Cavelier, Chem. – Eur. J., 2022, 28, e202201526 CrossRef PubMed.
- C. Wang, M. Taki, K. Kajiwara, J. Wang and S. Yamaguchi, ACS Mater. Lett., 2020, 2, 705–711 CrossRef CAS.
- H. Tsuji, K. Sato, Y. Sato and E. Nakamura, J. Mater. Chem., 2009, 19, 3364–3366 RSC.
- H. Chen, W. Delaunay, J. Li, Z. Wang, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Mathey, Z. Duan, R. Réau and M. Hissler, Org. Lett., 2013, 15, 330–333 CrossRef CAS PubMed.
- X. Chen, S. Liu, Y. Sun, D. Zhong, Z. Feng, X. Yang, B. Su, Y. Sun, G. Zhou, B. Jiao and Z. Wu, Mater. Chem. Front., 2023, 7, 1841–1854 RSC.
- Z. Wang, B. S. Gelfand and T. Baumgartner, Angew. Chem., Int. Ed., 2016, 55, 3481–3485 CrossRef CAS PubMed.
- E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS PubMed.
- Y. Matano, A. Saito, T. Fukushima, Y. Tokudome, F. Suzuki, D. Sakamaki, H. Kaji, A. Ito, K. Tanaka and H. Imahori, Angew. Chem., Int. Ed., 2011, 50, 8016–8020 CrossRef CAS PubMed.
- D. Wu, J. Zheng, C. Xu, D. Kang, W. Hong, Z. Duan and F. Mathey, Dalton Trans., 2019, 48, 6347–6352 RSC.
- T. Baumgartner, T. Neumann and B. Wirges, Angew. Chem., Int. Ed., 2004, 43, 6197–6201 CrossRef CAS PubMed.
- Y. Matano, A. Saito, Y. Suzuki, T. Miyajima, S. Akiyama, S. Otsubo, E. Nakamoto, S. Aramaki and H. Imahori, Chem. – Asian J., 2012, 7, 2305–2312 CrossRef CAS PubMed.
- Y. Dienes, S. Durben, T. Kárpáti, T. Neumann, U. Englert, L. Nyulászi and T. Baumgartner, Chem. – Eur. J., 2007, 13, 7487–7500 CrossRef CAS PubMed.
- N. Inai, S. Yamaguchi and T. Yanai, ACS Phys. Chem. Au, 2023, 3, 540–552 CrossRef CAS PubMed.
- N. König, Y. Godínez-Loyola, H. Weiske, S. Naumov, P. Lönnecke, R. Tonner-Zech, C. A. Strassert and E. Hey-Hawkins, Chem. Mater., 2023, 35, 8218–8228 CrossRef.
- N. König, Y. Godínez-Loyola, F. Yang, C. Laube, M. Laue, P. Lönnecke, C. A. Strassert and E. Hey-Hawkins, Chem. Sci., 2023, 14, 2267–2274 RSC.
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 11504–11508 CrossRef CAS PubMed.
- W. Ma and L. Ackermann, Synthesis, 2014, 2297–2304 CAS.
- Y.-R. Chen and W.-L. Duan, J. Am. Chem. Soc., 2013, 135, 16754–16757 CrossRef CAS PubMed.
- Y. Unoh, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 12975–12979 CrossRef CAS PubMed.
- Y.-L. Zhao, G.-J. Wu, Y. Li, L.-X. Gao and F.-S. Han, Chem. – Eur. J., 2012, 18, 9622–9627 CrossRef CAS PubMed.
- A. Sillen and Y. Engelborghs, Photochem. Photobiol., 1998, 67, 475–486 CrossRef CAS.
- F. Bu, E. Wang, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 4440–4449 CrossRef CAS PubMed.
- R. Kundu and C. Kulshreshtha, RSC Adv., 2015, 5, 77460–77468 RSC.
- T. Umeno, K. Usui and S. Karasawa, Asian J. Org. Chem., 2021, 10, 1123–1130 CrossRef CAS.
- S. Redon, G. Eucat, M. Ipuy, E. Jeanneau, I. Gautier-Luneau, A. Ibanez, C. Andraud and Y. Bretonnière, Dyes Pigm., 2018, 156, 116–132 CrossRef CAS.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS PubMed.
- L. A. Rodríguez-Cortés, A. Navarro-Huerta and B. Rodríguez-Molina, Matter, 2021, 4, 2622–2624 CrossRef.
- Q. Qiu, P. Xu, Y. Zhu, J. Yu, M. Wei, W. Xi, H. Feng, J. Chen and Z. Qian, Chem. – Eur. J., 2019, 25, 15983–15987 CrossRef CAS PubMed.
- J. L. Belmonte-Vázquez, Y. A. Amador-Sánchez, L. A. Rodríguez-Cortés and B. Rodríguez-Molina, Chem. Mater., 2021, 33, 7160–7184 CrossRef.
- A. Huber, J. Dubbert, T. D. Scherz and J. Voskuhl, Chem. – Eur. J., 2023, 29, e202202481 CrossRef CAS PubMed.
- Y. Sugihara, N. Inai, M. Taki, T. Baumgartner, R. Kawakami, T. Saitou, T. Imamura, T. Yanai and S. Yamaguchi, Chem. Sci., 2021, 12, 6333–6341 RSC.
- E. Yamaguchi, A. Fukazawa, Y. Kosaka, D. Yokogawa, S. Irle and S. Yamaguchi, Bull. Chem. Soc. Jpn., 2015, 88, 1545–1552 CrossRef CAS.
- L. Viglianti, N. L. C. Leung, N. Xie, X. Gu, H. H. Y. Sung, Q. Miao, I. D. Williams, E. Licandro and B. Z. Tang, Chem. Sci., 2017, 8, 2629–2639 RSC.
- Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS.
- Y. R. Poh, S. Pannir-Sivajothi and J. Yuen-Zhou, J. Phys. Chem. C, 2023, 127, 5491–5501 CrossRef CAS.
- L. Tu, Y. Xie, Z. Li and B. Tang, SmartMat, 2021, 2, 326–346 CrossRef CAS.
- M. Durko-Maciag, A. Popczyk, M. Rémond, Z. Zheng, Y. Bretonniere, C. Andraud and J. Mysliwiec, ChemPhotoChem, 2022, 6, e202200008 CrossRef CAS.
- Y.-T. Lee, C.-L. Chiang and C.-T. Chen, Chem. Commun., 2007, 217–219 Search PubMed.
- J. Li, P. Shen, Z. Zhao and B. Z. Tang, CCS Chem, 2019, 1, 181–196 CrossRef CAS.
- J. Liu, H. Zhang, L. Hu, J. Wang, J. W. Y. Lam, L. Blancafort and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 7901–7910 CrossRef CAS PubMed.
- T. Han, H. Deng, Z. Qiu, Z. Zhao, H. Zhang, H. Zou, N. L. C. Leung, G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 5588–5598 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- H. Folkerts, D. Nußhär, F. Weller, K. Dehnicke, J. Magull and W. Hiller, Z. Anorg. Allg. Chem., 1994, 620, 1986–1991 CrossRef CAS.
- X. Wu, X. Peng, L. Chen, B. Z. Tang and Z. Zhao, ACS Mater. Lett., 2023, 5, 664–672 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods, synthesis, crystallographic data, compound spectra, optical properties and theoretical calculations. CCDC 2302468–2302476. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc04519f |
| This journal is © The Royal Society of Chemistry 2024 |