
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of nanocrystalline high-entropy oxide CoNiFeCrMnOx thin film electrodes by dip-coating for oxygen evolution electrocatalysis†
Qingyang
Wu
a,
Achim
Alkemper
 a,
Stefan
Lauterbach
b,
Jan P.
Hofmann
a and
Marcus
Einert
*a
a,
Stefan
Lauterbach
b,
Jan P.
Hofmann
a and
Marcus
Einert
*a
aSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany. E-mail: meinert@surface.tu-darmstadt.de
bInstitute for Applied Geosciences, Geomaterial Science, Technical University of Darmstadt, Schnittspahnstrasse 9, 64287 Darmstadt, Germany
First published on 25th March 2024
Abstract
The variation of calcination conditions allows for adjusting the oxidation states and coordination sites of near-surface elements in sol–gel-derived high-entropy oxides. Optimized synthesis conditions resulted in fabrication of nanocrystalline CoNiFeCrMnOx thin films with 1–2 nm pores, showing low OER overpotentials of 258 mV VS. RHE at 10 mA cm−2.
For catalyzing the water splitting reaction under alkaline conditions, earth-abundant, cheap and catalytically active elements have to be selected for tailoring the chemical composition of novel catalysts. Transition metal-based spinel high-entropy oxides are a promising class of electrocatalysts owing to manifold synergistic effects of the interacting constituting cations in the crystal structure. By implementation of five or more elements in (nearly) equiatomic concentrations at a single crystallographic site, the configurational entropy of a system can be drastically increased leading to an entropy-based stabilization effect with respect to formation of a solid solution.1,2 The principal concept of entropy-stabilized alloys was expanded to multicationic oxide systems in 2015.3 Novel and unpredictable properties of high entropy materials have been observed and investigated, such as exceptionally stable electrochemical performance4,5 and high electrocatalytic activities.6–9 In the field of electrocatalysis, the interaction of numerous cations in transition metal (TM)-based high entropy oxides has been demonstrated to affect the electronic structure and thus the adsorption energetics of oxygen evolution reaction (OER) intermediates, such as OH* and OOH*.10 This can be understood on the basis of binding energy distribution patterns, for which unary catalysts possess just one distinct binding energy (assuming an infinite planar crystal surface).10 In contrast, for high-entropy materials, the configuration of neighbouring atoms at the surface with respect to the binding atom various to a much larger extend resulting in an almost continuous distribution of binding energies, and thus, formation of active sites with ideal binding energy.10 In this context, transition metal (TM)-based high-entropy oxides in spinel structure, firstly prepared by Dabrowa et al.,11 were presented to show enhanced OER activity.2,12–20 Furthermore, improvement of OER performance was reported by modification of the electronic structure in high-entropy ferrite thin films by synthetic tailoring of the chemical composition leading to enhanced charge carrier transport properties owing to an facilitated electron hopping mechanisms through d–d states of the transition metals.21 Multiatomic TM-based spinel oxide OER electrocatalysts with overpotentials ranging predominantly from 220 mV to 495 mV and with long-term stability up to 120 hours were reported recently12,13 and collectively prove the beneficial effect of increasing the configurational disorder/entropy within a compound.
In a systematic comparison between compact and mesoporous HEO thin films it was shown that the OER performance was substantially impacted by the overall surface area predetermining the absolute number of available catalytically active reaction sites.22 Ludwig et al. have recently determined the most OER active cantor alloy oxide (CoNiFeCrMn)3O4 by high-throughput analysis presenting an accelerated discovery of novel electrocatalysts by screening of sputter-deposited thin film libraries.23 However, there is still a lack of knowledge about the impact of calcination temperature and holding time on sol–gel derived TM-based high-entropy oxide thin films, which would be advantageous towards optimization of the OER activity since the content of oxygen vacancies as well as coordination sites and oxidation states of catalytically reactive cations (such as Co3+, Ni2+, Cr6+ and Mn4+) can be adjusted/varied in this way.
Therefore, HEO thin films were directly deposited on conductive substrates via an easy-to-handle sol–gel-based dip-coating approach and subsequently calcined at comparatively low annealing temperatures of 400 °C for either 10 min or 1 h. By control of the holding time, the impact of the nanostructure and surface area, crystallinity, charge transfer resistance, and surface composition on the OER performance was studied in alkaline solution. Based on the structural and electrochemical characterization, the HEO thin films were proven to be nanocrystalline, rich in defective oxygen sites and showed a dependence of the Cr6+ and Mn4+ concentration on the temperature holding time. Those control parameters were identified to be the reason for the observed second-highest OER activity (overpotential of 258 mV at 10 mA cm−2) and, to the best of our knowledge, lowest OER onset potential of only +1.33 V vs. RHE yet reported for CoNiFeCrMn-based HEOs.
The HEO thin films were prepared by a sol–gel-based dip-coating process recently reported by our group for preparation of mesoporous spinel HEO thin film electrodes utilizing the same (initial) concentration of transition metal nitrate precursors and structure directing agent (Pluronic F-127) in the dip-coating solution and keeping experimental conditions constant (see Experimental section in the ESI†).22 However in the underlying work, the HEO samples were annealed at 400 °C for ether 10 min or 1 h (resulting samples referred to as HEO-400-10min and HEO-400-1h, respectively) in order to study the impact of calcination time on the micro- and nanostructure, crystallography, and chemical surface composition and how these material properties influence the electrocatalytic OER performance. The microscopic surface morphology of both HEO-400-10min and HEO-400-1h investigated by scanning electron microscopy (SEM), was initially found to be compact in structure and crack-free even on the micro- and millimeter length scale (Fig. 1 and Fig. S1A, B, ESI†). Decreasing the calcination temperature of HEO to 350 °C for 10 min (HEO-350-10 min) also lead to compact thin film structures with a tendency for agglomeration of particles (island formation at the surface, Fig. S1C, ESI†). At temperatures of 600 °C, the formation of a developed mesoporous HEO network can be observed (Fig. S1D, ESI†), similar as reported in literature.22 Scanning transmission electron microscopy (STEM)-based energy dispersive X-ray spectroscopy (EDS) elemental mappings confirmed the presence of Cr (green), Mn (violet), Fe (red), Co (blue), and Ni (yellow) in HEO-400 thin films as illustrated in Fig. 1D. To verify the homogenous distribution of the transition metals throughout the entire thin film structure, EDS analyses were conducted on multiple distinct spots on the HEO samples (see Fig. S2, S3 and Table S1, ESI†). The results prove that all initially added transition metals are implemented homogenously throughout the HEO thin film in near-equiatomic concentrations (see Fig. S2–S4 and Table S1, ESI†), thus allowing for classification of the samples as high-entropy oxides.3 The selected area electron diffraction (SAED) patterns present diffuse and broad diffraction rings, wherefore the samples can be considered as crypto-crystalline since the nanostructures/-crystallites were too minute to relate the diffraction pattern to a specific crystal phase and to receive any structural information by X-ray-based analysis (Fig. 1C). The SAED patterns can be interpreted as preliminary stage for the spinel phase since several Debye-rings in the SAED can be assigned to this crystallographic structure.
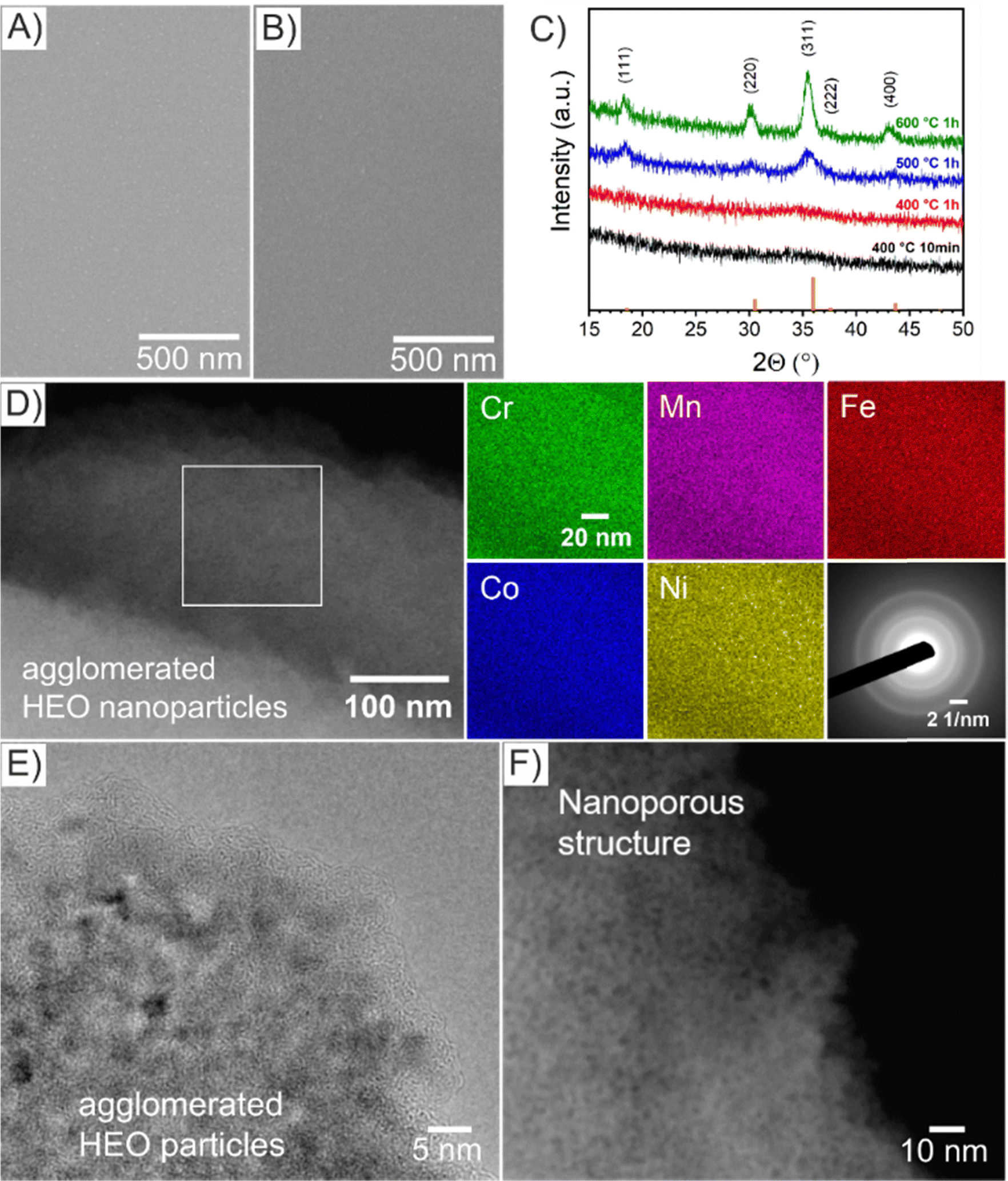 | ||
| Fig. 1 Top-view SEM images of (A) HEO-400-10min and (B) HEO-400-1h thin films. (C) GIXRD of HEO thin films calcined at various temperature of 400 °C (black), 500 °C (blue), and 600 °C (green) with line patterns (orange) representing the diffraction signals of the cubic spinel phase. (D) STEM image of HEO-400-1h with the corresponding EDS-based elemental mapping of Cr (green), Mn (violet), Fe (red), Co (blue), and Ni (yellow). SAED patterns from bulk areas (see Fig. S5, ESI†) revealing a diffraction pattern typical for crypto-crystalline materials. (E) HRTEM and (F) STEM images of selected areas of HEO-400-1h thin films. | ||
For further analysis of the morphology on the nanoscale, high-resolution transmission electron microscopy (HR-TEM, Fig. 1E) and STEM (Fig. 1F) was performed. Importantly, the HEO sample calcined at 400 °C demonstrates nanopores and nanoparticles both approximately between 1 nm to 2 nm in size (derived and measured from Fig. 1F). The contrast in the image (Fig. 1F) can be considered as verification of the nanoporous nature of the thin films. Furthermore, amorphous features were identified at the surface of HEO agglomerates (Fig. 1E), which can most likely be attributed to residual carbon originating from the porogen (Pluronic F-127) agreeing with the XSP data (Fig. S7B, ESI†). For comparison, spinel HEO thin films calcined at 600 °C for 1 h exhibit much larger mesopores due to thermally induced nanocrystal growth affecting the pore size.22 Temperature-dependent grazing-incidence X-ray diffraction (GIXRD) analysis was conducted to study the impact of annealing temperature and time on the crystallization process of the HEO thin films (Fig. 1C). Interestingly, the diffraction patterns of HEO-400-10min and HEO-400-1h exhibit a broad and low-intensity peak at 2θ = 36°, which coincides with the strongest diffraction signal of the (311) plane of the cubic spinel phase (Fd3m, space group no. 227). The (111)-, (220)-, (311)-, and (400)-peak become more pronounced when heating the HEO samples to 500 °C indicating thermally induced crystallite growth and formation of the spinel phase. At 600 °C, the HEO sample possess a crystallographic structure which can be consistently indexed to the cubic spinel phase in accordance with literature.21,22 Based on XRD data, the sol–gel-derived HEO thin films calcined at 400 °C show weak diffraction signals, suggesting a short-range order of atoms as also visualized by the diffuse Debye-rings in the SAED patterns (Fig. 3D). TEM analysis together with the GI-XRD data both demonstrate broad diffraction signals of the HEO-400 samples (in the XRD patterns at 2θ = 32°–38°, corresponding to most intense diffraction of (311)-plane of the cubic spinel phase), which is indicative for the presence of ultra-small nanocrystals/-particles since the integral width of a peak correlates reciprocally with the average crystallite size. Hence, it can be expected that the nanoparticles, only 1–2 nm in diameter, already start to crystallize between 300 °C and 400 °C in the spinel phase, which is consistent with TEM data. Due to the extremely low nanosizes of the pores and crystallites, the thermal decomposition of Pluronic F-127 in the HEO thin films can be expected to be in its initial stage with the compound still being present (compare XPS Fig. 2). In contrast, Pluronic F-127 templated Co3O4 thin films synthesized under comparable conditions were reported to possess well-developed nanoporous structures and a progressed degradation of the block-copolymer, also in accordance with the thermodynamic decomposition behavior of Pluronic F-127 in synthetic air (main mass loss derivative at 292 °C and less than 5% of total mass remain after 400 °C).24
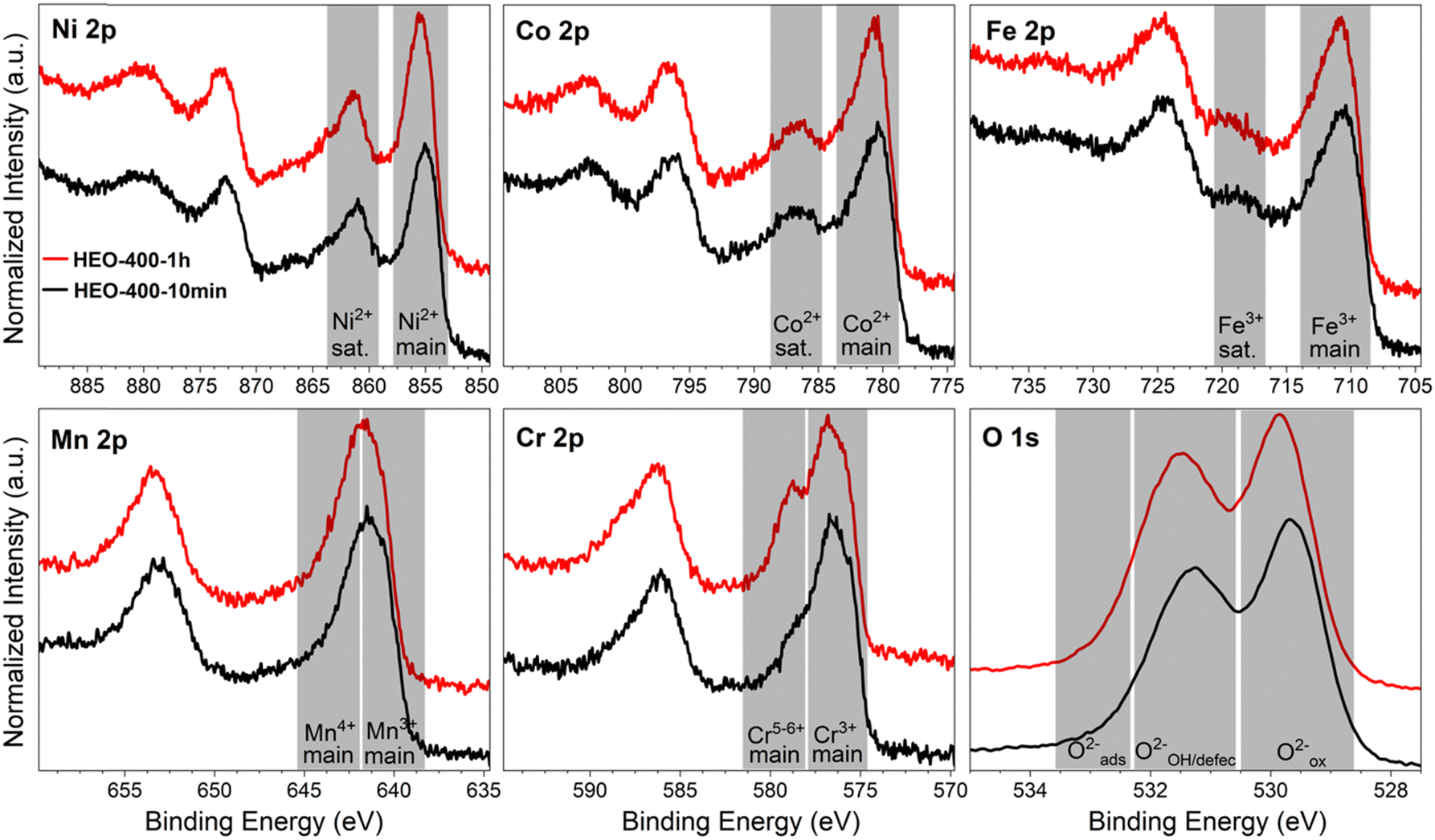 | ||
| Fig. 2 2p core level photoemission spectra of Ni, Co, Fe, Mn, Cr, and O for the HEO-400-10min (black) and HEO-400-1h (red) thin films acquired at a constant kinetic energy of 610 eV with grey areas indicating commonly assigned speciation/oxidation state ranges. | ||
The absolute thicknesses of the HEO-400-10min and HEO-400-1h were analyzed by profilometry (Fig. S6, ESI†) and determined to be (180 ± 10) nm and (210 ± 10) nm, respectively. These values are higher compared to the thickness of HEO thin films calcined at 600 °C for 1 h, a reasonable finding, since shrinkage of the amorphous (composite) film proceeds with increasing calcination time due to crystallization of the nanoporous framework and decomposition of the organic micelles (densification).25 The chemical surface composition and oxidation states of the constituting metal cations were evaluated by X-ray photoelectron spectroscopy (XPS) and measured at beamline PM3 of BESSY (Berlin, Germany).26 The survey spectrum (Fig. S7A, ESI†) shows that Ni, Co, Fe, Mn, Cr, O and C are present within the first atomic layers of the HEO-400-10min and HEO-400-1h thin films, thus validating the successful insertion of all cations, which were added in form of their nitrates upon dip-coating throughout the crypto-crystalline surface phase structure. The carbon signal, presented in Fig. S7B (ESI†), can be attributed to the residual porogen Pluronic F-127 as well as adventitious carbon. This observation agrees with the TEM data (compare Fig. 1E). The amount of detected carbon decreases for prolonged calcination at 400 °C (HEO-400-10min > HEO-400-1h). Fig. 2 illustrates the 2p core level spectra of the transition metals Ni, Co, Fe, Mn, Cr, and the O 1s spectrum analyzed for HEO-400-10min and HEO-400-1h. In general, 2p transition metal oxide spectra exhibit complex main line and satellite structures instead of a simple Gaussian–Lorentzian peak each. A straightforward fit based on one or two symmetric peaks per species would be incorrect. The overall shape is indicative of the oxidation state and coordination environment of the cations due to strong charge transfer and multiplet effects.27 Consequently, only the dominating species were assigned based on the 2p3/2 peak position and the qualitative line shape in comparison with literature references. An upshift of all photoemission peaks by 0.3 ± 0.1 eV is observed for the HEO-400-1h when compared to HEO-400-10min, which is related to charging and/or a Fermi level shift rather than any chemical differences of the involved species. The Ni 2p level (Fig. 2) shows a symmetric main line at 855.1–855.5 eV and an intense satellite at +6 eV. This is indicative for Ni2+ as oxide or hydroxide with the next-nearest-neighbor not being nickel.28,29 The main Co 2p emission line was identified at 780.4–780.7 eV, with a pronounced satellite at a distance of 6 eV providing evidence for a 2+ oxidation state. Co3+ does not feature a satellite and can thus be excluded.28 The Fe 2p spectra depict a defined main peak at 710.6–710.9 eV and a weak satellite feature separated by 8 eV. Therefore, iron can be assigned as 3+ valent in the form of Fe–O and Fe–OOH species.22,30,31 In the Mn 2p spectra, the broad main emission line is located at 641.4–641.8 eV in the absence of any satellite features, thus excluding the formation of larger quantities of Mn2+.32,33 Hence, Mn can be expected to be mainly present in the oxidation states 3+ and 4+. Importantly, the Mn 2p3/2 peak of HEO-400-1h was identified as more asymmetric, specifically with more intensity at higher binding energies compared to HEO-400-10min implying that Mn4+ is present in higher concentrations at the surface, when the annealing time is prolonged from 10 min to 1 h. For the Cr 2p emission lines, Cr shows a complex main peak at 576.4–576.7 eV without the presence of any satellites suggesting the existence of Cr3+ cations being present as Cr–O and/or Cr–OH groups at the surface.34,35 Interestingly, a pronounced shoulder develops at 579.0 eV for the HEO-400-1h sample, which allows the assumption that Cr3+ is oxidized to Cr5+/6+ with prolonged calcination at 400 °C. The formation of higher oxidized chromium species was also observed for HEO spinel films calcined at 600 °C and other sol–gel derived La(CrMnFeCo2Ni)O3 perovskite HEOs.22,36,37 We note that – apart from the Mn4+ and Cr5+/6+ species – no significant differences/shifts in binding energies were observed for the two control samples allowing the prediction that the annealing time has little impact on the chemical state of the transition metal cations at the surfaces and can be considered to be comparable in terms of their chemical status. The O 1s photoemission line (Fig. 2) features three components dominated by lattice oxygen (OOx2− at 529.7–529.9 eV). Between 532.7 eV and 530.5 eV, hydroxyl groups accompanied with defective oxide species, i.e., undercoordinated or close to an undercoordinated site (OOH/defect2−) were identified. Oxygen-containing molecules adsorbed at the surface (Oad2−) exhibit binding energies below 530.5 eV.22,37 As illustrated in Fig. 3, the electrochemical properties of the HEO-400-10min and HEO-400-1h were analyzed and compared using cyclic voltammetry (CV, Fig. 3A), linear sweep voltammetry (LSV, Fig. 3B), electrochemical impedance spectroscopy (EIS, Fig. 3C), evaluation of the double layer capacitance (CDL, Fig. 3D), determination of the Tafel slope (Fig. 3E), and chronopotentiometry (CP, Fig. 3F) in 1 M KOH. For elucidating, if calcination below 400 °C has a beneficial effect on the OER performance of sol–gel HEO thin films, the control sample HEO-350-10 min was also investigated toward OER performance (see Fig. S8, ESI†). CVs for HEO-400-10min and HEO-400-1h were performed for 50 cycles from 0.9 eV to 1.5 eV vs. RHE (iR-corrected) with a scan rate of 100 mV s−1. The oxidation peak appearing between 1.2 eV to 1.4 eV is not precisely attributable to one specific oxidation process of a constituting cation, since several distinct cations theoretically may contribute to the overall signal. Therefore, the broad oxidation peak has to be ascribed to a superposition of several simultaneously proceeding oxidation reactions, which can be found for the transformation of Ni2+ to Ni3+, Co2+ to Co3+, Co3+ to Co4+, Cr3+ to Cr6+, and/or Fe2+ to Fe3+. Especially, the presence of transition metal cations in the oxidation state +3 suggests that electrocatalytically active oxyhydroxide species, such as NiOOH, CoOOH, CrOOH, and/or FeOOH were formed in situ upon CV cycling, which is in accordance with literature where, e.g., the presence of iron was demonstrated to favor the transformation of the spinel phase into catalytically active oxyhydroxide species.38 The two reduction peaks observed between 1.0 V and 1.4 V can be understood as the reverse reaction of the higher oxidized cations and can most likely be described as superposition of several reduction peaks of the constituting elements. Prolonged CV cycling (from the first to the 50th cycle, Fig. 3A) neither reveals any significant change in the redox peak positions nor in the current density both implying improved electrochemical stability. The stable electrochemical performance can most likely be explained in surface reconstruction developing a long-term-stable surface composition and structure.39,40 It is noteworthy that – at this stage of the study – no precise identification of the redox active species is possible owing to the overlap of several redox peaks of reactants/cations, for which a synchrotron-based follow-up study is currently in progress for a detailed in situ evaluation of the electrocatalytically active centers (such as determination of tetrahedrally and octahedrally coordinated occupation sites) in the HEO thin films. The overpotential (vs. RHE) reached at Jgeo = 10 mA cm−2 is a characteristic parameter typically utilized for comparison of electrocatalysts. The HEO-400-10min and HEO-400-1h exhibit relatively low (iR-corrected) overpotentials of 270 mV and 258 mV (Fig. S8A, ESI†), respectively, the last value being determined as second-best OER activity ever reported for cantor alloy-based (CoNiFeCrMn) high entropy oxides.12,13 For comparison, mesoporous Co3O4 thin films, also prepared by soft-templating and EISA process under comparable synthesis conditions,41 were adduced as reference. It is noteworthy that only structurally related materials can be utilized as reference catalyst since the nanostructure is known to have major impact on the OER activity.22 The HEO thin films showed higher OER activities compared to mesoporous Co3O4 thin films (η10 = 340 mV). The difference in OER performance between HEO-400-10min and HEO-400-1h can be mainly assigned to the increased surface area of the longer calcined samples providing more catalytically active reaction site and being in agreement with results observed for mesoporous and dense HEO thin films.22 Additionally, the electrochemical performances might also be affected by the variation of the catalytically active surface species Cr6+ and Mn4+ as demonstrated by XPS analysis (Fig. 2). Especially, Cr6+ and Mn4+ cations are known to facilitate the oxygen evolution reaction since electron transfer from water to higher oxidized states is generally more likely to occur (compared to lower oxidized cations).37 The presence of both Mn3+ and Mn4+ were reported to have synergistic effects in promoting the OER,42,43 in which Mn4+ acts as secondary supply for the formation of catalytically active Mn3+ due to comproportionation of Mn2+ and Mn4+ in alkaline media.37,44,45 Furthermore, it is also demonstrated that oxidation of Cr3+ to Cr6+ significantly enhances the OER activity,46,47 wherefore the presence of both Cr6+ and Mn4+ at the surface of the HEO-400-1h thin film can be considered as additional catalytically active species (Fig. 3A). Furthermore, Cr and Mn promote the formation of different oxidation states of catalytically active Fe, Ni, and Co in HEO.48 Beside Mn and Cr, Co3+ is reported as highly catalytically active species dominating the OER performance of HEO perovskite electrocatalysts.37 Since Fe3+ has a higher affinity for electrons (than Co3+) it is capable of capturing electrons from Co3+ leading to redistribution of the Co 3d electrons. In this way, Co3+ ions can be activated by adjacent Fe3+ in spinel oxides resulting in enhanced OER activity.49 Ni, especially as Ni3+,50 is known to form highly active NiOOH phases in situ,51,52 contributing synergistically and collectively with the other near-surface located transition metals to an overall high OER activity by providing suitable binding energetics for reaction intermediates on the nanoporous HEO surface.
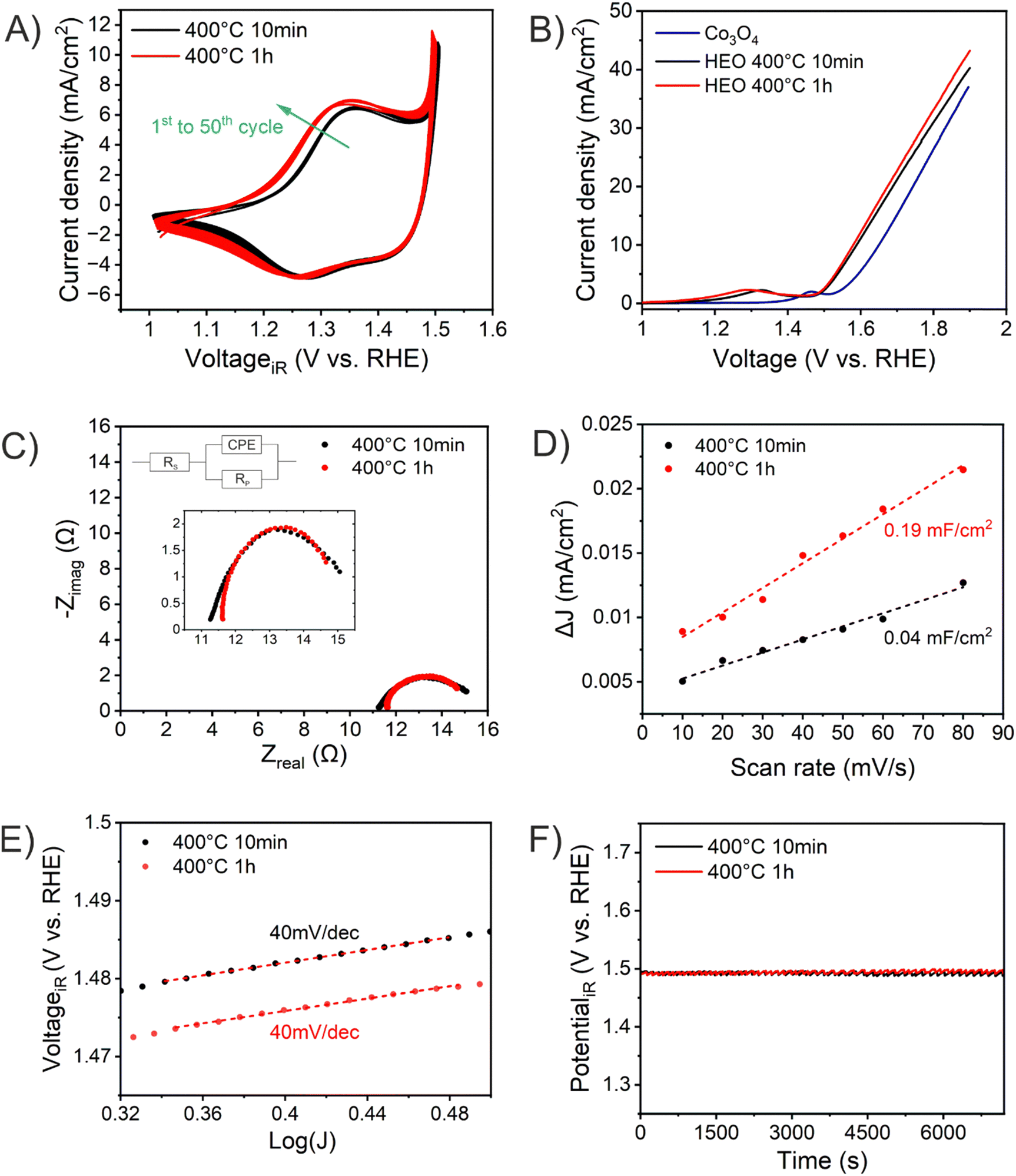 | ||
| Fig. 3 Electrochemical performance of HEO-400-10min (black) and HEO-400-1h (red): (A) cyclic voltammetry curves, (B) linear sweep voltammetry, (C) Nyquist plots measured at 1.6 V vs. RHE, (D) differential current densities and the corresponding the double-layer capacities, CDL, determined by linear fitting, (E) Tafel plots, and (F) chronopotentiometry analysis performed at 10 mA cm−2 for 2 hours. All experiments were conducted in 1 M KOH (pH ≈ 13.6). | ||
HEO-350-10 min showed with η10 = 272 mV no improvement of the OER performance related to the HEO-400 samples, wherefore no further (detailed) characterization was conducted. In general, the OER activity of TM oxides is correlated to the bond strength between the metal oxide surface and the reaction intermediate. Upon OER, a hydroxide anion electrochemically adsorbs at the surface and desorbs as hydroxide radical, ultimately oxidizing water to oxygen.53 The classic four-step proton-coupled electron transfer mechanism is described as adsorbate evolution mechanism (AEM). Since for TM oxides the formation and impact of oxygen vacancies are prevalent, the lattice oxygen mechanism (LOM) is frequently utilized to describe the OER evolution.54
To further explain this exceptional OER performance, EIS analyses were conducted to determine the charge transfer resistance (Rct) at the electrode–electrolyte interface. Rct is known to have major impact on the OER activity53 and was evaluated to be 4.6 Ω and 3.5 Ω for HEO-400-10min and HEO-400-1h, respectively, which are comparably small values related to other spinel oxide OER electrocatalysts from literature.12 The results imply that prolonged calcination at 400 °C has a beneficial impact on the charge carrier transfer at the interface of the HEO thin film and the electrolyte, thus contributing to an overall better OER performance of HEO-400-1h. To assess the electrochemical active surface area (ECSA), scan-rate dependent cycle voltammetry measurements in the non-faradaic region (0.5 V to 0.9 V, Fig. S9A and B, ESI†) were collected (Fig. S9C and D, ESI†) and linear fitting of the differential scan rates were carried out. As shown in Fig. 3C, the HEO-400-1h possess an almost five times larger specific double layer capacity (and thus ECSA) compared to HEO-400-10min, which is reasonable since the thermal degradation of the organic porogen proceeds with increased annealing time. The results are in accordance with SEM and TEM analysis (Fig. 1 and Fig. S1, S5, ESI†). Interestingly and in agreement with literature,22 the ECSA normalized LSV curves (Fig. S10, ESI†) show the opposite trend with JECSA HEO-400-10min > JECSA HEO-400-1h allowing the assumption that HEO-400-10min possess a higher intrinsic OER activity. This can be explained based on the chemical composition (in the bulk structure), which was determined to be Cr0.99Mn0.93Fe0.97Co1.09Ni1.03 for HEO-400-10min and Cr0.99Mn0.92Fe0.97Co1.06Ni1.06 through EDS analysis (Fig. S3 and Table S1, ESI†). In principal, the transition elements are present in near-equiatomic concentrations. However, while the Cr, Mn, and Fe content are almost similar for both samples, shows the HEO-400-10min an increased Co concentration, which likely contribute to the enhanced intrinsic (ECSA normalized) OER activity, since Co is a highly electrocatalytically active metal.37 However, for precise evaluation of the surface composition advanced surface characterization techniques will be required. The Tafel plots show slopes of 40 mV dec−1 for both samples, which are comparably low values for transition metal oxide electrocatalysts12,13,15 and in the same range as observed for mesostructured HEO thin films calcined at 600 °C.21,22 The presence of short diffusion paths provided by the nanoporous network can be considered as advantageous with respect to the activation energy for migration and transfer of charge carriers. To further assess the electrochemical stability of the HEO samples, chronopotentiometric measurements were performed at Jgeo = 10 mA cm−2vs. RHE for 2 hours (Fig. 3F) and for the best-performing HEO-400-1h sample for 10 hours. Interestingly, both HEO samples demonstrate stable electrochemical performance possessing a constant overpotential of solely (260 ± 2) mV over the entire analysis time. All experimentally obtained data are summarized in Table 1. After 10 hours of CP, the potential of HEO-400-1h decreases only 0.8% (Fig. S11A, ESI†). This stable performance of high-entropy spinel oxides can be ascribed to the reconstruction of the surface induced by in situ electrochemical activation through formation of an electrochemically stable oxyhydroxide surface layer.39 Furthermore, dissolution of the less stable elements Mn and Cr were reported also resulting in enhancement of the OER performance.48 SEM images taken after chronopotentiometry experiments illustrate slightly rougher surface morphologies (Fig. S11B and C, ESI†) clearly revealing the nanoporous structure of HEO-400-1h. The in-depth characterization of these surface reconstruction phenomena in nanoporous spinel HEO thin films will be scope of a perspective research study.
| HEO-400-10min | HEO-400-1h | |
|---|---|---|
| Composition (by EDS) | Cr0.99Mn0.93Fe0.97Co1.09Ni1.03 | Cr0.99Mn0.92Fe0.97Co1.06Ni1.06 |
| Charge states cations (by XPS) | Fe3+, Ni2+, Co3+, Cr3+ and Mn3+ and Mn4+ (low) | Fe3+, Ni2+, Co3+, Cr3+, Cr6+, Mn3+ and Mn4+ |
| (Geometrical) η10 | 258 mV vs. RHE | 270 mV vs. RHE |
| R ct at 1.6 V vs. RHE | 3.5 Ω | 4.6 Ω |
| C DL | 0.19 mF cm−2 | 0.04 mF cm−2 |
| Tafel slope | 40 mV dec−1 | 40 mV dec−1 |
| Stability (CP for 2 hours) | 100% at 10 mA cm−2 | 100% at 10 mA cm−2 |
Conclusions
In this study, it was shown that the chemical surface composition as well as crystallographic structure affect the oxygen evolution reaction performance of nanoporous CoNiFeCrMnOx thin films. Uniform and crack-free HEO thin films were fabricated by a sol–gel-based dip-coating method accompanied by soft-templating. The HEOs showed stable and low overpotentials of (260 ± 2) mV vs. RHE for driving the OER in alkaline solutions over at least 2 hours. The stable electrochemical performance was assigned to incorporation and formation of catalytically active Fe3+, Ni2+, Co3+, Cr6+ and Mn4+ species and the presence of defective oxygen sites collectively and synergistically interacting with each other. The concentration of Cr6+ and Mn4+ sites was increased by prolonged calcination at 400 °C. Due to development of a nanoporous network and an increase of the configurational disorder by implementation of five transition metals within the surface composition, an electrochemically stable surface structure was formed. This wet-chemical approach represents an inexpensive and facile way to prepare non-precious and nanoporous thin film electrocatalysts for efficiently driving the oxygen evolution reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Arslan Waheed for assistance with sample preparation, Dr Torsten Kachel for support during BESSY II beamtime under no. 222-11450-ST-1.1-P, and Ulrike Kunz for help with SEM-based EDS analysis. Marcus Einert acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, Walter Benjamin Program to ME) under project no. 469377211. Jan Philipp Hofmann and Achim Alkemper acknowledge financial support from DFG SPP2080 under project no. 493685339. Qingyang Wu acknowledges support from China Scholarship Council (CSC) under no. 202208320036.References
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, High-Entropy Oxides: Fundamental Aspects and Electrochemical Properties, Adv. Mater., 2019, 31, 1806236 CrossRef PubMed.
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, High-entropy energy materials: challenges and new opportunities, Energy Environ. Sci., 2021, 14, 2883–2905 RSC.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Entropy-stabilized oxides, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. de Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya and H. Hahn, High entropy oxides for reversible energy storage, Nat. Commun., 2018, 9, 3400 CrossRef PubMed.
- Y. Ma, Y. Hu, Y. Pramudya, T. Diemant, Q. Wang, D. Goonetilleke, Y. Tang, B. Zhou, H. Hahn and W. Wenzel, Resolving the Role of Configurational Entropy in Improving Cycling Performance of Multicomponent Hexacyanoferrate Cathodes for Sodium–Ion Batteries, Adv. Funct. Mater., 2022, 32, 2202372 CrossRef CAS.
- W. Huang, J. Zhang, D. Liu, W. Xu, Y. Wang, J. Yao, H. T. Tan, K. N. Dinh, C. Wu and M. Kuang, Tuning the electronic structures of multimetal oxide nanoplates to realize favorable adsorption energies of oxygenated intermediates, ACS Nano, 2020, 14, 17640–17651 CrossRef CAS PubMed.
- Z.-J. Chen, T. Zhang, X.-Y. Gao, Y.-J. Huang, X.-H. Qin, Y.-F. Wang, K. Zhao, X. Peng, C. Zhang and L. Liu, Engineering microdomains of oxides in high-entropy alloy electrodes toward efficient oxygen evolution, Adv. Mater., 2021, 33, 2101845 CrossRef CAS.
- Y. Sun, L. Yu, S. Xu, S. Xie, L. Jiang, J. Duan, J. Zhu and S. Chen, Battery-driven N2 electrolysis enabled by high-entropy catalysts: from theoretical prediction to prototype model, Small, 2022, 18, 2106358 CrossRef CAS PubMed.
- Q. Zhang, Y. Hu, H. Wu, X. Zhao, M. Wang, S. Wang, R. Feng, Q. Chen, F. Song and M. Chen, Entropy-Stabilized Multicomponent Porous Spinel Nanowires of NiFeXO4 (X = Fe, Ni, Al, Mo, Co, Cr) for Efficient and Durable Electrocatalytic Oxygen Evolution Reaction in Alkaline Medium, ACS Nano, 2023, 17, 1485–1494 CrossRef CAS PubMed.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, What makes high-entropy alloys exceptional electrocatalysts?, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef PubMed.
- J. Dąbrowa, M. Stygar, A. Mikuła, A. Knapik, K. Mroczka, W. Tejchman, M. Danielewski and M. Martin, Synthesis and microstructure of the (Co, Cr, Fe, Mn, Ni)3O4 high entropy oxide characterized by spinel structure, Mater. Lett., 2018, 216, 32–36 CrossRef.
- J. R. Esquius and L. Liu, High entropy materials as emerging electrocatalysts for hydrogen production through low-temperature water electrolysis, Mater. Futures, 2023, 2, 022102 CrossRef.
- X. Xu, Z. Shao and S. P. Jiang, High-Entropy Materials for Water Electrolysis, Energy Technol., 2022, 10, 2200573 CrossRef CAS.
- C. Triolo, S. Schweidler, L. Lin, G. Pagot, V. Di Noto, B. Breitung and S. Santangelo, Evaluation of electrospun spinel-type high-entropy (Cr0.2 Mn0.2Fe0.2Co0.2Ni0.2)3O4,(Cr0.2Mn0.2Fe0.2Co0.2Zn0.2)3O4 and (Cr0.2Mn0.2Fe0.2Ni0.2Zn0.2)3O4 oxide nanofibers as electrocatalysts for oxygen evolution in alkaline medium, Energy Adv., 2023, 2, 667–678 RSC.
- M. Anandkumar and E. Trofimov, Synthesis, Properties, and Applications of High-Entropy Oxide Ceramics: Current Progress and Future Perspectives, J. Alloys Compd., 2023, 170690 CrossRef CAS.
- H. Yu, Q. Li, Y. Hu, K. Li, K. M. Reddy, G. Xie, X. Liu and H.-J. Qiu, Self-Floating Nanoporous High-Entropy Oxides with Tunable Bandgap for Efficient Solar Seawater Desalination, Nano Lett., 2023, 23, 10554–10562 CrossRef CAS.
- R. Zhang, Z. Xu, Z. Du, Y. Wan, S. Yuan, F. Zeng, J. Xu, Z. Meng, X. Hu and H. Tian, Electrodeposition of Self-Supported High-Entropy Spinel Oxides for Stable Oxygen Evolution, Inorg. Chem., 2023, 62, 19052–19059 CrossRef CAS PubMed.
- B. Talluri, K. Yoo and J. Kim, High entropy spinel metal oxide (CoCrFeMnNi)3O4 nanoparticles as novel efficient electrocatalyst for methanol oxidation and oxygen evolution reactions, J. Environ. Chem. Eng., 2022, 10, 106932 CrossRef CAS.
- Z. Sun, Y. Zhao, C. Sun, Q. Ni, C. Wang and H. Jin, High entropy spinel-structure oxide for electrochemical application, Chem. Eng. J., 2022, 431, 133448 CrossRef CAS.
- C. Duan, X. Li, D. Wang, Z. Wang, H. Sun, R. Zheng and Y. Liu, Nanosized high entropy spinel oxide (FeCoNiCrMn)3O4 as a highly active and ultra-stable electrocatalyst for the oxygen evolution reaction, Sustainable Energy Fuels, 2022, 6, 1479–1488 RSC.
- M. Einert, A. Waheed, S. Lauterbach, M. Mellin, M. Rohnke, L. Q. Wagner, J. Gallenberger, C. Tian, B. M. Smarsly and W. Jaegermann, Sol–Gel-Derived Ordered Mesoporous High Entropy Spinel Ferrites and Assessment of Their Photoelectrochemical and Electrocatalytic Water Splitting Performance, Small, 2023, 19, 2205412 CrossRef CAS PubMed.
- M. Einert, M. Mellin, N. Bahadorani, C. Dietz, S. Lauterbach and J. P. Hofmann, Mesoporous high-entropy oxide thin films: electrocatalytic water oxidation on high-surface-area spinel (Cr0.2Mn0.2Fe0.2Co0.2Ni0.2)3O4 electrodes, ACS Appl. Energy Mater., 2022, 5, 717–730 CrossRef CAS.
- V. Strotkötter, O. A. Krysiak, J. Zhang, X. Wang, E. Suhr, W. Schuhmann and A. Ludwig, Discovery of High-Entropy Oxide Electrocatalysts: From Thin-Film Material Libraries to Particles, Chem. Mater., 2022, 34, 10291–10303 CrossRef PubMed.
- K. Kirchberg and R. Marschall, Sol–gel synthesis of mesoporous CaFe2O4 photocathodes with hierarchical pore morphology, Sustainable Energy Fuels, 2019, 3, 1150–1153 RSC.
- E. Celik, Y. Ma, T. Brezesinski and M. T. Elm, Ordered mesoporous metal oxides for electrochemical applications: correlation between structure, electrical properties and device performance, Phys. Chem. Chem. Phys., 2021, 23, 10706–10735 RSC.
- T. Kachel, F. Eggenstein and R. Follath, A soft X-ray plane-grating monochromator optimized for elliptical dipole radiation from modern sources, J. Synchrotron Radiat., 2015, 22, 1301–1305 CrossRef CAS PubMed.
- F. de Groot, Multiplet effects in X-ray spectroscopy, Coord. Chem. Rev., 2005, 249, 31–63 CrossRef CAS.
- N. S. McIntyre and M. G. Cook, X-ray photoelectron studies on some oxides and hydroxides of cobalt, nickel, and copper, Anal. Chem., 1975, 47, 2208–2213 CrossRef CAS.
- M. Lenglet, A. d'Huysser and C. K. Jørgensen, Optical spectra, X-ray photoelectron spectra and XANES of divalent nickel in mixed spinels NiFe2−xCrxO4, Inorg. Chim. Acta, 1987, 133, 61–65 CrossRef CAS.
- W. Temesghen and P. Sherwood, Analytical utility of valence band X-ray photoelectron spectroscopy of iron and its oxides, with spectral interpretation by cluster and band structure calculations, Anal. Bioanal. Chem., 2002, 373, 601–608 CrossRef CAS PubMed.
- M. Aronniemi, J. Sainio and J. Lahtinen, Chemical state quantification of iron and chromium oxides using XPS: the effect of the background subtraction method, Surf. Sci., 2005, 578, 108–123 CrossRef CAS.
- A. E. Bocquet, T. Mizokawa, T. Saitoh, H. Namatame and A. Fujimori, Electronic structure of 3d-transition-metal compounds by analysis of the 2p core-level photoemission spectra, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3771 CrossRef CAS PubMed.
- B. Gillot, S. Buguet, E. Kester, C. Baubet and P. Tailhades, Cation valencies and distribution in the spinels CoxCuyMnzFeuO4+δ (δ ≥ 0) thin films studied by X-ray photoelectron spectroscopy, Thin Solid Films, 1999, 357, 223–231 CrossRef CAS.
- Z. M. Hanafi, F. M. Ismail and A. K. Mohamed, X-ray photoelectron spectroscopy of chromium trioxide and some of its suboxides, Zeitschrift für Physikalische Chemie, 1996, 194, 61–67 CrossRef CAS.
- M. C. Biesinger, C. Brown, J. R. Mycroft, R. D. Davidson and N. S. McIntyre, X-ray photoelectron spectroscopy studies of chromium compounds, Surf. Interface Anal., 2004, 36, 1550–1563 CrossRef CAS.
- G. Wang, J. Qin, Y. Feng, B. Feng, S. Yang, Z. Wang, Y. Zhao and J. Wei, Sol–gel synthesis of spherical mesoporous high-entropy oxides, ACS Appl. Mater. Interfaces, 2020, 12, 45155–45164 CrossRef CAS PubMed.
- T. X. Nguyen, Y.-C. Liao, C.-C. Lin, Y.-H. Su and J.-M. Ting, Advanced high entropy perovskite oxide electrocatalyst for oxygen evolution reaction, Adv. Funct. Mater., 2021, 31, 2101632 CrossRef CAS.
- Y. Li, X. Lin and J. Du, Iron-Facilitated Transformation of Mesoporous Spinel Nanosheets into Oxyhydroxide Active Species in the Oxygen Evolution Reaction, Inorg. Chem., 2021, 60, 19373–19380 CrossRef CAS PubMed.
- S. Jiang, K. Tian, X. Li, C. Duan, D. Wang, Z. Wang, H. Sun, R. Zheng and Y. Liu, Amorphous High-entropy Non-precious metal oxides with surface reconstruction toward highly efficient and durable catalyst for oxygen evolution reaction, J. Colloid Interface Sci., 2022, 606, 635–644 CrossRef CAS PubMed.
- K. Huang, J. Xia, Y. Lu, B. Zhang, W. Shi, X. Cao, X. Zhang, L. M. Woods, C. Han and C. Chen, Self-Reconstructed Spinel Surface Structure Enabling the Long-Term Stable Hydrogen Evolution Reaction/Oxygen Evolution Reaction Efficiency of FeCoNiRu High-Entropy Alloyed Electrocatalyst, Adv. Sci., 2023, 10, 2300094 CrossRef CAS PubMed.
- Q. Wu, M. Mellin, S. Lauterbach, C. Tian, C. Dietz, J. P. Hofmann and M. Einert, Soft-templated, mesoporous Co3O4 thin films for electrocatalysis of the oxygen evolution reaction, Mater. Adv., 2024, 5, 2098–2109 RSC.
- M. Risch, K. A. Stoerzinger, B. Han, T. Z. Regier, D. Peak, S. Y. Sayed, C. Wei, Z. Xu and Y. Shao-Horn, Redox processes of manganese oxide in catalyzing oxygen evolution and reduction: an in situ soft X-ray absorption spectroscopy study, J. Phys. Chem. C, 2017, 121, 17682–17692 CrossRef CAS.
- M. F. Tesch, S. A. Bonke, T. E. Jones, M. N. Shaker, J. Xiao, K. Skorupska, R. Mom, J. Melder, P. Kurz and A. Knop-Gericke, Evolution of oxygen–metal electron transfer and metal electronic states during manganese oxide catalyzed water oxidation revealed with in situ soft X-ray spectroscopy, Angew. Chem., 2019, 131, 3464–3470 CrossRef.
- H. Ooka, T. Takashima, A. Yamaguchi, T. Hayashi and R. Nakamura, Element strategy of oxygen evolution electrocatalysis based on in situ spectroelectrochemistry, Chem. Commun., 2017, 53, 7149–7161 RSC.
- T. Takashima, K. Hashimoto and R. Nakamura, Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts, J. Am. Chem. Soc., 2012, 134, 1519–1527 CrossRef CAS PubMed.
- X. Bo, Y. Li, X. Chen and C. Zhao, High valence chromium regulated cobalt–iron-hydroxide for enhanced water oxidation, J. Power Sources, 2018, 402, 381–387 CrossRef CAS.
- R. N. Singh, J. P. Singh, B. Lal, M. J. Thomas and S. Bera, New NiFe2−xCrxO4 spinel films for O2 evolution in alkaline solutions, Electrochim. Acta, 2006, 51, 5515–5523 CrossRef CAS.
- A. Abdelhafiz, B. Wang, A. R. Harutyunyan and J. Li, Carbothermal shock synthesis of high entropy oxide catalysts: dynamic structural and chemical reconstruction boosting the catalytic activity and stability toward oxygen evolution reaction, Adv. Energy Mater., 2022, 12, 2200742 CrossRef CAS.
- X.-T. Wang, T. Ouyang, L. Wang, J.-H. Zhong, T. Ma and Z.-Q. Liu, Redox-inert Fe3+ ions in octahedral sites of Co–Fe spinel oxides with enhanced oxygen catalytic activity for rechargeable zinc–air batteries, Angew. Chem., 2019, 131, 13425–13430 CrossRef.
- X. Yang, S. Liping, L. Qiang, H. Lihua and Z. Hui, Co-prosperity of electrocatalytic activity and stability in high entropy spinel (Cr0.2Mn0.2Fe0.2Ni0.2Zn0.2)3O4 for the oxygen evolution reaction, J. Mater. Chem. A, 2022, 10, 17633–17641 RSC.
- S. Tao, Q. Wen, W. Jaegermann and B. Kaiser, Formation of Highly Active NiO(OH) Thin Films from Electrochemically Deposited Ni(OH)2 by a Simple Thermal Treatment at a Moderate Temperature: A Combined Electrochemical and Surface Science Investigation, ACS Catal., 2022, 12, 1508–1519 CrossRef CAS.
- J. Gallenberger, H. M. Fernández, A. Alkemper, M. Li, C. Tian, B. Kaiser and J. P. Hofmann, Stability and decomposition pathways of the NiOOH OER active phase of NiOx electrocatalysts at open circuit potential traced by ex situ and in situ spectroscopies, Catal. Sci. Technol., 2023, 13, 4693–4700 RSC.
- H. Wang, K. H. L. Zhang, J. P. Hofmann and F. E. Oropeza, The electronic structure of transition metal oxides for oxygen evolution reaction, J. Mater. Chem. A, 2021, 9, 19465–19488 RSC.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, A review on fundamentals for designing oxygen evolution electrocatalysts, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00026a |
| This journal is © The Royal Society of Chemistry 2024 |