
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anodic peroxide production for advanced oxidation processes with different metal oxide electrodes in carbonate electrolytes†
Tobias
Schanz
and
Jonathan Z.
Bloh
 *
*
DECHEMA-Forschungsinstitut, Theodor-Heuss-Allee 25, 60486 Frankfurt am Main, Germany. E-mail: jonathan.bloh@dechema.de
First published on 19th July 2024
Abstract
As an alternative to the anthraquinone process that can be used directly on site without storage and transport, electrochemical peroxide synthesis is a promising technology to produce reagents for water remediation via Advanced Oxidation Processes (AOP). The focus of research here is on anodic peroxide production, since cathodic synthesis is already at a high degree of maturity. Different materials and electrolytes have been reported for the anode reactions so far. It has also been shown that some electrolytes such as carbonate-based ones lead to the formation of secondary peroxides such as percarbonates which are well-suited as oxidants for AOP. Herein, these materials and electrolytes are evaluated under different conditions with particular focus on the actual oxidation power of the formed product mixtures.
Environmental significanceAmongst the most promising ways to combat water pollutants are the Advanced Oxidation Techniques. However, many of them require reagents on site such as hydrogen peroxide or iron salts, necessitating logistics and storage. Electrochemical methods present a promising technique to generate or regenerate these reagents directly on site or even in situ. Among these, electrochemical peroxide synthesis is particularly efficient as it can be generated both cathodically and anodically. Especially the latter is the focus of current research activities as the former is already quite mature. However, electrochemical peroxide production is typically only compared on the basis of the electrochemical performance and the rate of formation, not the actual oxidation power of the formed products. This is a relevant distinction as it is known that in certain electrolytes (such as the commonly used carbonate ones) also secondary peroxides such as percarbonate are formed with different oxidation power and kinetics. Thus, in our work, we evaluate the performance of different anodes and electrolytes with respect to not just the electrochemical performance but also the actual oxidation power against model substrates. Interestingly, we find that the ideal conditions for maximum oxidation power are not equivalent to those of maximum electrochemical performance, highlighting the importance of this approach. |
Introduction
One of the great challenges of our time is the provision of clean water to Earth's growing population. Yet, even in modern industrialized countries, drinking water is often polluted by many contaminants that may be harmful even in dilute concentration. One of the most promising technologies to combat these so-called micropollutants are Advanced Oxidation Processes (AOP) that usually rely on Reactive Oxygen Species (ROS) such as ozone, hydrogen peroxide, superoxide or hydroxyl radicals. Provision or generation of these ROS on site is often reliant on external reagents such as hydrogen peroxide or iron salts that need to be added, requiring supply chain and storage logistics. Direct on-site or even in situ generation techniques are therefore advantageous.This is particularly interesting for hydrogen peroxide as this is a dangerous and volatile compound which complicates shipping and storage. Today, hydrogen peroxide is almost exclusively produced by the anthraquinone process. Here, hydrogen gas, nowadays liberated from natural gas by steam reforming, is used to hydrogenate the anthraquinone which subsequently reacts with oxygen gas to form hydrogen peroxide. The disadvantage of this process is its reliance on large, centralized plants to work efficiently so the compound then must be distributed over large distances to the typically decentralized consumers.
Electrochemical peroxide synthesis, once the pioneering industrial process, can be efficiently deployed in smaller decentralized units at the point of use, thus saving on energy and costs for transportation and storage. In addition, electrochemical synthesis is more environmentally friendly than the anthraquinone process if powered by renewable energy sources.
At the cathode, oxygen can be converted to hydrogen peroxide via a two-electron reduction reaction (2e− ORR) (1).1 Cathodic peroxide production is very efficient with current efficiencies (CE) exceeding 90% achieved with different materials. Here, mostly carbon-based materials in various modifications2,3 with and without dopants4 have been used.
| O2 + 2H+ + 2e− → H2O2, E0 = 0.70 V | (1) |
Anodic hydrogen peroxide production proceeds via a two-electron oxidation of water (2e− WOR) to hydrogen peroxide. The redox potential for the oxidation of water to hydrogen peroxide is 1.76 V against the normal hydrogen electrode (2).1
| 2H2O → H2O2 + 2H+ + 2e−, E0 = 1.76 V | (2) |
The problem here is that the redox potentials of the oxidation of water to oxygen (3) and the oxidation of hydrogen peroxide to oxygen (4) are more negative, which automatically puts these reactions in competition with the desired one.1
| 2H2O → O2 + 4H+ + 4e−, E0 = 1.23 V | (3) |
| H2O2 → O2 + 2H+ + 2e−, E0 = 0.70 V | (4) |
For this reason, it is important to find materials that have a high CE for peroxide production with the appropriate conditions. In recent years, a wide variety of materials have been investigated in the field of anodic peroxide synthesis. Good results have already been documented by photoelectrochemical and electrochemical experiments.5 The first electrochemical experiments on anodic hydrogen peroxide synthesis were realized with different metal oxides. Izgorodin et al. used MnOx as a catalyst in an ionic liquid (butyl ammonium bisulfate (BAS)).6 In a later work, Shi et al. compared different metal oxides for their properties as electrochemical catalysts for peroxide synthesis. The metal oxides studied were TiO2, SnO2, WO3 and BiVO4. BiVO4 exhibited the highest CE.7 BiVO4 was further investigated in subsequent work.8–10 In most cases, these were not purely electrochemical experiments, but photoelectrochemical experiments. For the further work with bismuth vanadate, this was also doped or provided with a protective layer11 for increased stability and better catalyst properties.9,12 Another interesting catalyst material is calcium stannate (CaSnO3), which is stable over a longer polarization range and yields good CEs.13 A decisive disadvantage, however, is that CaSnO3 is highly carcinogenic, which makes it unsuitable for many applications. In addition to the previously mentioned metal oxides for anodic peroxide production, there is another very interesting mixed oxide that has yielded promising results. Miyase et al. have deposited a mixed oxide on fluorinated tin oxide (FTO) (InSbOx/CuSb2Ox/FTO). This is reported to be able to achieve near quantitative CEs in peroxide production and also to be much more stable than, for example, bismuth vanadate.14 Besides metal oxides, other materials are also capable of anodically generating peroxides. Carbon-based electrodes such as carbon fibre paper (CFP)15 and boron-doped diamond (BDD)16,17 have already achieved promising results. A very interesting phenomenon is that most of the anodic peroxide syntheses with high CEs were carried out in electrolytes containing carbonate. For example, Fuku et al. investigated various electrolytes for their properties for peroxide synthesis. Here, two molar potassium hydrogen carbonate solution (2 M KHCO3) exhibited the highest efficiency. The reason for this is probably that the electrolyte itself is oxidized. The potassium hydrogen carbonate is oxidized to peroxymonocarbonate (HCO4−), which is then able to convert water to peroxide, eqn (5) and (6).18 Recent work has even shown that various carbonate-based buffers exhibit high CE in anodic peroxide synthesis.16,19 The oxidation of the electrolyte itself can lead to the formation of peroxymonocarbonate, as already mentioned, and in the case of carbonate buffers with a high carbonate content, peroxydicarbonate can also be formed.20 The formation of this species possibly creates an equilibrium, stabilizing the peroxide and achieving a higher CE. This is interesting, since percarbonates have a high oxidation power and may thus contribute significantly to the overall oxidative action of the formed product mixture.
| HCO3− + H2O → HCO4− + 2e− + 2H+ | (5) |
| HCO4− + H2O → HCO3− + H2O2 | (6) |
We therefore studied different materials and electrolytes for the anodic peroxide production with the aim of not just characterizing the pure electrochemical performance but also the actual oxidative power of the formed product mixtures.
Results
Amongst the earliest modern work concerning the anodic hydrogen peroxide formation from water was a report by Izgorodin et al., who used MnOx as a catalyst in an ionic liquid (butyl ammonium bisulfate (BAS)) as electrolyte and reported up to 77% current efficiency albeit at only very short experiments and low product formation (0.08 μmol).6 Our own attempts to reproduce this system were unsuccessful. The catalyst preparation itself was successful, electrodepositing a thin MnOx layer on a gold foil substrate. However, only minute, barely quantifiable amounts of H2O2 were detected after employing these electrodes for anodic peroxide generation in the BAS-electrolyte. Moreover, the catalyst layer decomposed or delaminated after prolonged experiments. On top of the active layer decomposition, a further explanation may be that manganese oxides are known peroxide decomposition catalysts and thus prevent the formation of higher concentration.21 For these reasons, this catalyst system was discarded for further studies.Bismuth vanadate (BiVO4) is another interesting material which was explored in depth as photoanode material for photoelectrochemical water splitting. Recent calculations and experiments revealed that in the absence of other water oxidation catalysts, this material oxidizes water primarily via hydrogen peroxide as intermediate.22,23 In addition to its use as a photoanode for photoelectrochemical applications, this material may also be used a electrocatalyst without the use of light. Fuku et al. established that this electrode material works exceptionally well in bicarbonate electrolytes.23,24
However, BiVO4 is thermodynamically unstable under high applied potential, particularly at pH below 4 or above 11.23 This can partly be counteracted by doping with gadolinium (Gd) which increases the stability, as shown by Baek et al.12 Additionally, the poor conductivity of the semiconducting BiVO4 can be improved by doping with molybdenum (Mo) to reduce resistive losses.
Thus, films of 0.1% Mo, 10% Gd-doped BiVO4 on fluorine-doped tin oxide (FTO) substrates were synthesized and tested for anodic peroxide production in 2 M KHCO3 electrolyte (pH 8.3). The physico-chemical characterization showed that these samples are monoclinic BiVO4 with a relatively uniform film thickness of approximately 900 nm (Fig. S1–S3†). EDX analysis also confirmed the presence of the doping elements Mo and Gd (Fig. S4†). As shown in Fig. 1, these materials show a similar electrochemical behavior to their bare FTO substrate, albeit with 100 mV lower onset potential (0.1 mA cm−2) at 1.8 V vs. Ag/AgCl. This corresponds to an overpotential of approximately 730 mV. The current efficiency for peroxide formation varied between 10 to 20% for the BiVO4-based anodes over a wide potential range (Fig. S5†). This is significantly lower than some reports with this electrode and electrolyte system which claim up to 78% CE in 2 M KHCO3 (pH 8.3) at 3.1 V vs. RHE.12 However, there is a significant chance that this is related to the fact that percarbonate species are formed which may lead to overestimation of the peroxide content in many analytic methods and thus makes a direct comparison with other reports problematic.25
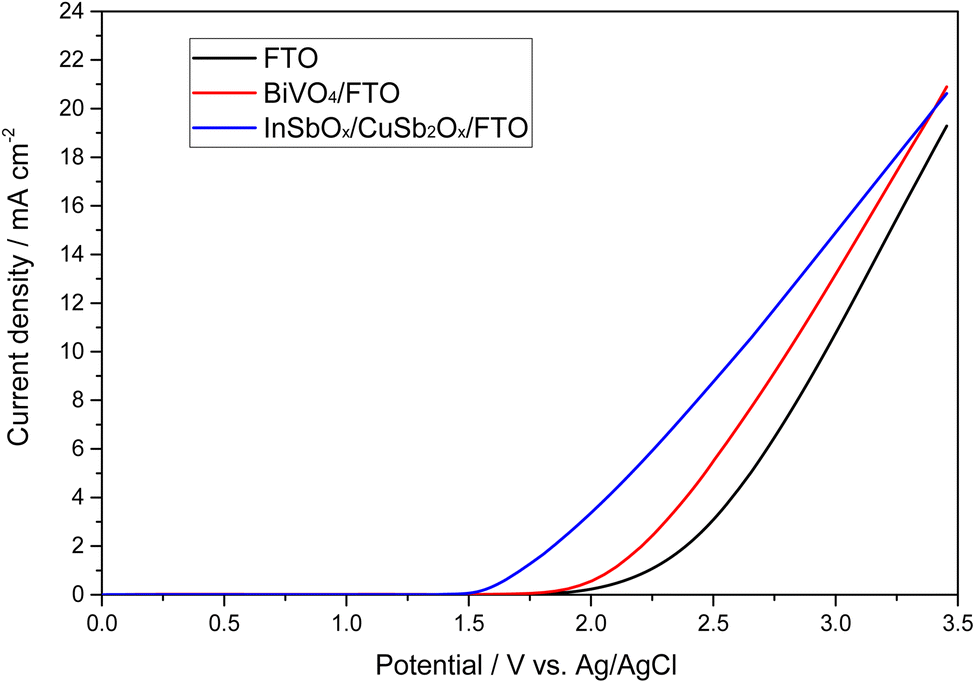 | ||
| Fig. 1 Linear sweep voltammogram of different anode materials for peroxide production in 2 M KHCO3 electrolyte (pH 8.3). | ||
Even though it is not pure hydrogen peroxide, it is highly interesting for the application, since even small amounts of peroxides or oxidative species are sufficient due to the strong oxidation properties.25 As such, even seemingly small concentrations may effect the same oxidation power as much higher concentrations of hydrogen peroxide alone. This also has impact on the analytical methods used for peroxide quantification. For example, the calorimetric test strips employed in many studies are oversensitive towards peroxomonocarbonate and thus give inflated values.25 When attempting to employ these in our experiments, they regularly yielded values well exceeding 100% current efficiency, which is implausible. Therefore, more specific tests based on enzymatic reactions or triiodide were employed in this study, which proved reliable even in the presence of peroxomonocarbonate.
The proposed mechanism leading to the increased peroxide selectively proceeds via direct oxidation of carbonate species which is in competition to the oxidation of water. Therefore, a high concentration of carbonates is critical for this mechanism,10,18 both to promote its oxidation as well as to reduce the water activity and thereby suppress the water oxidation. However, using bicarbonate, the solubility is limited to approximately 2 M using the most soluble potassium salt. However, the respective carbonate salts have significantly higher solubilities but require and/or lead to a higher pH value. Gill et al. found an electrolyte comprising 0.5 M KHCO3 and 3.5 M K2CO3 at pH 11.3 to be optimal for peroxide production over FTO anodes.19
By using a larger amount of total carbonate, and the correct ratio of carbonate to bicarbonate (3.5 M K2CO3/0.5 M KHCO3), the CEs could be more than doubled. Thus, with bismuth vanadate electrodes, CEs of over 40% could be achieved in longer measurements (Fig. 2). Here, the peroxide concentration increased almost linearly and reached over 6 mM of peroxide after a passed charge of 300 C, which corresponds to 42% CE. Interestingly, the CE continuously increased during polarization. However, the bismuth vanadate layer of the electrodes dissolved and detached during the reaction. This was not surprising, since bismuth vanadate is not stable at the electrolyte pH of 11.3.23 Since also the bare FTO used as the substrate for the BiVO4 film has also been demonstrated to show a similar performance under these conditions,19 it is likely that this experiment is just showing the response of the FTO substrate after a quick dissolution of the BiVO4. Therefore, using even Gd-stabilized BiVO4 appears to not be an option in higher pH carbonate electrolytes.
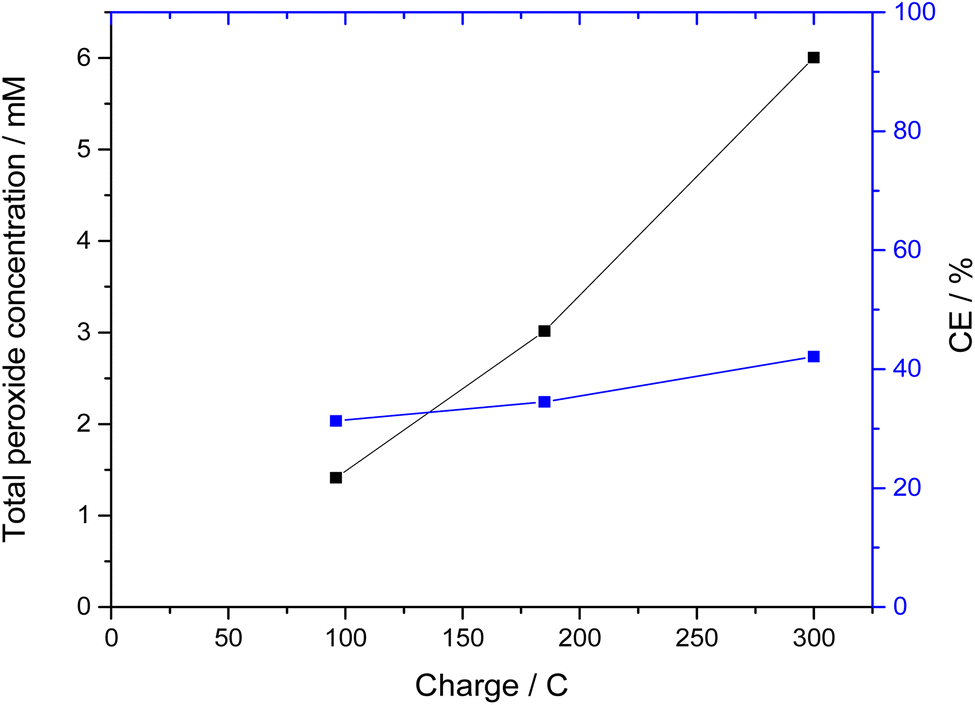 | ||
| Fig. 2 Peroxide formation during chronocoulometry at 2.385 V vs. Ag/AgCl using a BiVO4 anode. Total charge of 300 C passed during the experiment. Conditions: 3.75 cm2 BiVO4 working electrode (anode), 4.9 cm2 gas diffusion electrode (cathode), divided cell (cation exchange membrane), 110 mL 0.5 M KHCO3 and 3.5 M K2CO3 in each chamber. | ||
Recently, also another interesting anode material has been reported for anodic peroxide production by Miyase et al., who deposited indium–copper–antimony mixed oxides onto FTO (InSbOx/CuSb2Ox/FTO) and reported that these materials were much more stable than BiVO4-based variants.14 They achieved a CE of 53% in 0.5 M KHCO3, albeit at a low current density of 2 mA cm−2 and only in short experiments (1.8 C passed charge).14 Based on the promising data, we wanted to compare these electrodes to the other evaluated materials and therefore synthesized them according to the published procedure.14 For this purpose, the corresponding metal oxides were to be dissolved in butyl acetate and applied to the FTO substrates by means of spin coating. However, it was found that the metal oxides did not dissolve but only formed a dispersion. This dispersion was then applied to FTO substrates using spin coating and subsequently calcined. However, the resulting yellowish film was not bonded well to the substrate and easily removed or flaked off. Yet, when measuring the electrodes, an increased current compared to the pure FTO was observed. To ensure that the electrodes were not FTO that had changed structurally during calcination, an untreated FTO substrate was calcined under the same conditions. As shown in Fig. 3, there is a significant difference in the onset potential in the case of the coated FTO.
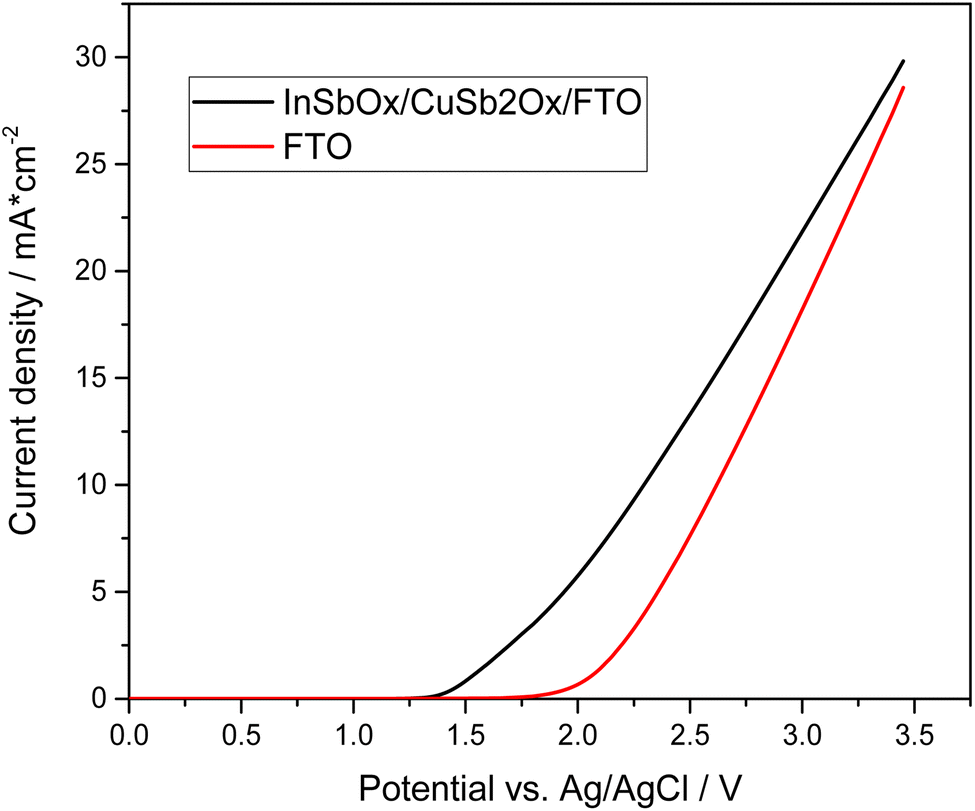 | ||
| Fig. 3 Linear sweep voltammetry from 0 to 3.5 V vs. Ag/AgCl. The diagram shows the corresponding current response of the InSbOx/CuSb2Ox/FTO electrode with increasing voltage compared to pure FTO substrate. Conditions: 4 cm2 InSbOx/CuSb2Ox/FTO working electrode (anode) and 3.6 cm2 InSbOx/CuSb2Ox/FTO working electrode (anode), 4.9 cm2 gas diffusion electrode (cathode), 110 mL 0.5 M KHCO3 and 3.5 M K2CO3 in the cell. | ||
In addition to the comparative measurements with pure FTO substrates, various analytical methods were used to detect and analyze the mixed oxide layer. However, due to the expected low thickness, this proved challenging. Thus, we were not able to observe the desired InSbOx/CuSb2Ox layer by scanning electron microscopy or even analyze the structure of the oxide layer in XRD, both of which just showed the FTO substrate (Fig. S11†). To detect the thin mixed oxide layer or the elements of it, XPS measurements were then performed. The results of these measurements showed that indeed the expected elements (In, Sb, Cu) were found on the surface of the FTO substrates (Fig. S7†). This confirmed our assumption that a thin layer of the mixed oxides has been baked into the FTO surface by the calcination process. Due to the small amount of InSbOx/CuSb2Ox, no further conclusions can be made about the catalyst layer at this point. However, we can see that the appropriate elements are present, and the peroxide formation performance is improved significantly compared to pure FTO substrate. Not only do these electrodes show lower onset potential of 1.35 V vs. Ag/AgCl compared to 1.80 V vs. Ag/AgCl of bare FTO and thus lowered overpotential by 450 mV, but our measurements also show a higher current efficiency of 50–57% (see Fig. S8†) compared to pure FTO, which achieves lower efficiency of 34% under these conditions (Fig. S9†). These show a CE of 52% at 3 V vs. Ag/AgCl as the highest efficiency. Of particular interest here is that the high efficiencies were achieved over a wide potential range. Thus, at a potential of 2.385 V vs. Ag/AgCl, CEs of over 50% could be achieved. At a higher potential of 3 V vs. Ag/AgCl, even up to 71% CE was observed. Interestingly, the current efficiency increased over the duration of the experiments (Fig. 4). While initially below 50%, it increased to a plateau of up to 71% in prolonged experiments (500 C). This suggests an initial period of activation is required for this catalyst material.
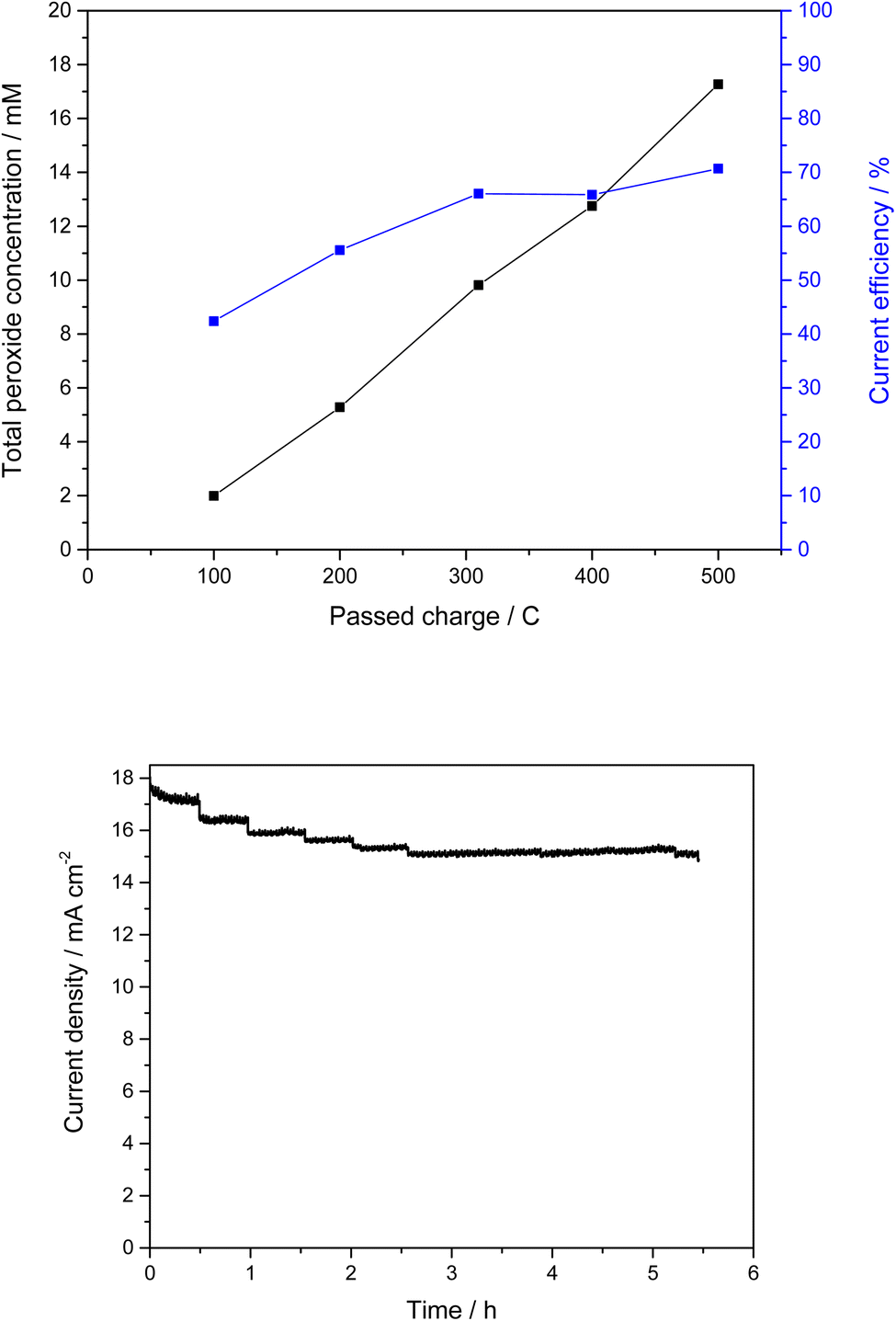 | ||
| Fig. 4 Total peroxide concentration and CE plotted against charge quantity. Chronocoulometry using InSbOx/CuSb2Ox/FTO as the working electrode. Polarization was performed at 3 V vs. Ag/AgCl in carbonate buffer under stirring with 0.5 M KHCO3 and 3.5 M K2CO3 at a pH of 11.3. The anode compartment contained 110 mL of electrolyte and was separated from the cathode compartment by a cation exchange membrane. A GDE was used as the cathode. In the gas compartment of the GDE, oxygen was passed through water for high humidity, which was then offered to the GDE at 15 mbar overpressure. | ||
Fig. 4 shows that the current density changes slightly over the test period of more than 5 hours. The jumps were caused by the sampling. Subsequently, the current density decreased by only 0.5 mA cm−2 over a period of 3 hours, which may be due to gas evolution. In this case, gas bubbles accumulate on the membrane, displacing electrolyte and decreasing the current density. The stability of the electrodes showed no reason for this, even after several prolonged polarizations. In addition to measurement durations of a few hours, even measurements over 24 hours were carried out. We were able to generate a peroxide concentration of 49.0 mM in a measurement over 24 hours with a charge quantity of 3.7 kC. At the beginning of the measurement and up to a peroxide concentration of just under 20 mM, it was even possible to achieve CEs between 74 and 83%. Subsequently, the reaction settled more and more into an equilibrium between peroxide generation and its degradation. As a result, the peroxide concentration did not continue to increase linearly, and the efficiency decreased. The corresponding data can be found in Fig. S10.†
However, while the absolute concentrations of peroxides formed as well as the current efficiency was highest at pH 11.3, this does not necessarily mean that this is the optimal condition to produce oxidizing agents. As we previously reported, anodic peroxide formation in carbonate containing electrolytes always leads to stable concentrations of percarbonate species such as peroxomonocarbonate (PMC, HCO4−) and peroxodicarbonate (PDC, C2O62−).25 Particularly the former has been shown to have very favorable oxidation kinetics in comparison to hydrogen peroxide.25–28
| HCO3− + H2O → HCO4− + 2e− + 2H+ | (7) |
| 2CO32− → C2O62− + 2e− | (8) |
The chemical equilibrium between PMC, PDC and hydrogen peroxide is strongly dependent on the concentration of bicarbonate and carbonate in the electrolyte solution and consequently the electrolyte pH. Therefore, we determined an oxidant efficiency factor of the different electrolyte systems. For this, we determined the discoloration rate of methylene blue as a model pollutant using fixed concentrations of hydrogen peroxide in the respective electrolyte systems. These were subsequently normalized to the discoloration rate observed in a carbonate-free electrolyte of the same pH (using KOH to adjust pH). The resulting enhancement factors, i.e., how much faster the carbonate-containing electrolytes are oxidizing in comparison to carbonate-free, are shown in Fig. 5 as relative oxidation power. It should be noted here that the points at pH 10.4 and above were obtained at 4 M total carbonate concentration whereas only 2 M were employed at pH 9.4 and below due to solubility constrains.
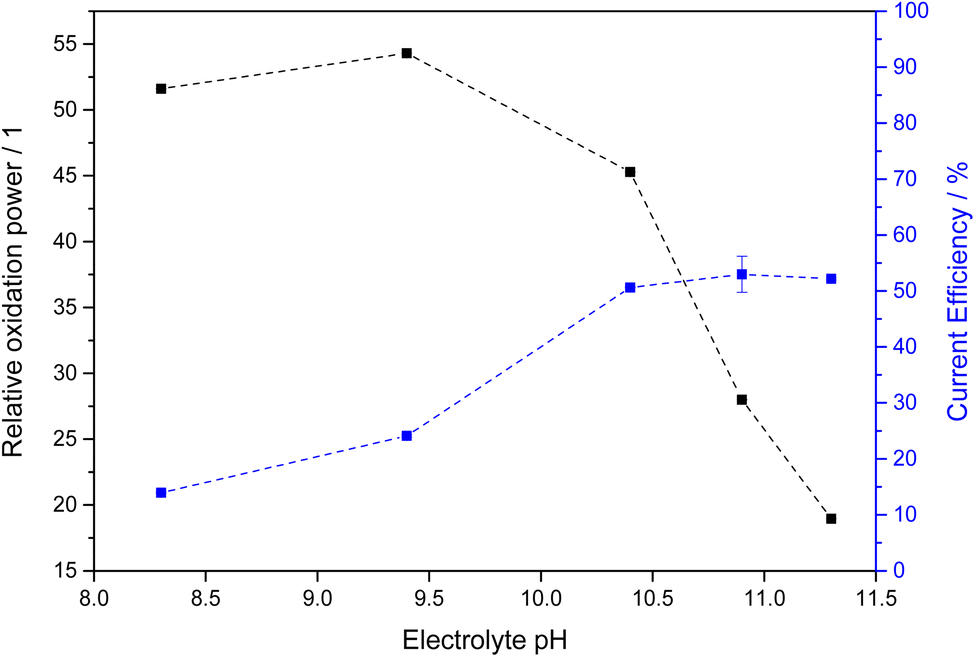 | ||
| Fig. 5 Relative oxidation power and current efficiency for different carbonate electrolytes. The electrolytes at pH 9.4 and below are 2 M total carbonate while those above are 4 M total carbonate. The relative oxidation power was obtained by measuring the methylene blue degradation rate constant and normalizing it for the one obtained in a carbonate-free electrolyte. | ||
In all studied cases, the oxidation power was much higher with carbonate present, confirming earlier studies.25 However, the highest enhancement was observed at lower pH, where the predominant species is bicarbonate. With increasing pH and thereby decreasing bicarbonate concentration, the oxidation power vanes in turn. This suggests that PMC (formed from bicarbonate) is a more effective oxidant than PDC (formed from carbonate).
When factoring in both this relative oxidation power and the current efficiency, the most favorable condition is found at pH 10.4 (1.5 M KHCO3 and 2.5 M K2CO3), where both the oxidation power is still relatively strong owing to the significant bicarbonate concentration but also the current efficiency is notably increased, presumably due to the carbonate present and/or the higher overall carbonate concentration (Fig. 6). At this point, the effective oxidant current efficiency reaches 23, meaning that a pure hydrogen peroxide forming process would need to have 2300% current efficiency to produce the same oxidation power (vs. methylene blue). This is of course an arbitrary metric, but it should serve to illustrate that for the purpose of generating oxidants, not just the current efficiency but also the nature of the formed products should be considered.
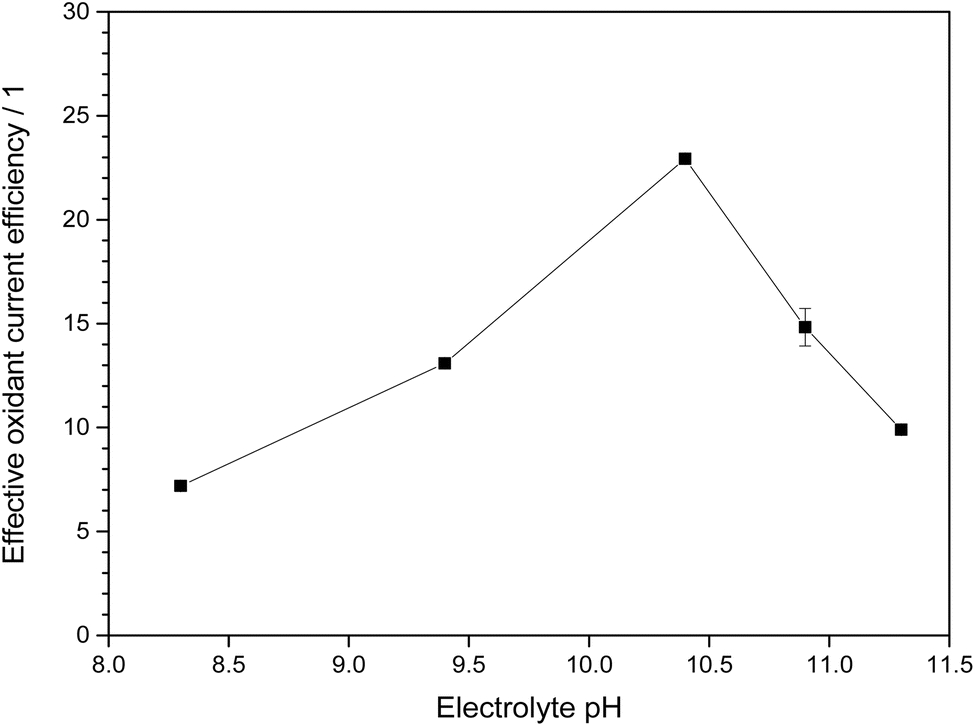 | ||
| Fig. 6 Effective oxidant current efficiency, obtained from the product of current efficiency and relative oxidation power shown in Fig. 5, for different electrolytes. The electrolytes at pH 9.4 and below are 2 M total carbonate while those above are 4 M total carbonate. | ||
Thus, for processes in which the generated peroxides are meant for directly coupled or in situ oxidation reactions without intermediate isolation and purification, the ideal electrolyte is a 4 M carbonate solution at pH 10.4.
Conclusion
Electrochemical generation of peroxides is a promising technique for in situ or on-site generation of ROS for water remediation. Herein, different materials were evaluated for electrochemical anodic peroxide production. The previously as promising material reported MnOx did not produce significant peroxide concentrations or oxidation power, proved unstable in longer experiments and was thus deemed unsuitable for the envisioned application.Much better results were obtained with BiVO4-based electrodes in carbonate-containing electrolytes. In 2 M KHCO3 electrolyte, the current efficiency was up to 20%. It was also shown, that in this electrolyte, there is always a significant proportion of peroxomonocarbonate present, which was a much higher oxidation power than hydrogen peroxide itself and is thus very beneficial for environmental remediation purposes. However, attempts to further increase the carbonate concentration in the electrolyte for a better performance proved unsuccessful with these electrodes, as they are not stable in the resulting higher pH.
This problem was solved by using a different electrode material consisting of a mixed oxide layer (InSbOx/CuSb2Ox/FTO), which is stable at a more basic pH and also exhibits very good peroxide formation. Compared to pure FTO substrates, both a higher current density and a higher current efficiency (up to 71%) were achieved in the 4 M KHCO3/K2CO3 buffer.
However, at this higher pH value, the fraction of bicarbonate in the electrolyte and with it the amount of highly oxidizing peroxomonocarbonate is also lower. This begs the question if these high pH electrolytes are really ideal for the most efficient generation of oxidation power. An analysis of the oxidation power and current efficiency over the pH range from 8.3 to 11.3 revealed that an intermediate pH value of 10.4 is ideal as it both benefits from an already elevated current efficiency while at the same time still having a high specific oxidation power. Under these conditions, the effective oxidant current efficiency in comparison to ordinary hydrogen peroxide is as high as 2300%, illustrating nicely the immense potential of electrochemical generation of ROS in carbonate electrolytes.
Experimental details
Peroxide quantification method
For the quantification of hydrogen peroxide, a previously established enzymatic assay based on horseradish peroxidase (HRP) was employed.29,30 This technique is based on the HRP-catalyzed stoichiometric dimerization of p-hydroxyphenylacetic acid (POHPAA) which yields a fluorescent product.29 In defined temporal intervals samples were taken from the electrolyte. 1 mg lyophilized powder of HRP (163 U mg−1 type II, Sigma Aldrich) and 4 mg freshly recrystallized POHPAA (Alfa Aesar) were both dissolved in 12.5 mL TRIS buffer (1.0 M, pH 8.8, Carl Roth) each. 100 μL of the sample was then mixed with 12.5 μL of each solution for 30 min and the fluorescence signal (λex = 315 nm, λem = 406 nm, 25 °C) was measured in a microplate reader (SynergyMx, BioTek). The concentrations were calculated according to calibration with an authentic H2O2 standard diluted in 2 M KHCO3 (Fig. S6†). Based on the specific interaction of the enzyme with the oxidant inside the active site, this technique is assumed to be specific for H2O2.As peroxide quantification method we using a colorimetric iodometry method.31 For this purpose, 100 μL of a sample taken from the electrolyte was mixed with 135 μL pH 4.1 potassium phosphate (Carl Roth) buffer, 10 μL 1.2 M potassium iodide (Alfa Aesar) solution and 5 μL of a 35 mM MoVI solution (ammonium molybdate(VI) tetrahydrate, Acros Organics) into a 96-well plate. The absorption of the resulting triiodide could then be measured at 350 nm and compared to that of a calibration using an H2O2 standard in this electrolyte (Fig S7†).
Preparation of BiVO4 anodes
The BiVO4 anodes (doped with 12% gadolinium and 0.1% molybdenum) were produced by dip coating onto a FTO (7 Ω sq.−1, Sigma Aldrich) substrate. First, the FTO was cleaned with acetone and water. A precursor solution was prepared by dissolving 0.616 M bismuth 2-ethylhexanoate (Alfa Aesar) and 0.7 M vanadium(V) oxytriethoxide (Acros) in chloroform (Carl Roth). For the doping, 84 mM gadolinium(III) isopropoxide (abcr) and 0.7 mM molybdenum(VI) oxide bis(2,4-pentanedionate) (Alfa Aesar) were added. The anodes were subsequently prepared by dip-coating the cleaned FTO slides from this precursor solution at a drawing speed of 100 or 700 mm min−1. The coated electrodes were then first heated to 100 °C for 10 hours in an oven. Then the temperature was increased to 450 in 10 hours and then held for two hours.Preparation of InSbOx/CuSb2Ox/FTO anodes
The fabrication of the InSbOx/CuSb2Ox/FTO electrodes was realized via spin coating followed by calcination.14 First, the FTO substrates were cleaned by ultrasonic bath in an acetone/water mixture (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50). Then, the CuSb2Ox precursor solution was first spin-coated (1000 rpm, 15 s) onto the substrate (30 μL cm−2). After applying the precursor solution, the substrates were calcined at 973 K for 30 min. After the application of the CuSb2Ox layer, the InSbOx layer was applied using the same parameters. However, the subsequent calcination ran for 30 min at 923 K. The preparation of the CuSb2Ox precursor solution was carried out by dispersing the respective metal oxides (CuO and Sb2O5) with a concentration of 0.2 M total concentration in an Sb/Cu molar ratio of 2.0, in butyl acetate. The InSbOx precursor solution was prepared in the same way with In2O3 and Sb2O5, but the ratio of the two metals to each other is 1:1.
:50). Then, the CuSb2Ox precursor solution was first spin-coated (1000 rpm, 15 s) onto the substrate (30 μL cm−2). After applying the precursor solution, the substrates were calcined at 973 K for 30 min. After the application of the CuSb2Ox layer, the InSbOx layer was applied using the same parameters. However, the subsequent calcination ran for 30 min at 923 K. The preparation of the CuSb2Ox precursor solution was carried out by dispersing the respective metal oxides (CuO and Sb2O5) with a concentration of 0.2 M total concentration in an Sb/Cu molar ratio of 2.0, in butyl acetate. The InSbOx precursor solution was prepared in the same way with In2O3 and Sb2O5, but the ratio of the two metals to each other is 1:1.
Electrochemical experiments
The electrochemical experiments were carried out in an H-cell in which the anode and cathode chambers both contained 100–120 mL of the electrolyte each when the cell was separated. The two chambers were separated by a Fumatech cation exchange membrane. The materials described above were used as the respective anode while a gas diffusion electrode (Gaskatel, 4.9 cm2) was used as the cathode. The experiments were run with a Zahner Zennium-E4 potentiostat at a constant potential vs. Ag/AgCl (chronocoulometry). The electrochemical experiments were all performed at room temperature and with stirring.Methylene blue discoloration
The methylene blue decolorization experiments were carried out in a 96-well plate. For this 100 μL of a 40 μM methylene blue solution was added to each well. Subsequently, 100 μL of the corresponding peroxide solution was added, whereby the final concentration in the well was then half the initial concentration of the peroxide solution and 20 μM methylene blue. The peroxide solution was the result of diluting a stock solution in the respective electrolyte to 10 mM. For the measurements with water, a 10 mM hydrogen peroxide solution was prepared. The corresponding pH value was adjusted using KOH. After adding the peroxide solution, the kinetic measurement was started immediately. The absorbance at 665 nm was monitored over time. In the first 10 minutes the measurements were carried out every 30 seconds, then for a further measured every minute for a further 10 min, followed by 40 min with measurements every 2 minutes. Finally, the measurements were every 10 minutes for a further 15 hours. Before each measurement, the plate was shaken for 1 s. The discoloration profile was then analyzed using first-order kinetics and the obtained rate constant was used as a figure of merit. Enhancement factors for the different electrolytes were obtained by dividing the rate constant obtained in the electrolyte by that of an aqueous peroxide solution at the same pH. The degradation over time profiles of methylene blue and the corresponding first-order degradation rates are shown in Fig. S13.†Surface characterization
All XPS spectra have been corrected for the C1s peak of the environment at 284.5 eV. The radiation source was a Mg Kα (1253.6 eV) and the power was 120 W with a pass energy of 10 eV. Further characterization included X-ray diffraction (XRD, Bruker D8 XRD Advance with Cu-Kα radiation, 40 kV, 30 mA), scanning electron microscopy (SEM, Hitachi FlexSEM 1000, 20 kV) and compositional profiles were measured with energy dispersive X-ray spectroscopy (20 kV, resolution 129.9 eV).Data availability
Additional information about the physico-chemical characterization of the electrodes, additional results from electrochemical experiments, calibration data as well as the raw data of the methylene blue degradation can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support by the German Ministry of Education and Research (BMBF) within the project “CO2SimO” (grant no. 033RC029C).References
- S. Siahrostami, S. J. Villegas, A. H. Bagherzadeh Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, A Review on Challenges and Successes in Atomic-Scale Design of Catalysts for Electrochemical Synthesis of Hydrogen Peroxide, ACS Catal., 2020, 10(14), 7495–7511, DOI:10.1021/acscatal.0c01641
.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction, Science, 2009, 323(5915), 760–764, DOI:10.1126/science.1168049
- V. L. Kornienko, G. A. Kolyagin, G. V. Kornienko, V. A. Parfenov and I. V. Ponomarenko, Electrosynthesis of H2O2 from O2 in a Gas-Diffusion Electrode Based on Mesostructured Carbon CMK-3, Russ. J. Electrochem., 2018, 54(3), 258–264, DOI:10.1134/S1023193518030060
- V. S. Antonin, M. H. M. T. Assumpcao, J. C. M. Silva, L. S. Parreira, M. R. V. Lanza and M. C. Santos, Synthesis and Characterization of Nanostructured Electrocatalysts based on Nickel and Tin for Hydrogen Peroxide Electrogeneration, Electrochim. Acta, 2013, 109, 245–251, DOI:10.1016/j.electacta.2013.07.078
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Electrochemical and Photoelectrochemical Water Oxidation for Hydrogen Peroxide Production, Angew. Chem., Int. Ed., 2021, 60(19), 10469–10480, DOI:10.1002/anie.202011215
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Low Overpotential Water Oxidation to Hydrogen Peroxide on a MnOx Catalyst, Energy Environ. Sci., 2012, 5(11), 9496–9501, 10.1039/c2ee21832a
- X. Shi, S. Siahrostami, G. L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Understanding Activity Trends in Electrochemical Water Oxidation to Form Hydrogen Peroxide, Nat. Commun., 2017, 8(1), 1–12, DOI:10.1038/s41467-017-00585-6
- S. Wan, C. Dong, J. Jin, J. Li, Q. Zhong, K. Zhang and J. H. Park, Tuning the Surface Wettability of a BiVO4 Photoanode for Kinetically Modulating Water Oxidative H2O2 Accumulation, ACS Energy Lett., 2022, 3024–3031, DOI:10.1021/acsenergylett.2c01078
- T. H. Jeon, H. Kim, H. I. Kim, H. I. Kim, W. Choi and W. Choi, Highly Durable Photoelectrochemical H2O2 Production: Via Dual Photoanode and Cathode Processes under Solar Simulating and External Bias-Free Conditions, Energy Environ. Sci., 2020, 13(6), 1730–1742, 10.1039/c9ee03154e
- H. Kusama, M. Kodera, K. Yamashita and K. Sayama, Insights into the Carbonate Effect on Water Oxidation over Metal Oxide Photocatalysts/Photoanodes, Phys. Chem. Chem. Phys., 2022, 24(10), 5894–5902, 10.1039/d1cp05797a
- K. Fuku, Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, WO3/BiVO4 Photoanode Coated with Mesoporous Al2O3 Layer for Oxidative Production of Hydrogen Peroxide from Water with High Selectivity, RSC Adv., 2017, 7(75), 47619–47623, 10.1039/c7ra09693c
- J. H. Baek, T. M. Gill, H. Abroshan, S. Park, X. Shi, J. Nørskov, H. S. Jung, S. Siahrostami and X. Zheng, Selective and Efficient Gd-Doped BiVO4 Photoanode for Two-Electron Water Oxidation to H2O2, ACS Energy Lett., 2019, 4(3), 720–728, DOI:10.1021/acsenergylett.9b00277
- S. Y. Park, H. Abroshan, X. Shi, H. S. Jung, S. Siahrostami and X. Zheng, CaSnO3: An Electrocatalyst for Two-Electron Water Oxidation Reaction to Form H2O2, ACS Energy Lett., 2019, 4(1), 352–357, DOI:10.1021/acsenergylett.8b02303
- Y. Miyase, Y. Miseki, T. Gunji and K. Sayama, Efficient H2O2 Production via H2O Oxidation on an Anode Modified with Sb-Containing Mixed Metal Oxides, ChemElectroChem, 2020, 7(11), 2448–2455, DOI:10.1002/celc.202000276
- D. Pangotra, L. I. Csepei, A. Roth, C. Ponce de León, V. Sieber and L. Vieira, Anodic Production of Hydrogen Peroxide Using Commercial Carbon Materials, Appl. Catal., B, 2022, 303, 120848, DOI:10.1016/j.apcatb.2021.120848
- S. Mavrikis, M. Göltz, S. Rosiwal, L. Wang and C. Ponce de León, Carbonate-Induced Electrosynthesis of Hydrogen Peroxide via Two-Electron Water Oxidation, ChemSusChem, 2022, 15(4), 1–9, DOI:10.1002/cssc.202102137
- K. Wenderich, B. A. M. Nieuweweme, G. Mul and B. T. Mei, Selective Electrochemical Oxidation of H2O to H2O2 Using Boron-Doped Diamond: An Experimental and Techno-Economic Evaluation, ACS Sustain. Chem. Eng., 2021, 9(23), 7803–7812, DOI:10.1021/acssuschemeng.1c01244
- T. M. Gill, L. Vallez and X. Zheng, The Role of Bicarbonate-Based Electrolytes in H2O2 Production through Two-Electron Water Oxidation, ACS Energy Lett., 2021, 6(8), 2854–2862, DOI:10.1021/acsenergylett.1c01264
- T. M. Gill, L. Vallez and X. Zheng, Enhancing Electrochemical Water Oxidation toward H2O2 via Carbonaceous Electrolyte Engineering, ACS Appl. Energy Mater., 2021, 4(11), 12429–12435, DOI:10.1021/acsaem.1c02258
- C. P. Chardon, T. Matthée, R. Neuber, M. Fryda and C. Comninellis, Efficient Electrochemical Production of Peroxodicarbonate Applying DIACHEM® Diamond Electrodes, ChemistrySelect, 2017, 2(3), 1037–1040, DOI:10.1002/slct.201601583
- S. H. Do, B. Batchelor, H. K. Lee and S. H. Kong, Hydrogen Peroxide Decomposition on Manganese Oxide (Pyrolusite): Kinetics, Intermediates, and Mechanism, Chemosphere, 2009, 75(1), 8–12, DOI:10.1016/j.chemosphere.2008.11.075
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, One- or Two-Electron Water Oxidation, Hydroxyl Radical, or H2O2 Evolution, J. Phys. Chem. Lett., 2017, 8(6), 1157–1160, DOI:10.1021/acs.jpclett.6b02924
- X. Shi, S. Back, T. M. Gill, S. Siahrostami and X. Zheng, Electrochemical Synthesis of H2O2 by Two-Electron Water Oxidation Reaction, Chem, 2021, 7(1), 38–63, DOI:10.1016/j.chempr.2020.09.013
- K. Fuku and K. Sayama, Efficient Oxidative Hydrogen Peroxide Production and Accumulation in Photoelectrochemical Water Splitting Using a Tungsten Trioxide/Bismuth Vanadate Photoanode, Chem. Commun., 2016, 52(31), 5406–5409, 10.1039/C6CC01605G
- T. Schanz, B. O. Burek and J. Z. Bloh, Fate and Reactivity of Peroxides Formed over BiVO4 Anodes in Bicarbonate Electrolytes, ACS Energy Lett., 2023, 8(3), 1463–1467, DOI:10.1021/acsenergylett.3c00227
- H. Pan, Y. Gao, N. Li, Y. Zhou, Q. Lin and J. Jiang, Recent Advances in Bicarbonate-Activated Hydrogen Peroxide System for Water Treatment, Chem. Eng. J., 2021, 408, 127332, DOI:10.1016/j.cej.2020.127332
- X. Liu, S. He, Y. Yang, B. Yao, Y. Tang, L. Luo, D. Zhi, Z. Wan, L. Wang and Y. Zhou, A Review on Percarbonate-Based Advanced Oxidation Processes for Remediation of Organic Compounds in Water, Environ. Res., 2021, 200, 111371, DOI:10.1016/j.envres.2021.111371
- X. Yang, Y. Duan, J. Wang, H. Wang, H. Liu and D. L. Sedlak, Impact of Peroxymonocarbonate on the Transformation of Organic Contaminants during Hydrogen Peroxide in Situ Chemical Oxidation, Environ. Sci. Technol. Lett., 2019, 6(12), 781–786, DOI:10.1021/acs.estlett.9b00682
- G. G. Guilbault, P. J. Brignac and M. Juneau, New Substrates
for the Fluorometric Determination of Oxidative Enzymes, Anal. Chem., 1968, 40(8), 1256–1263, DOI:10.1021/ac60264a027
- B. O. Burek, S. R. de Boer, F. Tieves, W. Zhang, M. van Schie, S. Bormann, M. Alcalde, D. Holtmann, F. Hollmann, D. W. Bahnemann and J. Z. Bloh, Photoenzymatic Hydroxylation of Ethylbenzene Catalyzed by Unspecific Peroxygenase: Origin of Enzyme Inactivation and the Impact of Light Intensity and Temperature, ChemCatChem, 2019, 11(13), 3093–3100, DOI:10.1002/cctc.201900610
- J. Xiao, M. Wang, Z. Pang, L. Dai, J. Lu and J. Zou, Simultaneous Spectrophotometric Determination of Peracetic Acid and the Coexistent Hydrogen Peroxide Using Potassium Iodide as the Indicator, Anal. Methods, 2019, 11(14), 1930–1938, 10.1039/c8ay02772b
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4va00176a |
| This journal is © The Royal Society of Chemistry 2024 |