
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the design rules of electrides†
Zhikun
Yao
a,
Yanzhen
Zhao
a,
Wenjun
Zhang
 a and
Lee A.
Burton
*b
a and
Lee A.
Burton
*b
aInternational Centre for Quantum and Molecular Structures, Department of Physics, Shanghai University, Shanghai 200444, China
bDepartment of Materials Science and Engineering, The Ilby and Aladar Fleischman Faculty of Engineering, Tel Aviv University, Ramat Aviv, Tel Aviv 6997801, Israel. E-mail: leeburton@tauex.tau.ac.il
First published on 6th May 2024
Abstract
There are three heuristic criteria commonly used to identify electrides: an apparent valence of plus one, empty space in the crystal structure and the presence of a strongly electron-donating cation. We evoke and explore these criteria by mapping probable charges to a database of all known materials and isolating around around 4000 compounds that are likely to exhibit an oxidation state of +1. Of these, we identify peaks in off-atom electron density by density functional theory and discuss the validity with which the design rules can be applied to these likely electrides. In doing so, we recover 4 experimentally confirmed electrides among 51 candidates identified as potential new electrides that were not considered previously. All results for each candidate are provided but perhaps of special significance is the material Ba3AlO4 that has similar composition and chemistry to the stable electride catalyst Ca12Al14O32. Overall, we find that the valence and void space rules are surprisingly useful but that there is a breadth of chemistry to electrides that can be overlooked by considering only alkili and alkali earth metal compounds.
Introduction
Electrides are materials in which the electrons detach from the valence shell of ions and reside in the crystal structure, acting as anions.1,2 Because these anionic electrons are not bound to a nucleus, they are easier to migrate and extract compared to electrons in conventional materials, leading to relatively low work functions and high conductivities.1,3 These properties are desirable for several important technological applications to which it is expected that electrides can be successfully applied. For example electron emitters,4 ion sources,5 light emitting diodes (LED) devices,6 nonlinear optical switchers,7 superconductors,8 and catalysts,9,10 for e.g. the synthesis of ammonia,11 are all areas of active investigation for electrides.The first reports of anionic electrons were detailed for solutions of alkali metals, however attempts to crystallise these systems with the electron remaining off-atom proved difficult.12 The first successful solid system containing anionic electrons was an organic complex condensed from such a solution by Dye et al. in 1983, and given the name electride.13 However, the material was extremely unstable at room temperature. It was not until the first purely inorganic electride was synthesised around 20 years later, that it was demonstrated that electrides could be stable with respect to temperature and pressure.14 The inorganic electride was synthesised by chemical treatment of the mineral mayenite (12CaO·7Al2O3) to remove the oxygen ions from crystallographic cages, creating [Ca24Al28O64]4+(4e−) often written as (C12A7:e−). This material can be considered a zero-dimensional (0D) electride,14 as the anion electrons are trapped in a cavity in the crystal structure. There are also 1D electrides,15 where anion electrons are confined in a channel of the structure and 2D electrides,16 in which anion electrons reside as layers.
In general, electrides are expected to meet specific conditions that distinguish them from conventional materials.
First, they must have excess electrons,17 which can be inferred from the apparent oxidation state of the chemical formula. Second, the material must have free space in which the excess electron can reside, hence the 0D, 1D, 2D nomenclature mentioned earlier. Finally, it must contain a cation from which the excess electron will be pushed into the crystal structure and not recombine. For example, Ca2N the first compound to be confirmed as 2D electride material,16 conforms to all three criteria simultaneously, see Fig. 1. Ca2N has 2Ca2+ cations which provide 4 electrons and 1 N3− anion which can only accept 3 electrons; thus one electron remains unassigned. Together, the [Ca2N]+e− system is electroneutral but the chemical formula alone denotes an apparent valence of +1. Secondly, the structure of the Ca2N is layered allowing space for the anionic electrons between the bonded sheets. And finally, the donor ion calcium is extremely electropositive.
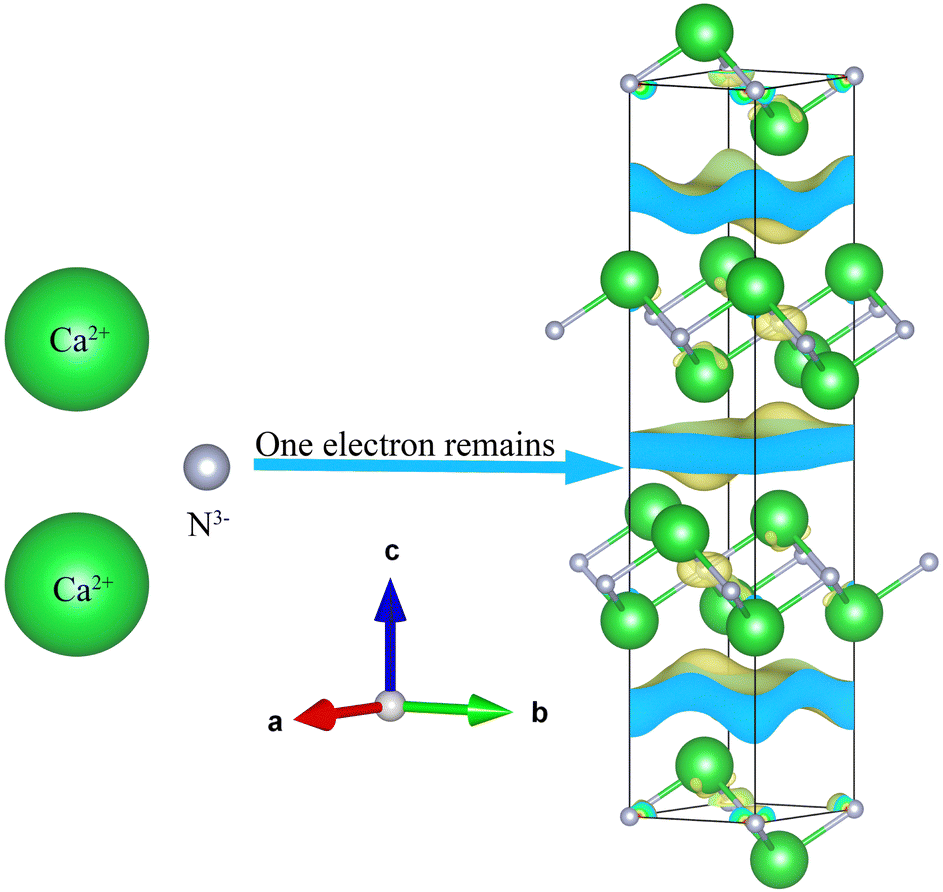 | ||
| Fig. 1 Illustration of the archetypal electride Ca2N that conforms to all three heuristic rules of electrides: apparent valence of +1, a crystal void space and an electropositive cation. | ||
Several screening studies have successfully employed the design principles in seeking known or designing new electrides.18–22 However, the degree to which these design principles can be relied upon to define an electride in the general sense has recently been called in to question with the emergence of so-called intermetallic electrides, such as La3Cu2Si4,23 Y3Pd2,24 and LaRuSi25 reported in the literature. If the design principles are not adhered to, what distinguishes an electride from a metal or mixed alloy? On the other hand, to what extent are the design principles limiting the identification and use of these fascinating systems?
In this paper we investigate the design rules of electrides using a high-throughput approach. We find that the plus one valence rule is a simple yet surprisingly powerful motif in finding and designing new electrides by applying probable valence charge to all stable materials in the Materials Project database. In doing so, we identify 51 candidates as potential new electrides that were not reported previously and are able to use these to further comment on the relevance of the remaining two design principles. Overall, we conclude that electrides encompass a richer and broader swathe of chemistries than was thought possible until now, while confirming the effectiveness of at least two of the existing design rules.
Methods
The process of high-throughput screening for +1 valence electrides in this work is shown in Fig. 2.26 Our screening starts from the Materials Project database27 which includes more than 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 inorganic compounds and uses the Vienna ab initio simulation package (VASP) to calculate them.28 As the definition of electrides requires an excess electron, in the first step, we identify the materials with the total valence as +1, such as Ca2N. This is accomplished by mapping the most common oxidation states for each element, identified in a previous study,29 to chemical formulae in the database and summing the component values. In total this screening provides 3804 candidates.
000 inorganic compounds and uses the Vienna ab initio simulation package (VASP) to calculate them.28 As the definition of electrides requires an excess electron, in the first step, we identify the materials with the total valence as +1, such as Ca2N. This is accomplished by mapping the most common oxidation states for each element, identified in a previous study,29 to chemical formulae in the database and summing the component values. In total this screening provides 3804 candidates.
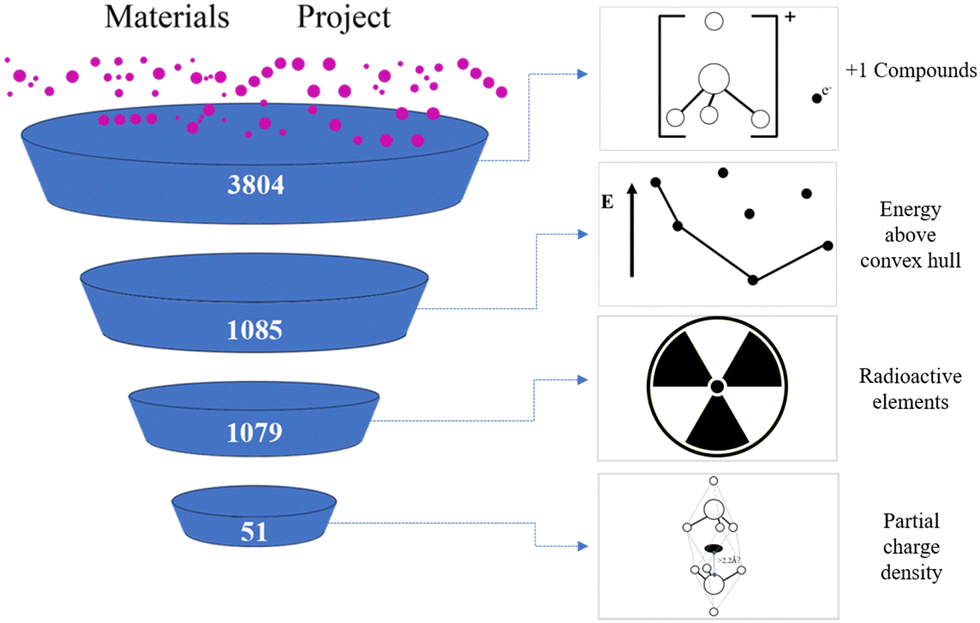 | ||
| Fig. 2 High-throughput screening process for +1 valence electrides, starting from the Material Projects. The decreasing size of the candidate pools on the left represents the number of compounds remaining after each step elimination process. | ||
Subsequently, we eliminate the unstable materials among the plus one valence candidates by cross-referencing the formulae against their “energy above convex hull” parameter in the Materials Project. If the energy is greater than zero eV, the candidates are discarded, leaving 1085 candidates remaining. Then, we also removed any compounds that contain radioactive elements (Np, U, Pa, Th, Pm, Ac) to arrive at a final number of 1079 materials, which were subject to ab initio analysis.
Ab initio calculations were performed using the Vienna ab initio simulation package (VASP) for the candidates. Density functional theory (DFT)30 implementing the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) function was used in each case.31 Spin polarization and magnetism was considered. All VASP input files for the structure optimization were automatically generated by pymatgen32 “io.vasp.sets” module with the default parameters provided by Materials Project. Bader charge33–36 analysis was performed to identify the electron distribution of the partial charge density within 0.2 eV around Fermi Energy level. Finally, we use Bader charge analysis to identify maxima in electron density 2.2 Å or more away from the nearest atom. Where present we consider the candidates as electrides, which was found to be effective in this regard in a previous study.2
Results and discussion
The goal of this work is to assess the validity of the three most common design principles for discovering new or uncovering known electrides. Naturally, this goal requires evoking at least one of these design rules first to gain a handle on the electrides against which to benchmark said rules. At the time of writing the Materials Project hosts over 530000 nanoporous materials, which is a prohibitively large candidate pool and includes families of materials that as yet have produced no confirmed electrides, such as metal–organic-frameworks or MOFs. Thus, the feature of void space within the crystal structure remains intractable as an early approach to high-throughput analysis for the time being. The heuristic rule of strongly electron-donating constituent elements is one feature to which virtually all confirmed electrides conform and one that could in theory be the starting point for an electride search. At the time of writing however, there are 60933 compounds in the Materials Project database containing at least one of the alkali- or alkali-earth metals, which is already unmanageable, without also accounting for electrides with cations outside of this group such as the confirmed electride Y5Si3.37 By contrast, the rule of total valence summing to plus one has recently become a viable screening tool for the first time. Ding et al.29 reported scanning the inorganic crystal structure database (ICSD)38 containing 169800 materials, to find the oxidation states of 108 elements. They found that just 84 oxidation states could cover more than 90% of all reported charges in the ICSD. The small number of the probable oxidation states effectively reduces the combinatorial explosion of possibilities when matching elements to make compounds and allows for a most probably overall valence value to be calculated for a given chemical formula. Fig. 3 is found by taking the most probable oxidation states reported by Ding et al. to calculate the overall charge of all compounds in the Materials Project (including those not thermodynamically stable). The x-axis is the total valence of compounds, and the y axis is the number of the compounds corresponding to the total valence. As can be seen, there are around 4000 compounds with an overall oxidation state of +1, which is a surprisingly large candidate pool for finding electrides but still much more manageable than the candidates approximated by the other two design rules. Naturally however, the overall largest assigned valence is zero by a wide margin, corroborating the results of the previous study and perhaps explaining the perceived rarity of electrides. As for the compounds that present neither an overall charge of zero or plus one, we see reported entries that have very unique chemistries. For example, the material SF6 (mp-8560)39 returns an overall charge of −8 with this method. The large and broad array of materials with charges greater than +1 or indeed greater than +8 predominantly consist of alloys and mixed metals systems, for example PuGe3 (mp-21484) has a nominative charge of +16 from this approach.40 We refer the interested reader to the original study of Ding et al. for more information,29 but overall consider these instances as heuristic outliers beyond the scope of this study.
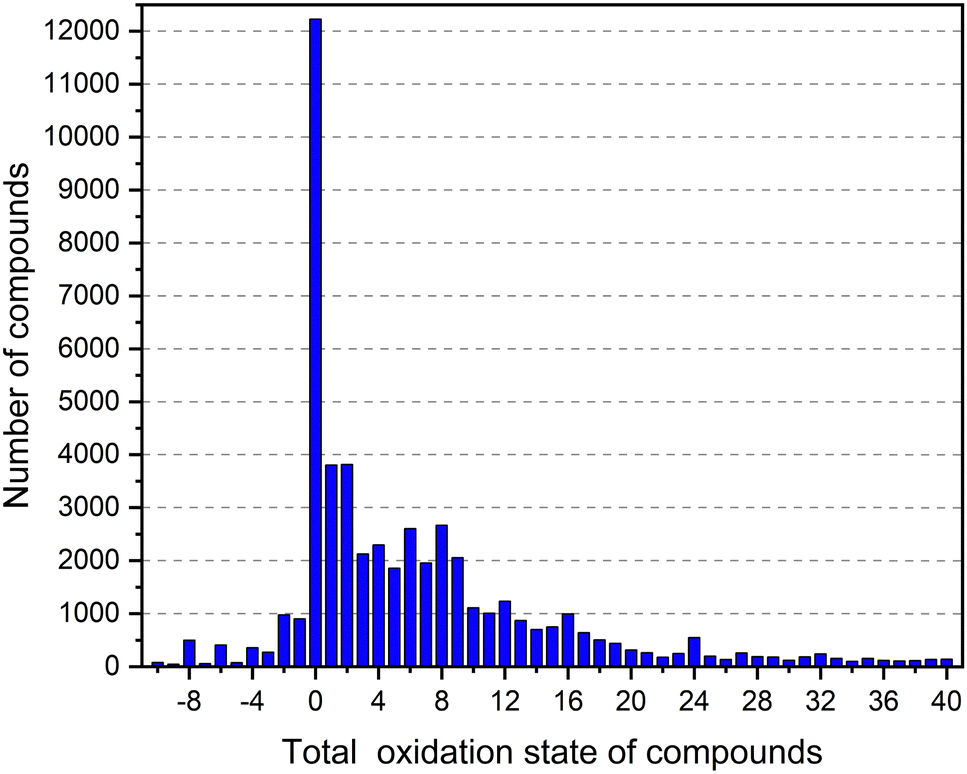 | ||
| Fig. 3 The distribution of overall oxidation state for compounds in the Materials Project using the most probable valence found by Ding et al. The x axis is the total oxidation state of compounds, and the y axis is the number of the compounds corresponding to the total valence. | ||
After obtaining the compounds that have an overall valence of +1, an energy above convex hull not greater than 0 eV and no radioactive elements, we simulate the material with density functional theory (DFT). Further analysis using the Bader charge method (described in the Methods section) identifies 51 candidates, which include 4 compounds that have been confirmed as electrides previously in experiment (Ca2N, Sr2N, Ba2N and Ca12Al14O32) and one previously predicted to be an electride (Cs3O).41 It is of course unsurprising to find many of the archetypal candidates with the +1 valence design principle, since that principle was derived mainly from these well-known examples, but their presence does lend confidence to the ab initio screening approach. The full list of the 51 electride candidates, their structure and partial charge density can be found in the ESI.†
We conduct analysis on all of the predicted electride materials that were screened using our method. Firstly, we can consider the design principle of electropositive elements; thus the statistics on the composed elements of these 51 candidates are shown in Fig. 4. The electrides are mostly composed of Lanthanide (26.6%) and transition metal (26.6%) elements. For the lanthanides this is perhaps unsurprising as they are relatively electropositive and are unlikely to take back the anionic electron. The prevalence of transition metals is surprising by contrast as transition metals can often present multiple valences and therefore should be able to accept the electron back from the crystal structure. It was only recently that the first redox active electride was reported with the transition metal chromium in the compound Sr3CrN3.42 In the Sr3CrN3 electride the Cr exhibits a +4 charge but it is commonly reported as stable at +3, thus it defies expectations by not reclaiming the free electron in the crystal structure. Furthermore 16% and 10.6% of electrides are composed of alkali metal and alkali earth metal, respectively, which is a return to expected behaviour in line with existing design principles.
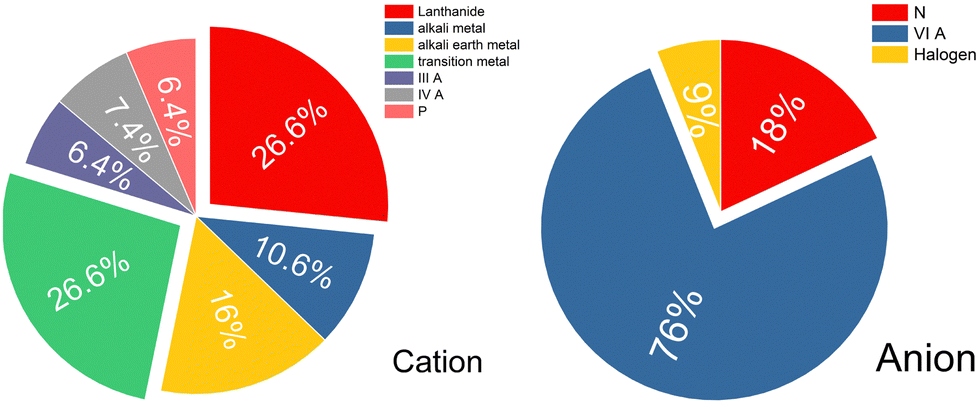 | ||
| Fig. 4 Percentage distribution of cations (left panel) and anions (right panel) in the identified candidates. The labels IIIA, IVA, P, N, VIA, Halogen correspond to group IIA elements, group IVA elements, Phosphorus, group VIA elements and Halogens, respectively. | ||
We go further than the usual design rule of only considering the cation to also consider the present anions. For the negatively charged ions, group six elements make up the majority of the compostion, accounting for 76%. Such a high prevalence of chalcogenides is quite surprising, especially as oxygen is known to be very electronegative; second only to fluorine in its ability to attract and hold electron density. That said, perhaps the most well-known and well applied electride is the oxide Ca12Al14O32. Hopefully this result indicates that yet more air and water stable electrides can be made if the systems are already oxidised in their native form. Only 6% of the total results set is composed of halogens. Halogen electrides have been predicted to exists,26 but are not prevalent among experimentally demonstrated electrides. It is however, once again not necessarily in line with expectations as a strongly electronegative ion could draw the anionic electron out of the structure. The final surprising result is the relatively small proportion of nitrides among these candidates, considering that nitride compounds comprise the majority of experimentally confirmed electrides. Overall, these findings indicate a surprising breadth to potential electride chemistry that has barely begun to be accessed, the lanthanide and transition-metal combinations with group VIA elements especially represent a promising but relatively unexplored research area that merits further investigation.
To combine considerations of cation and anion components we consider the general property of electronegativity. Electronegativity is a measure of an atom's ability to attract electrons towards itself when it forms a chemical bond with another atom.43 We saw that the formation of electrides is often associated with the presence of highly electropositive elements, such as alkali metals, alkaline earth metals, and rare earth metals. These elements have a low electronegativity and can easily donate electrons to form electrides thus it may be the case that chemical formulae with apparent +1 valence also exhibit unusual values in their average electronegativity. The average electronegativity is calculated by multiplying the electronegativity of each element by the corresponding number of atoms in the chemical formula, summing and dividing by the total number of atoms. The average electronegativity of the final 51 candidates can be found in Fig. 5. The full list of all the electronegativies of electrides can be seen in the ESI† (Table S1). As the figure shows, some of the electrides do have a relatively high average electronegativity. For comparison, the materials CaCO3, Fe2O3 and NaCl have average electronegativities of 1.4, 2.8 and 2.0 respectively and are certainly not considered electrides. However, Cs3O has the smallest average electronegatively of all candidates of 1.45 and can confidently be counted as an electride based upon our and other published studies.41,44 Given that the majority (over 80%) of our candidates have electronegativity values between the 1.4 and 2.8 of the non-electrides CaCO3 and Fe2O3 respectively, it is unlikely that average electronegativity derived from the chemical formula can be used to predict electrides in the same way that total valence appears able.
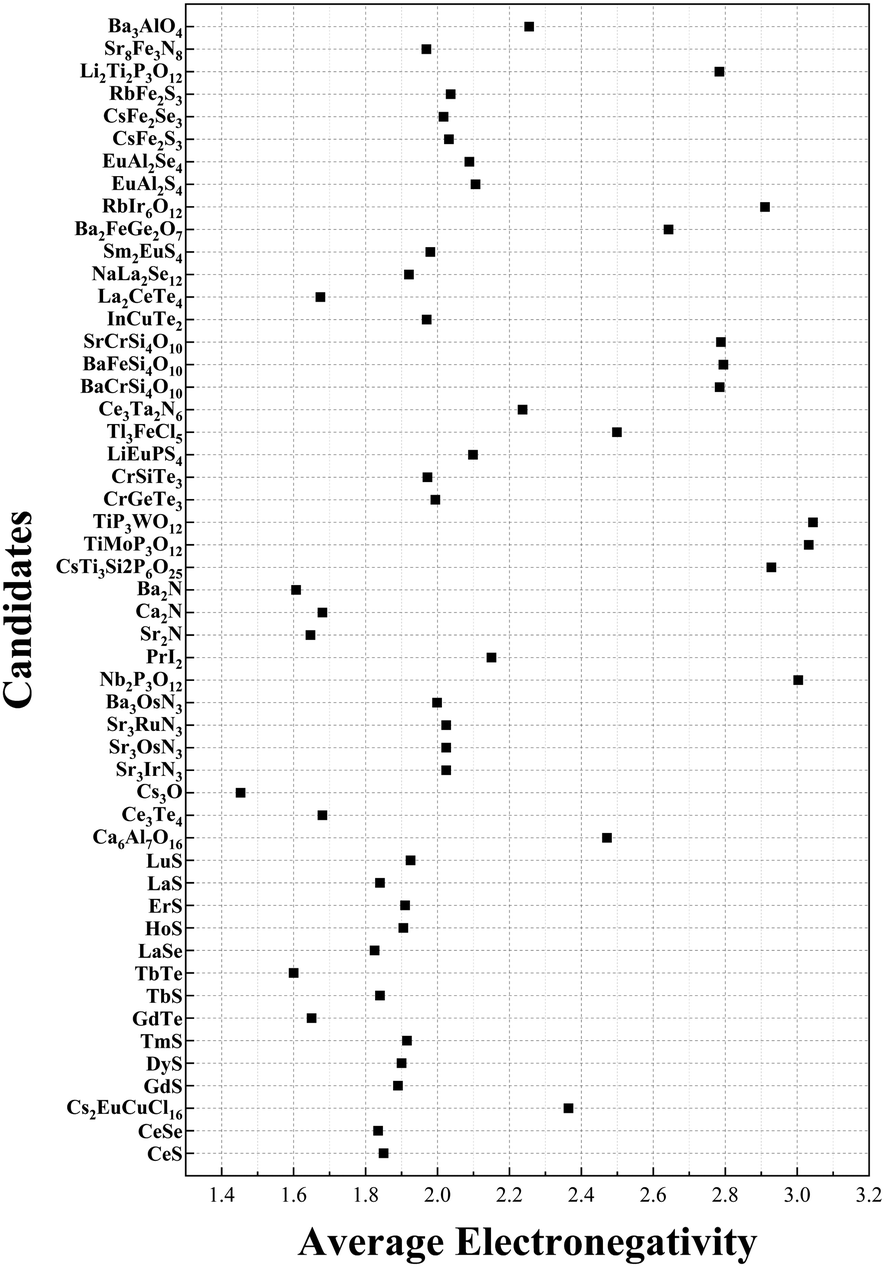 | ||
| Fig. 5 The average electronegativity for each of the 51 electride candidates. | ||
We also investigate the design principle of internal space for hosting the anionic electron in candidate compounds. In order to assess the influence of internal distance on the identified electrides, we treat the nearest neighbour distance from the off-atom peak in electron density as a variable; the results of which are shown in Fig. 6. The x-axis is the minimum distance we set and the y-axis is the number of candidates containing electron density maxima greater than that distance from the nearest atom. The electride candidates at each distance are shown in the Table S3 (ESI†). The figure shows the perhaps unsurprising trend that, as the cut-off distance is increased, the number of the candidates decrease quite sharply. However, it is interesting to observe that at 2.8 Å cutoff, all five of the known electrides are present in the 9 candidates alongside Sr3RuN3, Sr3OsN3, EuAl2S4 and Ba3AlO4. In these, Sr3RuN3 and Sr3OsN3 have the same prototype structure and consistent formula with the experimentally confirmed electride Sr3CrN3 (previously discussed as the first redox active electride)42 and Ba3AlO4 which has a similar composition, component elements and acquisition methods to the confirmed electride Ca12Al14O32. Increasing the cutoff further to 2.9 Å results in the three confirmed electrides Ca2N, Ba2N, Ca12Al14O32 being eliminated among others and leaving the following compounds remaining: Sr2N, Cs3O and EuAl2S4. Sr2N is a previously confirmed electride,45 Cs3O is a previously predicted electride,41 but to the knowledge of the authors EuAl2S4 has not yet been considered as an electride. Given that every other candidate is all but confirmed to be an electride we have a high degree of confidence that that EuAl2S4 will be as well.
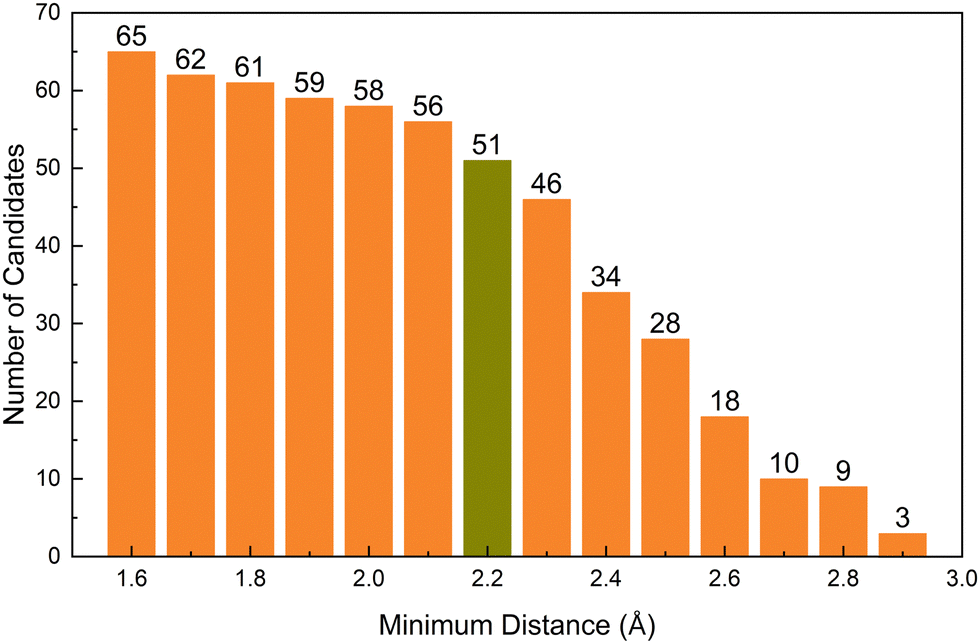 | ||
| Fig. 6 The number of electride candidates as a function of minimum distance (in Angstroms) from the nearest atom to the peak in off-atom electron density. | ||
The most common structure among the candidates is cubic with space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (No. 225). These correspond to LaS, LaSe, CeS, CeSe, GdS, GdTe, TbS, DyS, HoS, ErS, TmS, LuS, and Cs2EuCuCl6. The anionic electron for these compounds is located in the centre of the cubic coordination environment (see Fig. 7 as an example). Of these 14 cubic systems, 13 are binary compounds composed of f-block elements and group five elements. Those candidates are particularly interesting as they are rare-earth-based electrides, which have been predicted to have high chemical stability and unique electronic properties previously,46 making them promising candidates for various applications. There are also two candidates that have cubic structure but with the space group I
m (No. 225). These correspond to LaS, LaSe, CeS, CeSe, GdS, GdTe, TbS, DyS, HoS, ErS, TmS, LuS, and Cs2EuCuCl6. The anionic electron for these compounds is located in the centre of the cubic coordination environment (see Fig. 7 as an example). Of these 14 cubic systems, 13 are binary compounds composed of f-block elements and group five elements. Those candidates are particularly interesting as they are rare-earth-based electrides, which have been predicted to have high chemical stability and unique electronic properties previously,46 making them promising candidates for various applications. There are also two candidates that have cubic structure but with the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d (No. 220), including the first room temperature stable inorganic electride C12A7:e−, which has been well studied before, and the other is Ce3Te4. To the knowledge of the authors, it is also the first time for Ce3Te4 to be considered as an electride.47
3d (No. 220), including the first room temperature stable inorganic electride C12A7:e−, which has been well studied before, and the other is Ce3Te4. To the knowledge of the authors, it is also the first time for Ce3Te4 to be considered as an electride.47
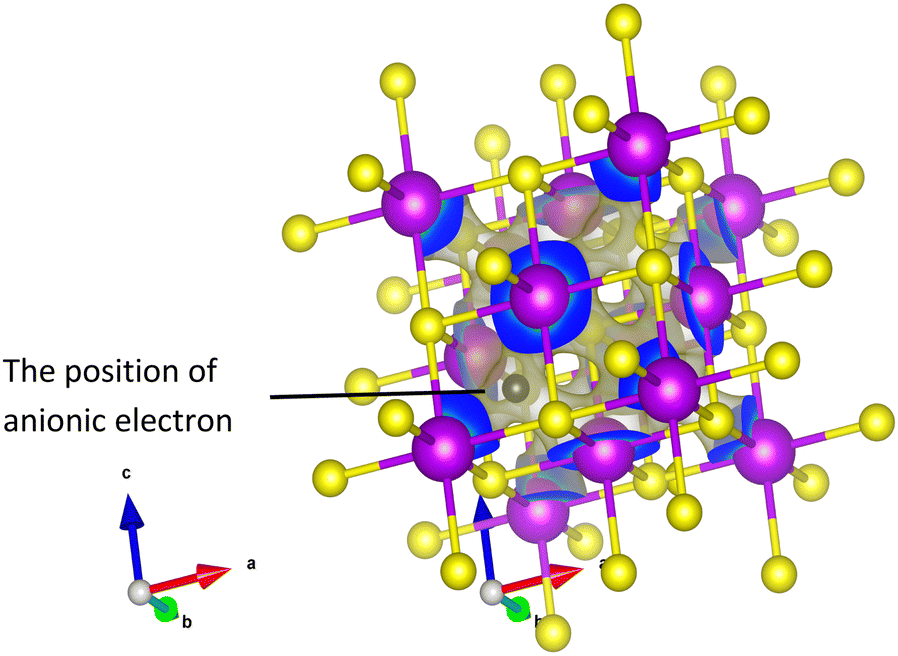 | ||
| Fig. 7 Electron density around the Fermi level for the material CeS as an example of the candidate electrides. The peak in electron density is in the centre of the cubic coordination environment. | ||
In our candidate pool, there are also 4 candidates that match the structure of Sr3CrN3, which as mentioned previously was recently reported to be the first electride with partially filled 3d-shell. These are Sr3IrN3, Sr3OsN3, Ba3OsN3, Sr3RuN3, all of them are hexagonal with the space group P63/m (No. 176). All of them are also ternary transition metal nitride compounds. The anion is located in a one-dimensional (1D) channel formed by a series of oriented cavities throughout the c axis of the material, see Fig. 8. Sr3CrN3 itself was found to be an electride in a previous screening study,2 however it is not included in our analysis because the oxidation of Cr element in Sr3CrN3 was allocated as the most common +3 valence in the study of Ding et al., which causes the assigned total valence of Sr3CrN3 to be equal to zero. However, this result highlights that there is likely a larger family of these 1D electrides compounds either already known or yet to be made and also lends significant credence to the design principle of +1 valence; even after so much research in the area it is possible to find many likely new electrides using such a primitive, heuristic approach.
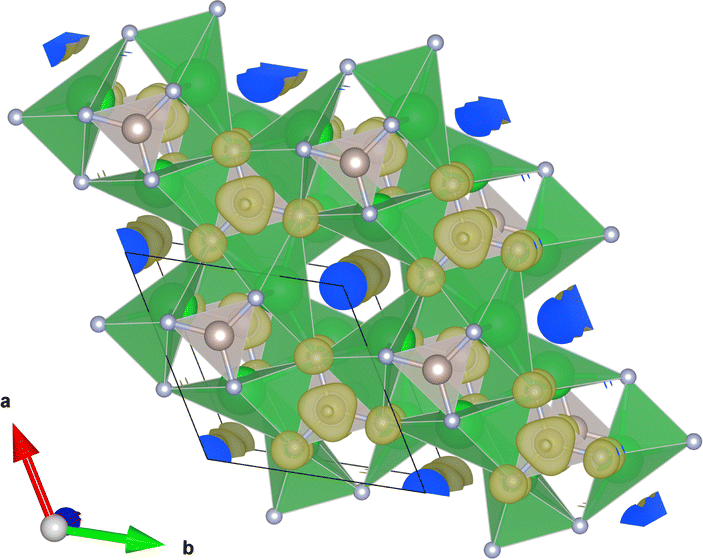 | ||
| Fig. 8 The electron density channel for the Sr3CrN3 type compounds (Sr3RuN3 in this case). | ||
Additionally, our analysis identifies the interesting candidate Ba3AlO4, which derives from the original compound Ba3AlO4H.48 It has similar composition ratio, component elements and chemistry to Ca12Al14O32 – the first electride stable at room temperature. Ba3AlO4 is orthorhombic with the space group Pnma (No. 62) and is shown alongside Ca12Al14O32 in Fig. 9. Both cases exhibit similar 0D electride behaviour. We calculate the band structures of Ba3AlO4 and Ba3AlO4H, as shown in Fig. 10. As we can see, the band gap in Ba3AlO4H disappears in Ba3AlO4 with the removal of hydrogen atoms, just as is observed for other electrides upon removal of an anion species, e.g. H in of Sr3CrN3 and O in Ca12Al14O32.49,50 The bands also show the strongly dispersive nature around the Fermi level, similar to the electronic band structures calculated for other electrides.2 Finally, while not identical, the analogous compound BaAl2O4−xHy system has been reported as an electride that can enhance ammonia synthesis when incorporated as a catalyst,51 again like Ca12Al14O32,11 which boosts the confidence in our findings.
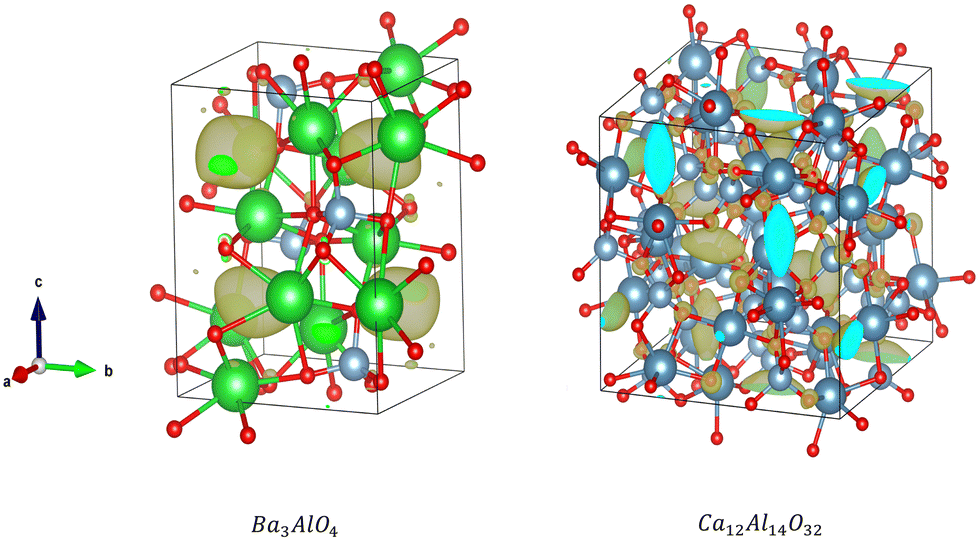 | ||
| Fig. 9 Electron density around the Fermi level for Ba3AlO4 (left) and Ca12Al14O32 (right). | ||
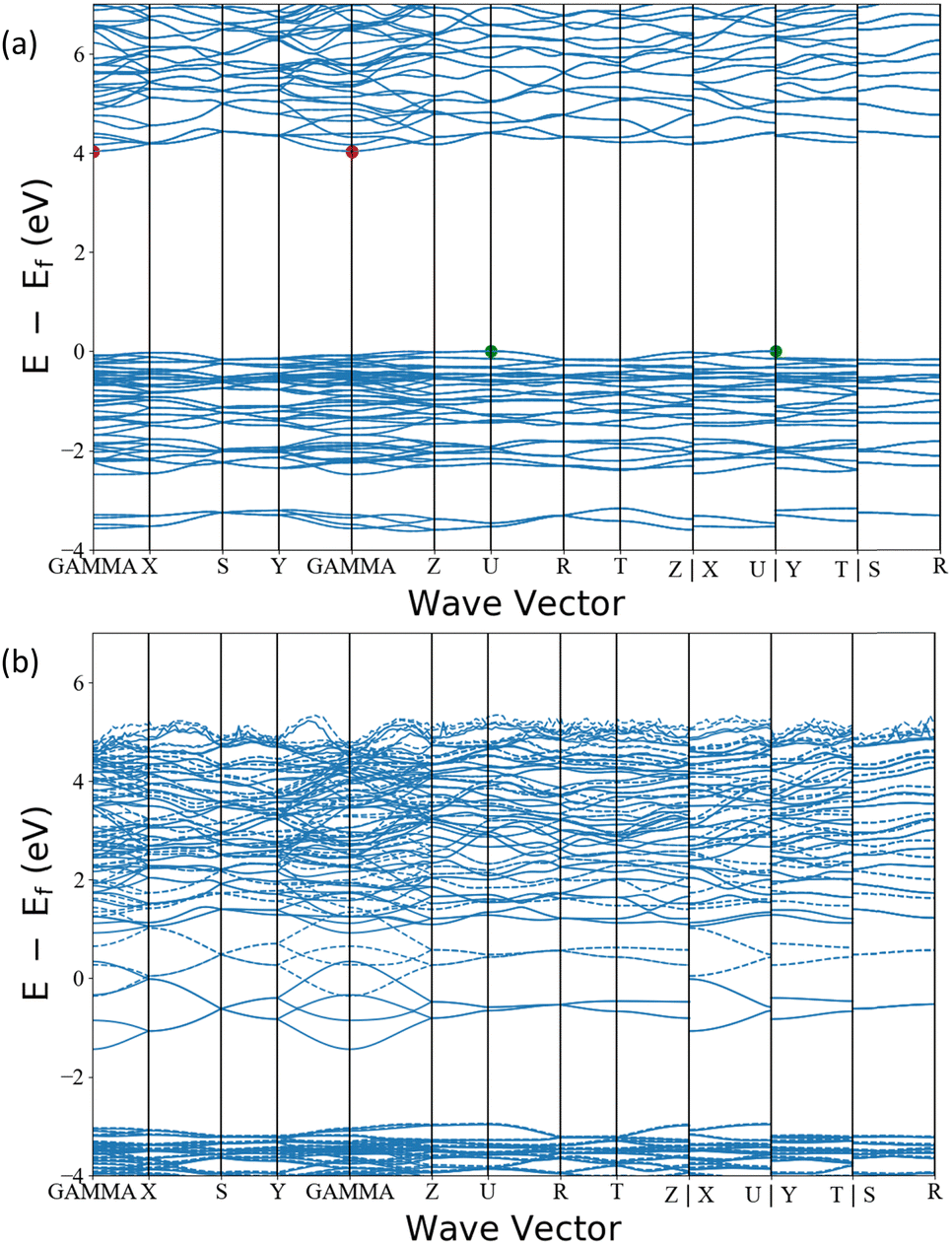 | ||
| Fig. 10 The electronic band structure of (a) Ba3AlO4H and (b) Ba3AlO4. | ||
A few other candidates that are worthy of note are Nb2P3O12, which is the only instance of niobium present in an electride in our candidate set and, to the knowledge of the authors, the first predicted niobium containing electride (see Fig. S17 of the ESI† for the chemical structure and electron density plot for this material). This compound contains no strongly electron positive species, only the niobium, which is of course a transition metal element capable of multiple oxidation states. GdS and GdTe are also worthy of further exploration as isostructural cubic 0D gadolinium electrides, given that Gd2C is a known electride that exhibits ferromagnetism above room temperature.52 The structure and electron density for these compounds is shown in Fig. 7.
In this work and previous works, candidates have tended to be grouped based on their structure. Among the electrides we identified, there are many electrides with same prototype structure and consistent formula, such as Sr3IrN3, Sr3OsN3, Sr3RuN3, and Ba3OsN3; Ca2N, Sr2N, and Ba2N; BaCrSi4O10, BaFeSi4O10 and SrCrSi4O10etc. which indicates that chemical substitution from confirmed electrides is a good way to design new electrides. However, we note that electride behaviour is not solely defined by structure. For instance, BaFeSi4O10 and SrCrSi4O10 are candidates that have the identical structure but have discernibly different electron distributions around the Fermi energy, as show in Fig. 11.
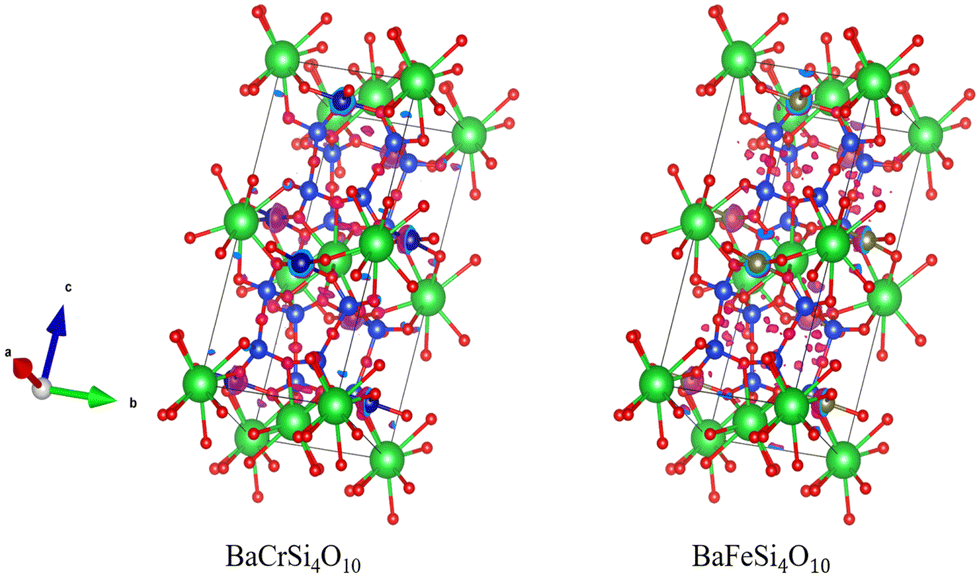 | ||
| Fig. 11 Electron density around the Fermi level of BaCrSi4O10 (left) and BaFeSi4O10 (right) showing different isosurfaces despite identical chemical structure. | ||
Finally, we highlight potential shortcomings in our method. Firstly, we enforce a criteria of energy above convex hull of 0 eV. This has previously been reported as too strict to account for the errors of DFT and it has previously suggested that a cut-off of 24 meV above convex hull would better capture all candidates instead.53 Our method also only captures peaks in electron density within a window of 0.2 eV around the Fermi Energy level. This implicitly means that our method will not capture any potential semiconductor electrides, the first of which was reported recently.54 These drawbacks mean that our results set is likely retrieving the bare minimum candidates and there are many more yet to be uncovered.
Conclusions
In summary, we have performed a high-throughput ab initio screening of all potentially stable electrides by automatically assigning common oxidation states to the constituent elements of compounds that are on their convex hull. Of the >1000 stable materials with a valence of +1 we identify 51 candidate electrides, 4 of which were already proven in experiment-demonstrating the validity of our approach.We used our results set to effectively explore the established design principles commonly used to identify electrides. We discuss the element composition with respect to cations, anions and overall electronegativity and find a surprising breadth of chemistries on display. The heuristic rule of necessitating strongly donating cations applies to many cases but not all, see Nb2P3O12 for example (Fig. S17, ESI†). Similarly, we see a strong presence of transitional metals and variable oxidation state cations in our candidates, see for example CrSiTe3 (Fig. S27, ESI†), which have been previously eschewed in screening studies seeking electrides.19 We also find that the anions can and are often surprisingly electronegative, with nitrides being a minority of candidates unlike the case for confirmed electrides in experiment. This observation imparts hope for further discovery of more air and water stable electrides such as Ca12Al14O32 that can find use in industrial applications. To this end, we recommend the analogous Ba3AlO4 as a promising candidate for future study (Fig. S11, ESI†). Unfortunately, we find that average electronegativity does not correlate with electride-like tendencies for compounds.
We also considered the internal space present in the crystal structure of the candidate electrides and find that the design principle of a large pore for hosting anionic electrons appears valid up to a cut-off of around 3 Å. Several of the confirmed electrides conform to this rule at the upper end of the scale. Whether the electrides with this internal distance are disproportionately confirmed in experiment because the largest pore sizes are easier to characterise in experiment or this distance has an intrinsic significance to anionic electron species warrants further investigation.
Finally, our study inherently relies on the design principle of +1 valence. This was a necessity in order to limit the number of candidates to the realm of feasibility but our method of mapping overall valence on to chemical formulae also shows ample positive valence states higher than +1. We believe it likely that more strongly electropositive electrides reside in this space and encourage the exploration of these interesting compounds.
Overall, we believe we have uncovered yet more electrides to contribute to the ever-widening pool of these interesting materials. We have high-confidence in all of our candidates but Ba3AlO4, Sr3IrN3 and Sr3RuN3 especially are remarkably similar to confirmed electrides.
The compounds discussed in this work are anticipated to have significance for a diverse range of applications and contribute to the fundamental understanding of materials. Each of them are detailed by materials project ID, chemical formula, structure, symmetry and valence electron density in the ESI.†
Conflicts of interest
There are no conflicts to declare.References
- C. Liu, S. A. Nikolaev, W. Ren and L. A. Burton, Electrides: A Review, J. Mater. Chem. C, 2020, 8(31), 10551–10567, 10.1039/D0TC01165G.
- L. A. Burton, F. Ricci, W. Chen, G.-M. Rignanese and G. Hautier, High-Throughput Identification of Electrides from All Known Inorganic Materials, Chem. Mater., 2018, 30(21), 7521–7526, DOI:10.1021/acs.chemmater.8b02526.
- C. Wang, M. Xu, K. T. Butler and L. A. Burton, Ultralow Work Function of the Electride Sr 3 CrN 3, Phys. Chem. Chem. Phys., 2022, 24(15), 8854–8858, 10.1039/D1CP05623A.
- R. H. Huang and J. L. Dye, Low Temperature (−80 °C) Thermionic Electron Emission from Alkalides and Electrides, Chem. Phys. Lett., 1990, 166(2), 133–136, DOI:10.1016/0009-2614(90)87265-S.
- T. Eguchi, M. Sasao, Y. Shimabukuro, F. Ikemoto, M. Kisaki, H. Nakano, K. Tsumori and M. Wada, A Compact Electron Cyclotron Resonance Negative Hydrogen Ion Source for Evaluation of Plasma Electrode Materials, Rev. Sci. Instrum., 2020, 91(1), 013508, DOI:10.1063/1.5128610.
- H. Yanagi, K.-B. Kim, I. Koizumi, M. Kikuchi, H. Hiramatsu, M. Miyakawa, T. Kamiya, M. Hirano and H. Hosono, Low Threshold Voltage and Carrier Injection Properties of Inverted Organic Light-Emitting Diodes with [Ca24Al28O64]4+ (4e−) Cathode and Cu2− xSe Anode, J. Phys. Chem. C, 2009, 113(42), 18379–18384, DOI:10.1021/jp906386q.
- H.-M. He, Y. Li, H. Yang, D. Yu, S.-Y. Li, D. Wu, J.-H. Hou, R.-L. Zhong, Z.-J. Zhou, F.-L. Gu, J. M. Luis and Z.-R. Li, Efficient External Electric Field Manipulated Nonlinear Optical Switches of All-Metal Electride Molecules with Infrared Transparency: Nonbonding Electron Transfer Forms an Excess Electron Lone Pair, J. Phys. Chem. C, 2017, 121(1), 958–968, DOI:10.1021/acs.jpcc.6b11919.
- H. Hosono, S.-W. Kim, S. Matsuishi, S. Tanaka, A. Miyake, T. Kagayama and K. Shimizu, Superconductivity in Room-Temperature Stable Electride and High-Pressure Phases of Alkali Metals, Philos. Trans. R. Soc., A, 2015, 373(2037), 20140450, DOI:10.1098/rsta.2014.0450.
- T.-N. Ye, J. Li, M. Kitano and H. Hosono, Unique Nanocages of 12CaO·7Al2O3 Boost Heterolytic Hydrogen Activation and Selective Hydrogenation of Heteroarenes over Ruthenium Catalyst, Green Chem., 2017, 19(3), 749–756, 10.1039/C6GC02782B.
- Y. Toda, H. Hirayama, N. Kuganathan, A. Torrisi, P. V. Sushko and H. Hosono, Activation and Splitting of Carbon Dioxide on the Surface of an Inorganic Electride Material, Nat. Commun., 2013, 4(1), 2378, DOI:10.1038/ncomms3378.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S.-W. Kim, M. Hara and H. Hosono, Ammonia Synthesis Using a Stable Electride as an Electron Donor and Reversible Hydrogen Store, Nat. Chem., 2012, 4(11), 934–940, DOI:10.1038/nchem.1476.
- J. L. Dye and M. G. DeBacker, Physical and Chemical Properties of Alkalides and Electrides, Annu. Rev. Phys. Chem., 1987, 38(1), 271–299, DOI:10.1146/annurev.pc.38.100187.001415.
- A. Ellaboudy, J. L. Dye and P. B. Smith, Cesium 18-Crown-6 Compounds. A Crystalline Ceside and a Crystalline Electride, J. Am. Chem. Soc., 1983, 105(21), 6490–6491, DOI:10.1021/ja00359a022.
- S. Matsuishi, Y. Toda, M. Miyakawa, K. Hayashi, T. Kamiya, M. Hirano, I. Tanaka and H. Hosono, High-Density Electron Anions in a Nanoporous Single Crystal: [Ca24Al28O64]4+ (4e−), Science, 2003, 301(5633), 626–629, DOI:10.1126/science.1083842.
- Y. Zhang, Z. Xiao, T. Kamiya and H. Hosono, Electron Confinement in Channel Spaces for One-Dimensional Electride, J. Phys. Chem. Lett., 2015, 6(24), 4966–4971, DOI:10.1021/acs.jpclett.5b02283.
- K. Lee, S. W. Kim, Y. Toda, S. Matsuishi and H. Hosono, Dicalcium Nitride as a Two-Dimensional Electride with an Anionic Electron Layer, Nature, 2013, 494(7437), 336–340, DOI:10.1038/nature11812.
- J. L. Dye, Electrides: Ionic Salts with Electrons as the Anions, Science, 1990, 247(4943), 663–668, DOI:10.1126/science.247.4943.663.
- Y. Zhang, H. Wang, Y. Wang, L. Zhang and Y. Ma, Computer-Assisted Inverse Design of Inorganic Electrides, Phys. Rev. X, 2017, 7(1), 011017, DOI:10.1103/PhysRevX.7.011017.
- T. Tada, S. Takemoto, S. Matsuishi and H. Hosono, High-Throughput Ab Initio Screening for Two-Dimensional Electride Materials, Inorg. Chem., 2014, 53(19), 10347–10358, DOI:10.1021/ic501362b.
- C. J. Pickard and R. J. Needs, Dense Low-Coordination Phases of Lithium, Phys. Rev. Lett., 2009, 102(14), 146401, DOI:10.1103/PhysRevLett.102.146401.
- Y. Ma, M. Eremets, A. R. Oganov, Y. Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle and V. Prakapenka, Transparent Dense Sodium, Nature, 2009, 458(7235), 182–185, DOI:10.1038/nature07786.
- Z. Wang, Y. Gong, M. L. Evans, Y. Yan, S. Wang, N. Miao, R. Zheng, G.-M. Rignanese and J. Wang, Machine Learning-Accelerated Discovery of A2BC2 Ternary Electrides with Diverse Anionic Electron Densities, J. Am. Chem. Soc., 2023, 145(48), 26412–26424, DOI:10.1021/jacs.3c10538.
- T.-N. Ye, Y. Lu, J. Li, T. Nakao, H. Yang, T. Tada, M. Kitano and H. Hosono, Copper-Based Intermetallic Electride Catalyst for Chemoselective Hydrogenation Reactions, J. Am. Chem. Soc., 2017, 139(47), 17089–17097, DOI:10.1021/jacs.7b08252.
- T.-N. Ye, Y. Lu, Z. Xiao, J. Li, T. Nakao, H. Abe, Y. Niwa, M. Kitano, T. Tada and H. Hosono, Palladium-Bearing Intermetallic Electride as an Efficient and Stable Catalyst for Suzuki Cross-Coupling Reactions, Nat. Commun., 2019, 10(1), 1–10, DOI:10.1038/s41467-019-13679-0.
- J. Wu, J. Li, Y. Gong, M. Kitano, T. Inoshita and H. Hosono, Intermetallic Electride Catalyst as a Platform for Ammonia Synthesis, Angew. Chem., Int. Ed., 2019, 58(3), 825–829, DOI:10.1002/anie.201812131.
- J. Zhou, L. Shen, M. Yang, H. Cheng, W. Kong and Y. P. Feng, Discovery of Hidden Classes of Layered Electrides by Extensive High-Throughput Material Screening, Chem. Mater., 2019, 31(6), 1860–1868, DOI:10.1021/acs.chemmater.8b03021.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation, APL Mater., 2013, 1(1), 011002, DOI:10.1063/1.4812323.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed (accessed 2021-11-23).
- Y. Ding, Y. Kumagai, F. Oba and L. A. Burton, Data-Mining Element Charges in Inorganic Materials, J. Phys. Chem. Lett., 2020, 11(19), 8264–8267, DOI:10.1021/acs.jpclett.0c02072.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136(3B), B864 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (Pymatgen): A Robust, Open-Source Python Library for Materials Analysis, Comput. Mater. Sci., 2013, 68, 314–319, DOI:10.1016/j.commatsci.2012.10.028.
- W. Tang, E. Sanville and G. Henkelman, A Grid-Based Bader Analysis Algorithm without Lattice Bias, J. Phys.: Condens. Matter, 2009, 21(8), 084204, DOI:10.1088/0953-8984/21/8/084204.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, Improved Grid-Based Algorithm for Bader Charge Allocation, J. Comput. Chem., 2007, 28(5), 899–908, DOI:10.1002/jcc.20575.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36(3), 354–360, DOI:10.1016/j.commatsci.2005.04.010.
- M. Yu and D. R. Trinkle, Accurate and Efficient Algorithm for Bader Charge Integration, J. Chem. Phys., 2011, 134(6), 064111, DOI:10.1063/1.3553716.
- Y. Lu, J. Li, T. Tada, Y. Toda, S. Ueda, T. Yokoyama, M. Kitano and H. Hosono, Water Durable Electride Y5Si3: Electronic Structure and Catalytic Activity for Ammonia Synthesis, J. Am. Chem. Soc., 2016, 138(12), 3970–3973, DOI:10.1021/jacs.6b00124.
- Inorganic Crystal Structure Database – ICSD | FIZ Karlsruhe. https://www.fiz-karlsruhe.de/de/produkte-und-dienstleistungen/inorganic-crystal-structure-database-icsd (accessed 2021-11-23).
- J. K. Cockcroft and A. N. Fitch, The solid phases of sulphur hexafluoride by powder neutron diffraction, Z. Kristallogr. - Cryst. Mater., 1988, 184(1–4), 123–146, DOI:10.1524/zkri.1988.184.14.123.
- S. M. Baizaee and A. Pourghazi, Structural Stability, Electronic Structure and f Hybridization of PuM3 and Pu3M (M = Ge, Sn, Pb) Intermetallic Compounds, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 387(1), 287–291, DOI:10.1016/j.physb.2006.04.034.
- H. Hosono, Electron Transfer from Support/Promotor to Metal Catalyst: Requirements for Effective Support, Catal. Lett., 2022, 152(2), 307–314, DOI:10.1007/s10562-021-03648-y.
- P. Chanhom, K. E. Fritz, L. A. Burton, J. Kloppenburg, Y. Filinchuk, A. Senyshyn, M. Wang, Z. Feng, N. Insin, J. Suntivich and G. Hautier, Sr3CrN3: A New Electride with a Partially Filled d-Shell Transition Metal, J. Am. Chem. Soc., 2019, 141(27), 10595–10598, DOI:10.1021/jacs.9b03472.
- L. Pauling, The Nature of the Chemical Bond. IV. The Energy of Single Bonds and the Relative Electronegativity of Atoms, J. Am. Chem. Soc., 1932, 54(9), 3570–3582, DOI:10.1021/ja01348a011.
- C. Park, S. W. Kim and M. Yoon, First-Principles Prediction of New Electrides with Nontrivial Band Topology Based on One-Dimensional Building Blocks, Phys. Rev. Lett., 2018, 120(2), 026401, DOI:10.1103/PhysRevLett.120.026401.
- N. E. Brese and M. O’Keeffe, Synthesis, Crystal Structure, and Physical Properties of Sr2N, J. Solid State Chem., 1990, 87(1), 134–140, DOI:10.1016/0022-4596(90)90074-8.
- Y. Lu, J. Li, T.-N. Ye, Y. Kobayashi, M. Sasase, M. Kitano and H. Hosono, Synthesis of Rare-Earth-Based Metallic Electride Nanoparticles and Their Catalytic Applications to Selective Hydrogenation and Ammonia Synthesis, ACS Catal., 2018, 8(12), 11054–11058, DOI:10.1021/acscatal.8b03743.
- T. Vo, P. von Allmen, C.-K. Huang, J. Ma, S. Bux and J.-P. Fleurial, Electronic and Thermoelectric Properties of Ce3Te4 and La3Te4 Computed with Density Functional Theory with on-Site Coulomb Interaction Correction, J. Appl. Phys., 2014, 116(13), 133701, DOI:10.1063/1.4896670.
- B. Huang and J. D. Corbett, Ba3AlO4H: Synthesis and Structure of a New Hydrogen-Stabilized Phase, J. Solid State Chem., 1998, 141(2), 570–575, DOI:10.1006/jssc.1998.8022.
- M. Xu, C. Wang, B. J. Morgan and A. Burton, L. Hydride Ion Intercalation and Conduction in the Electride Sr3CrN3, J. Mater. Chem. C, 2022, 10(17), 6628–6633, 10.1039/D1TC05850A.
- R. P. S. M. Lobo, N. Bontemps, M. I. Bertoni, T. O. Mason, K. R. Poeppelmeier, A. J. Freeman, M. S. Park and J. E. Medvedeva, Optical Conductivity of Mayenite: From Insulator to Metal, J. Phys. Chem. C, 2015, 119(16), 8849–8856, DOI:10.1021/acs.jpcc.5b00736.
- Y. Jiang, R. Takashima, T. Nakao, M. Miyazaki, Y. Lu, M. Sasase, Y. Niwa, H. Abe, M. Kitano and H. Hosono, Boosted Activity of Cobalt Catalysts for Ammonia Synthesis with BaAl2O4–xHy Electrides, J. Am. Chem. Soc., 2023, 145(19), 10669–10680, DOI:10.1021/jacs.3c01074.
- S. Y. Lee, J.-Y. Hwang, J. Park, C. N. Nandadasa, Y. Kim, J. Bang, K. Lee, K. H. Lee, Y. Zhang, Y. Ma, H. Hosono, Y. H. Lee, S.-G. Kim and S. W. Kim, Ferromagnetic Quasi-Atomic Electrons in Two-Dimensional Electride, Nat. Commun., 2020, 11(1), 1526, DOI:10.1038/s41467-020-15253-5.
- G. Hautier, S. P. Ong, A. Jain, C. J. Moore and G. Ceder, Accuracy of Density Functional Theory in Predicting Formation Energies of Ternary Oxides from Binary Oxides and Its Implication on Phase Stability, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(15), 155208, DOI:10.1103/PhysRevB.85.155208.
- L. M. McRae, R. C. Radomsky, J. T. Pawlik, D. L. Druffel, J. D. Sundberg, M. G. Lanetti, C. L. Donley, K. L. White and S. C. Warren, Sc2C, a 2D Semiconducting Electride, J. Am. Chem. Soc., 2022, 144(24), 10862–10869, DOI:10.1021/jacs.2c03024.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc01468e |
| This journal is © The Royal Society of Chemistry 2024 |