
Recent advances and future prospects in oxidative-reduction low-triggering-potential electrochemiluminescence strategies based on nanoparticle luminophores
Li
Fu
 a,
Tianyuan
Song
a,
Qi
Li
a,
Guizheng
Zou
*b,
Fuwei
Zhang
a,
Zongchao
Li
a,
Haotian
Guan
a and
Yingshu
Guo
*a
a,
Tianyuan
Song
a,
Qi
Li
a,
Guizheng
Zou
*b,
Fuwei
Zhang
a,
Zongchao
Li
a,
Haotian
Guan
a and
Yingshu
Guo
*a
aSchool of Chemistry and Chemical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan, 250100, China. E-mail: yingshug@126.com
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: zouguizheng@sdu.edu.cn
First published on 14th November 2024
Abstract
The oxidative-reduction electrochemiluminescence (ECL) potential of a luminophore is one of the most significant parameters during light generation processes when considering the growing demand for anti-interference analysis techniques, electrode compatibility and the reduction of damage to biological molecules due to excessive excitation potential. Nanoparticle luminophores, including quantum dots (QDs) and metal nanoclusters (NCs), possess tremendous potential for forming various ECL sensors due to their adjustable surface states. However, few reviews focused on nanoparticle luminophore-based ECL systems for low-triggering-potential (LTP) oxidative-reduction ECL to avoid the possible interference and oxidative damage of biological molecules. This review summarizes the recent advances in the LTP oxidative-reduction ECL potential strategy with nanoparticle luminophores as ECL emitters, including matching efficient coreactants and nanoparticle luminophores, doping nanoparticle luminophores, constructing donor–acceptor systems, choosing suitable working electrodes, combining multiplex nanoparticle luminophores, and employing surface-engineering strategies. In the context of the different LTP ECL systems, potential-lowering strategies and bio-related applications are discussed in detail. Additionally, the future trends and challenges of low ECL-triggering-potential strategies are discussed.
Introduction
Electrochemiluminescence (ECL) is an analytical tool to electrochemically light up luminophores with simple device requirements, low background, and space resolution of the signal.1,2 ECL research has been performed with an annihilation or coreactant ECL route, and ECL evolution was mainly achieved with the latter path for oxidizing or reducing luminophores and coreactant synchronously in one potential direction.3,4 According to different CV scanning modes, the coreactant ECL routes are typically categorized into oxidative-reduction and reduction-oxidative ECL types.5,6 In the process of reduction-oxidative ECL, the coreactant needs to inject holes into the valence band (HOMO) of the nanoparticles,7 and common reduction-oxidative cathode ECL coreactants involve H2O2,8 persulfate (S2O82−),9,10 and dibenzoyl peroxide (BPO). In the process of oxidative-reduction ECL, the coreactant needs to inject electrons into the conduction band (LUMO) of the nanoparticles, and oxidative-reduction anode ECL coreactants involve tri-n-propylamine (TPrA),11 2-(dibutylamino)ethanol (DBAE),12,13 triethylamine (TEA),14,15 triethanolamine (TEOA),16 and oxalate (C2O42−).17 The ECL of specific nanoparticle luminophores can be captured at the cathode in a coreactant-free buffer, attributed to the presence of dissolved oxygen which undergoes reduction to H2O2 at 0.4 to 0.8 V, commonly utilized as a reduction-oxidative ECL coreactant.18,19 The oxidative-reduction ECL effectively eliminates interference from dissolved oxygen, allowing for the conventional operation of the ECL system in the anodic direction.20 Effective ECL luminophores based on molecules were first reported by Bard's group in 1984.21 The traditional oxidative-reduction ECL system, Ru(bpy)32+/tri-n-propylamine (TPrA), has occupied an important position in ECL evolution since it was extensively exploited in the ECL bioassay in 1991,22 which eventually dominated all commercially available ECL bioassays worldwide.23Besides molecular luminophores,24,25 such as luminol derivatives,11 Ru(bpy)32+,4 iridium(III) complexes,26 and BODIP,27 a series of nanoparticle luminophores, including quantum dots (QDs) and metal nanoclusters (NCs),28–34 such as Si35–37 and carbon dots (CDs),38,39 binary II–VI materials,40 ternary CuInS2 (CIS) QDs,41 CdSe/CdS/ZnS QDs,42,43 multinary QDs,44 and AuNCs45–47 have been proposed for ECL. The development of both luminophores and coreactants plays an important role in basic ECL research.4,48,49 On the one hand, the high triggering ECL potential of the traditional ECL system Ru(bpy)32+/TPrA (+1.2 V, vs. Ag/AgCl) is susceptible to interference,21,50,51 which can contribute to irreversible oxidative damage of biological molecules due to excessive excitation potential (e.g., oligonucleotide sequences are destroyed at voltages higher than +1.0 V).52 On the other hand, low-triggering-potential (LTP) ECL is beneficial for the development of potential-resolved ECL for multiplexed bioassay, decreasing the crosstalk of ECL between luminophores and enhancing the electrode compatibility.53 Therefore, it is clear that extended investigations of LTP ECL progress with a suitable ECL wave window are necessary.
This review provides a comprehensive summary of recent advancements in oxidative-reduction LTP ECL based on nanoparticle luminophores, including illustrative examples and perspectives. The LTP ECL strategies based on nanoparticle luminophores are classified into various methods including selecting efficient coreactants, doping of nanoparticle luminophores, adjusting the synthetic process and ratio of elements, building donor–acceptor systems, choosing suitable working electrodes, combining multiplex martials, and surface-engineering strategies (Fig. 1). Multiple strategies have shown great potential throughout the literature to decrease the ECL potential for fewer interfaces. It is anticipated that this review will serve as a valuable reference for researchers interested in understanding the current trends and unresolved challenges in oxidative-reduction LTP ECL.
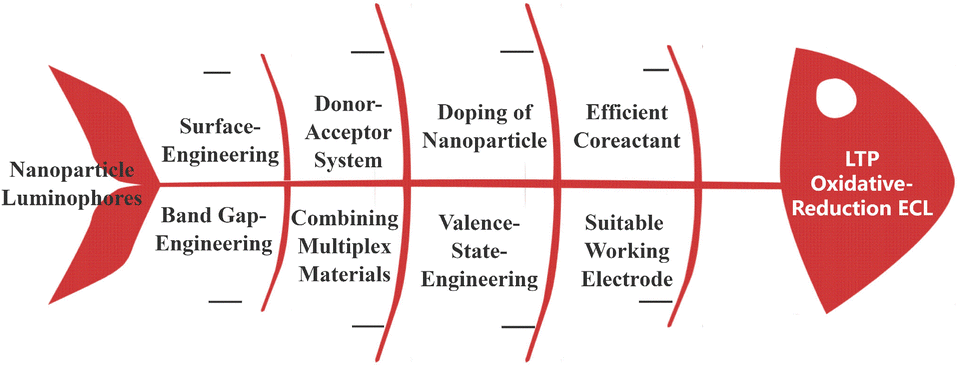 | ||
| Fig. 1 Schematic diagram of the strategies used for LTP ECL. | ||
Matching efficient coreactant and nanoparticle luminophores for oxidative-reduction ECL
ECL is sensitive to the surface states of nanoparticle luminophores, and the key step is the electron transfer between the nanoparticle luminophores and the intermediate involving the chosen coreactant (coreactant route).54–57 Traditional, II–VI QDs have occupied an important position in the development of ECL. Oxidative-reduction ECL at approximately 0.9 V was realized with CdSe QDs as models and sulfite/dissolved oxygen as a coreactant at an indium tin oxide (ITO) electrode (Fig. 2A & D), enabling sensitive detection of dopamine or nitrite (Fig. 2B & C).58,59 | ||
| Fig. 2 (A) ECL (a) and cyclic voltammetry (b) profiles of CdSe QDs. Inset: ECL profiles of QDs (solid line) and 0.6 mM sulfite (dashed line) in the solution; (B) ECL of CdSe QDs in HCl-tris buffer containing sulfite and (a) 0, (b) 0.5, (c) 50 and (d) 100 M DA, and (C) linear calibration plot for dopamine detection;58 (D) ECL profiles of TGA@CdSe QDs in air-saturated pH 7.0 phosphate (a) and Tris-HCl (b) buffer in the presence of 0.6 mM sulfite;59 (E) ECL and (F) cyclic voltammetry profiles of MPA@CdTe QDs (black), 1.0 mM Na2SO3 (blue), 0.2 μM QDs + 0.4 mM Na2SO3 (red), and 0.2 μM QDs + 1.0 mM Na2SO3 (green) in air-saturated pH 7.5 PBS; (G) linear calibration plot for tyrosine detection and (H) ECL curves of 0, 0.1, 1.0, and 100 pM tyrosine with MPA@CdTe QDs as the luminophore;61 (I) cyclic voltammetry and (J) ECL of (a) CdTe QDs in pH 7.5 PBS, (b) (a) +1.5 × 10−4 M DBAE; (K) ECL of CdTe QDs with different kinds of amines with the same concentration of 3 mM: (1) diethylamine, (2) TEA, (3) TPrA, (4) DBAE, (5) TEOA, (6) ethanolamine; (L) ECL of CdTe QDs with different concentrations of DBAE in PBS.65 | ||
Moreover, solution components show a clear effect on ECL emission. Thioglycolic acid (TGA) modified CdSe (TGA@CdSe) QDs exhibited an anodic ECL emission peak at around 1.16 V in air saturated pH 7.0 phosphate buffer containing 0.6 mM sulfite, showing a value around 0.23 V greater than in tris(hydroxymethyl)aminomethane hydrochloride-HCl (Tris-HCl) buffer (Fig. 2D).59 This difference can be attributed to the ITO surface being full of negative charge for the absorption of phosphate, so a lower triggering potential for oxidative-reduction ECL was obtained in the presence of Tris-HCl buffer. ECL from 3-mercaptopropionic acid (MPA) capped CdTe (MPA@CdTe) QDs displayed two ECL processes at around 1.17 and 1.74 V (vs. Ag/AgCl), involving superoxide radical generation by reduction of dissolved oxygen at a lower potential or water splitting at a higher potential, respectively.60
Beneficial to the reaction of an amine group with a superoxide radical, MPA@CdTe QDs/dissolved oxygen can be utilized to detect melamine. Upon carefully selecting the appropriate coreactant, i.e., sulfite, the excited MPA@CdTe QDs released a coreactant anodic ECL at around 0.90 V in air-saturated pH 7.5 PBS (phosphate buffer solution) (Fig. 2E & F).61 The ECL intensity could be reduced with an increasing concentration of tyrosine, which was attributed to the interaction of the excited QDs and quencher o-quinone from oxidized tyrosine (Fig. 2G & H).62 Zhang et al. found that both aliphatic alkyl and hydroxy groups could lead to the enhancement of ECL intensity. Thus, the ECL of mercaptosuccinic acid protected CdTe QDs at around 0.90 V was achieved with DBAE as a coreactant (Fig. 2I & G). The effect of different coreactant amines with the same concentration was also explored in this research, and it was found that increased ECL intensity could be realized via increasing the number and length of coreactant amines attached to the nitrogen atoms, enhancing the stability of the radical ions (Fig. 2K).63,64 The ECL sensor utilized for DBAE detection operated at approximately 0.90 V with CdTe QDs as the luminophore (Fig. 2L). The ECL intensity exhibited excellent linearity with the DBAE concentration, covering a wide linear range from 1.7 × 10−8 to 1.5 × 10−4 M (R2 = 0.995) with a remarkable detection limit (S/N = 3) of 8 nM.65
Long et al. synthesized and verified the ECL nature of water-soluble CuInS2/ZnS (CIS/ZnS) QDs for the first time with the participation of TPrA. Impressively, the L-glutathione and sodium citrate-stabilized CIS/ZnS QDs exhibited two hole-injected potentials at around 0.55 and 0.94 V, respectively.66 The hole-injected potential at around 0.94 could combine with the injected electron from TPrA to generate an efficient oxidative-reduction ECL. Although both the hole-injected potentials of 0.55 and 0.94 V can be involved in efficient annihilation ECL by directly stepping CIS/ZnS QDs from the electron-injected potential to hole-injected potential. CIS/ZnS QDs fail to result in a coreactant ECL process at around 0.55 V, indicating that the oxidation potential of TPrA lags behind the hole-injected potential (Fig. 3A).35,67 Coreactant ECL at around 1.1 V of CIS/ZnS QDs was eventually adopted for determining VEGF165 in this case, with a linear range from 0.10 to 1000 pM and a limit of detection of 0.050 pM (S/N = 3) (Fig. 3B). Fu et al. proposed an LTP ECL system by employing glutathione (GSH) and citrate-stabilized CIS/ZnS (GSH/Cit@CIS/ZnS) QDs with low hole-injected potential as a luminophore and novel N2H4·H2O with low oxidation potential as a coreactant. The CIS/ZnS QDs are able to inject holes at around 0.48 and 0.87 V, resulting in LTP ECL at around 0.55 V through a unique internal Cu(I)/Cu(II) couple cycling (Fig. 3C).68 N2H4·H2O undergoes electrochemical oxidation at approximately 0.10 V under physiological conditions, yielding two additional reducing species N2H3˙ and N2H2, which exhibit enhanced ECL of CIS/ZnS QDs.69 The LTP ECL of CIS/ZnS QDs/N2H4·H2O exhibits a decreased tendency upon introducing Cu2+(Fig. 3D). Li et al. established another LTP ECL system based on the 4-mercaptobenzoic acid (MBA) and citrate capped CIS/ZnS (MBA-Cit@CIS/ZnS) QDs with N2H4·H2O as a coreactant, resulting in a distinct LTP ECL peak at approximately 0.45 V (Fig. 3C)70,71 Moreover, both MBA-Cit@CIS/ZnS QDs and 6-aza-2-thiothymine and citrate capped CIS/ZnS (ATT-Cit@CIS/ZnS) QDs could enable a low charge-transfer process between the QDs and traditional coreactant TPrA, proving that the conjugated structure in the capping agent of CIS/ZnS QDs is beneficial to a well-defined hole-injection process at low potential.72 Based on the MBA-Cit@CIS/ZnS QDs/N2H4·H2O LTP system, potential-resolved ECL multiplexing with good resolution was realized with MBA-Cit@CIS/ZnS QDs/[Ru(bpy)2(dcbpy)]2+ as luminophores and N2H4·H2O as the coreactant, which could determine human prostate specific antigen and carcinoma antigen 125 simultaneously.71 Fu et al. built a LTP ECL system universally by employing the environmental-friendly carbohydrazide as a coreactant, leading to a similar LTP oxidative-reduction ECL of 0.55 V, including Ru(bpy)32+ and CdTe, CdSe, CIS/ZnS QDs, and Au NCs (Fig. 3D). Thanks to the LTP and eight-electron releasing nature of carbohydrazide, LTP ECL from a series of luminophores could be realized via a successive electron-injecting process from carbohydrazide to reduce luminophores with an onset around 0.25 V.67,73 The traditional accumulated ECL spectra of surface confined Au NCs, CdSe, and CdTe QDs were very close to the PL spectra of the corresponding QDs, while the accumulated ECL spectra of surface confined CIS/ZnS QDs exhibited ∼80 nm red-shift compared to the PL spectrum, indicating that the surface states of these QDs had a large effect on the ECL spectrum (Fig. 3G). Spooling ECL spectra of surface confined Ru(bpy)32+ and Au NCs verified that the luminophore could be pre-reduced by the reductive radicals that were generated through the electrochemical oxidation of carbohydrazide, which was responsible for LTP ECL (Fig. 3H & I).
 | ||
| Fig. 3 Scheme for (A) coreactant ECL of GSH/Cit@CIS/ZnS QDs with TPrA as coreactant; (B) ECL profiles for VEGF165 detection with a sensor formed with (a) 0.1, (b) 0.5, (c) 1, (d) 5, (e) 10, (f) 50, (g) 100, (h) 500, or (i) 1000 pM VEGF165; (inset) calibration curve for determining VEGF165;66 (C) LTP ECL from GSH/Cit@CIS/ZnS QDs with N2H4·H2O as coreactant; (D) ECL profiles for Cu2+ detection; inset: calibration curve for determining Cu2+;68 (E) LTP ECL from MBA-Cit@CIS/ZnS QDs with N2H4·H2O as coreactant; (F) LTP ECL of surface-confined luminophores with carbohydrazide as the coreactant;70 (G) traditional accumulated ECL spectra of Nafion surface confined (a) Au NCs, (b) CdSe QDs, (c) Ru(bpy)32+, (d) CIS/ZnS QDs, (e) CdTe QDs, and (f) bare GCE with CON4H6 as coreactant; (H) spooling ECL spectra of Nafion surface confined Au NCs with CON4H6 as coreactant (the spectra were recorded at 1.0 s time intervals); (I) accumulated ECL spectra of Nafion surface confined Ru(bpy)32+, collecting all the ECL photons with successive scanning of the potential from (a) 0 to 0.7 V and (b) from 0.7 to 1.4 V at 50 mV s−1;74 (J) molecular structure of the three co-reactants of amines, DIPEA-OH, DIPEA, and TEA; (K) cyclic voltammetry and (L) ECL of 0.1 mM Au NCs at a Au electrode in 0.1 M PBS containing 0.2 M DIPEA-OH, DIPEA, and TEA.75 | ||
Recently, Fu et al. explored the effect of hydrazine compounds with various structures as coreactants on the ECL triggering potential and efficiency in detail. The hydrazine based coreactant with a N–N single bond and symmetrical structure preferentially results in LTP ECL, while the hydrazine based coreactants with –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, –CO or strong electronegative atoms are prone to generate higher triggering potential ECL with ternary CIS/ZnS QDs as luminophores. Due to the fact that a hydrazine based coreactant with –CS would competitively bond the Cu element within CIS/ZnS QDs, CIS/ZnS QDs fail to produce ECL. Furthermore, the LTP of CIS/ZnS QDs around 0.11 V could be achieved via adjusting the hole injection processes of QDs and selecting pasoniazide as a coreactant.74 The structure of the coreactant could mediate and induce to regulate the ECL behavior of QD based luminophores. Hong et al. designed N,N diisopropylethylamine (DIPEA) and 2-(diisopropylamino)-ethanol (DIPEA-OH), similar to a classical coreactant structure of TEA, as coreactants to explore the ECL mechanism. Bovine serum albumin (BSA) capped Au (BSA@Au) NCs exhibited a sole ECL process at around 0.75 V with DIPEA-OH as a coreactant, while BSA@Au NCs displayed a higher ECL process at around 1.3 V with the oxidation of classical coreactant TEA,75 proving the enhanced electron releasing ability via replacing the ethyl group in TEA with the isopropyl group of DIPEA and the enhanced ECL efficiency via forming a hydroxyl group to catalyze the oxidation of DIPEA-OH (Fig. 3J–L). Later, Peng et al. designed a LTP system based on molecular orbital theory, consisting of β-CD-protected Au NCs as luminophores and DIPEA as a new coreactant with a glassy carbon electrode (GCE) as the working electrode. The level matching between Au NCs and DIPEA is beneficial, and β-CD-protected Au NCs exhibit unprecedented ECL efficiency (145-fold higher than that of the classic Ru(bpy)32+/TPrA perchlorate system) and a LTP ECL of 0.76 V. Interestingly, other ligand capped Au NCs including N-acetyl-L-cysteine (NAC), L-proline (L-Pro), cysteamine hydrochloride/NAC (Cys/NAC), BSA, and carboxylated chitosan/dithiothreitol (CC/DTT) also exhibited LTP ECL at around 0.70 V with DIPEA as a coreactant (Fig. 4C), demonstrating the electron transfer reaction between the Au NCs and the oxidized DIPEA (DIPE˙+) based on the matching energy levels (Fig. 4A & D).76
N, –CO or strong electronegative atoms are prone to generate higher triggering potential ECL with ternary CIS/ZnS QDs as luminophores. Due to the fact that a hydrazine based coreactant with –CS would competitively bond the Cu element within CIS/ZnS QDs, CIS/ZnS QDs fail to produce ECL. Furthermore, the LTP of CIS/ZnS QDs around 0.11 V could be achieved via adjusting the hole injection processes of QDs and selecting pasoniazide as a coreactant.74 The structure of the coreactant could mediate and induce to regulate the ECL behavior of QD based luminophores. Hong et al. designed N,N diisopropylethylamine (DIPEA) and 2-(diisopropylamino)-ethanol (DIPEA-OH), similar to a classical coreactant structure of TEA, as coreactants to explore the ECL mechanism. Bovine serum albumin (BSA) capped Au (BSA@Au) NCs exhibited a sole ECL process at around 0.75 V with DIPEA-OH as a coreactant, while BSA@Au NCs displayed a higher ECL process at around 1.3 V with the oxidation of classical coreactant TEA,75 proving the enhanced electron releasing ability via replacing the ethyl group in TEA with the isopropyl group of DIPEA and the enhanced ECL efficiency via forming a hydroxyl group to catalyze the oxidation of DIPEA-OH (Fig. 3J–L). Later, Peng et al. designed a LTP system based on molecular orbital theory, consisting of β-CD-protected Au NCs as luminophores and DIPEA as a new coreactant with a glassy carbon electrode (GCE) as the working electrode. The level matching between Au NCs and DIPEA is beneficial, and β-CD-protected Au NCs exhibit unprecedented ECL efficiency (145-fold higher than that of the classic Ru(bpy)32+/TPrA perchlorate system) and a LTP ECL of 0.76 V. Interestingly, other ligand capped Au NCs including N-acetyl-L-cysteine (NAC), L-proline (L-Pro), cysteamine hydrochloride/NAC (Cys/NAC), BSA, and carboxylated chitosan/dithiothreitol (CC/DTT) also exhibited LTP ECL at around 0.70 V with DIPEA as a coreactant (Fig. 4C), demonstrating the electron transfer reaction between the Au NCs and the oxidized DIPEA (DIPE˙+) based on the matching energy levels (Fig. 4A & D).76
 | ||
| Fig. 4 (A) Energy level positions of different ligand (NAC, L-Pro, β-CD, Cys/NAC, BSA, and CC/DTT) modified Au NCs and DIPE˙+; (B) differential pulse voltammetry profile of the bare GCE in 70 mM pH 11.5 DIPEA (a) and differential pulse voltammetry profiles of surface-confined different ligand modified Au NCs in N2 saturated 0.1 M pH 11.5 PBS; (C) ECL intensities of surface-confined different ligand modified Au NCs in 70 mM pH 11.5 DIPEA; (D) energy level-related electron transfer reactions between Au NCs and DIPEA.76 | ||
Suitable working electrode
The selection of a suitable working electrode could improve the ECL properties of nanoparticle luminophores sufficiently. A sandwich-typed NIR immunoassay was prepared with dual-stabilizer-capped NIR CdTe QDs as ECL labels and DBAE as coreactant. The maximum ECL potential is around 0.85 V with an anodic Au electrode as the working electrode, which is clearly lower than that of the ECL system with other working electrodes.40Dihydrolipoic acid (DHLA) capped sliver nanoclusters (DHLA@Ag NCs) were synthesized by Ai et al. and employed as a LTP ECL luminophore at around 0.82 V with N2H4 as a coreactant at the Au electrode surface (Fig. 5A).77 Capture HPV gene (c-DNA) could link the Au electrode to form Au|c-DNA via an Au–S bond. Meanwhile, NH2-probe HPV gene (p-DNA) could link DHLA@Ag NCs with the assistance of N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride and 1-hydroxypyrrolidine-2,5-dione. A target HPV gene (t-DNA) incubated with Au|c-DNA to form Au|c-DNA|t-DNA could be incubated with p-DNA|DHLA-AgNC tags to detect t-DNA human papillomavirus (Fig. 5A).
 | ||
| Fig. 5 (A) Bioconjugates and LTP and narrow-potential-window ECL sensing with DHLA@Ag NCs as ECL tags;77 (B) schematic illustration for the ECL mechanism of GSH@CIS/ZnS/N2H4·H2O with luminophores of (left) the monodispersed and (right) surface-confined states in the Au electrode;53 (C) the ECL maximum-emission-potential of AIS/ZnS NCs on different electrodes with different co-reactants; (D) schematic illustration of the ECL mechanism of the AIS/ZnS QDs/N2H4 system on the Au electrode.78 | ||
Li et al. designed a potential-selective ECL system using AgInS2/ZnS (AIS/ZnS) QDs as luminophores for LTP ECL at around 0.3 V. Both the working electrode and coreactants have significant impacts on the ECL potential of the luminophore. The maximum emission potential of the ECL from AIS/ZnS QDs can be adjusted from 0.95 to 0.30 V by optimizing the working electrode, as well as the coreactant, indicating that selecting a suitable working electrode is an effective method for achieving a good ECL performance (Fig. 5C). AIS/ZnS QDs can be electrochemically injected with holes at 0.30 V, suggesting that the ECL potential of the AIS/ZnS QDs/N2H4 system is primarily governed by the oxidative potential of the ECL luminophore (Fig. 5D).78 The state of the luminophore is significant for the ECL potential. Dong et al. proposed strong LTP ECL from GSH capped CIS/ZnS QDs (GSH@CIS/ZnS) compared to other thiol ligand capped CIS/ZnS QDs with a Au electrode as a working electrode. A large shift in potential was obtained between the monodispersed state and surface-confined state CIS/ZnS QDs with N2H4 as a coreactant. Sandwich DNA hybridization products were constructed by extensively utilizing the Au–S bond and detected via oxidative-reduction ECL at around 0.32 V for CIS/ZnS QDs (Fig. 5B).53
Doping of nanoparticle luminophores
The favorable photophysical and electrochemical properties of CDs have led to their emergence in a wide range of applications.The ECL performance of CDs was promoted by introducing surface emissive sites through N and S element doping. Ding et al. synthesized CDs using citric acid and high/low thiourea concentrations, which exhibited aggregation induced emission (AIE) and aggregation caused quenching (ACQ) PL effects, respectively, due to different S doping concentrations of the CDs. For oxidation-reduced ECL, the S-rich CDs exhibited two ECL processes at around 1.0 and 1.2 V with TPrA as coreactant, which clearly differs from the ECL of S-scarce CDs, indicating that the ECL potential could be adjusting by employing an element doping strategy.79
Zhang et al. prepared water-solution nitrogen doped graphene QDs (N-GQDs) with green photoinduced luminescence, which are promising ECL labels with low oxidative-reduction ECL at around 0.84 V with DBAE as a coreactant. Carbon nanotubes (MWCNTs) could enhance the LTP ECL intensity with a high surface-to-volume ratio and accelerated electron transport based on the prepared N-GQDs system.80 N-GQDs with green-luminescence were promising ECL labels, which can facilitate ECL bioassays for α-fetoprotein antigen (AFP) detection. Nitrogen doped hydrazide conjugated CDs (NHCDs) also display a LTP effect at around 0.65 V with H2O2 as a coreactant compared to the ECL of pure hydrazide conjugated CDs (HCDs) at around 0.75 V, demonstrating that nitrogen doping induced a shift of the highest occupied molecular orbital to the upper energy level. A biosensor was constructed with NHCDs for cell-secreted H2O2 detection.81 Multinary QDs are liable to exhibit significant ECL properties upon element doping. Zhang et al. investigated the ECL of Zn–Ag–In–S QDs for the first time with a LTP at around 0.95 V. The ZnS shell coating in the surface of ZAIS could reduce the surface defects and enhance the ECL efficiency.44 Otherwise, ZAIS exhibited an ECL process at around 1.1 V with TEA as a coreactant.
Surface-engineering strategy
Due to the pathway of major ECL systems being induced by surface defects and traps, surface-engineering strategies have been extensively exploited to modulate ECL intensity, waveband and QD potential. Considering the above principles, the ECL potential and potential windows of CdTe QDs were modulated via modifying their synthesis and purification procedure, verifying that CdTe QDs linked with sulfhydryl-RNA result in only one ECL process at around 0.32 V with a narrow triggering potential window of 0.35 V, while CdTe QDs linked with amino-RNA/DNA via an amide bond exhibit only one ECL peak at around 0.82 V (Fig. 6B & C).82 This strategy provides an interesting method to adjust the ECL potential via linking biological substances to form conjugates, which could determine open reading frame 1 ab (ORF1ab) and nucleoprotein (N) genes simultaneously. As shown in Fig. 6A, a CdTe NC labeled SH-modified probe RNA for ORF1ab (i.e., PORF-SH|CdTe) and CdTe NC labeled NH2-modified probe RNA for N gene (i.e., PN-NH2|CdTe) are achieved via Cd–S bonds and carboxylic groups of CdTe QDs, respectively. The Au electrode is incubated with 3′-phosphate-modified capture ORF1ab gene (CORF) and capture N gene (CN) to form Au|CORFCNvia Au–S bonds, and then target ORF1ab (TORF) and N gene (TN) with various concentrations are casted on Au|CORFCN to form Au|CORFCN < TORFTN. The resultant electrode is allowed to react with the mixture of PORF-SH|CdTe and PN-NH2|CdTe to form an ECL biosensor, i.e.,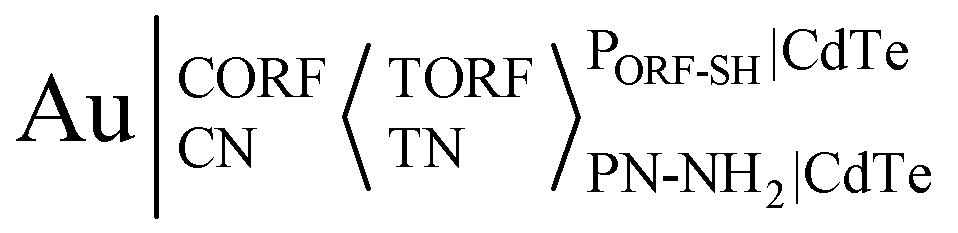 . The corresponding ECL spectra for PORF-SH|CdTe and PN-NH2|CdTe closely resemble that of CdTe QDs, suggesting that the ECL from PORF-SH|CdTe and PN-NH2|CdTe share the same excited state of CdTe QDs (Fig. 6D). Similar to PORF-SH|CdTe, PHBV-3′-SH|CdTe and PHPV-5′-SH|CdTe also exhibit a single ECL process at around 0.32 V (Fig. 6E, curve a and b). Similar to PN-NH2|CdTe, PHPV-3′-NH2|CdTe and PHBV-5′-NH2|CdTe only display one ECL process at around 0.82 V (Fig. 6E, curve c and d).
. The corresponding ECL spectra for PORF-SH|CdTe and PN-NH2|CdTe closely resemble that of CdTe QDs, suggesting that the ECL from PORF-SH|CdTe and PN-NH2|CdTe share the same excited state of CdTe QDs (Fig. 6D). Similar to PORF-SH|CdTe, PHBV-3′-SH|CdTe and PHPV-5′-SH|CdTe also exhibit a single ECL process at around 0.32 V (Fig. 6E, curve a and b). Similar to PN-NH2|CdTe, PHPV-3′-NH2|CdTe and PHBV-5′-NH2|CdTe only display one ECL process at around 0.82 V (Fig. 6E, curve c and d).
 | ||
| Fig. 6 Schematic illustration of the preparation of (A) PORF-SH|CdTe and PN-NH2|CdTe conjugates, as well as the dual-potential encoded ECL sensor for ORF1ab and N gene detection by using CdTe QDs as a tag in a labeling-bond engineered way; (B) DPV, (C) ECL profiles, and (D) ECL spectra of 1.0 μM (a) PORF-SH|CdTe and (b) PN-NH2|CdTe on a Au electrode; (E) ECL profiles of 1.0 μM (a) PHBV-3′-SH|CdTe NCs, and (b) PHPV-5′-SH|CdTe NCs, (c) PHPV-3′-NH2|CdTe NCs, and (d) PHBV5′-NH2|CdTe NCs on a bare Au electrode;82 (F) scheme showing ECL potential regulation of CdTe QDs with differing protein-weights of the Ab2;83 (G) schematic of ECL regulation using thioglycol/GSH dual ligand protected Au NCs.84 | ||
Zou's group also proposed that the bioconjugates of the CdTe QDs and the secondary antibody (Ab2) with differing protein-weights (Ab2|CdTe) displayed clearly different ECL potentials. The formation of Ab2|CdTe with the Ab2 below 30 kDa only displayed coreactant-free ECL at around 0.24 V. Otherwise, an individual coreactant-free ECL at around 0.84 V occurred when the bioconjugate was constructed with the Ab2 exceeding 30 kDa (Fig. 6F).83
Wang et al. found that the ECL potential at around 1.8 V with only GSH stabilized Au NCs as models was apparently higher-shifted in the anodic direction compared with the thioglycol/GSH capped Au NCs as models. Meanwhile, the ECL wavelength was also blue shifted from 630 nm to 580 nm (Fig. 6G).84
Jia introduced a dual ligand strategy for the surface of Au NCs, forming the dual-thiol bond and limiting the intramolecular motion and nonradiative processes, which contributed to the red shift of the ECL wavelength and low shift of the ECL potential in the anodic direction.85 Zou et al. proposed NIR ECL from CdTe QDs capped with a dual-stabilizer, achieving two LTP ECL waves on a GCE at around 0.63 and 0.88 V, respectively.86 All the aforementioned examples confirm that ECL is sensitive to the surface structure of the QDs and serves as an ideal tool for probing their surface structure and related mechanisms.
Valence-state-engineered LTP ECL
Changing the valence state of the body element by adjusting the capping agent would conveniently tune the potential of the nanoparticle luminophores. Wang et al. explored the effect of Au valence on ECL based on prepared BSA@Au NCs of different Au(I)/Au(0) via adjusting the incubation time of the precursors (Fig. 7A & B). The BSA@Au NCs could produce three ECL anodic processes at around 0.37 V, 0.85 V and 1.45 V (Fig. 7C) in the presence of N2H4.H2O, proving that the LTP ECL at around 0.37 V was separate from Au(0), while the higher triggering potentials at around 0.85 V and 1.45 V were attributed to Au(I). Finally, a stable ECL with efficient LTP emission at approximately 0.37 V can be conveniently achieved by incubating BSA@Au NCs for 24 h in the synthesis process. Furthermore, the ECL wavelength can also be successfully altered through valence engineering of the Au element, resulting from the regulation of a band-gap-engineered route or a surface-defect involved route on the surface of the BSA@Au NCs (Fig. 7D & F).87 The LTP ECL system utilizing BSA@Au NCs/N2H4·H2O was employed to conduct sandwich-type immunoassays for the selective detection of CA125 (Fig. 7E).88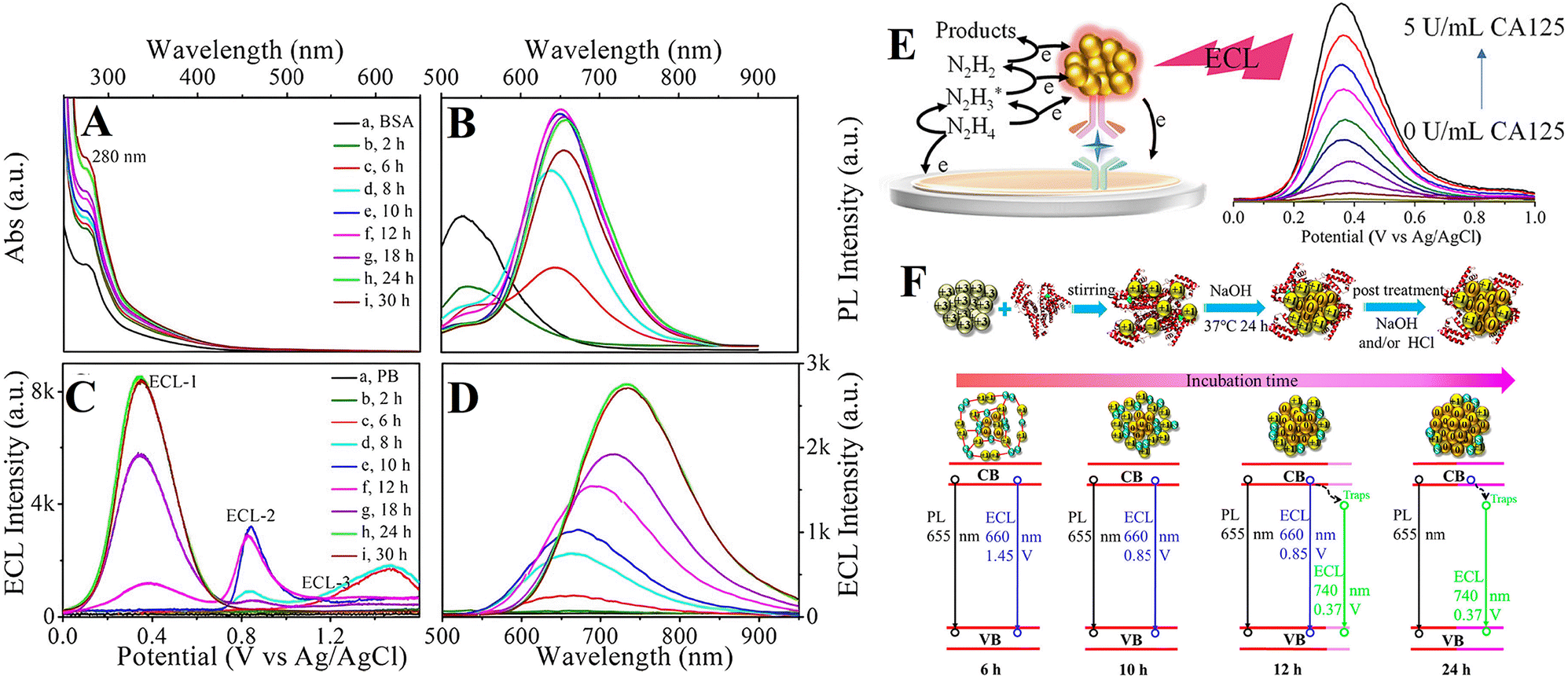 | ||
| Fig. 7 (A) UV-vis absorption and (B) PL spectra of (a) BSA and (b–i) BSA@Au NCs with an incubation times of (b) 2, (c) 6, (d) 8, (e) 10, (f) 12, (g) 18, (h) 24, and (i) 30 h. (C) ECL potential profiles and (D) accumulated ECL spectra of bare Au in (a) 0.1 M pH 7.4 phosphate buffer containing 10 mM N2H4 and (b–i) 0.5 mg mL−1 BSA@Au NCs with incubation times of (b) 2, (c) 6, (d) 8, (e) 10, (f) 12, (g) 18, (h) 24, and (i) 30 h;87 (E) schematic illustration of the LTP-ECL immunosensor with BSA@Au NCs as tags;88 (F) schematic illustration of the valence-state-engineered ECL of BSA@Au NCs: effect of incubation time on the valence state of body element Au and internal structure of BSA@Au NCs.87 | ||
Donor–acceptor system
The ECL properties could be modulated through intramolecular charge transfer of the donor–acceptor (D–A) moieties.89 Wang et al. proposed two aggregation-induced emission (AIE)-active conjugated polymer dots (Pdots) containing fluorene, carbazole, TPE, and BODIPY moieties with a D–A system. The Pdots with electron-donating carbazole moieties (P-2 dots, 1.028 V) decreased the anodic ECL peak potential by 553 mV compared with the Pdots with fluorene moieties (P-1 dots, 1.515 V) with the traditional TPrA as a coreactant, proving that the ECL potential and efficiency could be regulated via adjusting the D–A electron structure.90 Wang et al. designed a luminophore by covalently coupling carboxylated Pdots and the luminol analogue with ECL resonant energy transfer for good spectral overlap, promisingly resulting in strong anodic ECL emission at a low peak potential of about 0.45 V with H2O2 as a coreactant.91Combining multiplex materials
Hybrids of different QDs could produce unexpected ECL behavior. By hybridizing hydrazide-modified graphene QDs with AuCl4−, nano-hybrids (HM-GQDs-AuNPs) were obtained. Impressively, the surface confined nano-hybrid HM-GQDs-AuNPs on GCE (GCE|HM-GQD-AuNPs) demonstrated a significantly lower potential LTP ECL at approximately 0.60 V with the assistance of H2O2, surpassing that of the traditional Ru(bpy)23+/TPrA ECL system by far.92Future perspectives
With the continuous advancement of nanotechnology, it is anticipated that a greater variety of novel ECL nanoparticle luminophores will be developed in the future, potentially exhibiting lower potential requirements. For instance, emerging materials such as perovskite demonstrate significant promise for applications in the realm of ECL.34Various regulatory approaches can be employed for nanoparticle luminophores with LTP nature, encompassing size and shape regulation, engineering band-gap or surface states of nanoparticle luminophores, and fabrication of composite materials, as well as optimization of reaction conditions. These control strategies may be utilized individually or in combination to achieve precise regulation and enhancement of ECL performance. First, size regulation: by precisely adjusting the size of the nanoparticles, it is possible to control the ECL wavelength and optimize the ECL potential. Second, shape regulation: the ECL characteristics of the nanoparticles are subject to manipulation through morphological changes, such as transitioning from a spherical to rod-like or sheet shape. Third, engineering the band-gap or surface states of the nanoparticle luminophores, which can enhance the ECL intensity and biocompatibility of the nanoparticles and lower the ECL potential. For instance, the ECL performance of CdTe nanoclusters coated with double stabilizers can be optimized through the preparation of water-soluble Co2+-CdTe nanoclusters via Co2+ growth doping.93 Fourth, fabrication of composite materials, the combination of nanoparticles with other materials can lead to the collaborative optimization of their properties. By integrating nanoparticles with suitable substrate materials or carriers, it is possible to regulate the electron transfer of inter nanoparticle luminophores and lower their ECL potential. Fifth, in addition to the aforementioned regulatory measures, the ECL potential of nanoparticles can be further lowered through optimization of the reaction conditions for ECL. For instance, selecting the appropriate electrolyte solution, working electrode, and suitable coreactant with low oxidation potential and adjusting the pH level of the solution and other factors may impact the ECL potential (Table 1).
| Luminophores | Coreactant | Working electrode | Reference electrode | Electrolyte | ECL triggering potential | Ref. |
|---|---|---|---|---|---|---|
| TGA@CdSe QDs | Sulfite | ITO | Ag/AgCl | pH 7.0 Tris-HCl | 0.926 V | 58 |
| TGA@CdSe QDs | Sulfite | ITO | Ag/AgCl | pH 7.0 Tris-HCl | 0.927 V | 59 |
| MPA@CdTe QDs | Dissolved oxygen | ITO | Ag/AgCl | pH 7.4 PBS | 1.17 and 1.74 V | 60 |
| MPA@CdTe QDs | Na2SO3 | ITO | Ag/AgCl | pH 7.5 PBS | 0.90 V | 61 |
| CdTe QDs | DBAE | GCE | Ag/AgCl | pH 7.5 PBS | 0.90 V | 65 |
| GSH/Cit@CIS/ZnS QDs | TPrA | GCE | Ag/AgCl | pH 7.4 PBS | 0.94 V | 66 |
| GSH/Cit@CIS/ZnS QDs | N2H4·H2O | GCE | Ag/AgCl | pH 7.4 Tris-HCl | 0.55 and 0.87 V | 68 |
| MBA-Cit@CIS/ZnS QDs | N2H4·H2O or TPrA | GCE | Ag/AgCl | pH 7.4 Tris-HCl | 0.45 V | 70 |
| MBA-Cit@CIS/ZnS QDs | N2H4·H2O | GCE | Ag/AgCl | pH 7.4 PBS | 0.30 V | 71 |
| GSH/Cit@CIS/ZnS QDs | Pasoniazide | GCE | Ag/AgCl | pH 11.0 Tris-HCl | 0.11 V | 74 |
| Nanoparticulate luminophores | CON4H6 | GCE | Ag/AgCl | pH 7.4 Tris-HCl | 0.55 V | 50 |
| BSA@Au NCs | DIPEA/DIPEA-OH | Au electrode | Ag/AgCl | pH 11.0 PBS | 0.70 V | 75 |
| Au NCs | DIPEA | GCE | Ag/AgCl | pH 11.5 PBS | 0.75 V | 76 |
| CdTe QDs | TPrA | GCE | Ag/AgCl | pH 10.2 PBS | 0.63 and 0.82 V | 86 |
| CdTe QDs | DBAE | Au electrode | Ag/AgCl | pH 9.2 PBS | 0.85 V | 40 |
| GSH@CIS/ZnS QDs | N2H4·H2O | Au electrode | Ag/AgCl | pH 7.4 PBS | 0.32 V | 53 |
| CDs | TPrA | GCE | Ag/AgCl | pH 7.48 PBS | 1.0 and 1.2 V | 79 |
| N-GQDs | DBAE | Gold nanoflower modified paper | Ag/AgCl | pH 7.4 PBS | 0.84 V | 80 |
| NHCDs | H2O2 | GCE | Ag/AgCl | pH 7.4 PBS | 0.65 V | 81 |
| Zn–Ag–In–S QDs | TPrA | GCE | Ag/AgCl | pH 7.4 PBS | 0.95 V | 44 |
| CdTe QDs | Coreactant-free | Au electrode | Ag/AgCl | pH 7.0 PBS | 0.32 V | 82 |
| CdTe QDs | Coreactant-free | Au electrode | Ag/AgCl | pH 9.0 PBS | 0.24 V | 83 |
| Au NCs | TEA | GCE | Ag/AgCl | pH 7.0 PBS | 1.4 and 1.8 V | 84 |
| Polyfluorene derivatives | CA125 | ITO | Ag/AgCl | pH 7.4 PBS | 0.3 V | 94 |
| Au NCs | N2H4·H2O | Au electrode | Ag/AgCl | pH 7.4 PBS | 0.37 V | 88 |
| Ag NCs | N2H4·H2O | Au electrode | Ag/AgCl | pH 7.4 PBS | 0.82 V | 77 |
| AgInS2/ZnS QDs | N2H4·H2O | Au electrode | Ag/AgCl | pH 7.4 Tris-HCl | 0.30 V | 78 |
| Au NCs | tert-Butylamine borane | GCE | Ag/AgCl | pH 7.4 PBS | 0.57 V | 95 |
| Pdots | TPrA | GCE | Ag/AgCl | pH 7.4 PBS | 1.028 V | 90 |
| Pdots | H2O2 | GCE | Ag/AgCl | pH 11.0 carbonate buffer solution | 0.45 V | 91 |
| HM-GQDs-AuNPs | H2O2 | GCE | Ag/AgCl | pH 9.0 PBS | 0.60 V | 92 |
Researchers are continuously exploring and refining ECL technology with a low triggering potential. By improving nanoparticle design and optimizing reaction conditions, it is expected that a more stable and sensitive ECL phenomenon at low potential can be achieved, thus paving the way for new applications of electrochemical luminescence technology.
Conclusions
LTP ECL has been eagerly anticipated for its potential-resolved and minimal interference biosensing. Nanoparticle luminophores are attractive in the development of human biological science and ECL evolution. The benefits of the ability of nanoparticle luminophores to be regulated via adjusting their body element or surface state, LTP ECL development and strategies with nanoparticles as luminophores resulting in less electrochemical interference and enhanced electrode compatibility are reviewed in this article. The LTP ECL potential commonly involves three categories of strategies. First, adjusting the electron transfer of the inter ECL system, including choosing a suitable coreactant with low oxidation potential, working electrode and pH of electrolyte. Second, engineering the band-gap or surface states of the nanoparticle luminophores themselves, including doping of nanoparticle luminophores, surface-engineering, and valence-state-engineering. Third, regulating the electron transfer of inter nanoparticle luminophores, enclosing donor–acceptor systems and combining multiplex nanoparticle luminophores. Further exploration should focus on establishing a correlation between ECL potential and efficient ECL luminophore through deliberate manipulation of suitable surface states and precise management of charge transfer. LTP ECL technology is anticipated to drive progress in the fields of ECL and photoelectrochemical devices, as well as beyond, with a deepening understanding of the mechanism. Additionally, with the aid of novel analytical technology, nanoparticle luminophores utilizing LTP ECL can be applied to a wide range of biosensing and clinical assay applications.Author contributions
Li Fu: writing – original draft, review & editing, validation, supervision, investigation, formal analysis, conceptualization. Tianyuan Song: formal analysis, conceptualization. Qi Li: investigation. Guizheng Zou: visualization, validation, supervision, investigation, resources. Fuwei Zhang: investigation, formal analysis. Zongchao Li: investigation, formal analysis. Haotian Guan: investigation, formal analysis. Yingshu Guo: visualization, validation, supervision, investigation, resources.Data availability
Data availability is not applicable to this review article as no new data were created or analyzed in this manuscript. If necessary, we can provide any missing statement.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22276102, 22476103, 22304093), the Young Taishan Scholar Program of Shandong Province of China (tsqn202211212), the Shandong Provincial Natural Science Foundation (ZR2023QB158, ZR2023JQ004), the University Institute Innovation Team of Jinan (202228027), the Qilu University of Technology (Shandong Academy of Sciences) Youth Outstanding Talent Program (2024QZJH05), the Science, Education and Industry Integration Pilot Project Plan of Qilu University of Technology (Shandong Academy of Sciences) (2024RCKY016, 2023PYI002), Talent Research Project of Qilu University of Technology (Shandong Academy of Sciences) (2023RCKY105), and the Shandong Province Higher Education Undergraduate Teaching Reform Research Project Key Project (Z2023022).References
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef.
- B. Mohan, S. Kumar, V. Kumar, T. Jiao, H. K. Sharma and Q. Chen, TrAC, Trends Anal. Chem., 2022, 157, 116735 CrossRef.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef.
- E. Sobhanie, F. Salehnia, G. B. Xu, Y. Hamidipanah, S. Arshian, A. Firoozbakhtian, M. Hosseini, M. R. Ganjali and S. Hanif, TrAC, Trends Anal. Chem., 2022, 157, 116727 CrossRef PubMed.
- Q. Zhang, X. Zhang and Q. Ma, J. Anal. Test., 2020, 4, 92–106 CrossRef.
- D. Yaman, M. Jimenez, S. F. Gonzalez and D. Corrigan, Analyst, 2024, 149, 5156–5164 RSC.
- I. L. K. Sivakumar, V. B. Shettya, S. Paramasivam, M. K. Rao, S. Muthu and S. S. Kumar, J. Mater. Chem. C, 2024, 12, 10390–10407 RSC.
- K. Kuang, Y. Chen, Y. Li, Y. Ji and N. Jia, Biosens. Bioelectron., 2023, 247, 115914 CrossRef PubMed.
- L. Song, G. R. Kuang, G. Zhang, J. Guo and Y. Z. Fu, Chem. Eng. J., 2023, 472, 144923 CrossRef.
- J. Wang, Z. Han, T. Shang, Y. Feng, R. Liu and X. Lu, Chem. Sci., 2024, 15, 5581–5588 RSC.
- W. J. Dai, G. X. Chen, X. Y. Wang, S. J. Zhen, C. Z. Huang, L. Zhan and Y. F. Li, Biosens. Bioelectron., 2024, 246, 115863 CrossRef CAS PubMed.
- J. L. Liu, K. Yu, H. Zhang, J. He, J. Jiang and H. Luo, Chem. Sci., 2021, 12, 9494–9499 RSC.
- X. Ren, D. Zhang, C. Li, J. Zhao, R. Feng, Y. Zhang, R. Xu and Q. Wei, Anal. Chem., 2024, 96, 3898–3905 CrossRef CAS PubMed.
- X. L. Zhang, H. Dong, H. Zhou, Y. Y. Li, Q. Liu, H. Y. Cui, S. J. Wang, Y. Y. Li and Q. Wei, Sens. Actuators, B, 2024, 400, 134911 CrossRef CAS.
- Y. M. Lei, D. Wu, M. C. Pan, X. L. Tao, W. J. Zeng, L. Y. Gan, Y. Q. Chai, R. Yuan and Y. Zhuo, Chem. Sci., 2024, 15, 3255–3261 RSC.
- M. Jiang, M. Wang, W. Lai, X. Song, J. Li, D. Liu, Z. Wei and C. Hong, Anal. Chim. Acta, 2023, 1272, 341476 CrossRef CAS.
- M. Saqib, S. Bashir, S. A. Kitte, H. J. Li and Y. D. Jin, Chem. Commun., 2020, 56, 5154–5157 RSC.
- Y. He, S. Hou, L. Yang, F. Zhang and G. Zou, Chem. – Eur. J., 2018, 24, 9592–9597 CrossRef CAS PubMed.
- Y. P. He, L. Q. Yang, F. Zhang, B. Zhang and G. Z. Zou, J. Phys. Chem. Lett., 2018, 9, 6089–6095 CrossRef.
- P. Wu, X. D. Hou, J. J. Xu and H. Y. Chen, Chem. Rev., 2014, 114, 11027–11059 CrossRef.
- D. Ege, W. G. Becker and A. J. Bard, Anal. Chem., 1984, 56, 2413–2417 CrossRef.
- G. F. Blackburn, H. P. Shah, J. H. Kenten, J. Leland, R. A. Kamin, J. Link, J. Peterman, M. J. Powell, A. Shah, D. B. Talley, S. K. Tyagi, E. Wilkins, T. G. Wu and R. J. Massey, Clin. Chem., 1991, 37, 1534–1539 CrossRef.
- H. Yang, J. K. Leland, D. Yost and R. J. Massey, Nat. Biotechnol., 1994, 12, 193–194 CrossRef PubMed.
- Y. Cao, R. Wu, Y.-Y. Gao, Y. Zhou and J.-J. Zhu, Nano-Micro Lett., 2023, 16, 37 CrossRef.
- K. Maru, A. Singh, R. Jangir and K. K. Jangir, J. Mater. Chem. B, 2024, 12, 4553–4573 RSC.
- E. Kerr, D. J. Hayne, L. C. Soulsby, J. C. Bawden, S. J. Blom, E. H. Doeven, L. C. Henderson, C. F. Hogan and P. S. Francis, Chem. Sci., 2022, 13, 469–477 RSC.
- Z. Y. Xu, F. Wu, D. Zhu, H. M. Fu, Z. Shen and J. P. Lei, Chem. Commun., 2022, 58, 11515–11518 RSC.
- X. Zhang, P. Wang, Y. Nie and Q. Ma, TrAC, Trends Anal. Chem., 2021, 143, 116410 CrossRef.
- E. R. Yang, Y. J. Zhang and Y. F. Shen, Anal. Chim. Acta, 2022, 1209, 339140 CrossRef.
- H. Sun, Z. Cao, H. Qin, X. Peng and B. Su, Nano Res., 2024, 7776–7785 CrossRef.
- Q. Cai, H. Li, B. Wang and G. Jie, Chem. Eng. J., 2023, 476, 146799 CrossRef CAS.
- G. Awiaz, J. Lin and A. Wu, Exploration, 2023, 3, 1–33 CrossRef PubMed.
- H. Liu, H. Jiang, X. Liu and X. Wang, Exploration, 2023, 3, 1–20 Search PubMed.
- H.-G. Li, Y.-M. Zhu, X.-L. Liu, Z.-Y. Guo, Y.-N. Huang and X. Chen, J. Anal. Test., 2023, 7, 1–7 CrossRef CAS.
- Z. F. Ding, B. M. Quinn, S. K. Haram, L. E. Pell, B. A. Korgel and A. J. Bard, Science, 2002, 296, 1293–1297 CrossRef CAS PubMed.
- J. Descamps, Y. Zhao, B. Goudeau, D. Manojlovic, G. Loget and N. Sojic, Chem. Sci., 2024, 15, 2055–2061 RSC.
- Y. Zhao, Y. Leger, J. Descamps, N. Sojic and G. Loget, Small, 2024, 20, e2308023 CrossRef PubMed.
- F. Arcudi, L. Dordevic, S. Rebeccani, M. Cacioppo, A. Zanut, G. Valenti, F. Paolucci and M. Prato, Adv. Sci., 2021, 8, 2100125 CrossRef PubMed.
- Y.-Z. Guo, R. Liu, Y. Zeng, Y.-Y. Liao, J.-L. Liu, Y.-Q. Chai and R. Yuan, Chem. Eng. J., 2024, 488, 151057 CrossRef.
- G. Liang, S. Liu, G. Zou and X. Zhang, Anal. Chem., 2012, 84, 10645–10649 CrossRef PubMed.
- X. Y. Long, X. Tan, Y. P. He and G. Z. Zou, J. Mater. Chem. C, 2017, 5, 12393–12399 RSC.
- Z. Cao, Y. Shu, H. Qin, B. Su and X. Peng, ACS Cent. Sci., 2020, 6, 1129–1137 CrossRef PubMed.
- Z. Cao, C. Li, Y. Shu, M. Zhu, B. Su, H. Qin and X. Peng, J. Am. Chem. Soc., 2023, 145, 26425–26434 CrossRef PubMed.
- B. Zhang, F. Zhang, P. Zhang, D. Z. Shen, X. W. Gao and G. Z. Zou, Anal. Chem., 2019, 91, 3754–3758 CrossRef PubMed.
- L. Q. Yang, B. Zhang, L. Fu, K. N. Fu and G. Z. Zou, Angew. Chem., Int. Ed., 2019, 58, 6901–6905 CrossRef PubMed.
- H. P. Peng, Z. N. Huang, H. H. Deng, W. H. Wu, K. Y. Huang, Z. L. Li, W. Chen and J. W. Liu, Angew. Chem., Int. Ed., 2020, 59, 9982–9985 CrossRef PubMed.
- T. Y. Wang, D. C. Wang, J. W. Padelford, J. Jiang and G. L. Wang, J. Am. Chem. Soc., 2016, 138, 6380–6383 CrossRef PubMed.
- M. K. Zhang and X. D. Guo, Coord. Chem. Rev., 2022, 465, 214578 CrossRef.
- C. Wang, S. Liu and H. Ju, Bioelectrochemistry, 2022, 149, 108281 CrossRef PubMed.
- L. Fu, B. Zhang, K. Fu, X. Gao and G. Zou, Anal. Chem., 2020, 92, 6144–6149 CrossRef PubMed.
- S. Wang, S. Zhu, Z. Kang, Y. Chen, X. Liu, Z. Deng, K. Hu, G. Wang, Y. Zhang and G. Zang, Theranostics, 2022, 12, 6779–6808 CrossRef.
- Q.-M. Feng, Y.-Z. Shen, M.-X. Li, Z.-L. Zhang, W. Zhao, J.-J. Xu and H.-Y. Chen, Anal. Chem., 2015, 88, 937–944 CrossRef.
- S. T. Dong, X. W. Gao, L. Fu, J. N. Jia and G. Z. Zou, Anal. Chem., 2021, 93, 12250–12256 CrossRef PubMed.
- Y. M. Long, L. Bao, J. Y. Zhao, Z. L. Zhang and D. W. Pang, Anal. Chem., 2014, 86, 7224–7228 CrossRef PubMed.
- Y. P. Dong, T. T. Gao, Y. Zhou, L. P. Jiang and J. J. Zhu, Sci. Rep., 2015, 5, 15392 CrossRef PubMed.
- H. J. Wang, Y. L. Yuan, Y. Q. Chai and R. Yuan, Biosens. Bioelectron., 2015, 68, 72–77 CrossRef PubMed.
- L. Zhou, J. Huang, B. Yu and T. You, Sci. Rep., 2016, 6, 22234 CrossRef PubMed.
- X. Liu, L. X. Cheng, J. P. Lei and H. X. Ju, Analyst, 2008, 133, 1161–1163 RSC.
- X. Liu, L. Guo, L. Cheng and H. Ju, Talanta, 2009, 78, 691–694 CrossRef PubMed.
- T. Hu, T. Li, L. Yuan, S. Liu and Z. Wang, Nanoscale, 2012, 4, 5447–5453 RSC.
- X. Liu and H. X. Ju, Anal. Chem., 2008, 80, 5377–5382 CrossRef PubMed.
- X. Liu, H. Jiang, J. P. Lei and H. X. Ju, Anal. Chem., 2007, 79, 8055–8060 CrossRef PubMed.
- X. Q. Liu, L. H. Shi, W. X. Niu, H. J. Li and G. B. Xu, Angew. Chem., Int. Ed., 2007, 46, 421–424 CrossRef PubMed.
- A. W. Knight and G. M. Greenway, Analyst, 1996, 121, 101R–106R RSC.
- L. H. Zhang, X. Q. Zou, E. Ying and S. J. Dong, J. Phys. Chem. C, 2008, 112, 4451–4454 CrossRef.
- X. Long, F. Zhang, Y. He, S. Hou, B. Zhang and G. Zou, Anal. Chem., 2018, 90, 3563–3569 CrossRef PubMed.
- W. J. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef PubMed.
- L. Fu, B. Zhang, X. Long, K. Fu, X. Gao and G. Zou, Anal. Chem., 2019, 91, 10221–10226 CrossRef PubMed.
- D. Hercules, Acc. Chem. Res., 1969, 2, 301–307 CrossRef.
- Z. P. Li, S. Wu, B. Zhang, L. Fu and G. Z. Zou, J. Phys. Chem. Lett., 2019, 10, 5408–5413 CrossRef PubMed.
- Z. P. Li, S. Wu and G. Z. Zou, J. Electroanal. Chem., 2021, 888, 115173 CrossRef.
- J. Dai, J. Xi, L. Li, J. Zhao, Y. Shi, W. Zhang, C. Ran, B. Jiao, X. Hou, X. Duan and Z. Wu, Angew. Chem., Int. Ed., 2018, 57, 5754–5758 CrossRef PubMed.
- F. E. Lytle, Photochem. Photobiol., 1971, 13, 123–133 CrossRef.
- L. Fu, Z. Liu, P. Dong and Y. Guo, J. Phys. Chem. C, 2023, 127, 22827–22832 CrossRef.
- G. L. Hong, C. P. Su, M. C. Lai, Z. N. Huang, Z. M. Weng, Y. L. Chen, H. H. Deng, W. Chen and H. P. Peng, Anal. Chem., 2022, 94, 12500–12506 CrossRef PubMed.
- H. Peng, M. Lai, H. Wang, Z. Weng, Y. Yang, Z. Huang, W. Sun, J. Liu and W. Chen, Anal. Chem., 2023, 95, 11106–11112 CrossRef PubMed.
- Y. Ai, X. Gao, X. Ren, M. Li, B. Zhang and G. Zou, Anal. Chem., 2024, 96, 6652–6658 CrossRef PubMed.
- M. Li, X. Gao, X. Ren, Y. Ai, B. Zhang and G. Zou, Chem. Commun., 2024, 60, 4958–4961 RSC.
- J. R. Adsetts, S. Hoesterey, C. Gao, D. A. Love and Z. Ding, Langmuir, 2020, 36, 14432–14442 CrossRef PubMed.
- L. Zhang, L. Li, C. Ma, S. Ge, M. Yan and C. Bian, Sens. Actuators, B, 2015, 221, 799–806 CrossRef.
- A. Chen, W. Liang, H. Wang, Y. Zhuo, Y. Chai and R. Yuan, Anal. Chem., 2019, 92, 1379–1385 CrossRef PubMed.
- X. Gao, X. Ren, Y. Ai, M. Li, B. Zhang and G. Zou, Biosens. Bioelectron., 2023, 236, 115418 CrossRef PubMed.
- X. Gao, X. Ren, Y. Ai, M. Li, B. Zhang and G. Zou, Anal. Chem., 2023, 95, 6948–6954 CrossRef PubMed.
- H. Jiang, L. Liu and X. Wang, Nanoscale, 2017, 9, 9792–9796 RSC.
- H. Y. Jia, L. Yang, X. Dong, L. M. Zhou, Q. Wei and H. X. Ju, Anal. Chem., 2022, 94, 2313–2320 CrossRef PubMed.
- G. Z. Zou, G. D. Liang and X. L. Zhang, Chem. Commun., 2011, 47, 10115–10117 RSC.
- D. Wang, X. Gao, J. Jia, B. Zhang and G. Zou, ACS Nano, 2022, 17, 355–362 CrossRef PubMed.
- D. Y. Wang, X. C. Liu, Y. Zeng, Q. Q. Zhang, B. Zhang and G. Z. Zou, Anal. Chem., 2022, 94, 11688–11694 CrossRef CAS PubMed.
- C. Wang, Y. Pei, P. Liu, Y. Li and Z. Wang, ACS Sustainable Chem. Eng., 2022, 10, 14665–14670 CrossRef.
- Z. Wang, Y. Feng, N. Wang, Y. Cheng, Y. Quan and H. Ju, J. Phys. Chem. Lett., 2018, 9, 5296–5302 CrossRef PubMed.
- Z. Wang, H. Guo, Z. Luo, Y. Duan and Y. Feng, Anal. Chem., 2022, 94, 5615–5623 CrossRef PubMed.
- Y. Q. Dong, H. Wu, P. X. Shang, X. T. Zeng and Y. W. Chi, Nanoscale, 2015, 7, 16366–16371 RSC.
- X. W. Gao, K. N. Fu, L. Fu, H. S. Wang, B. Zhang and G. Z. Zou, Biosens. Bioelectron., 2020, 150, 111880 CrossRef PubMed.
- M. Yan, W. Gao, S. Ge, L. Ge, C. Chu, J. Yu, X. Song and S. Hou, J. Mater. Chem., 2012, 22, 5568–5573 RSC.
- L. Fu, P. Dong, Z. Liu, Q. Li and Y. Guo, Anal. Chem., 2024, 96(45), 18254–18261 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |