
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling a non-flammable methyl(2,2,2-trifluoroethyl) carbonate electrolyte in NMC622–graphite Li-ion cells by electrode pre-passivation†
Matilde
Longhini
 ab,
Florian
Gebert
a,
Fosca
Conti
*b and
Andrew J.
Naylor
*a
ab,
Florian
Gebert
a,
Fosca
Conti
*b and
Andrew J.
Naylor
*a
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, SE-75121, Uppsala, Sweden. E-mail: andy.naylor@kemi.uu.se
bDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131, Padova, Italy. E-mail: fosca.conti@unipd.it
First published on 5th April 2024
Abstract
Novel lithium-ion battery electrolytes often exhibit poor electrochemical stability against typical commercial layered oxide and graphite electrodes. Pre-passivating the electrodes prior to cell assembly with an electrically insulating, ionically conductive solid–electrolyte interphase (SEI) is one innovative strategy for stabilising systems with otherwise unusable electrolytes. Here, methyl(2,2,2-trifluoroethyl) carbonate (FEMC), a promising non-flammable electrolyte solvent that is generally unstable against graphite, is utilised after pre-passivation of electrodes with a state-of-the-art carbonate-based electrolyte. A significant improvement in performance is observed compared with the untreated electrodes. Hard X-ray photoelectron spectroscopy was used to probe the interphase layer composition.
Introduction
Safety is a critical aspect of lithium-ion batteries (LIBs), not only for adoption in the transportation sector. In a typical LIB, the most flammable component is the liquid electrolyte. For this reason, reducing the flammability of the electrolyte is central to improving overall LIB safety. A wide variety of novel liquid electrolytes for LIBs have been proposed, such as organophosphates, highly fluorinated molecules, and locally highly concentrated electrolytes.1–5 A key consideration when substituting electrolyte components is the stability of the solid electrolyte interphase (SEI). The SEI is a surface layer on the graphite anode resulting from the passivation of the electrode surface. It should ideally be electrically insulating and ionically conductive, which prevents the electrolyte from continually decomposing while enabling the passage of Li+ ions.6 For traditional electrolytes, which usually consist of LiPF6 dissolved in mixtures of ethylene carbonate (EC) and a linear carbonate like ethyl methyl carbonate (EMC), the SEI has been well-characterised and optimized over the past 30 years of LIB commercialisation. The SEI is generally formed during the energy-intensive and time-consuming formation and ageing process near the end of production.The SEI formed by novel electrolytes often exhibits very different characteristics and may have undesirable properties like poor electrical insulation or low ionic conductivity. Several strategies have been used to improve performance with electrolytes that form poor SEIs. These include artificial SEIs,7,8 core–shell structures of electrode materials,9 and film-forming electrolyte additives.10 However, many such strategies offer poor scalability and control, and can be costly to implement. An innovative approach that has received comparatively little attention is electrochemically passivating the electrodes prior to assembly of the cell. This potentially rapid and cost-effective procedure involves immersing electrodes in an electrolyte known to produce stable SEIs, applying a voltage or current, and subsequently assembling a battery with an alternative ‘long-term’ electrolyte. The method can combine the advantages of two different electrolyte systems: the SEI-forming ability of traditional carbonate esters, and the improved safety of a non-flammable solvent for long-term operation. Electrochemical pre-passivation has been used to stabilize anodes based on lithium-titanium oxide,11 silicon,12 hard carbon,13 and graphite.14,15 Variations of the technique, mainly with the aim of anode pre-lithiation, have been integrated into roll-to-roll LIB assembly processes, demonstrating its potential in commercial settings.16,17 Here, however, we demonstrate the first known instance of electrode pre-passivation to enable the use of safer non-flammable electrolyte during long-term battery operation.
In the present study, electrode pre-passivation is used to enable the use of methyl(2,2,2-trifluoroethyl) carbonate (FEMC), one of the most promising non-flammable solvents, but with poor electrochemical stability against graphite anodes and poor SEI-forming ability.18 FEMC typically requires combination with co-solvents and/or additives to be used,19–21 potentially compromising its non-flammable properties and increasing the complexity of the electrolyte formulation. Here, the high-voltage LIB electrodes NMC622 and graphite are pre-passivated by subjecting them to four charge–discharge cycles in LP57, a state-of-the-art flammable electrolyte consisting of 1 M LiPF6 in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 vol% ethylene carbonate (EC) and ethyl methyl carbonate (EMC). The electrodes are subsequently assembled into cells containing 1 M LiPF6 in FEMC for long-term operation. Pre-passivating the electrodes substantially improves the performance of the FEMC electrolyte, while cell resistance measurements and surface analysis reveal further details of the passivation layer.
:7 vol% ethylene carbonate (EC) and ethyl methyl carbonate (EMC). The electrodes are subsequently assembled into cells containing 1 M LiPF6 in FEMC for long-term operation. Pre-passivating the electrodes substantially improves the performance of the FEMC electrolyte, while cell resistance measurements and surface analysis reveal further details of the passivation layer.
Results and discussion
Fig. 1 shows the substantial improvement in cycling stability achieved when LiPF6/FEMC is used with pre-passivated rather than pristine electrodes. With pristine electrodes, the initial charge and discharge capacities are ca. 249 and 86 mA h g−1, respectively (Fig. 1a), corresponding to a coulombic efficiency of 35%. The poor reversibility indicates a substantial loss of lithium inventory during the first charge step, indicating that the LiPF6/FEMC electrolyte does not form an effective (i.e., highly electrically insulating) SEI. When using electrodes pre-passivated with LP57, the improvement in performance is striking: the first charge capacity is reduced to 173 mA h g−1 and the first discharge capacity is increased to 161 mA h g−1 (Fig. 1b). The pre-passivated electrodes also retain considerably more of their initial capacities during cycling (Fig. 1c). The interfacial layers formed by LP57 are clearly able to prevent the extensive decomposition of the LiPF6/FEMC electrolyte, unlike in the case of the untreated electrodes.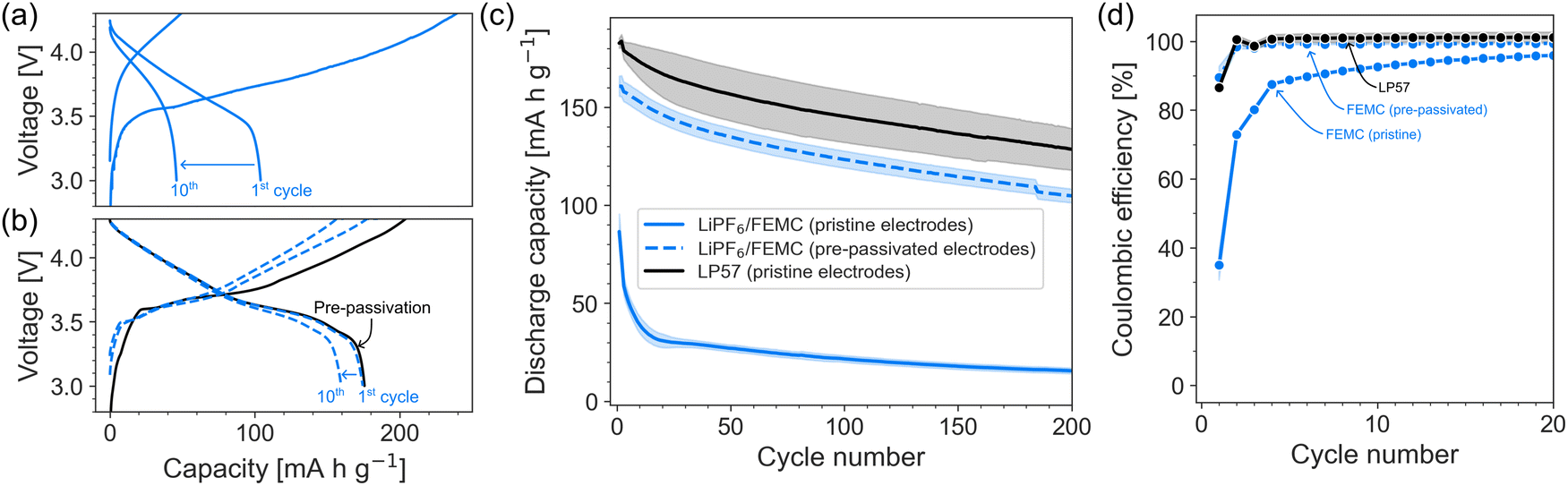 | ||
| Fig. 1 Charge–discharge curves of LiPF6/FEMC in (a) pristine and (b) pre-passivated NMC622|graphite cells; (c) galvanostatic cycling performance and (d) coulombic efficiency of FEMC in NMC622–graphite cells at C/5. Solid lines: electrolytes tested with pristine electrodes. Dashed lines electrolytes tested using the pre-passivated electrodes. LP57 is used as a benchmark. The data represent the average of three cells for each dataset; the shaded regions represent the standard error. | ||
Unsurprisingly, the cell with pre-passivated electrodes displays the highest coulombic efficiency, demonstrating an effective passivation of the surface by the pre-passivation technique, and protection against extensive FEMC decomposition. However, the initial coulombic efficiency of 93% does indicate some irreversible capacity and thereby some electrolyte decomposition even with the pre-passivated electrodes (for comparison, a standard cell with LP57 has a coulombic efficiency near 100% in the 5th cycle). However, this small capacity loss only occurs in the first cycle. In subsequent cycles, the coulombic efficiency of the pre-passivated LiPF6/FEMC system is close to 100%, comparable to that of LP57 itself (Fig. 1d). The initial discharge capacity when LP57 is used as the electrolyte (with pristine electrodes) is 180 mA h g−1, with a corresponding 1st-cycle coulombic efficiency of 86%. Overall, the capacity evolution during cycling is nearly identical for FEMC with pre-passivated electrodes and LP57 with pristine electrodes (Fig. 1c), with the only difference being that the pre-passivated/FEMC system is ca. 10% lower in capacity. This may demonstrate the need for optimisation of the pre-passivation method, which would be the subject of a follow-up study. The long-term cycling stability is unaffected, indicating that the electrodes are effectively passivated with no significant on-going side-reactions.
The improvement in performance is also reflected in the cell resistance, which was tracked via the intermittent current interruption (ICI) method developed by Lacey et al.22,23 (Fig. 2). The resistance of the FEMC cells containing pristine electrodes is more than twice that of the cells containing pre-passivated electrodes. (The jump in resistance after the 2nd cycle is likely an artefact of increasing the C-rate from C/10 to C/5.) It is notable that even the pre-passivated electrodes result in a significantly higher cell resistance than the commercial electrolyte LP57 used with pristine cells. This indicates, as the 1st-cycle coulombic efficiency also suggests, a certain degree of electrolyte decomposition on top of the pre-formed passivation layers, and can explain the slightly lower capacity observed for the pre-passivated system. While the superior performance of LP57 is not surprising, given that this electrolyte is the product of decades of focused optimization, the improvement in performance of FEMC after pre-passivation of the electrodes demonstrates that the investigated methodology is particularly promising enabling the development of novel electrolytes.
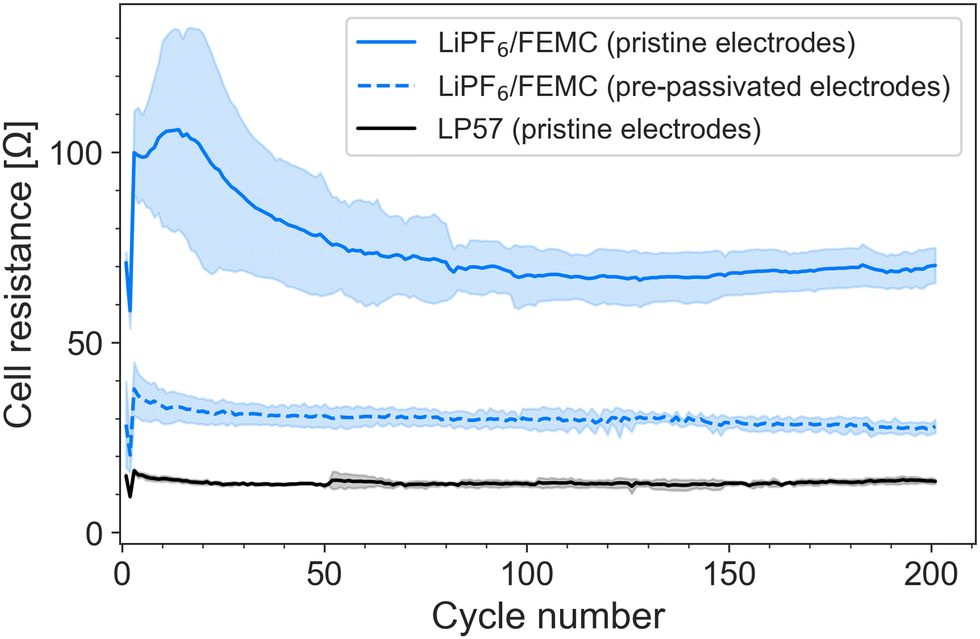 | ||
| Fig. 2 Internal resistance of cells cycled at C/5 with 1 M LiPF6 in FEMC with both pristine and pre-passivated electrodes and with LP57 as a reference. The data represent the average of three cells for each dataset; the shaded regions represent the standard error of 3 cells. | ||
Half-cell data make clear that the root of FEMC's poor cycling stability is on the graphite side (Fig. S1, ESI†), although it is worth noting that FEMC is known to have compatibility issues with lithium metal,24–26 as the sudden decrease in capacity at around the 20th cycle of the NMC622|Li cells shows.
Considering the implications a pre-passivation method may have on electrode fabrication and cell assembly, it is of interest to consider at what voltage LP57 forms interfacial layers capable of protecting against FEMC decomposition. This is both of practical and theoretical interest, as it sheds light on the reduction potential of FEMC on graphite. To that end, cells containing pristine electrodes and LP57 were charged to three separate voltages: 3.6, 3.8 and 4.0 V, followed by a CV step until the current dropped to 10% of the initial current (see charge curves in Fig. S2, ESI†). The current was stopped, and the partially passivated electrodes transferred to cells containing LiPF6/FEMC. It was found that the passivation layers formed by LP57 at 3.6 V are already sufficient to significantly improve the performance of the FEMC electrolyte (Fig. 3).
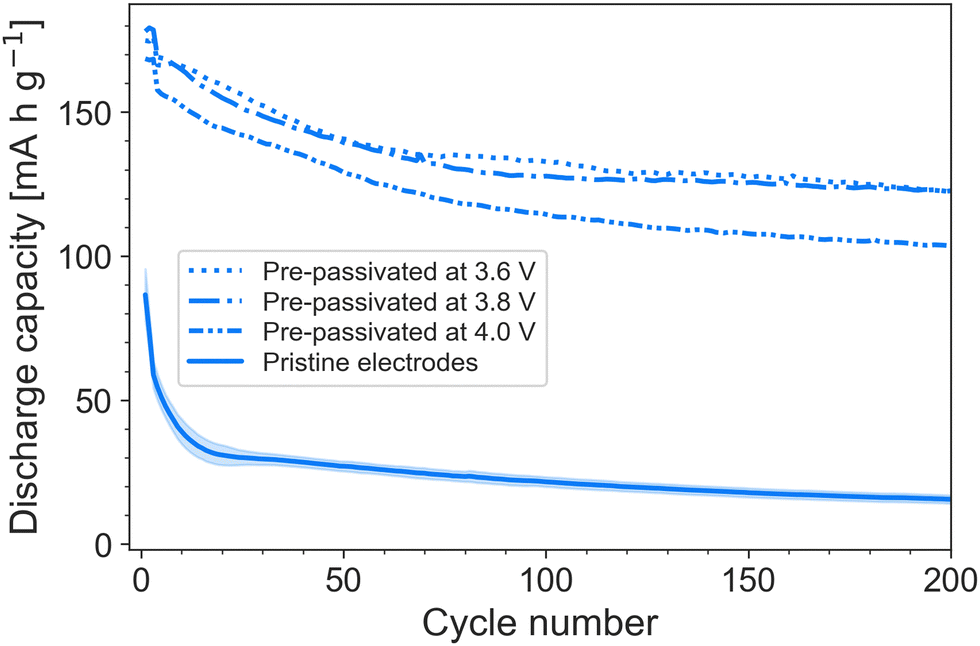 | ||
| Fig. 3 Cycling performance at C/5 of FEMC in electrodes pre-passivated at various voltages, compared to pristine electrodes. | ||
To understand the function of the pre-passivation layers, the evolution of the pre-formed SEI on graphite electrodes upon cycling in LiPF6/FEMC was studied using hard X-ray photoelectron spectroscopy (HAXPES). Pre-passivated electrodes were subjected to the following electrochemical aging in full cells: a single charge to 4.3 V, one full charge–discharge cycle, 5 cycles and 30 cycles. HAXPES spectra of the graphite electrodes from these cells are shown in Fig. 4. In these spectra, the peak originating from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonding environment of carbon in graphite appears between 281–283 eV, and is observed to shift negative with aging. This likely originates from the relative shift between surface layer components and bulk components often observed for anode materials, a phenomenon that has been described elsewhere.27
C bonding environment of carbon in graphite appears between 281–283 eV, and is observed to shift negative with aging. This likely originates from the relative shift between surface layer components and bulk components often observed for anode materials, a phenomenon that has been described elsewhere.27
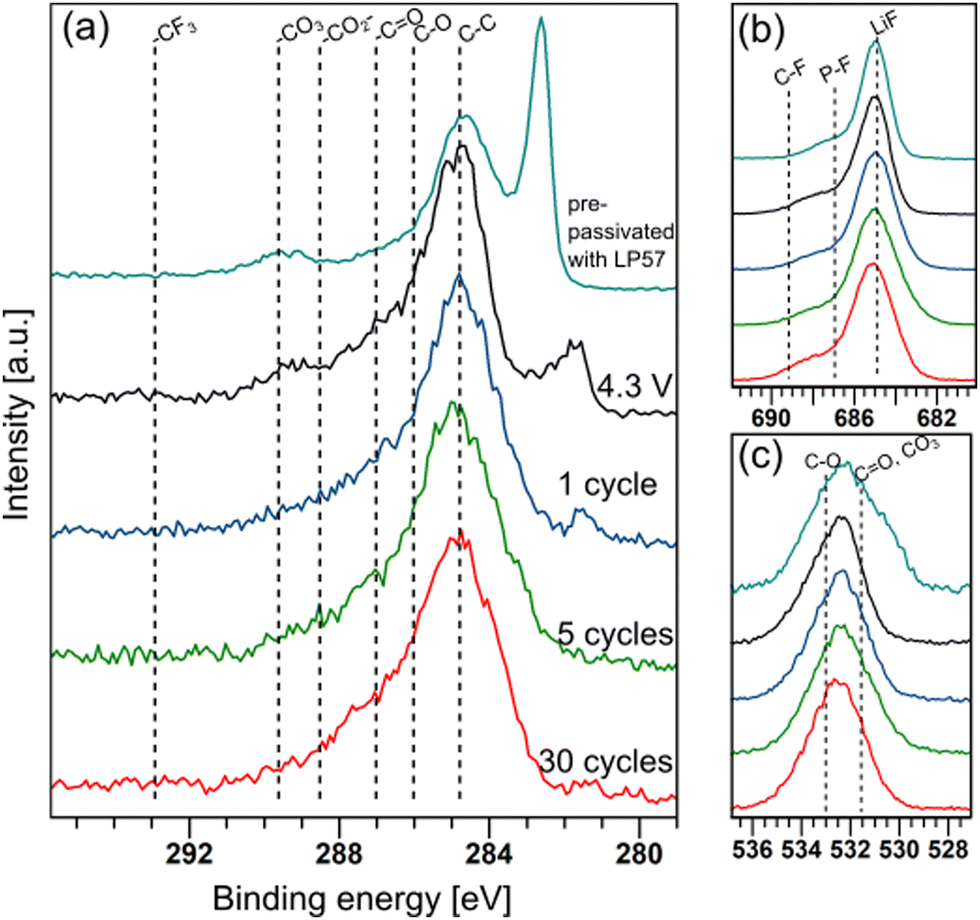 | ||
| Fig. 4 (a) C 1s, (b) F 1s and (c) O 1s XPS spectra of graphite electrodes after pre-passivation with LP57, after changing the electrolyte to LiPF6/LP57 and charging to 4.3 V, and 1, 5 and 30 cycles in NMC622|graphite full cells. | ||
The thickness of the SEI can be qualitatively tracked by comparing the intensity of the C 1s peak associated with graphite (CC) with the intensity of the rest of the spectrum (Fig. 4a). This suggests that cycling the electrodes in LiPF6/FEMC results in considerable additional SEI growth. Even after a single cycle, the graphite peak is considerably diminished compared to the spectrum of the electrode pre-passivated by LP57; after 30 cycles, it is practically indiscernible. The thickness of the SEI after 5 cycles can be estimated to be greater than the probing depth of the measurement, calculated as approximately 50 nm. This thicker SEI is consistent with the higher cell resistance observed from ICI (Fig. 2).
The SEI pre-formed by LP57 consists of ca. 47% Li, 30% C, 16% F, 6% O and 1% P (by atomic percentage). Upon replacing the electrolyte with LiPF6/FEMC and charging the cell to 4.3 V, the SEI becomes more inorganic, with higher F and O contents at the expense of C. The lack of new peaks in the 291–293 eV region of the C 1s spectrum (Fig. 4a) suggests that this increase in fluorine content does not originate from the incorporation of the –CF3 moiety of FEMC in the SEI. Instead, the increasing relative intensity of the shoulder in the F 1s spectra (Fig. 4b) at ca. 687 eV indicates an increased presence of LiPF6 and its decomposition products LiPFxOy.28 LiF remains the dominant species. After the first charge step, there are only slight changes in the SEI's chemical makeup upon further cycling (see ESI†). It is consistently composed of ca. 45–55% Li, 25% F, 12–18% C, 8–10% O and 2–4% P.
The biggest peak in the C 1s spectra is at ca. 284.8 eV, which is associated with C–C groups. This is most likely originating from polymerised electrolyte components.29 A peak originating from carbonate groups is also present, likely from both Li2CO3 and semicarbonates (organic carbonate salts). However, this environment is seen to decrease in intensity even from the first charge in both C 1s and O 1s (Fig. 4c). The composition of the additional SEI layers deposited by LiPF6/FEMC is richer in alkoxides (C–O), esters (–CO2–) and ketones (–CO).
The strikingly low coulombic efficiencies of the LiPF6/FEMC system (Fig. 1b) suggest that the rapid decline in capacity accompanied by it is caused by significant lithium inventory loss. However, it is not straightforward from the XPS data to draw conclusions about what specific decomposition reactions the LiPF6/FEMC system undergoes. The SEI formed by LiPF6/FEMC on pristine graphite is currently the subject of a follow-up study. It is notable that the SEI formed by LP57 does not fully prevent the decomposition of the LiPF6/FEMC electrolyte. However, the decomposition that does take place does not seem to significantly affect the electrochemical properties of the cells, suggesting that the extra SEI layers deposited are reasonably ionically conductive, or at least not detrimental to performance.
Conclusions
In conclusion, this study explored the potential of pre-passivating NMC622 and graphite electrodes when transitioning to novel electrolytes, which are often electrochemically unstable or form poor interfacial layers. FEMC performs extremely poorly when used with pristine electrodes, but very well when the electrodes are pre-passivated, prior to cell assembly, using LP57. Substantial improvements in coulombic efficiency and cycling stability were achieved, and the cell resistance was reduced by a factor of more than 2. This protective effect was already achieved by charging LP57-containing NMC622–graphite cells to 3.6 V. Hard X-ray photoelectron spectroscopy (HAXPES) analysis indicated that cycling the pre-passivated electrodes in the LiPF6/FEMC electrolyte resulted in the deposition of further layers on the pre-formed SEI. The new layers were different in composition to the SEI formed by LP57, containing overall more PF6−-derivatives, fewer carbonates and more alkoxides, ketones and esters. Although the newly deposited layers add to the SEI thickness, the effect on performance is minor. This study has demonstrated a promising strategy for enabling the use of novel electrolytes, which requires continued further investigation particularly in the effects of pre-passivation conditions, longer-term operation, and interfacial layer stability.Experimental methods
Prefabricated electrode sheets of NMC622 (LiNi0.6Mn0.2Co0.2O2) coated on 20 μm-thick aluminium foil and artificial graphite coated onto 14 μm-thick copper foil were purchased from Custom Cells. Circular electrodes 13 mm in diameter were punched out and dried in vacuum at 120 °C for 12 hours before use. Both electrodes contained conducting additives and binders. The active material contents of the cathode and anode was 93.5% and 95.0%, respectively. The capacity ratio between the negative and positive electrodes (N/P ratio) was 1.2. Samples were prepared in an argon-filled MBraun glove box. Cells were assembled using polymer-laminated aluminium pouches. 35 μl of electrolyte (LiPF6 in FEMC, or LP57) were placed on each electrode. Celgard® 2400 was used as the separator. LP57 (1 M LiPF6 in 3:7 vol% EC:EMC) was purchased from Solvionic and used without further purification. The FEMC electrolyte was prepared in a glove box by dissolving 1 M LiPF6 (99.99%, Sigma Aldrich) in commercial FEMC (98.0%, TCI Chemicals). LiPF6 was dried at 70 °C in vacuum before using. FEMC was used without further purification.
Pre-passivated electrodes were prepared by assembling NMC622–graphite full cells and cycling them twice at C/10 (0.265 mA, based on a cathode capacity of 180 mA h g−1) and twice at C/5 (0.53 mA) in a voltage window of 3.0–4.3 V, as per manufacturer recommendations. All reported full-cell specific capacities in this work are based on the amount cathode active material they contain. The electrolyte used for pre-passivation was LP57. The electrodes were removed from the cells in an argon-filled glove box and washed with 50 μl of FEMC each, to remove excess LP57 electrolyte. The solvent was allowed to evaporate for ca. 5 minutes and the electrodes were placed in new cells, which were assembled using 1 M LiPF6 in FEMC electrolyte.
Constant current–constant voltage (CCCV) cycling was used for cycling experiments: the cell was charged at constant current (C/5, 0.53 mA) to 4.3 V, then the voltage was held until the current reached 1/10 of the current used in the previous CC step; finally, the cell was discharged at CC to 3.0 V. Cycling was carried out on Neware testers.
Intermittent current interruption was carried out following the procedure reported by Lacey et al.22,23 Briefly, cells were charged and discharged at low C-rates (C/10 or C/5) and rested for 1 second every 5 minutes. From the voltage response after 1 second of rest, the cell resistance Rcell was calculated viaeqn (1) where ΔV1s is the voltage drop and I is the current:
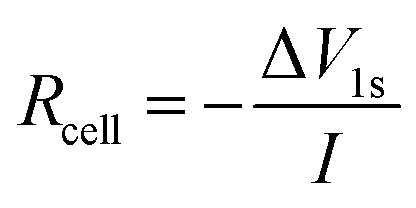 | (1) |
 | (2) |
Author contributions
Matilde Longhini: methodology, investigation, writing – original draft, writing – review & editing. Florian Gebert: methodology, investigation, writing – review & editing. Fosca Conti: resources, writing – original draft, writing – review & editing, supervision. Andrew J. Naylor: conceptualization, methodology, resources, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from the Swedish Energy Agency (projects P2021-90019 and P2022-00045) and STandUP for Energy for support to the Ångström Advanced Battery Centre. We acknowledge Diamond Light Source for time on Beamline I09 under Proposal SI30357. Dr Tien-Lin Lee, Dr Pardeep Thakur Kumar and Dr Deepnarayan Biswas at beamline I09 are thanked for their assistance.References
- R. Gond, W. van Ekeren, R. Mogensen, A. J. Naylor and R. Younesi, Mater. Horiz., 2021, 8, 2913–2928 RSC.
- F. Gebert, M. Longhini, F. Conti and A. J. Naylor, J. Power Sources, 2023, 556, 232412 CrossRef CAS.
- L. Qin, N. Xiao, J. Zheng, Y. Lei, D. Zhai and Y. Wu, Adv. Energy Mater., 2019, 9, 1902618 CrossRef CAS.
- Z. Wang, R. Han, H. Zhang, D. Huang, F. Zhang, D. Fu, Y. Liu, Y. Wei, H. Song, Y. Shen, J. Xu, J. Zheng, X. Wu and H. Li, Adv. Funct. Mater., 2023, 33, 2215065 CrossRef CAS.
- D. Zhu, Y. Ren, Y. Yu, L. Zhang, Y. Wan, H. Wang, W. Gao, B. Han and L. Zhang, ChemElectroChem, 2023, 10, e202300009 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322 CrossRef CAS.
- J. Zhou, K. Ma, X. Lian, Q. Shi, J. Wang, Z. Chen, L. Guo, Y. Liu, A. Bachmatiuk, J. Sun, R. Yang, J.-H. Choi and M. H. Rümmeli, Small, 2022, 18, 2107460 CrossRef CAS PubMed.
- S. Menkin, D. Golodnitsky and E. Peled, Electrochem. Commun., 2009, 11, 1789 CrossRef CAS.
- Y.-S. Hu, R. Demir-Cakan, M.-M. Titirici, J.-O. Müller, R. Schlögl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645 CrossRef CAS PubMed.
- A. L. Michan, B. S. Parimalam, M. Leskes, R. N. Kerber, T. Yoon, C. P. Grey and B. L. Lucht, Chem. Mater., 2016, 28, 8149 CrossRef CAS.
- V. Borgel, G. Gershinsky, T. Hu, M. G. Theivanayagam and D. Aurbach, J. Electrochem. Soc., 2013, 160, A650 CrossRef CAS.
- G. M. Overhoff, R. Nölle, V. Siozios, M. Winter and T. Placke, Batteries Supercaps, 2021, 4, 1163 CrossRef CAS.
- E. de la Llave, V. Borgel, K.-J. Park, J.-Y. Hwang, Y.-K. Sun, P. Hartmann, F.-F. Chesneau and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 1867 CrossRef CAS PubMed.
- P. K. Nayak, T. R. Penki, B. Markovsky and D. Aurbach, ACS Energy Lett., 2017, 2, 544 CrossRef CAS.
- K. Matsumoto, K. Nakahara, K. Inoue, S. Iwasa, K. Nakano, S. Kaneko, H. Ishikawa, K. Utsugi and R. Yuge, J. Electrochem. Soc., 2014, 161, A831 CrossRef CAS.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282 CrossRef CAS PubMed.
- R. W. Grant, M. Sweetland, A. M. Acharige and Nanoscale Components Inc, Method for alkaliating anodes, US pat. 9598789B2, 2017 Search PubMed.
- C.-C. Su, M. He, J. Shi, R. Amine, J. Zhang, J. Guo and K. Amine, Nano Energy, 2021, 89, 106299 CrossRef CAS.
- G. J. Chung, J. Han and S.-W. Song, ACS Appl. Mater. Interfaces, 2020, 12, 42868 CrossRef CAS PubMed.
- Q. Zheng, Y. Yamada, R. Shang, S. Ko, Y.-Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291 CrossRef CAS.
- X. Fan, L. Chen, O. Borodin, X. Ji, J. Chen, S. Hou, T. Deng, J. Zheng, C. Yang, S.-C. Liou, K. Amine, K. Xu and C. Wang, Nat. Nanotechnol., 2018, 13, 715 CrossRef CAS PubMed.
- M. J. Lacey, ChemElectroChem, 2017, 4, 1997 CrossRef CAS.
- Z. Geng, T. Thiringer and M. J. Lacey, IEEE Trans. Transp. Electrif., 2022, 8, 2985 Search PubMed.
- C.-C. Su, M. He, M. Cai, J. Shi, R. Amine, N. D. Rago, J. Guo, T. Rojas, A. T. Ngo and K. Amine, Nano Energy, 2022, 92, 106720 CrossRef CAS.
- T. Yang, S. Li, W. Wang, J. Lu, W. Fan, X. Zuo and J. Nan, J. Power Sources, 2021, 505, 230055 CrossRef CAS.
- C.-C. Su, M. He, R. Amine, T. Rojas, L. Cheng, A. T. Ngo and K. Amine, Energy Environ. Sci., 2019, 12, 1249–1254 RSC.
- J. Maibach, F. Lindgren, H. Eriksson, K. Edström and M. Hahlin, J. Phys. Chem. Lett., 2016, 7, 1775 CrossRef CAS PubMed.
- N.-S. Choi, I. A. Profatilova, S.-S. Kim and E.-H. Song, Thermochim. Acta, 2008, 480, 10 CrossRef CAS.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255 CrossRef CAS.
- C. J. Powell and A. Jablonski, NIST Electron Inelastic-Mean- Free-Path Database, Version 1.2, SRD 71, National Institute of Standards and Technology, Gaithersburg, MD, 2010 Search PubMed.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00052h |
| This journal is © The Royal Society of Chemistry 2024 |