
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnetic amino-functionalized graphene oxide nanocomposite for PFAS removal from water†
Shokouh
Mahpishanian
,
Muchu
Zhou
 and
Reza
Foudazi
*
and
Reza
Foudazi
*
School of Sustainable Chemical, Biological, and Materials Engineering, The University of Oklahoma, Norman, Oklahoma 73019, USA. E-mail: rfoudazi@ou.edu
First published on 13th August 2024
Abstract
The “forever chemicals”, per- and polyfluoroalkyl substances (PFAS), have become a threat to public health and environment because of their toxicity and bioaccumulation. Addressing this critical issue, we develop a state-of-the-art nanocomposite adsorbent by covalently grafting amine functional groups onto graphene oxide (GO) surfaces and making them magnetic with iron-oxide (Fe3O4) nanoparticles. This process results in the creation of magnetic amine-functionalized graphene oxide (MAGO). The efficiency of MAGO is evaluated in the adsorptive removal of perfluorooctanoic acid (PFOA), perfluorooctane sulfonate (PFOS), perfluorohexane sulfonate (PFHxS), and perfluorobutane sulfonate (PFBS) as model long-chain and short-chain PFAS under different experimental conditions. Our findings reveal that MAGO achieves remarkable removal rates—exceeding 95% for long-chain PFAS and 85% for short-chain PFAS within just 30 minutes—demonstrating not only rapid kinetics but also a resilience across pH levels from 4 to 7. These results are indicative of the synergistic effects of GO and amine groups, harnessing both electrostatic and hydrophobic interactions to adsorb PFAS molecules. MAGO not only shows potent pollutant removal but also has impressive regeneration capabilities. Moreover, we demonstrate a novel liquid phase extraction method for PFAS detection, utilizing a colored methylene blue-PFAS complex for spectrophotometric analysis.
Environmental significancePer- and polyfluoroalkyl substances (PFAS), commonly referred to as “forever chemicals,” pose a significant threat to public health and the environment due to their persistent nature, toxicity, and ability to bioaccumulate. Our development of a cutting-edge nanocomposite adsorbent, magnetic amine-functionalized graphene oxide (MAGO), addresses this critical environmental issue by offering an effective and rapid method for PFAS removal from contaminated water sources. MAGO's high removal efficiencies—over 95% for long-chain PFAS and 85% for short-chain PFAS—highlight its potential as a robust solution for mitigating PFAS pollution. The nanocomposite's stability across various pH levels and its regenerative capabilities further enhance its practical application in diverse environmental settings. Additionally, our innovative liquid phase extraction method for PFAS detection simplifies and improves the accuracy of monitoring these pollutants. Overall, this research contributes significantly to the development of sustainable technologies for environmental remediation and public health. |
1. Introduction
The increasing utilization of chemicals threatens both human health and the environment, and it presents an obstacle to meeting the growing need for clean drinking water supplies.1 Per- and polyfluoroalkyl substances (PFAS) consist of a family of highly stable, man-made chemicals featuring a carbon–fluorine backbone, typically including carboxylate or sulfonate headgroups. PFAS have found applications across various industries, including fire-fighting foams, non-stick cookware, and semiconductors owing to their unique physicochemical properties, i.e., possessing high chemical and thermal stability and reducing surface tension.2–5 However, the global issue of PFAS contamination arises from their persistent discharge into the environment, leading to the contamination of surface and groundwater. In the United States (U.S.), there are more than 2800 identified sites contaminated by PFAS,6 and PFAS-polluted drinking water affects over 200 million people.7–9Even minimal PFAS exposure (at the part per trillion level, ng L−1) can lead to bioaccumulation in the body and bloodstream, causing a variety of health problems such as reproductive and pregnancy complications, immune system suppression, cancer, and elevated cholesterol.10 PFAS has been associated with immunotoxic effects, including reduced vaccine efficacy and potentially exacerbated COVID-19 outcomes.11,12 In reaction to these health concerns, the U.S. Environmental Protection Agency (EPA) set Maximum Contaminant Levels (MCLs) for two common PFAS substances, perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS), at 4 ng L−1, and Maximum Contaminant Level Goals (MCLGs) for them at 0 ng L−1.13 This heightened regulatory focus on PFAS underscores the need for advanced methods to treat and remediate PFAS-contaminated water.
Various destructive and non-destructive techniques have been used for PFAS removal. Most current destructive technologies, such as advanced oxidation processes, UV irradiation, sonochemical degradation, and electrochemical degradation, have proven to be ineffective for PFAS removal.14,15 Thermal decomposition of PFAS at high temperatures (>500 °C) is energy intensive and generates toxic degradation gases or short-chain fluorinated by-products, which are more persistent than long-chain PFAS.16–18 Biological treatments are also ineffective since PFAS are highly resistant to biodegradation due to microbial inaccessibility of the carbon atoms on PFAS and their poor electron-donating ability.19,20 Although reverse osmosis and nanofiltration membranes can remove PFAS efficiently, they are expensive for the environmentally occurring concentrations of PFAS and suffer from fouling problems.1,21,22 Other conventional treatments including coagulation, sedimentation, and flocculation have also exhibited low remediation efficiency.23,24
Adsorption is one of the most practical and effective technologies for PFAS removal.22,25,26 PFAS adsorption is influenced by electrostatic, hydrogen bonding, and hydrophobic interactions,22 depending on the adsorbent, the nature of the adsorbates, and the solution chemistry.27,28 Hydrophobic interactions are entropically driven. In other words, the orderly packed water molecules around the hydrophobic parts (i.e., PFAS tail and hydrophobic sorbent) are released back into the solution upon PFAS adsorption.29,30 In addition, hemimicelles/micelles can be formed at the surface of adsorbent when the PFAS concentration is much lower than their critical micelle concentration due to strong hydrophobic interactions.22 Resins,31 polymers,32 minerals,33 and carbonaceous materials (i.e., activated carbon, biochar, carbon nanotubes)34–37 are the most widely used adsorbents for PFAS removal. However, these conventional sorbents have critical deficiencies, such as non-selectivity, slow adsorption kinetics, and low affinity toward short-chain PFAS. Numerous efforts have been made to modify carbonaceous adsorbents to enhance their adsorption capacity, selectivity toward PFAS, and regeneration ability.38 For negatively charged PFAS, they can effectively bind with adsorbates that have a positive charge. Amine functional groups have shown superior removal efficiencies for PFAS as they can create positively charged active sites on the adsorbent surface.39,40
Graphene oxide (GO), a two-dimensional carbonaceous nanomaterial, has demonstrated significant potential for contaminant removal.41–47 For instance, Zhao et al.45 studied PFOS removal by using GO in the presence of Mg2+ because of its ability to form a bridge between the GO and PFOS. The adsorbent exhibited a high PFOS removal efficiency of 98%; however, similar demonstrations for other PFAS are not available. In another study, the sorption of PFOA on GO and an iron oxide-modified reduced-GO composite (FeG) was investigated.46 FeG demonstrated a notably higher efficiency in removing PFOA (>90%) compared to GO (60%), attributed to the additional active sites of FeG and the benefit of employing multiple sorption mechanisms (combining mineral and C-phases). Pure GO has a negatively charged surface that can repel negatively charged PFOA. Sorption reached equilibrium within 3–4 h when using FeG, in contrast to the minimum of 48 h required with GO. Nevertheless, graphene-based adsorbents exhibit low removal efficiencies for short-chain PFAS. Wang et al.44 utilized molecular dynamics simulation to study the adsorption of PFOA onto the GO. Their finding showed that favorable adsorption takes place when the O/C ratio at the GO surface is 16.7%. In Ali et al.'s work,47 water-dispersible hybrid capsules, which were prepared from oil-in-water emulsions stabilized by GO and an outer layer of amino-functionalized mesoporous silicate, were used to adsorb PFOA from water. They reported an adsorption capacity of >60 mg g−1 dependent on pH and PFOA concentration. However, the experiments indicated slow removal kinetics since the capsules needed 2–3 days to reach a maximum removal of >99.9%. Tunioli et al.43 incorporated β-cyclodextrin onto the GO to improve the PFAS removal efficiency from water. Their work demonstrated that by using β-cyclodextrin-modified GO, the removal efficiency of PFAS was enhanced; for example, <20% for adsorbing PFOS by using GO compared to >90% by using β-cyclodextrin-modified GO. Moreover, surfactants can be used to modify the surface of GO to enhance PFAS removal efficiency. Pervez et al.'s work42 showed that the surface modification of GO by using cetyltrimethylammonium chloride (CTAC) surfactant effectively increased the PFAS adsorption efficiency due to a highly positive surface charge of the CTAC-GO adsorbents.
Measuring PFAS concentrations poses a challenge due to the complexity of the compounds. Several analytical techniques have been employed such as liquid chromatography-mass spectrometry (LC-MS),48,49 gas chromatography-mass spectrometry (GC-MS),50,51 and fluorine-19 nuclear magnetic resonance spectroscopy (19F NMR).52,53 Although these methods provide a low detection limit and can measure multiple PFAS simultaneously, operators with specialized skills are needed to perform the tests. Also, the analytes and analysis are expensive. Therefore, developing simpler and more cost-effective methods that are widely available is required. The Methylene Blue active substances (MBAS) assay is a non-specific method to determine the concentration of total anionic surfactants (AS) in water samples.54,55 This method is based on the formation of ion-pairs between anionic surfactants and the cationic methylene blue (MB) dye, which are then extracted into an organic solvent (chloroform) and spectrophotometrically measured.56 Nonetheless, tedious extraction steps and consumption of large volumes of organic solvents limit the widespread application of this method.
This study aimed to develop an effective GO-based adsorbent to efficiently remove PFAS from water. To accomplish this, a magnetic amine-functionalized graphene oxide (MAGO) nanocomposite was prepared to provide multiple binding sites with high affinity and selectivity toward both long-chain and short-chain PFAS. Ethylenediamine (EDA) was covalently grafted on the surface of the GO nanosheets followed by the introduction of iron oxide magnetic nanoparticles (Fe3O4). The electrostatic interactions with amine groups along with hydrophobic interactions with the carbon basal plan of GO enhanced PFAS removal efficiencies and provided rapid removal kinetics. Furthermore, after PFAS adsorption, the adsorbent can be easily separated from the water in the magnetic field without additional centrifugation or filtration, making separation faster and improving the recovery and reuse of the sorbent. This is a great advantage when considering the low levels of PFAS contamination in the environment. To be able to study the adsorbent efficiency for PFAS removal in-house, we also developed a simple and rapid miniaturized method based on the formation of MB-PFAS colored complex. We investigated the influence of different experimental conditions including pH, adsorbent amount, initial PFAS concentration, and contact time on adsorptive removal efficiencies for both long-chain and short-chain PFAS.
2. Materials and methods
2.1. Materials
Graphite flake (CAS: 7782-42-5) and potassium permanganate (KMnO4, CAS: 7722-64-7, ACS reagent, ≥99.9%) were purchased from Sigma-Aldrich. Hydrogen peroxide solution (H2O2, CAS: 7722-84-1, ACS reagent, 30 wt% in H2O) was purchased from Honeywell. Concentrated sulfuric acid (H2SO4, CAS: 7664-93-9, Certified ACS Plus) and hydrochloric acid (HCl, CAS: 7647-01-0, Certified ACS Plus, 36.5–38.0%) were purchased from Fisher Scientific. Ethanol (CAS: 64-17-5) was obtained from Sigma-Aldrich. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, CAS: 25952-53-8, 98+%) was purchased from Thermo Scientific and used to activate carboxyl groups. N-Hydroxysuccinimide (NHS, CAS: 6066-82-6, 98%) and EDA (CAS: 107-15-3, ≥99.9%) were purchased from Sigma-Aldrich. Ferric chloride (FeCl3, CAS: 7705-08-0, 97%) and ferrous chloride tetrahydrate (FeCl2·4H2O, CAS: 13478-10-9, 99+%) were purchased from Sigma-Aldrich and Acros Organics, respectively. Ammonium hydroxide (CAS: 1336-21-6, ACS, 28.0–30.0% NH3) was purchased from Alfa Aesar. Chloroform and dichloromethane were purchased from Alfa Aesar and Fisher Scientific, respectively. Methylene blue (MB) was purchased from Thermo Scientific. PFAS compounds including PFOA (95%, CAS: 335-67-1), PFOS (≥98.0%, CAS: 2795-39-3), perfluorohexanesulphonic acid (PFHxS, ≥98.0%, CAS: 3871-99-6), and perfluorobutane sulfonate (PFBS, 98%, CAS: 29420-49-3) were purchased from Sigma-Aldrich. All chemicals used in the study were at least reagent grade and were used as received without further purification.All samples were prepared by using ultrapure water (>18 MΩ cm) at room temperature (∼24 °C). The MB stock solution with a concentration of 200 ppm was stored in an amber glass bottle in the dark, underlaid with a 1/10 volume ratio of chloroform. Because of the adsorption of PFAS onto the glass surface, all experiments in this study were conducted using polypropylene containers and pipette tips.
2.2. Preparation of the adsorbent
GO was prepared from graphite flakes using Hummers' method57 with slight modifications. Briefly, concentrated H2SO4 was added to the graphite flake, followed by the slow addition of KMnO4. The mixture was covered and kept under stirring for 12 h. Next, deionized water was added to the mixture, and it was heated up to 95 °C for 15 min. After cooling down to room temperature, H2O2 solution (30%) was slowly added to the mixture. Then, the resulting mixture (identified as mixture 1) was centrifuged (Eppendorf Centrifuge 5804, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min) and the supernatant was removed. Fig. S1† shows the mixture 1 before and after the centrifugation. The remaining solid product was successively washed with HCl (10% v/v), and deionized water. Finally, the wet GO, shown in Fig. S2,† was vacuum-dried at 60 °C to obtain dried GO powder (Fig. S3a† shows the dried GO before crushing it to the powder).
000 rpm for 10 min) and the supernatant was removed. Fig. S1† shows the mixture 1 before and after the centrifugation. The remaining solid product was successively washed with HCl (10% v/v), and deionized water. Finally, the wet GO, shown in Fig. S2,† was vacuum-dried at 60 °C to obtain dried GO powder (Fig. S3a† shows the dried GO before crushing it to the powder).
To introduce amine functionality, the prepared GO powder was added to 100 mL ethanol (2 mg mL−1), and the mixture (Fig. S4†) was ultrasonicated for 1 h to form a homogeneous GO dispersion solution. Consequently, 0.05 g of EDC and NHS were added to the dispersion, respectively, followed by stirring at room temperature for 30 min. Next, 0.5 g EDA was added to the mixture and refluxed for 24 h at room temperature. The resulting solid, amino-functionalized GO (AGO), was separated from the solution by centrifugation and washed with deionized water. The dried AGO is shown in Fig. S3b.†
To further modify the adsorbents by incorporating the Fe3O4, 0.1 g of as-prepared AGO was dissolved in 100 mL ultrapure water and ultrasonicated for 30 min. Appropriate amounts of FeCl3 (1.8 mmol) and FeCl2 (1.2 mmol) were added to the solution. Next, ammonia solution was added dropwise to the mixture to increase the pH to 11. The mixture was stirred at 65 °C for 1 h (identified as mixture 2, shown in Fig. S5a†). The resulting material (MAGO) was washed with ultrapure water until the pH was close to 7, followed by drying in a vacuum oven at 60 °C. The separation of MAGO from water can easily be done by using a magnet, as shown in Fig. S5b.† The steps for MAGO preparation are schematically shown in Fig. 1. It should be noted that as control samples, magnetic GO without amine functionalization step (MGO) and Fe3O4 nanoparticles were also synthesized by the same method and studied for PFAS removal.
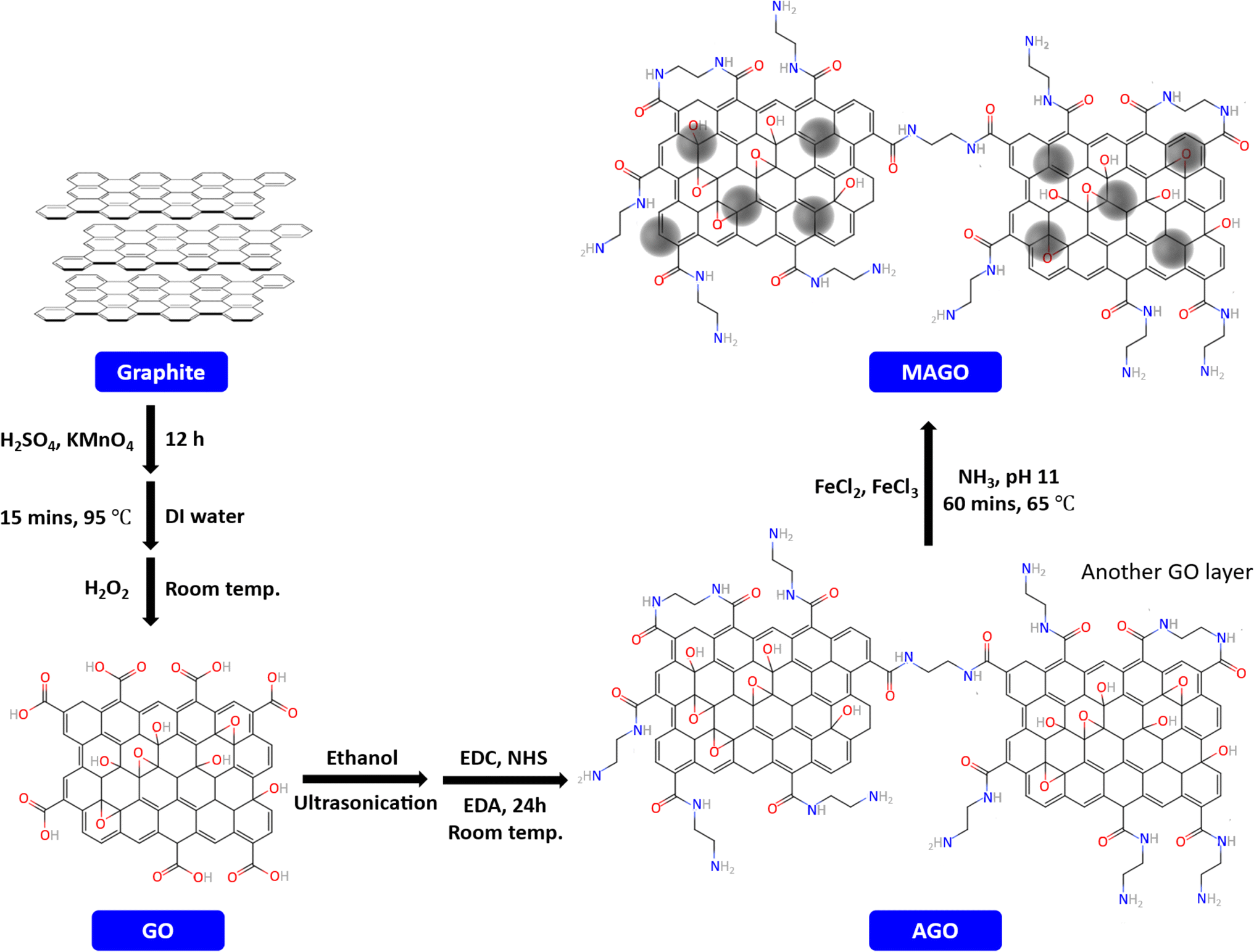 | ||
| Fig. 1 The preparation procedure of the adsorbent. | ||
2.3. Adsorbent characterization
The morphology of the as-synthesized adsorbents was observed using a Zeiss Neon 40 EsB high-resolution scanning electron microscope (FE-SEM) under an acceleration voltage of 20 kV. An energy-dispersive X-ray spectroscopy (EDS) system (Oxford Instruments INCA Energy 250) was used to investigate the elemental composition of the prepared materials. The Fourier Transform Infrared (FTIR) spectrometer (Thermo Scientific, Nicolet iS50R-IR, USA) was used to analyze the functional groups of synthesized adsorbent materials. The 0.01 w/w% particle dispersions (shown in Fig. S6†) were prepared through 1 h of ultrasonication to perform the ultraviolet-visible (UV-vis) spectroscopy in the range of 325 to 1100 nm by using UV-vis spectrophotometer (Thermo Scientific, Genesys 50), and measure the zeta-potential by using a zeta potential analyzer (Malvern ZetaSizer Nano ZS). The thermogravimetric analyzer (TA Instrument, TGA 55) was employed to obtain the TGA curves of GO, AGO, MAGO, and Fe3O4 particles in the air atmosphere within the temperature range of 40 °C to 900 °C.2.4. Adsorption experiments
The PFAS removal efficiency of the MAGO adsorbent was evaluated by conducting the adsorption experiments using synthetic PFAS-contaminated water. The tests were performed in polypropylene centrifuge tubes to minimize PFAS sorption to secondary surfaces (confirmed by controlled experiments). Different experiments were conducted to investigate and optimize the conditions affecting removal efficiencies, including contact time, pH, amount of the adsorbent, and the initial concentration of compounds. For each experiment, the adsorbent was exposed to PFAS aqueous solution (ultrapure water containing PFAS with a concentration between 5 and 500 µg L−1), and the experiments were repeated in triplicate. Briefly, a specific weight of MAGO (as optimized in Section 3.3.3) was added to 40 mL of aqueous solution of PFAS for each experiment. The mixture was then shaken continuously using a shaker at 200 rpm, at room temperature, and for a specific duration of time (Section 3.3.4). The adsorbent was then magnetically collected from the sample solution. The concentration of the residual PFAS in the solution was measured using a spectrophotometric method developed in Section 2.6. The removal efficiency (%) was obtained as follows: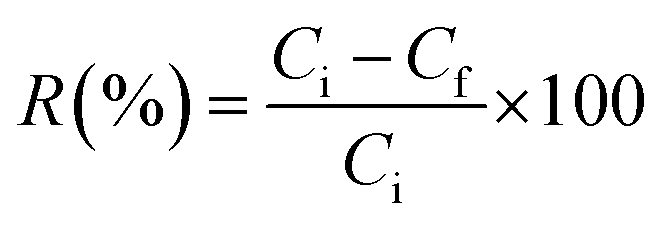 | (1) |
The adsorption capacities (q, µg g−1) of PFAS compounds were calculated as follows:
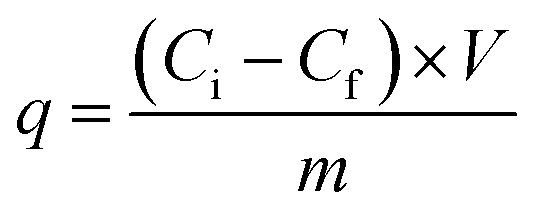 | (2) |
2.5. Adsorption kinetics
The kinetics of PFAS adsorption onto the MAGO were investigated by pseudo-first-order, pseudo-second-order, and intra-particle diffusion models. The linearized forms can be represented as:| Pseudo-first-order ln(qe − qt) = lnqe − k1t | (3) |
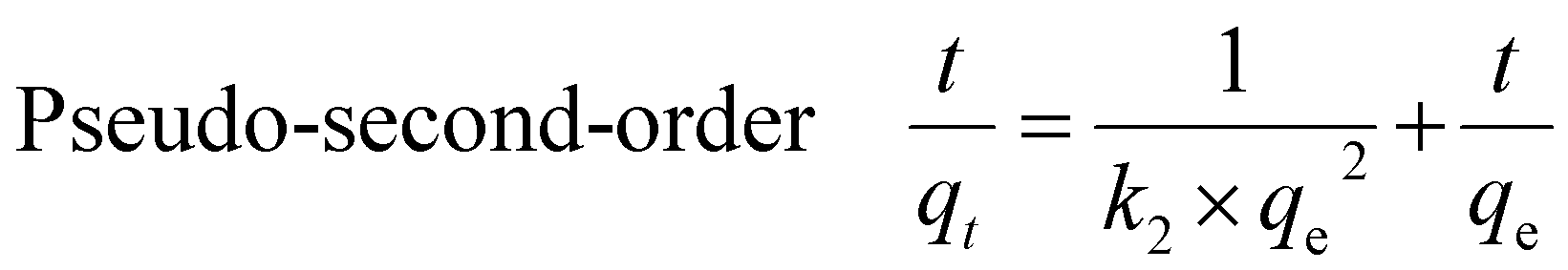 | (4) |
| Weber-Morris intraparticle diffusion qt = kidt0.5 + C | (5) |
2.6. Spectrophotometry detection of PFAS compounds
A simple spectroscopic detection method based on a modified liquid-phase extraction (LPE) was developed to determine the concentration of PFAS in aqueous solutions. The method was based on the formation of a water-insoluble ion pair between the PFAS and the cationic dye (methylene blue), which was extracted by dichloromethane. For this purpose, 100 µL methylene blue solution (200 ppm) and 0.5 mL phosphate buffer (1 mM; Na2HPO4/NaH2PO4, pH 7.1) were added to the sample solution containing PFAS molecules followed by shaking for a few seconds. Next, 2 mL of dichloromethane was added to the solution and mixed for 30 s using a vortex mixer. The dichloromethane phase (containing PFAS-MB ion pairs) was then separated from the aqueous solution using centrifugation at 5000 rpm for 5 min. The organic phase was transferred into a microcuvette (0.7 mL volume) using a pipette. To determine the concentration of the PFAS, the color intensity of the organic phase (which was proportional to the PFAS concentration) was analyzed using a spectrophotometer at 652 nm. Fig. 2 depicts a schematic showing the PFAS adsorption by using the MAGO adsorbent and detection of remaining PFAS in water by extracting and analyzing the MB-PFAS ion pair.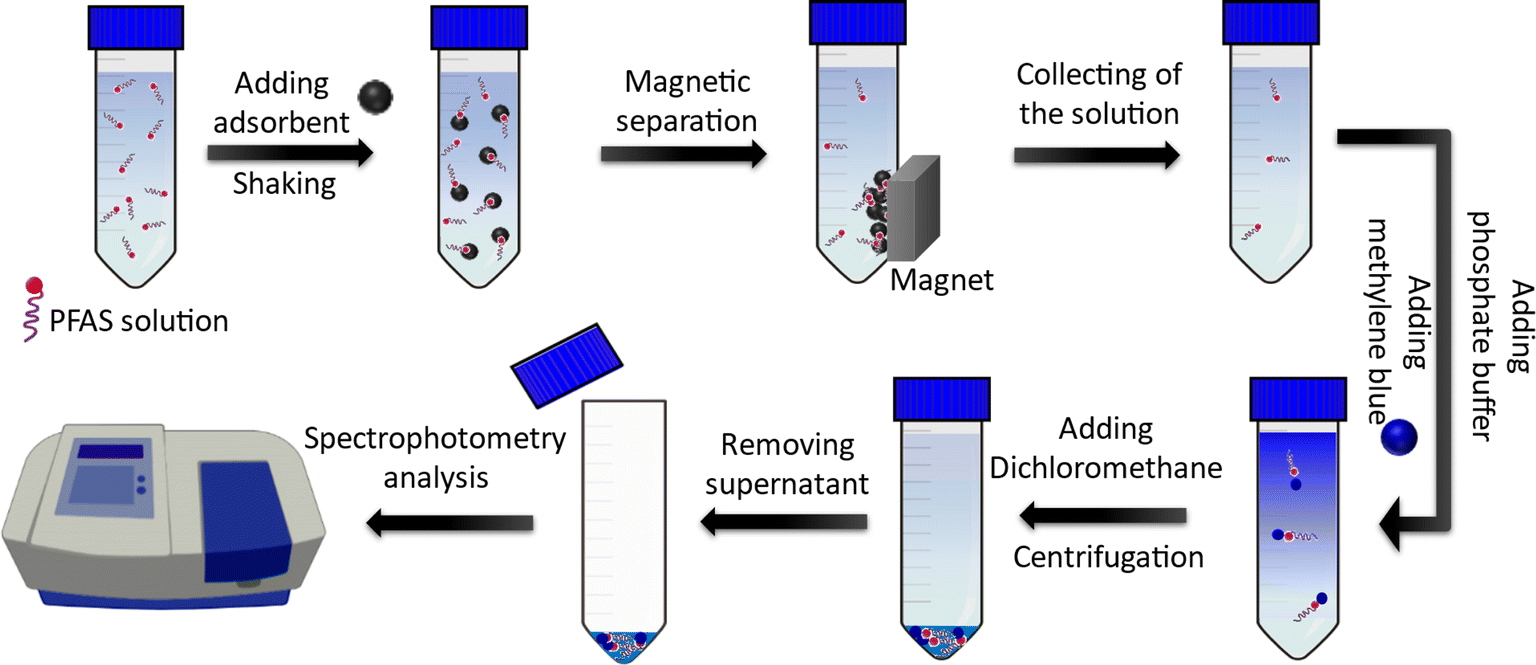 | ||
| Fig. 2 A schematic representation of the adsorption and detection steps of PFAS. | ||
3. Results and discussion
3.1. Spectrophotometry detection of PFAS
Anionic surfactants (AS), such as PFOA and PFOS, can combine with cationic dyes such as methylene blue or ethyl violet to create an ion pair.54,56 The electrostatic interaction between them makes the ion pair hydrophobic and insoluble in water. Therefore, it can be extracted into an organic phase to measure associated color intensity at visible spectrum ranges. In a recent study, Fang et al. used a modified version of the MBAS test called astkCARE™, which replaces chloroform with ethyl acetate and methylene blue with ethyl violet, to detect the color of AS using a smartphone camera.58 The astkCARE™ kit requires 10 mL of water sample mixed with 7 mL of the astkCARE™ reagent, shaken for approximately 10 s, and left to stand for 1–2 min to extract the ion-pair of the cationic dye and AS in the top layer (i.e., the organic phase). Like the MBAS test, the color intensity of the organic phase corresponds to the ion-pair concentration, allowing for AS concentration testing with a detection limit of 0.1 ppm for visual testing or 10 ppb for the smartphone app test, as recently reported.The LPE method developed herein involved mixing reagents (2 mL of dichloromethane, 0.5 mL of phosphate buffer, and 100 µL of MB) with 40 mL of a PFAS aqueous solution. The resulting mixture was shaken to extract the MB-PFAS complex from the sample solution into dichloromethane. Finally, spectrophotometric detection was used to analyze the extracted complex. Fig. 3 typically shows the organic phase precipitated from samples containing various concentrations of PFOS (ranging from 5–500 µg L−1) as a representative example. As demonstrated, the intensity of the color or hue is correlated with the PFAS concentration level in the initial aqueous solutions. The difference in the color intensity of the organic phase was visibly different at concentrations as low as 50 µg L−1, which highlights the high sensitivity of the method for detecting low concentration levels of PFAS compounds. This sensitivity is particularly important in the analysis of environmental samples, where low PFAS concentrations are often present. Additionally, the ability to visually detect the color change of the organic phase provides a convenient and rapid screening tool for the presence of PFAS in aqueous solutions.
 | ||
| Fig. 3 The color intensity of the organic phase (lower phase) at different PFAS concentrations in the primary aqueous solutions. | ||
To achieve the highest efficiency of LPE process, the type and volume of organic phase, pH, and MB concentration were optimized. However, the optimization process was out of the scope of this publication and not presented here. After optimization, a series of standard solutions with different concentrations of PFAS were prepared and analyzed using the developed method. By plotting the absorption intensity of the MB-PFAS ion pair in the organic phase as a function of the concentration of the PFAS standard solutions, the calibration curve was obtained. We obtained linear calibration curves up to 5000 µg L−1 for PFOS, PFOA, and PFHxS and up to 1000 µg L−1 for PFBS. This method showed excellent linearities with correlation coefficients (R2) between 0.9951 and 0.9991. As a common practice in analytical chemistry, the limit of detection (LOD) for the developed method was determined from 3SB/m ratio, where SB is the standard deviation of the blank signal and m is the slope of the calibration curve.59,60 The LODs are presented in Table 1. The relative standard deviations (RSDs) were below 4.4% for 5 replicate determinations of 100 µg L−1 of PFAS. The pre-concentration, the ratio of the final volume of the extracted analyte (∼2 mL) to the initial volume of the sample (40 mL), was approximately 20. Compared to the tedious standard method requiring at least 50 mL of chloroform and multistage extraction, the extraction procedure took less than one minute using 25 times less solvent. Moreover, the developed method is simple and cost-effective as it does not require expensive instrumentation or complicated procedures.
It should be noted that coexisting anions can form ion-pairs with MB, which can potentially be extracted into the chloroform, leading to a positive interference. In addition, cations can compete with MB to form ion-pairs with anionic surfactants, thereby hindering their extraction and causing a negative interference. Anionic surfactants can also compete with PFAS molecules to form ion-pairs with MB. However, MAGO can effectively adsorb and remove a wide range of impurities and interfering ions, including organic compounds, heavy metal ions, and anions61–63 prior to spectrophotometric detection of residual PFAS in water. Therefore, it minimizes the potential interferences in the subsequent LPE stage for PFAS detection. Overall, the developed LPE method offers a practical and reliable approach for the rapid and simple detection of PFAS in aqueous solutions for on-site application and common laboratory tests. Some preliminary data on the effect of background anions on PFAS removal efficiency are shown in Fig. S7.† However, additional experiments are required to evaluate the influence of a broader range of competing species.
3.2. Adsorbent characteristics
The adsorbent morphology was observed using SEM as shown in Fig. 4. As presented in Fig. 4a and b, the GO and AGO represented the overlap of some folded layers and wrinkles. GO has a 2D structure composed of carbon atoms arranged in a hexagonal lattice. The formation of wrinkles and folded layers can be attributed to the introduction of oxygen-containing functional groups, such as hydroxyl and carboxyl groups. Navaee et al. reported that the AGO sheets have more informally folded areas with more ripples, and thus, forming tangled patches.64Fig. 4c indicates the successful introduction of magnetite Fe3O4 nanoparticles onto the surface of GO since the as-synthesized MAGO retained the folded structure of GO with a much higher overall roughness. The EDS analyses (Fig. 5) indicate the presence of nitrogen and iron in the structure of the adsorbent, confirming the successful incorporation of amine functionalities and Fe3O4 nanoparticles onto the GO structure. | ||
| Fig. 4 SEM images of (a) GO, (b) AGO, and (c) MAGO. | ||
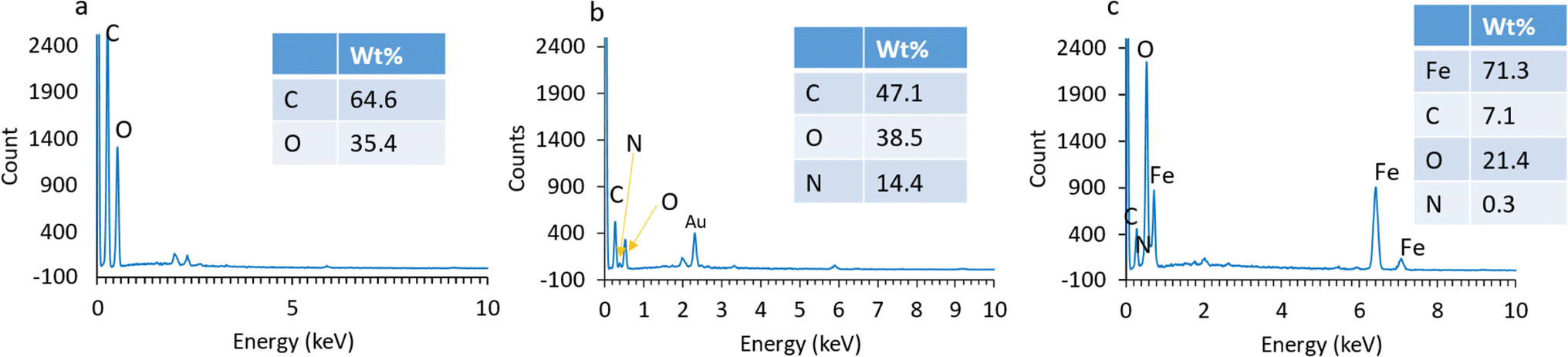 | ||
| Fig. 5 The EDS spectra of (a) GO, (b) AGO, and (c) MAGO. | ||
Fig. 6a shows the FTIR spectra of GO and MAGO. For GO, the broad absorption band at 3420 cm−1 is due to the O–H stretching vibration of the carboxylic acid groups at the surface.64 The characteristic bands located at 1723 cm−1 and 1611 cm−1 can be ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O strong carbonyl stretching and vibration of CC aromatic groups, respectively.64 The peaks at 1389 and 1161 cm−1 are associated with C–OH and C–O–C stretching vibrations, respectively. The peak around 1030 cm−1 can be due to the C–O stretching vibrations of the epoxide groups.64 For the spectrum of MAGO, the peak at 580 cm−1 belongs to the Fe–O bond vibration of Fe3O4 nanoparticles in the adsorbent. The characteristic peak at 1650 cm−1 can be attributed to the amide groups due to the reaction between amine groups and carboxylic groups of GO during the functionalization process. The peak at 1100 cm−1 is from the C–N stretching.64 The peak at 3450 cm−1 is attributed to the N–H stretching, overlapping with the peak due to the carboxylic acid stretching.64 Moreover, the bandwidth of the peaks associated with oxygen-functionalized groups seems to be reduced for MAGO, indicating the replacement by amino groups.64 The results confirm the successful incorporation of amine groups and Fe3O4 magnetic nanoparticles onto the GO.
O strong carbonyl stretching and vibration of CC aromatic groups, respectively.64 The peaks at 1389 and 1161 cm−1 are associated with C–OH and C–O–C stretching vibrations, respectively. The peak around 1030 cm−1 can be due to the C–O stretching vibrations of the epoxide groups.64 For the spectrum of MAGO, the peak at 580 cm−1 belongs to the Fe–O bond vibration of Fe3O4 nanoparticles in the adsorbent. The characteristic peak at 1650 cm−1 can be attributed to the amide groups due to the reaction between amine groups and carboxylic groups of GO during the functionalization process. The peak at 1100 cm−1 is from the C–N stretching.64 The peak at 3450 cm−1 is attributed to the N–H stretching, overlapping with the peak due to the carboxylic acid stretching.64 Moreover, the bandwidth of the peaks associated with oxygen-functionalized groups seems to be reduced for MAGO, indicating the replacement by amino groups.64 The results confirm the successful incorporation of amine groups and Fe3O4 magnetic nanoparticles onto the GO.
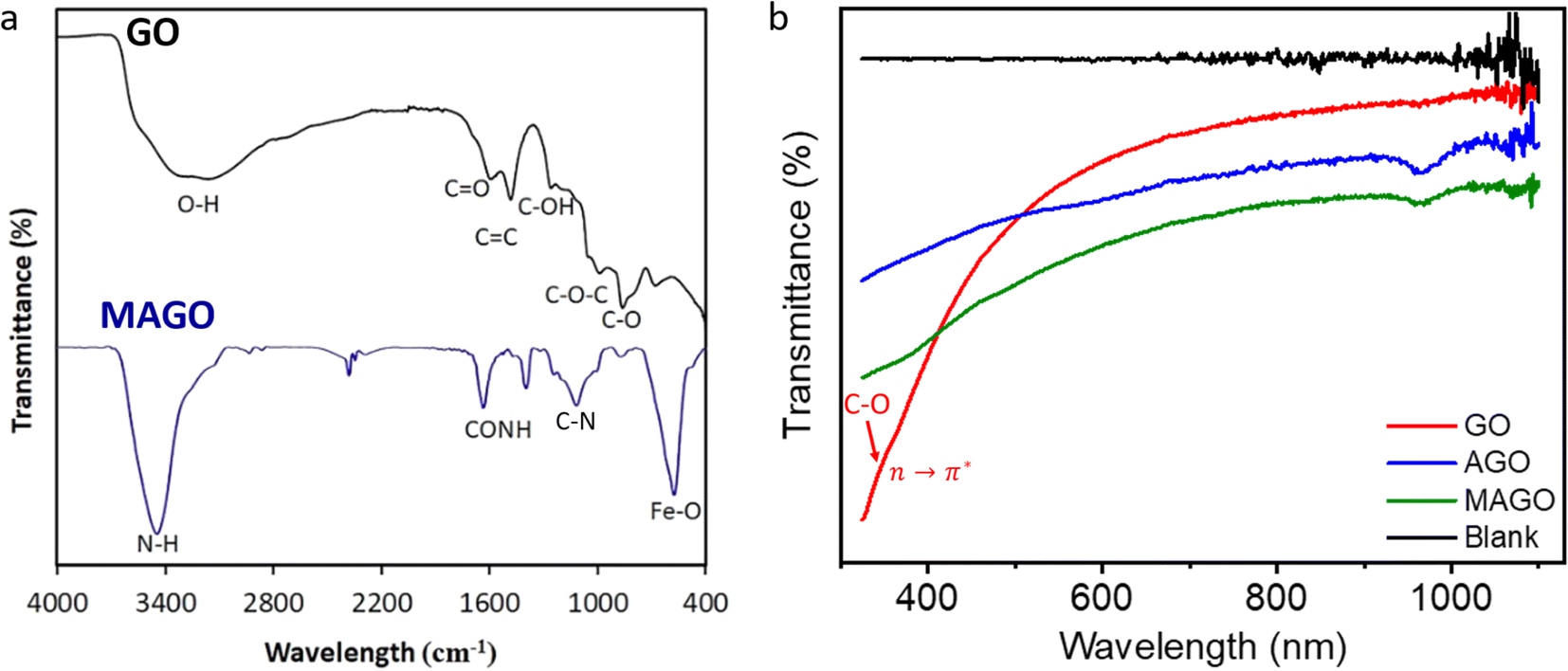 | ||
| Fig. 6 (a) FTIR spectra of GO and MAGO. And (b) UV-vis absorption spectra of GO, AGO, and MAGO. | ||
Fig. 6b is the UV-vis absorption spectra of GO, AGO, and MAGO. After amine functionalization, the dispersibility and solubility of GO decreased (Fig. S6†), which is in agreement with the observation reported by Navaee et al.64 The black line in Fig. 6b represents the UV-vis absorption spectrum of the ultrapure water. In the wavelength range of 325 to 1100 nm, a shoulder at ∼350 nm appears (red arrow in Fig. 6b) due to n → π* transitions because of the C–O–C and R–O–O–R groups.65–67 The peak will become more observable for a GO film on a quartz substrate, attributed to the large number of peroxides linking the graphene oxide layers.66 Due to the instrument limitation, the peak due to π → π* transitions for CC groups (at ∼230 nm) was not observed.65,66 One can see that a new peak at 955 nm appears for AGO and MAGO, suggesting significant conjugation or the formation of a charge-transfer complex. Amine groups can interact with the oxygen functionalities on GO, potentially leading to new conjugated structures or charge-transfer complexes. Charge transfer complexes can absorb at significantly longer wavelengths. Charge transfer takes place when there is an electron density shift between the donor (i.e., amine groups) and acceptor (i.e., oxygenated groups of GO) molecules, leading to new absorption bands at longer wavelengths. Chen et al.68 also reported the UV-vis spectrum of AGO from 200 nm to 900 nm, and their result also showed the strong absorbance in the entire visible and near infrared regions.
The TGA curves of the adsorbents are shown in Fig. 7. The black line shows the thermal property of pristine graphite, which shows a high thermal stability in the air atmosphere. The slight weight loss when the temperature is below ∼700 °C is due to the loss of moisture and adsorbed oxygen atoms.69 Above ∼700 °C, the weight of pristine graphite dramatically decreases, indicating the start of oxidization and decomposition.69 When the temperature is 900 °C, there is still 40% of weight remaining for pristine graphite. The TGA curves of GO and AGO show that they were completely decomposed in the air atmosphere before ∼900 °C, leaving no residue behind. GO starts to lose weight when the temperature is below 100 °C and a weight loss of 6.8% is obtained at ∼150 °C due to the loss of water.70 A dramatic weight loss occurs from ∼150 °C to ∼220 °C, attributed to the degradation of oxygen-containing groups, i.e., –COOH and –OH, releasing CO and CO2.70–72 At this temperature range, the highly conductive reduced GO is expected to be obtained.71 A weight loss of 48.8% is obtained at ∼460 °C, where the carbon skeleton of GO starts to combust. The combustion ends at around 524 °C (ref. 70 and 71) with a weight loss of 40.7%. Alam et al.71 reported an exothermic peak at around 456 °C, corresponding to the carbon skeleton combustion. Compared to pristine graphite, GO decomposes more easily since it has a high level of oxygen, giving rise to lower energy required for decomposition.72
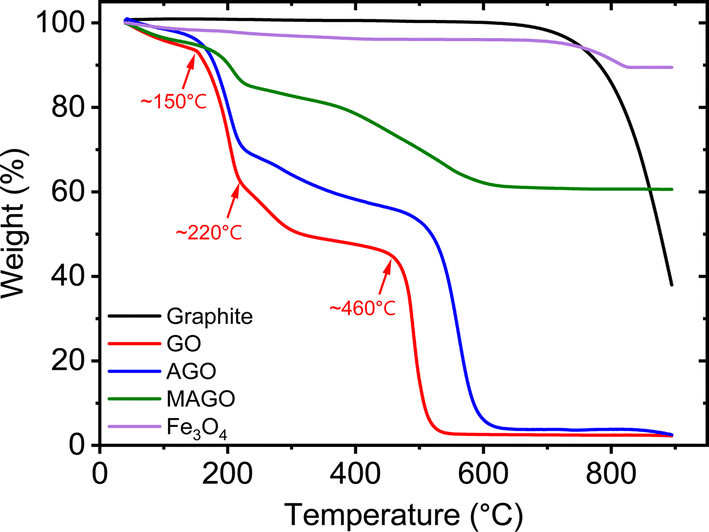 | ||
| Fig. 7 TGA curves of pristine graphite, GO, AGO, MAGO, and Fe3O4. | ||
The TGA curve of AGO also exhibits similar weight losses but at slightly higher temperatures. The first weight loss again is due to the loss of the adsorbed water. At ∼223 °C, a dramatic weight loss occurs, which can be attributed to the degradation of oxygen-containing and amine functional groups. According to Alzate-Carvajal et al.,70 the decomposition of covalently attached EDA molecules takes place at ∼300 °C. EDA is a bifunctional amine, which offers two general bonding possibilities, as shown in Fig. 1, by involving only one or both NH2 groups. The latter case can result in cross-linking of adjacent GO sheets.70 At ∼506 °C, the individual GO sheets start to decompose followed by the decomposition of cross-linked GO sheets at ∼600 °C.70 In addition, for MAGO, the thermal stability in the air atmosphere is enhanced due to the incorporated Fe3O4, which starts to decompose at ∼826 °C. The modified GO adsorbents show a higher thermal stability than the one found for GO, which is in agreement with the literature.70
The zeta potential of adsorbents is summarized in Table 2. The zeta potential of GO is closely linked to its functional groups and is crucial for assessing its colloidal stability. The measured potentials show the negative charge around the double layer surrounding the colloidal particle, stemming from the ionization of various functional groups at the surface of particles. Typically, particles displaying a zeta potential smaller than −30 mV or larger than +30 mV are considered stable due to sufficient electrostatic repulsion preventing aggregation.73 The GO has a zeta potential of −33.9 ± 1.2 mV, indicating that the GO adsorbents are colloidally stable. For AGO and MAGO, the zeta potential increased due to the addition of protonated amine groups and the loss of carboxyl groups during the amine functionalization.74 This result confirms the successful incorporation of amine groups. Furthermore, the increase in zeta potential for the adsorbents after functionalization suggests that their colloidal stability is slightly reduced, which is in agreement with our observation. While colloidal stability of adsorbents may affect their application in water treatment, our results as will be discussed in the next sections confirm that the adsorption capacity of samples is not adversely influenced. According to Bao et al.'s study,75 the Fe3O4/SiO2-GO adsorbents became to some extent unfunctional for adsorbing heavy metal ions when the zeta potential is positive. Therefore, even though the particles are considered to be colloidally stable when their zeta potential is higher than +30 mV, the adsorption capacity can be changed depending on factors such as pH (as will be shown in the next sections).
| Adsorbent type | Zeta potential (mV) |
|---|---|
| GO | −33.9 ± 1.2 |
| AGO | −24.2 ± 1.0 |
| MAGO | −27.8 ± 0.3 |
3.3. Effect of adsorption experimental conditions
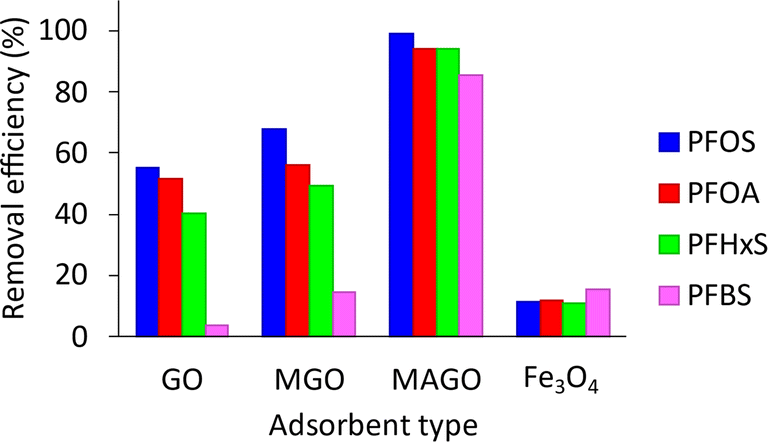 | ||
| Fig. 8 Removal efficiency of different adsorbents for PFAS compounds. Conditions: 40 mL of 500 µg L−1 PFAS, pH 6.5, 20 mg adsorbent, and 60 min extraction time. | ||
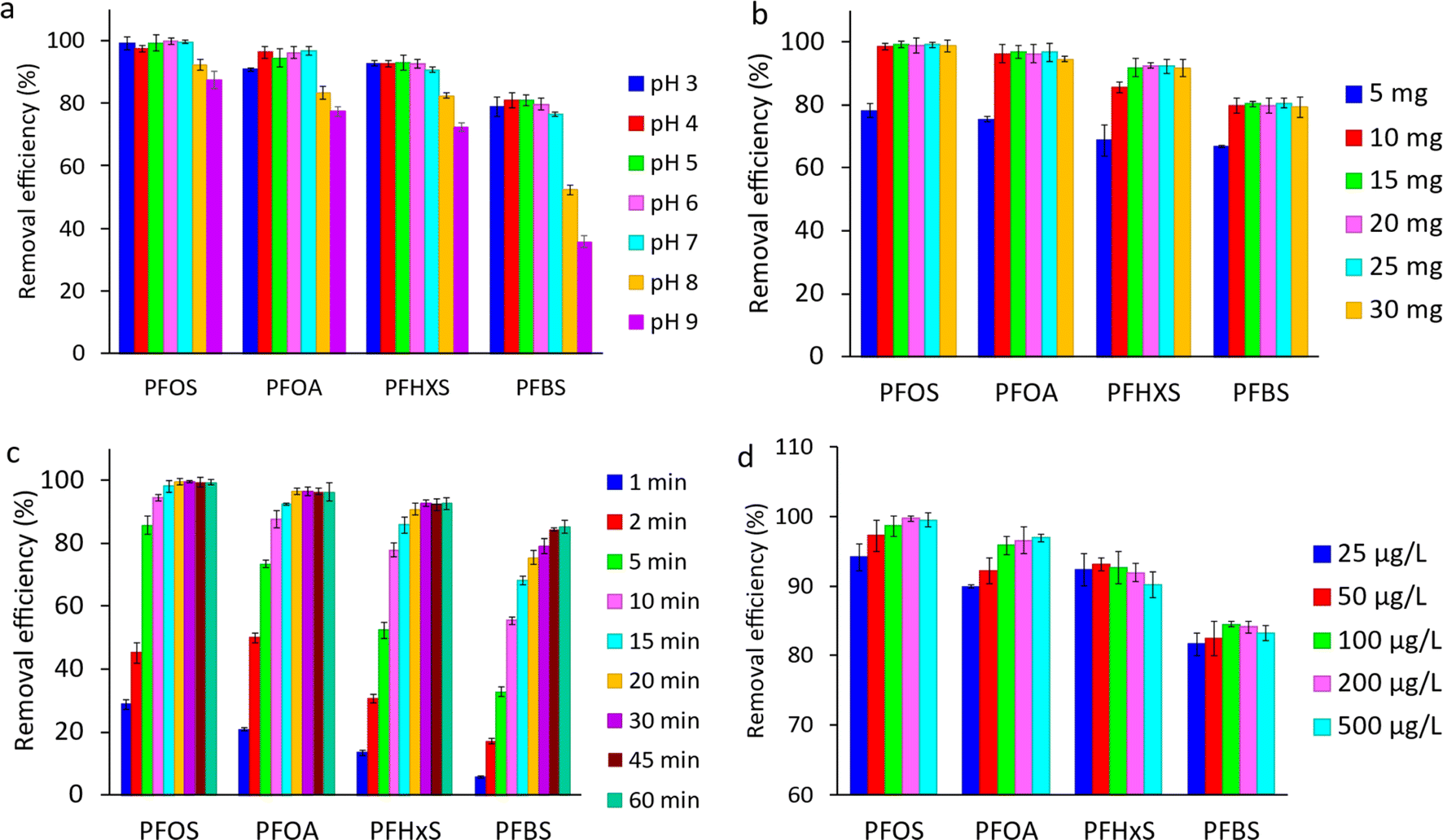 | ||
| Fig. 9 (a) Effect of the solution pH on PFAS removal efficiency (conditions: 40 mL of 500 µg L−1 PFAS, 20 mg adsorbent, and 30 min extraction time); (b) effect of adsorbent amount on PFAS removal efficiency (conditions: 40 mL of 500 µg L−1 PFAS, pH 6.5, and 30 min extraction time); (c) effect of contact time on the removal efficiency of PFAS (conditions: 40 mL of 500 µg L−1 PFAS, pH 6.5, and 15 mg adsorbent); and (d) effect of initial PFAS concentration on the removal efficiency (conditions: 40 mL PFAS solution, pH 6.5, 15 mg adsorbent, and 30 min extraction time). | ||
Hydrophobic interactions are present at the carbon basal plane of graphene, while positive amine active sites can electrostatically attract negatively charged PFAS. The hydrophobic interactions become more dominant, especially for long-chain PFAS, when the electrostatic attractions become negligible. The reduction in the number of positive-charge active sites on the adsorbent surface at elevated pH values results in a decrease in removal efficiencies, especially for short-chain PFBS. The observations indicated that both electrostatic and hydrophobic interactions contribute to the adsorption of PFAS on MAGO, and pH variation can only affect the electrostatic interactions. Our results are in agreement with recent studies showing that the hydrophobic interactions play a significant role in the PFAS adsorption on different adsorbents such as activated carbons,24,76 cationic polymers,32 and molecularly imprinted polymers.77 Since MAGO has the highest efficiency for adsorbing PFAS within 3 to 7 pH range, it is ideal for treating water sources, which usually have a pH in the range of 5.8–6.5.
000 or higher),5,18 the observed trend is not due to micelle formation. Overall, the impact of initial PFAS concentration on removal efficiency is complex, and further research is needed to fully understand the relationship between initial PFAS concentration and removal efficiency.
It should be noted that most of the previous studies have investigated the adsorptive removal of PFAS compounds at high initial concentration levels, often several hundred mg L−1, which is much higher than the environmentally relevant concentrations by a factor of 105–107.32 In this study, the adsorption experiments have been performed at µg L−1 concentration levels. Compared to previous reports, this is closer to PFAS concentrations typically found in contaminated water and wastewater.
3.4. Kinetics study
Adsorption kinetics of PFOS, PFOA, PFHxS, and PFBS onto the MAGO nanocomposite were investigated to evaluate the adsorption mechanisms. Fig. 10a–l displays the linearized kinetic models that were fitted to the experimental results, and Table 3 summarizes the parameters corresponding to each model. The findings indicate that the adsorption of all PFAS onto MAGO is better represented by pseudo-second-order modeling, as evidenced by higher correlation coefficients (R2 values), compared to the pseudo-first-order model. These results suggest that the sorption rate is primarily governed by the chemical sorption, of which the capacity is linearly related to the number of active sites on the sorbent. This result was in agreement with the previous studies, which also reported that the pseudo-second-order kinetic model describes the PFAS adsorption kinetics better.35,40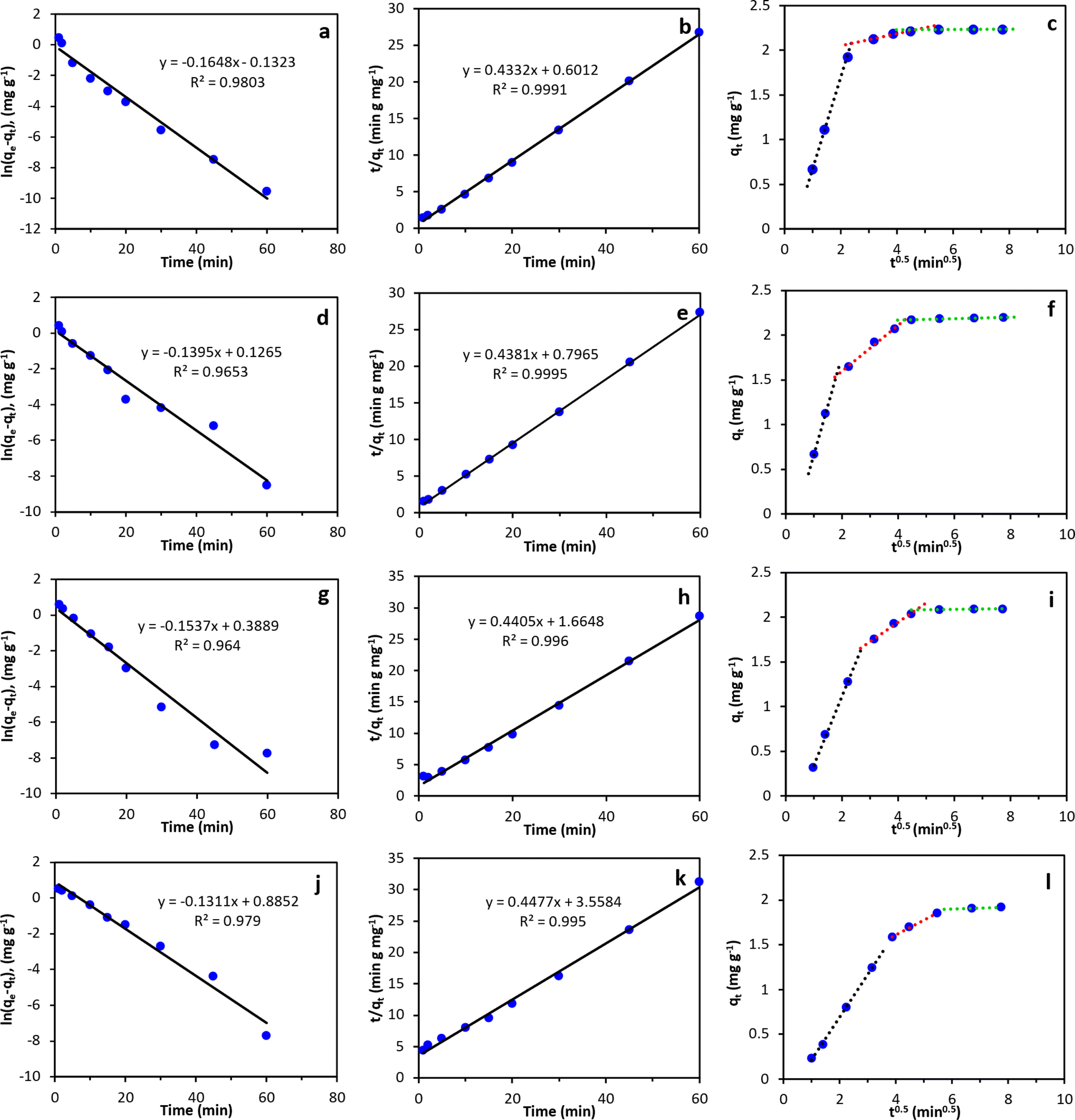 | ||
| Fig. 10 The linear plots of kinetics models: (a, d, g and j) pseudo-first-order, (b, e, h and k) pseudo-second-order, and (c, f, i and l) Weber Morris intra-particle diffusion model for (a–c) PFOS, (d–f) PFOA, (g–i) PFHxS, and (j–l) PFBS. | ||
| Model | Parameter | Value | |||
|---|---|---|---|---|---|
| PFOS | PFOA | PFHxS | PFBS | ||
| Pseudo first order | q e (µg g−1) | 0.88 | 1.13 | 1.47 | 2.42 |
| K 1 (min−1) | 0.16 | 0.14 | 0.15 | 0.13 | |
| R 2 | 0.9803 | 0.9653 | 0.964 | 0.9790 | |
| Pseudo second order | q e (µg g−1) | 2.31 | 2.28 | 2.27 | 2.23 |
| K 2 (g µg−1 min−1) | 0.31 | 0.24 | 0.12 | 0.056 | |
| R 2 | 0.9991 | 0.9996 | 0.9960 | 0.9950 | |
| Intraparticle diffusion | C i | 1.12 | 1.00 | 0.60 | 0.23 |
| k i (µg g−1 min−0.5) | 0.19 | 0.20 | 0.25 | 0.26 | |
| R 2 | 0.58 | 0.6927 | 0.7302 | 0.8561 | |
The intra-particle diffusion model was also used to study the adsorption processes of PFAS on MAGO nanocomposite. The plot of qt against t0.5 should exhibit a strong linear relationship with the line passing through the origin if intraparticle diffusion is the primary rate-controlling step. The results indicated that PFAS adsorption occurs in three distinct steps. A rapid PFAS adsorption is observed within the first 10 min of the experiment. After that, the rate of adsorption gradually decreases and eventually stabilizes at equilibrium within 30 min. As the uptake curves do not pass through the origin, adsorption is not controlled by intra-particle diffusion.
3.5. Regeneration of the adsorbent
The successful regeneration of adsorbents is crucial for their practical applications, as it can significantly reduce the cost of the process and improve the sustainability of the treatment method. The desorption of the adsorbent can be achieved using different solvents or methods (such as regeneration at high temperatures), depending on the nature of the adsorbate and the adsorbent. In the present study, to find a proper regeneration method, we tested several solutions for regeneration, including water, methanol, hot water + NaCl, methanol + NaOH, and hot water/methanol (30:70 v/v%), among which the latter showed the highest regeneration rate. The PFAS desorption was carried out by soaking the spent adsorbent in 10 mL of a hot water (50 °C)/methanol mixture (30:70 v/v) and shaking for 30 min followed by magnetically collecting the adsorbent from the mixture. The regenerated MAGO was then used for the next PFAS removal run.
The results in Fig. 11 demonstrate that the regenerated MAGO adsorbent maintains its performance and removal efficiency for PFAS even after five consecutive adsorption/desorption cycles. Therefore, the MAGO is a highly stable and efficient adsorbent for PFAS removal, and its regeneration capability makes it a promising candidate for practical applications. While we cannot completely rule out the possibility of incomplete desorption of PFAS from MAGO during regeneration, the negligible differences between results in subsequent cycles suggest that it is insignificant.
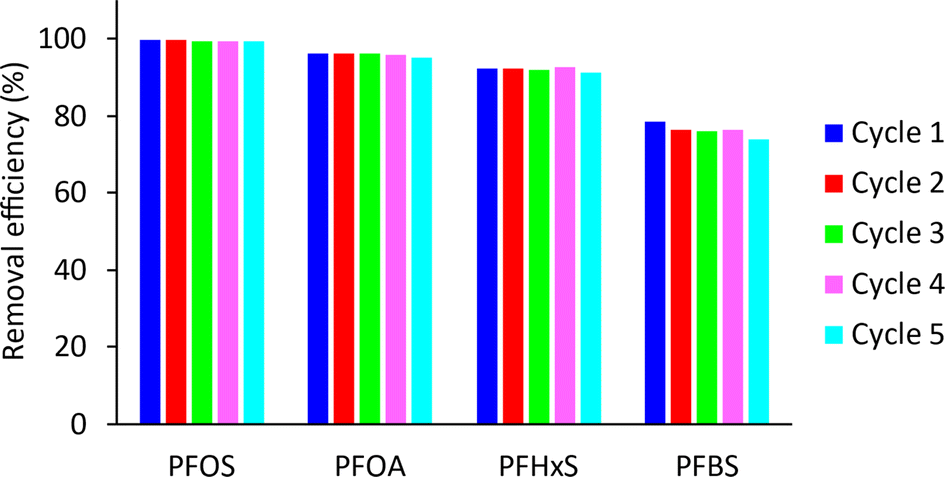 | ||
| Fig. 11 The performance of the regenerated adsorbent for PFAS removal using the regenerated MAGO at different adsorption/desorption analysis cycles. | ||
4. Conclusion
In the present study, MAGO nanocomposite was prepared as a promising adsorbent to remove different PFAS compounds at µg L−1 concentration levels from water samples. The as-prepared specimens were characterized using FTIR, TGA, and SEM/XPS to confirm the successful surface functionalization and incorporation of magnetic particles. The PFAS (PFOS, PFOA, PFHxS, and PFBS) adsorption tests were performed under different conditions. The influences of various experimental conditions were also investigated to enhance the adsorption capabilities of the adsorbent for all PFAS. The best removal efficiencies were observed in the acidic to neutral pH ranges (3–7) and the efficiencies improved by increasing the adsorbent amount and contact time, reaching equilibrium within 30 min. The results from adsorption kinetics of PFAS onto the adsorbent followed the pseudo-second-order kinetic model. Additionally, MAGO showed good regeneration properties using a hot water/methanol (30:70 v/v%) mixture.
Our findings indicated that the presence of amine groups is crucial for the adsorption of negatively charged PFAS compounds, particularly short-chain PFAS, through electrostatic interactions, although the introduction of amine group and iron oxide nanoparticles slightly reduced the colloidal stability. Most importantly, our results indicated that MAGO holds great promise as an efficient adsorbent for eliminating PFAS from water matrices, adding a new possibility to the current materials for effective PFAS removal from water sources. In addition, a straightforward spectrophotometric method was developed to easily detect PFAS compounds by forming a colored complex between MB and PFAS molecules, providing a simple way to detect the PFAS at parts per billion levels.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Shokouh Mahpishanian: investigation, methodology, validation, writing – original draft. Muchu Zhou: investigation, methodology, validation, writing – review & editing. Reza Foudazi: conceptualization, project administration, resources, supervision, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was partially supported by the Bureau of Reclamation (BoR) under the Grant of R21AC10418-00. We thank Dr Galizia at OU for providing access to the UV-vis and FT-IR spectrometers and Seyed Mostafa Tabatabaei at OU for measuring the zeta potential.References
- T. F. Mastropietro, R. Bruno, E. Pardo and D. Armentano, Reverse osmosis and nanofiltration membranes for highly efficient PFASs removal: overview, challenges and future perspectives, Dalton Trans., 2021, 50(16), 5398–5410, 10.1039/d1dt00360g.
- H. Sharifan, M. Bagheri, D. Wang, J. G. Burken, C. P. Higgins, Y. Liang, J. Liu, C. E. Schaefer and J. Blotevogel, Fate and transport of per- and polyfluoroalkyl substances (PFASs) in the vadose zone, Sci. Total Environ., 2021, 771, 145427, DOI:10.1016/j.scitotenv.2021.145427.
- S. P. Lenka, M. Kah and L. P. Padhye, A review of the occurrence, transformation, and removal of poly- and perfluoroalkyl substances (PFAS) in wastewater treatment plants, Water Res., 2021, 199, 117187, DOI:10.1016/j.watres.2021.117187.
- J. Zhang, H. Pang, S. Gray, S. Ma, Z. Xie and L. Gao, Pfas removal from wastewater by in-situ formed ferric nanoparticles: Solid phase loading and removal efficiency, J. Environ. Chem. Eng., 2021, 9(4), 105452, DOI:10.1016/j.jece.2021.105452.
- M. Zhou, Z. Abbasian Chaleshtari, B.-J. Shiau, B. P. Grady and R. Foudazi, Air-water interfacial properties of perfluorosulfonic acid salts with different chain lengths, Colloids Surf., A, 2024, 694, 134129, DOI:10.1016/j.colsurfa.2024.134129.
- https://www.ewg.org/interactive-maps/pfas_contamination/ .
- C. Death, C. Bell, D. Champness, C. Milne, S. Reichman and T. Hagen, Per- and polyfluoroalkyl substances (PFAS) in livestock and game species: A review, Sci. Total Environ., 2021, 774, 144795, DOI:10.1016/j.scitotenv.2020.144795.
- R. Drew, T. G. Hagen and D. Champness, Accumulation of PFAS by livestock–determination of transfer factors from water to serum for cattle and sheep in Australia, Food Addit. Contam.: Part A, 2021, 38(11), 1897–1913, DOI:10.1080/19440049.2021.1942562.
- J. B. Brown, J. M. Conder, J. A. Arblaster and C. P. Higgins, Assessing Human Health Risks from Per- And Polyfluoroalkyl Substance (PFAS)-Impacted Vegetable Consumption: A Tiered Modeling Approach, Environ. Sci. Technol., 2020, 54(23), 15202–15214, DOI:10.1021/acs.est.0c03411.
- S. Pilli, A. K. Pandey, V. Pandey, K. Pandey, T. Muddam, B. K. Thirunagari, S. T. Thota, S. Varjani and R. D. Tyagi, Detection and removal of poly and perfluoroalkyl polluting substances for sustainable environment, J. Environ. Manage., 2021, 297, 113336, DOI:10.1016/j.jenvman.2021.113336.
- P. Grandjean, C. A. Gade Timmermann, M. Kruse, F. Nielsen, P. J. Vinholt, L. Boding, C. Heilmann and K. Mølbak, Severity of COVID-19 at elevated exposure to perfluorinated alkylates, PLoS One, 2020, 15, e0244815, DOI:10.1371/journal.pone.0244815.
- Q. Wu, X. Coumoul, P. Grandjean, R. Barouki and K. Audouze, Endocrine disrupting chemicals and COVID-19 relationships: A computational systems biology approach, Environ. Int., 2021, 157, 106232, DOI:10.1016/j.envint.2020.106232.
- PFAS National Primary Drinking Water Regulation, Environmental Protection Agency, 40 CFR Parts 141 and 142. Federal Register / Vol. 89, No. 82 / Friday, April 26, 2024 / Rules and Regulations.
- M. M. Schultz, D. F. Barofsky and J. A. Field, Fluorinated alkyl surfactants, Environ. Eng. Sci., 2003, 20(5), 487–501, DOI:10.1089/109287503768335959.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review, Water Res., 2014, 50, 318–340, DOI:10.1016/j.watres.2013.10.045.
- A. Alinezhad, P. Challa Sasi, P. Zhang, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and F. Xiao, An Investigation of Thermal Air Degradation and Pyrolysis of Per- and Polyfluoroalkyl Substances and Aqueous Film-Forming Foams in Soil, ACS ES&T Eng., 2022, 2(2), 198–209, DOI:10.1021/acsestengg.1c00335.
- B. B. Baker and D. J. Kasprzak, Thermal degradation of commercial fluoropolymers in air, Polym. Degrad. Stab., 1993, 42(2), 181–188, DOI:10.1016/0141-3910(93)90111-U.
- F. Li, J. Duan, S. Tian, H. Ji, Y. Zhu, Z. Wei and D. Zhao, Short-chain per- and polyfluoroalkyl substances in aquatic systems: Occurrence, impacts and treatment, Chem. Eng. J., 2020, 380, 122506, DOI:10.1016/j.cej.2019.122506.
- J. Liu and S. Mejia Avendaño, Microbial degradation of polyfluoroalkyl chemicals in the environment: A review, Environ. Int., 2013, 61, 98–114, DOI:10.1016/j.envint.2013.08.022.
- J. S. C. Liou, B. Szostek, C. M. DeRito and E. L. Madsen, Investigating the biodegradability of perfluorooctanoic acid, Chemosphere, 2010, 80(2), 176–183, DOI:10.1016/j.chemosphere.2010.03.009.
- V. Franke, P. McCleaf, K. Lindegren and L. Ahrens, Efficient removal of per- And polyfluoroalkyl substances (PFASs) in drinking water treatment: Nanofiltration combined with active carbon or anion exchange, Environ. Sci.: Water Res. Technol., 2019, 5(11), 1836–1843, 10.1039/c9ew00286c.
- Z. Abbasian Chaleshtari and R. Foudazi, A Review on Per-and Polyfluoroalkyl Substances (PFAS) Remediation: Separation Mechanisms and Molecular Interactions, ACS ES&T Water, 2022, 2(12), 2258–2272 Search PubMed.
- P. Westreich, R. Mimna, J. Brewer and F. Forrester, The removal of short-chain and long-chain perfluoroalkyl acids and sulfonates via granular activated carbons: A comparative column study, Remediation, 2018, 29(1), 19–26, DOI:10.1002/rem.21579.
- P. N. Omo-Okoro, C. J. Curtis, A. M. Marco, L. Melymuk and J. O. Okonkwo, Removal of per- and polyfluoroalkyl substances from aqueous media using synthesized silver nanocomposite-activated carbons, J. Environ. Health Sci. Eng., 2021, 19(1), 217–236, DOI:10.1007/s40201-020-00597-3.
- B. Shrestha, M. Ezazi, S. Ajayan and G. Kwon, Reversible adsorption and desorption of PFAS on inexpensive graphite adsorbents: via alternating electric field, RSC Adv., 2021, 11(55), 34652–34659, 10.1039/d1ra04821j.
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang, J. Huang and G. Yu, Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents-A review, J. Hazard. Mater., 2014, 274, 443–454, DOI:10.1016/j.jhazmat.2014.04.038.
- I. M. Militao, F. A. Roddick, R. Bergamasco and L. Fan, Removing PFAS from aquatic systems using natural and renewable material-based adsorbents: A review, J. Environ. Chem. Eng., 2021, 9(4), 105271, DOI:10.1016/j.jece.2021.105271.
- S. Garg, J. Wang, P. Kumar, V. Mishra, H. Arafat, R. Shyam Sharma and L. F. Dumée, Remediation of water from per-/poly-fluoroalkyl substances (PFAS) - Challenges and perspectives, J. Environ. Chem. Eng., 2021, 9(4), 105784, DOI:10.1016/j.jece.2021.105784.
- W. Cai, D. A. Navarro, J. Du, G. Ying, B. Yang, M. J. McLaughlin and R. S. Kookana, Increasing ionic strength and valency of cations enhance sorption through hydrophobic interactions of PFAS with soil surfaces, Sci. Total Environ., 2022, 817, 152975, DOI:10.1016/j.scitotenv.2022.152975.
- T. M. Mohona, Z. Ye, N. Dai and P. C. Nalam, Adsorption behavior of long-chain perfluoroalkyl substances on hydrophobic surface: A combined molecular characterization and simulation study, Water Res., 2023, 239, 120074, DOI:10.1016/j.watres.2023.120074.
- P. Chularueangaksorn, S. Tanaka, S. Fujii and C. Kunacheva, Adsorption of perfluorooctanoic acid (PFOA) onto anion exchange resin, non-ion exchange resin, and granular-activated carbon by batch and column, Desalin. Water Treat., 2014, 52(34–36), 6542–6548, DOI:10.1080/19443994.2013.815589.
- M. Ateia, M. Arifuzzaman, S. Pellizzeri, M. F. Attia, N. Tharayil, J. N. Anker and T. Karanfil, Cationic polymer for selective removal of GenX and short-chain PFAS from surface waters and wastewaters at ng/L levels, Water Res., 2019, 163, 114874, DOI:10.1016/j.watres.2019.114874.
- D. Cao, M. Hu, C. Han, J. Yu, L. Cui, Y. Liu, H. Wang, Y. Cai, Y. Kang and Y. Zhou, Proton sponge-functionalized silica as high performance adsorbents for solid-phase extraction of trace perfluoroalkyl sulfonates in the environmental water samples and their direct analysis by MALDI-TOF-MS, Analyst, 2012, 137(9), 2218–2225, 10.1039/c2an16190g.
- L. Liu, Y. Liu, B. Gao, R. Ji, C. Li and S. Wang, Removal of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) from water by carbonaceous nanomaterials: A review, Crit. Rev. Environ. Sci. Technol., 2019, 50, 2379–2414, DOI:10.1080/10643389.2019.1700751.
- D. Zhang, Q. He, M. Wang, W. Zhang and Y. Liang, Sorption of perfluoroalkylated substances (PFASs) onto granular activated carbon and biochar, Environ. Technol., 2021, 42(12), 1798–1809, DOI:10.1080/09593330.2019.1680744.
- M. Park, S. Wu, I. J. Lopez, J. Y. Chang, T. Karanfil and S. A. Snyder, Adsorption of perfluoroalkyl substances (PFAS) in groundwater by granular activated carbons: Roles of hydrophobicity of PFAS and carbon characteristics, Water Res., 2020, 170, 115364, DOI:10.1016/j.watres.2019.115364.
- D. Saha, S. Khan and S. E. Van Bramer, Can porous carbons be a remedy for PFAS pollution in water? A perspective, J. Environ. Chem. Eng., 2021, 9(6), 106665, DOI:10.1016/j.jece.2021.106665.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. A. Vagliasindi and P. Roccaro, Removal of poly- and perfluoroalkyl substances (PFAS) from water by adsorption: Role of PFAS chain length, effect of organic matter and challenges in adsorbent regeneration, Water Res., 2020, 171, 115381, DOI:10.1016/j.watres.2019.115381.
- M. Ateia, A. Alsbaiee, T. Karanfil and W. Dichtel, Efficient PFAS Removal by Amine-Functionalized Sorbents: Critical Review of the Current Literature, Environ. Sci. Technol. Lett., 2019, 6(12), 688–695, DOI:10.1021/acs.estlett.9b00659.
- D. Tian, D. Geng, W. Tyler Mehler, G. Goss, T. Wang, S. Yang, Y. Niu, Y. Zheng and Y. Zhang, Removal of perfluorooctanoic acid (PFOA) from aqueous solution by amino-functionalized graphene oxide (AGO) aerogels: Influencing factors, kinetics, isotherms, and thermodynamic studies, Sci. Total Environ., 2021, 783, 147041, DOI:10.1016/j.scitotenv.2021.147041.
- S. Mahpishanian, H. Sereshti and M. Ahmadvand, A nanocomposite consisting of silica-coated magnetite and phenyl-functionalized graphene oxide for extraction of polycyclic aromatic hydrocarbon from aqueous matrices, J. Electr. Syst., 2016, 55, 164–173, DOI:10.1016/j.jes.2016.02.023.
- M. N. Pervez, T. Jiang, J. K. Mahato, A. K. Ilango, Y. Kumaran, W. Zhang, H. Efstathiadis, J. I. Feldblyum, M. V. Yigit and Y. Liang, Surface Modification of Graphene Oxide for Fast Removal of Per- and Polyfluoroalkyl Substances (PFAS) Mixtures from River Water, ACS ES&T Water, 2024, 4, 2968–2980, DOI:10.1021/acsestwater.4c00187.
- F. Tunioli, T. D. Marforio, L. Favaretto, S. Mantovani, A. Pintus, A. Bianchi, A. Kovtun, M. Agnes, V. Palermo, M. Calvaresi, M. L. Navacchia and M. Melucci, Chemical Tailoring of β-Cyclodextrin-Graphene Oxide for Enhanced Per- and Polyfluoroalkyl Substances (PFAS) Adsorption from Drinking Water, Chem.–Eur. J., 2023, 29(60), e202301854, DOI:10.1002/chem.202301854.
- X. Wang, H. Zhang, S. Ham and R. Qiao, Graphene Oxide and Its Derivatives as Adsorbents for PFOA Molecules, J. Phys. Chem. B, 2023, 127(44), 9620–9629, DOI:10.1021/acs.jpcb.3c04762.
- C. Zhao, J. Fan, D. Chen, Y. Xu and T. Wang, Microfluidics-generated graphene oxide microspheres and their application to removal of perfluorooctane sulfonate from polluted water, Nano Res., 2016, 9(3), 866–875, DOI:10.1007/s12274-015-0968-7.
- S. Lath, D. A. Navarro, D. Losic, A. Kumar and M. J. Mclaughlin, Sorptive remediation of perfluorooctanoic acid (PFOA) using mixed mineral and graphene/carbon-based materials, Environ. Chem., 2018, 15(8), 472–480, DOI:10.1071/EN18156.
- M. Ali, S. P. Meaney, L. W. Giles, P. Holt, M. Majumder and R. F. Tabor, Capture of Perfluorooctanoic Acid Using Oil-Filled Graphene Oxide-Silica Hybrid Capsules, Environ. Sci. Technol., 2020, 54(6), 3549–3558, DOI:10.1021/acs.est.9b05469.
- M. Al Amin, Z. Sobhani, Y. Liu, R. Dharmaraja, S. Chadalavada, R. Naidu, J. M. Chalker and C. Fang, Recent advances in the analysis of per- and polyfluoroalkyl substances (PFAS)—A review, Environ. Technol. Innovation, 2020, 19, 100879, DOI:10.1016/j.eti.2020.100879.
- K. L. Rodriguez, J. H. Hwang, A. R. Esfahani, A. H. M. A. Sadmani and W. H. Lee, Recent developments of PFAS-detecting sensors and future direction: A review, Micromachines, 2020, 11(7), 667, DOI:10.3390/mi11070667.
- M. Ylinen, H. Hanhijärvi, P. Peura and O. Rämö, Quantitative gas chromatographic determination of perfluorooctanoic acid as the benzyl ester in plasma and urine, Arch. Environ. Contam. Toxicol., 1985, 14(6), 713–717, DOI:10.1007/BF01055778.
- R. Alzaga and J. M. Bayona, Determination of perfluorocarboxylic acids in aqueous matrices by ion-pair solid-phase microextraction-in-port derivatization-gas chromatography-negative ion chemical ionization mass spectrometry, J. Chromatogr. A, 2004, 1042(1–2), 155–162, DOI:10.1016/j.chroma.2004.05.015.
- A. O. Okaru, T. S. Brunner, S. M. Ackermann, T. Kuballa, S. G. Walch, M. Kohl-Himmelseher and D. W. Lachenmeier, Application of 19F NMR Spectroscopy for Content Determination of Fluorinated Pharmaceuticals, J. Anal. Methods Chem., 2017, 2017, 9206297, DOI:10.1155/2017/9206297.
- C. A. Moody, W. Chi Kwan, J. W. Martin, D. C. G. Muir and S. A. Mabury, Determination of perfluorinated surfactants in surface water samples by two independent analytical techniques: Liquid chromatography/tandem mass spectrometry and 19F NMR, Anal. Chem., 2001, 73(10), 2200–2206, DOI:10.1021/ac0100648.
- A. L. George and G. F. White, Optimization of the methylene blue assay for anionic surfactants added to estuarine and marine water, Environ. Toxicol. Chem., 1999, 18(10), 2232–2236, DOI:10.1002/etc.5620181016.
- S. Chitikela, S. K. Dentel and H. E. Allen, Modified method for the analysis of anionic surfactants as methylene blue active substances, Analyst, 1995, 120(7), 2001–2004, 10.1039/AN9952002001.
- R. Suárez, B. Horstkotte and V. Cerdà, In-syringe magnetic stirring-assisted dispersive liquid-liquid microextraction for automation and downscaling of methylene blue active substances assay, Talanta, 2014, 130, 555–560, DOI:10.1016/j.talanta.2014.06.063.
- W. S. Hummers and R. E. Offeman, Preparation of Graphitic Oxide, J. Am. Chem. Soc., 1958, 80(6), 1339–1339, DOI:10.1021/ja01539a017.
- C. Fang, X. Zhang, Z. Dong, L. Wang, M. Megharaj and R. Naidu, Smartphone app-based/portable sensor for the detection of fluoro-surfactant PFOA, Chemosphere, 2018, 191, 381–388, DOI:10.1016/j.chemosphere.2017.10.057.
- G. L. Long and J. D. Winefordner, Limit of detection. A closer look at the IUPAC definition, Anal. Chem., 1983, 55(7), 712A–724A, DOI:10.1021/ac00258a724.
- L. V. Rajaković, D. D. Marković, V. N. Rajaković-Ognjanović and D. Z. Antanasijević, Review: The approaches for estimation of limit of detection for ICP-MS trace analysis of arsenic, Talanta, 2012, 102, 79–87, DOI:10.1016/j.talanta.2012.08.016.
- A. Jeyaseelan, A. A. Ghfar, M. Naushad and N. Viswanathan, Design and synthesis of amine functionalized graphene oxide for enhanced fluoride removal, J. Environ. Chem. Eng., 2021, 9(4), 105384, DOI:10.1016/j.jece.2021.105384.
- D. Zhao, X. Gao, C. Wu, R. Xie, S. Feng and C. Chen, Facile preparation of amino functionalized graphene oxide decorated with Fe3O4 nanoparticles for the adsorption of Cr (VI), Appl. Surf. Sci., 2016, 384, 1–9, DOI:10.1016/j.apsusc.2016.05.022.
- N. Cai and P. Larese-Casanova, Application of positively-charged ethylenediamine-functionalized graphene for the sorption of anionic organic contaminants from water, J. Environ. Chem. Eng., 2016, 4(3), 2941–2951, DOI:10.1016/j.jece.2016.06.004.
- A. Navaee and A. Salimi, Efficient amine functionalization of graphene oxide through the Bucherer reaction: An extraordinary metal-free electrocatalyst for the oxygen reduction reaction, RSC Adv., 2015, 5(74), 59874–59880, 10.1039/c5ra07892j.
- R. Wijaya, G. Andersan, S. Permatasari Santoso and W. Irawaty, Green Reduction of Graphene Oxide using Kaffir Lime Peel Extract (Citrus hystrix) and Its Application as Adsorbent for Methylene Blue, Sci. Rep., 2020, 10(1), 667, DOI:10.1038/s41598-020-57433-9.
- S. Saxena, T. A. Tyson, S. Shukla, E. Negusse, H. Chen and J. Bai, Investigation of structural and electronic properties of graphene oxide, Appl. Phys. Lett., 2011, 99, 013104, DOI:10.1063/1.3607305.
- M. Shu, F. Gao, M. Zeng, C. Yu, X. Wang, R. Huang, J. Yang, Y. Su, N. Hu, Z. Zhou, K. Liu, Z. Yang, H. Tan and L. Xu, Microwave-Assisted Chitosan-Functionalized Graphene Oxide as Controlled Intracellular Drug Delivery Nanosystem for Synergistic Antitumour Activity, Nanoscale Res. Lett., 2021, 16(1), 75, DOI:10.1186/s11671-021-03525-y.
- J. Chen, L. Yang, S. Li and W. Ding, Various Physiological Response to Graphene Oxide and Amine-Functionalized Graphene Oxide in Wheat (Triticum aestivum), Molecules, 2018, 23(5), 1104, DOI:10.3390/molecules23051104.
- G. Zhang, M. Wen, S. Wang, J. Chen and J. Wang, Insights into thermal reduction of the oxidized graphite from the electro-oxidation processing of nuclear graphite matrix, RSC Adv., 2018, 8(1), 567–579, 10.1039/c7ra11578d.
- N. Alzate-Carvajal, D. A. Acevedo-Guzmán, V. Meza-Laguna, M. H. Farías, L. A. Pérez-Rey, E. Abarca-Morales, V. A. García-Ramírez, V. A. Basiuk and E. V. Basiuk, One-step nondestructive functionalization of graphene oxide paper with amines, RSC Adv., 2018, 8(28), 15253–15265, 10.1039/c8ra00986d.
- S. N. Alam, N. Sharma and L. Kumar, Synthesis of Graphene Oxide (GO) by Modified Hummers Method and Its Thermal Reduction to Obtain Reduced Graphene Oxide (rGO), Graphene, 2017, 06(01), 1–18, DOI:10.4236/graphene.2017.61001.
- F. Farivar, P. Lay Yap, R. U. Karunagaran and D. Losic, Thermogravimetric Analysis (TGA) of Graphene Materials: Effect of Particle Size of Graphene, Graphene Oxide and Graphite on Thermal Parameters, C, 2021, 7(2), 41, DOI:10.3390/c7020041.
- S. Kashyap, S. Mishra and S. K. Behera, Aqueous Colloidal Stability of Graphene Oxide and Chemically Converted Graphene, J. Nanopart., 2014, 2014, 640281, DOI:10.1155/2014/640281.
- C. Kim, J. Lee, W. Wang and J. Fortner, Organic functionalized graphene oxide behavior in water, Nanomaterials, 2020, 10(6), 1228, DOI:10.3390/nano10061228.
- S. Bao, W. Yang, Y. Wang, Y. Yu and Y. Sun, One-pot synthesis of magnetic graphene oxide composites as an efficient and recoverable adsorbent for Cd(II) and Pb(II) removal from aqueous solution, J. Hazard. Mater., 2020, 381, 120914, DOI:10.1016/j.jhazmat.2019.120914.
- Q. Yu, R. Zhang, S. Deng, J. Huang and G. Yu, Sorption of perfluorooctane sulfonate and perfluorooctanoate on activated carbons and resin: Kinetic and isotherm study, Water Res., 2009, 43(4), 1150–1158, DOI:10.1016/j.watres.2008.12.001.
- S. Deng, D. Shuai, Q. Yu, J. Huang and G. Yu, Selective sorption of perfluorooctane sulfonate on molecularly imprinted polymer adsorbents, Front. Environ. Sci. Eng. China, 2009, 3(2), 171–177, DOI:10.1007/s11783-009-0017-4.
- M. Shahabi Nejad, H. Soltani Nejad, S. Arabnejad and H. Sheibani, Enhanced adsorption of perfluorooctanoic acid using functionalized imidazolium iodide ionic liquid-based poly (glycidyl methacrylate), J. Appl. Polym. Sci., 2021, 138, e50962, DOI:10.1002/app.50962.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4va00171k |
| This journal is © The Royal Society of Chemistry 2024 |