
Reliable and precise lipoprotein detection based on a self-priming hairpin-triggered Cas12a/crRNA based signaling strategy†
Xiaoya
Liu‡
a,
Hai
Peng‡
bc,
Lisha
Gong‡
be,
Hong
Zhang
bc,
Chenglong
Zhao
bd,
Weiju
Lai
b,
Gang
An
*d and
Xianxian
Zhao
 *b
*b
aDepartment of Oncology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, China
bCentral Laboratory, Chongqing University FuLing Hospital, Chongqing, 408099, China. E-mail: zhaoxian_48hcu@163.com; Tel: +86-18875039819
cCenter of Smart Laboratory and Molecular Medicine, School of Medicine, Chongqing University, Chongqing, 400044, China
dDepartment of Orthopedics, The First Affiliated Hospital of Harbin Medical University, Harbin City, Heilongjiang Province 150000, China. E-mail: abc7231719@163.com; abc7231719@126.com; Tel: +86-0451-85552026
eDepartment of General Practice of Jiangbei Campus, The First Affiliated Hospital of Army Medical University (The 958th Hospital of Chinese People's Liberation Army), Chongqing, 400020, China
First published on 21st November 2024
Abstract
Cardiovascular disease, intimately linked to dyslipidemia, is one of the leading global causes of mortality. Dyslipidaemia often presents as an elevated concentration of low-density lipoprotein (LDL) and a decreased concentration of high-density lipoprotein (HDL). Therefore, accurately measuring the levels of LDL and HDL particles is crucial for assessing the risk of developing cardiovascular diseases. However, conventional approaches can commonly quantify HDL/LDL particles by detecting cholesterol or protein molecules within them, which may fail to reflect the number of intact particles. In addition, these approaches are sometimes tedious and time-consuming, highlighting the need for a novel method for precise and effective identification of intact HDL and LDL particles. We have devised a technique that allows accurately and sensitively determining the levels of intact HDL and LDL in a sample without the need for isolation. This method relies on antibody-based immobilization and a self-priming hairpin-triggered Cas12a/crRNA signaling strategy. Based on the elegant design, this technique can be employed to directly and precisely measure the concentration of “actual” HDL and LDL particles, rather than the cholesterol content inside HDL and LDL. The approach has detection limits of 12.3 mg dL−1 and 5.4 mg dL−1 for HDL and LDL, respectively, and is also suitable for analyzing lipoproteins in clinical samples. Hence, this platform exhibits immense potential for clinical applications and health management.
1. Introduction
Cardiovascular disease (CVD), encompassing heart and peripheral vascular diseases, is one of the primary causes of death globally.1,2 The escalating mortality and disability rates associated with CVD are mostly attributed to the exorbitant expenses associated with therapy, along with the absence of early diagnosis techniques, imposing a growing burden on public health and the economy.3,4 Therefore, it is crucial to improve the early diagnosis efficiency of CVD by developing a simple and accurate approach for effectively preventing the illness and reducing treatment expenses.Lipoproteins are a collection of compounds composed of lipids and proteins that transport cholesterol throughout the human body.5–7 Lipoproteins, particularly elevated levels of low-density lipoprotein (LDL) and reduced levels of high-density lipoprotein (HDL), are now widely acknowledged as significant risk factors for CVD.8,9 LDL and HDL particles are the primary vehicles for transporting cholesterol in the human bloodstream and play a crucial role in the transfer and metabolism of cholesterol.6,10,11 LDL, composed of the Apolipoprotein B-100 protein (ApoB) and lipid molecules such as cholesterol and triglycerides, is referred to as “bad” lipoprotein due to its tendency to accumulate in blood vessel walls, leading to narrowing of the passageways.12,13 This increases the risk of health issues such as heart attack or stroke. In contrast, HDL, which includes the Apolipoprotein A1 protein (ApoA1) and lipids on its surface, is a beneficial lipoprotein that carries cholesterol and specific phospholipids from the body's tissues to the liver through the bloodstream, where it aids in the elimination of other more detrimental types of cholesterol from the blood.14,15 Therefore, the risk of CVD can be evaluated by quantitatively detecting the LDL and HDL particles.
Currently, there are two major methods for measuring the concentration of LDL/HDL particles.16 One method involves detecting the concentration of cholesterol in LDL/HDL particles using a two-step protocol. In brief, the LDL and HDL particles are initially separated using specific substances like poly(ethylene glycol). Then, enzymes like cholesterol esterase, cholesterol oxidase, and peroxidase are introduced to measure the cholesterol concentration in the LDL/HDL particles (e.g., the Abell–Levy–Brodie–Kendall (ALBK) method17 or the cholesterol oxidase/p-aminophenazone (CHOD–PAP) assay18). This method has been fully integrated into automated analyzers and is extensively utilized in clinical applications to measure the cholesterol levels of LDL/HDL. An alternative method involves identifying the levels of ApoA1/ApoB, which indicate the levels of HDL/LDL concentrations. The enzyme-linked immunosorbent assay (ELISA) is a widely used method for measuring the levels of ApoA1/ApoB. As an illustration, HDL/LDL particles are typically mixed with biotin-conjugated ApoA1/ApoB antibodies and avidin-conjugated horseradish peroxidase (HRP), respectively. By following the prescribed procedure and adding 3,3′,5,5′-tetramethylbenzidine (TMB) solution, the quantity of HDL/LDL can be determined by measuring the absorbance at 450 nm. Both approaches indirectly measure the levels of HDL/LDL by identifying the cholesterol or proteins in HDL/LDL particles. These methods are sometimes tedious and time-consuming. Crucially, lipoproteins consist of both lipids and proteins, and the specific makeup of each lipoprotein differs. Consequently, there is a high demand for a precise and efficient approach for quantifying intact HDL/LDL particles, as opposed to just detecting cholesterol or lipoproteins, as it provides a more reliable assessment of CVD risks.
CRISPR/Cas systems, including CRISPR/Cas9, CRISPR/Cas12a, and CRISPR/Cas13a, have demonstrated a wide range of potential applications in the development of biosensors.19–21 This is due to their ability to accurately recognize genes and efficiently amplify signals. CRISPR/Cas12a systems can precisely identify DNA molecules and then trigger a trans-cleavage reaction, achieving a minimum of 104 turnovers per recognized DNA. However, the application of CRISPR/Cas systems in developing LDL detection approaches is rarely reported. Herein, we propose a novel platform for sensitive and accurate LDL or HDL detection by integrating the antibody-based first identification of LDL or HDL and cholesterol-binding protein-based secondary target check, coupled with self-priming initiated CRISPR-Cas12a assisted signal amplification. By the dual-check of the surface protein of LDL or HDL and the cholesterol molecule, the proposed method offers high selectivity for intact LDL or HDL particles. In addition, the self-priming initiated CRISPR-Cas12a assisted signal amplification enables highly sensitive target detection with a low limit of detection. Taking the merit of high sensitivity and accuracy, the proposed approach has great potential for clinical applications and health management.
2. Experimental section
2.1 Materials and reagents
The antibodies used in this research, including the anti-ApoA1 rabbit polyclonal antibody and the anti-ApoB rabbit polyclonal antibody (Ab), were purchased from Thermo Fisher Technology (China) Co., Ltd (Beijing, China). All oligonucleotides for the signal amplification process were purchased from Shanghai Sangon Biotechnology Co., Ltd (Shanghai, China). The Klenow fragment (3′-5′ exo-) DNA polymerase, Nb·BbvCI, 10× NEBuffer™ 2 (containing 100 mM Tris-HCl, 500 mM NaCl, 100 mM MgCl2, and 10 mM DTT, pH 7.9), and 10× CutSmart® buffer (containing 200 mM Tris-Ac, 500 mM KAc, 100 mM Mg(Ac)2, and 1 mg ml−1 BSA, pH 7.9) were acquired from New England Biolabs Inc. (Beverly, MA, USA). Furthermore, the fluorescence spectra were captured using a Hitachi fluorospectrophotometer. All solutions were prepared using Milli-Q integral purified deionized water.All experiments were performed in accordance with the “Guidelines for the ethical management of biological samples in clinical trials” (no. 2022CQSFLZXYYEC-007) and were approved by the ethics committee at “Chongqing University FuLing Hospital”. Informed consent was obtained from the human participants of this study.
2.2 Lipoprotein isolation
Human serum samples were collected from volunteers at Chongqing University Fuling Hospital. The gradient density ultracentrifugation method was employed to separate lipoprotein classes (LDL and HDL). The centrifugations were conducted at a speed of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C; the initial procedure involved placing 2 mL of a 0.195 M NaCl solution, with a density (ρ) of 1.006 g mL−1, on top of 4 mL of human serum in a centrifuge tube. After 8 h of centrifugation, the top layer included the fraction of very low-density lipoprotein (vLDL). The lowermost stratum comprising LDL, HDL, and other serum proteins was transferred to a fresh centrifuge tube and then overlaid with 2 mL of a solution containing 0.195 M NaCl and 2.44 M NaBr (with a density of 1.063 g mL−1). Following a 10-hour centrifugation, the LDL fraction was able to be extracted from the uppermost layer. The lower layer, which consisted of HDL and other serum proteins, was transferred to a separate tube containing a solution composed of 0.195 M NaCl and 7.65 M NaBr (with a density of 1.478 g mL−1) in a volume of 2 mL. Following 16 hours of mixing and centrifugation, the uppermost layer containing high-density lipoprotein (HDL) was collected.
000 rpm at 4 °C; the initial procedure involved placing 2 mL of a 0.195 M NaCl solution, with a density (ρ) of 1.006 g mL−1, on top of 4 mL of human serum in a centrifuge tube. After 8 h of centrifugation, the top layer included the fraction of very low-density lipoprotein (vLDL). The lowermost stratum comprising LDL, HDL, and other serum proteins was transferred to a fresh centrifuge tube and then overlaid with 2 mL of a solution containing 0.195 M NaCl and 2.44 M NaBr (with a density of 1.063 g mL−1). Following a 10-hour centrifugation, the LDL fraction was able to be extracted from the uppermost layer. The lower layer, which consisted of HDL and other serum proteins, was transferred to a separate tube containing a solution composed of 0.195 M NaCl and 7.65 M NaBr (with a density of 1.478 g mL−1) in a volume of 2 mL. Following 16 hours of mixing and centrifugation, the uppermost layer containing high-density lipoprotein (HDL) was collected.
2.3 Preparation of the “a” sequence with the cholesterol-binding protein
The conjugation of the “a” sequence with the cholesterol-binding protein relies on the robust interaction between streptavidin and biotin. Initially, 100 μL of cholesterol-binding protein (1 mg mL−1) was mixed with Sulfo-NHS-LC-Biotin (20 nmol) and the mixture was incubated for 2 h at room temperature. The mixture was then ultrafiltered using 30 kDa centrifugal filter units 3 times. Subsequently, 100 μL of streptavidin (1 mg mL−1) was added to the mixture, and the mixture was agitated for 1 h. 5 μL of a solution containing 0.1 mg mL−1 of biotin-conjugated cholesterol-binding protein was then added to the mixture and incubated at room temperature for 2 h. Afterwards, the mixtures were purified using centrifugal filter units 3 times.2.4 Procedures for LDL detection
Before the assay, 1 μM H1 probe and 1 μM H2 probe in 0.5× NEBuffer™ 2 and 0.5× CutSmart® buffer were subjected to heating at 95 °C for 5 min and then slowly cooled to 25 °C (at a rate of 0.1 °C per second) to create a stable hairpin structure independently. The various concentrations of the LDL solution were diluted to 50 μL and were added to the ApoB antibody on a plate at room temperature for 45 min. The liquid portion above the sediment was subsequently extracted, resulting in the formation of LDL-ApoB-antibody@plate. Next, 10 μL of the complex of “a”-cholesterol-binding protein was added to the plate and incubated at room temperature for 45 min. The liquid supernatant was subsequently extracted, resulting in “a”-LDL-ApoB-antibody@plate. The assay solution was generated by combining 50 μL of solution containing 500 nM H1 probe, 500 nM H2 probe, 250 μM dNTPs, 1 U μL−1 DNA polymerase, 0.375 U μL−1 Nb·BbvCI, 0.5× NEBuffer™ 2, and 0.5× CutSmart® buffer. The solution was then incubated at 37 °C for 30 min. After incubation, 2 μL of Cas12a and 2 μL of the crRNA sequence were added. Following a 20 min incubation at 25 °C, the fluorescence emission spectra were recorded. The first spectrum was recorded in the range of 500–600 nm with an excitation wavelength of 485 nm for FAM. The second spectrum was obtained in the range of 550–650 nm with an excitation wavelength of 534 nm for Cy3.3. Results and discussion
3.1 The working mechanism of the proposed method for LDL or HDL detection
The operational mechanism of the method, utilizing LDL as the detection target, is depicted in Scheme 1. To detect LDL, the ApoB antibody is immobilized on the plate surface, creating ApoB-antibody@plate. The cholesterol-binding protein is then connected to the “a” sequence (a-cholesterol-binding-protein), which initiates the signal amplification process. When LDL is present, the ApoB-antibody attaches to the ApoB protein on the surface of LDL, leading to the fixation of the LDL to the surface of the plate (LDL-ApoB-antibody@plate). After washing 3 times, the “a”-cholesterol-binding-protein is introduced onto the plate. The cholesterol-binding protein interacts with the cholesterol present on the LDL particles, forming the “a”-cholesterol-LDL-ApoB-antibody@plate complex. | ||
| Scheme 1 The working principle of the proposed approach for LDL particle detection. | ||
Following three rounds of washing, the H1 probe, H2 probe, Cas12a/crRNA, DNA polymerase, and Nb·BbvCI are introduced onto the plate, commencing the process of self-priming-triggered CRISPR-Cas12a-assisted signal amplification. Specifically, the “a” sequence forms a bond with the “a*” region of the H1 probe, unfolding the H probe (Fig. S1†). Thus, the “c” segment binds with the “c*” segment of the H probe, resulting in the formation of a self-primer. With the assistance of DNA polymerase, the “c” portion is elongated using the “a”, “d”, and “e” sections as a template. Consequently, a double-stranded DNA is created, and the “a”-cholesterol-binding-protein is released to unfold a subsequent H1 probe. The enzyme Nb·BbvCI specifically identifies and cleaves the “d*” region, resulting in the creation of a nicking site. By cooperation of DNA polymerase and Nb·BbvCI, a large number of “e*” sequences are produced. The “e*” sequences trigger a subsequent self-priming step by unfolding the H2 probe. The “f” portion in the H2 probe comprises the non-target strand (NTS) of LbCas12a, together with the 5′-TTTA-3′ protospacer adjacent motif (PAM). Following the activation of the “e*” self-priming signal amplification, the hairpin structure (c–c*–e–f) undergoes further extension to become a complete hairpin structure (f*–e*–c–c*–e–f). Subsequently, the recently produced hairpin, which includes the PAM and the Target Strand/Non-Target Strand (TS/NTS) double helix, can activate the LbCas12a/crRNA complex, resulting in the fragmentation of FQ reporters.
3.2 Preparation of ApoB-antibody@plate and its feasibility for LDL identification
The immobilization of the ApoB antibody on the plate was confirmed using a fluorescence assay, in which the ApoB antibody was tagged with a fluorescent moiety. The schematic illustration of the fluorescence assay is shown in Fig. 1A. The fluorescence fluctuations of the ApoB-antibody@plate with various concentrations and duration combinations were observed and are depicted in Fig. 1B. As the ApoB antibody concentrations increased from 0 to 1.2 μg mL−1, the signals also increased and eventually achieved a state of equilibrium. After 30 min of incubation, the signal enhancement reached its highest level, which was determined to be the optimal incubation time.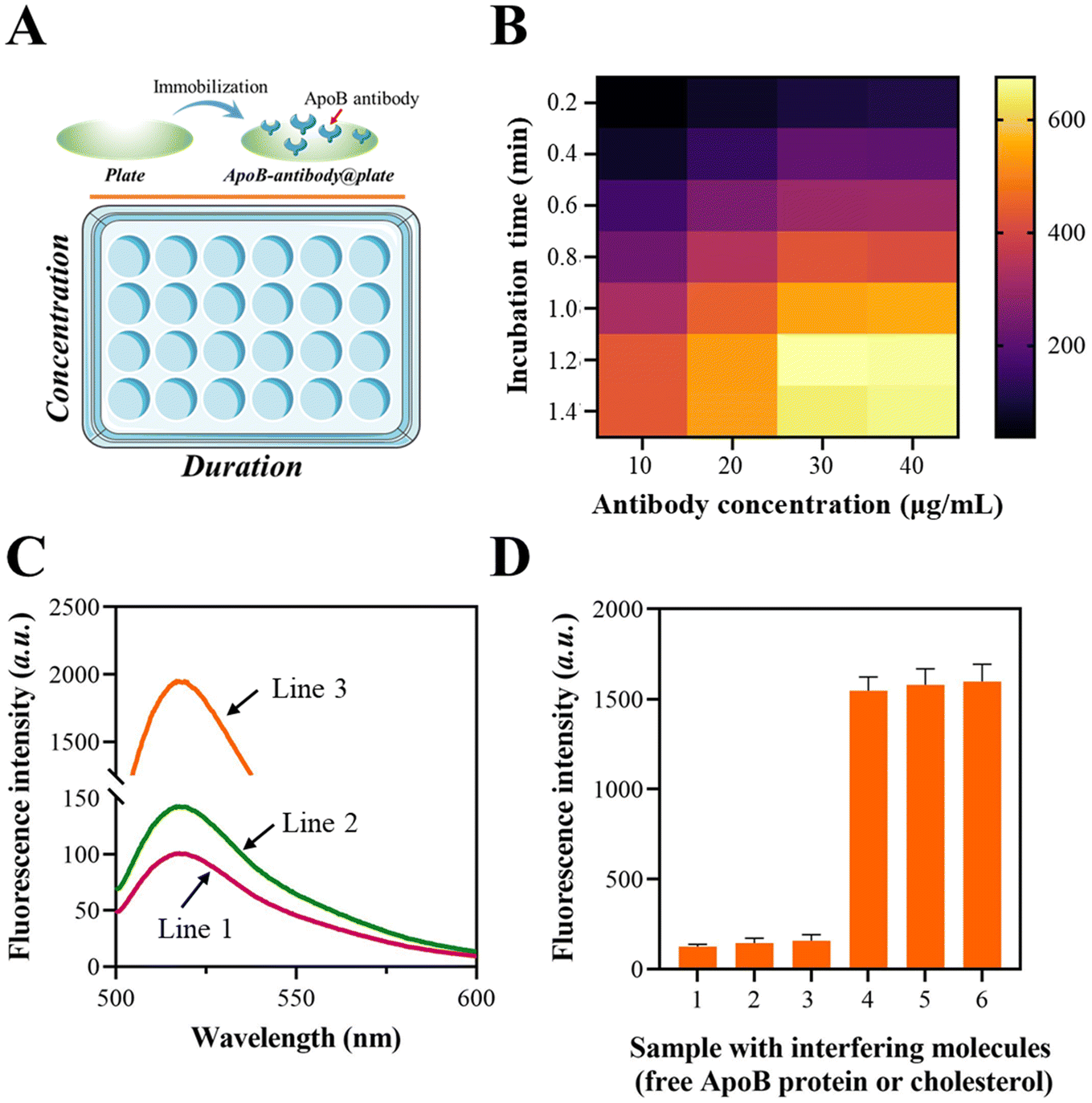 | ||
| Fig. 1 Preparation of ApoB-antibody@plate and LDL identification. (A) Schematic illustration of the immobilization of the ApoB antibody. (B) Heat map of ApoB antibody@plate. (C) Fluorescence spectrum of the FAM-labeled “a”-cholesterol-binding-protein. Line 1: ApoB antibody@plate; line 2: ApoB antibody@plate + “a”-cholesterol-binding protein; line 3: ApoB antibody@plate + “a”-cholesterol-binding protein + LDL. (D) Fluorescence intensity of LDL immobilization in the presence or absence of interfering molecules. 1, blank control; 2, ApoB protein; 3, cholesterol molecule; 4, LDL; 5, LDL + ApoB protein; 6, LDL + ApoB protein + cholesterol molecule. | ||
The FAM moiety is labeled at the terminus of the “a” sequence to validate the LDL-mediated fixation of the “a”-cholesterol-binding-protein. According to the data presented in Fig. 1C, the fluorescence intensities at 520 nm significantly increased when the LDL was pre-treated with ApoB-antibody@plate. This suggests that the intact LDL is capable of facilitating the binding of the “a”-cholesterol-binding-protein to the plate.
To characterize the selectivity of the platform for intact LDL, the free ApoB protein or cholesterol molecule was added to ApoB-antibody@plate, followed by mixing with the “a”-cholesterol-binding-protein after washing. The result in Fig. 1D showed that both the ApoB protein and cholesterol molecule could induce negligible elevation in fluorescence signals. The above results demonstrated that the established platform could only be applied for the fixation of intact LDL particles.
3.3 Feasibility of signaling mode
The viability of the self-priming triggered signal amplification was subsequently confirmed. For this experiment, the H1 probe was labeled with FAM at one end and BHQ at the other end, while the H2 probes were tagged with Cy3 at one end and BHQ at the other end. Fig. 2A illustrates the variations in the fluorescence signals of the H1 probe and H2 probe prior to and after their assembly into a hairpin form.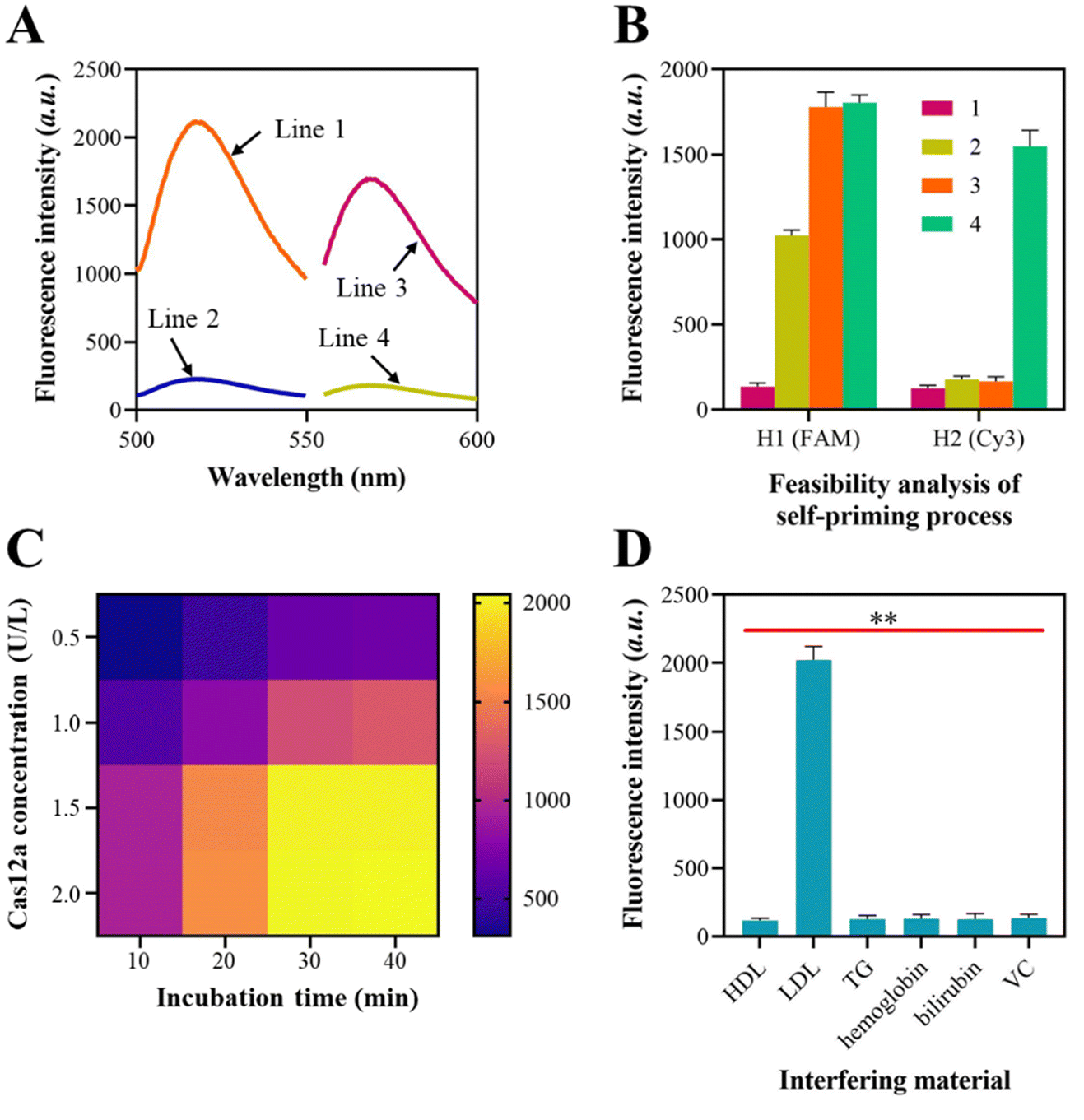 | ||
| Fig. 2 Feasibility analysis of the signaling mode. (A) Fluorescence spectra of the H1 probe and H2 probe before and after assembly. Line 1: FAM-labeled H1 probe before assembly; line 2: FAM-labeled H1 probe after assembly; line 3: Cy3-labeled H2 probe before assembly; line 4: Cy3-labeled H2 probe after assembly. (B) Fluorescence intensity of the FAM-labeled H1 and Cy3-labeled H2 probes during the self-priming process. 1, probe alone; 2, probe + “a”; 3, probe + “a” + polymerase; 4, probe + “a” + polymerase + Nb·BbvCI. (C) Heat map of the approach with different Cas12a protein concentrations and incubation times. (D) Fluorescence intensity of the approach in the presence or absence of clinical interfering materials. **, P < 0.05. | ||
In order to evaluate the recycling of the “a” sequence, a mixture containing 500 nM of FAM-tagged H1 probe and 5 nM of the “a” sequence was prepared. The fluorescence signals of the H1 probe in Fig. 2B increased when mixed with the “a” sequence, suggesting that the “a” sequence caused the unfolding of the H1 probe. The fluorescence signal was further amplified when the DNA polymerase was introduced into the mixture, suggesting that the addition of the polymerase caused the recycling of “a” sequences and the unfolding of more H1 probes by chain elongation. When the H2 probe was added, there were no notable improvements in the Cy3 signals. This suggests that the H2 probe was in a hairpin structure and did not produce any “e*” sequences. Upon the addition of Nb·BbvCI to the mixture, the intensity of the Cy3 signals significantly amplified, indicating the production of the “e*” sequences and confirming the viability of the self-priming process based on the H2 probe. The gel electrophoresis results also demonstrated the “a” sequence-mediated self-priming and DNA polymerase-assisted chain extension process (Fig. S2†).
The utilization of CRISPR-Cas12a to perform trans-cleavage of the FQ reporters was also examined using a synthetic dsDNA duplex that contained “f” and “f*”. The data presented in Fig. 2C demonstrate that the Cas12a/crRNA system achieves efficient cleavage of FQ reporters at a concentration of 1.5 U L−1, with an incubation time of 30 min.
Subsequently, we evaluated the resistance of the established approach to interference during lipoprotein detection. In clinical settings, the identification of HDL and LDL by traditional methods is often hindered by triglycerides (TG), hemoglobin, bilirubin, and vitamin C (VC) in human serum. Hence, we investigated the potential influence of TG, hemoglobin, bilirubin, and VC on the LDL detection performance of the approach. In addition, the possible inter-interferences between HDL and LDL during the detection process are verified since HDL and LDL have similar structures. We utilized HDL, LDL, TG, hemoglobin, bilirubin, and VC, with concentrations of 0.4 mg mL−1, 1.5 mg mL−1, 1.5 mg mL−1, 160 mg mL−1, 10 mg L−1, and 10 mg L−1, respectively, in healthy human serum as interferents to detect HDL and LDL utilizing the approach. The fluorescence spectroscopy results indicated that there was no significant change in the fluorescence intensity when the interferents were added during the detection of HDL and LDL (Fig. 2D). This suggests that the substances mentioned above do not interfere with the method's response. Furthermore, the detection of HDL and LDL will not be mutually affected, despite their similar structures.
3.4 Sensitivity of the approach for LDL and HDL detection
To enhance the efficacy of the self-priming assisted Cas12a/crRNA-based signal amplification, we conducted the optimization of essential factors including reaction time, H1 and H2 probe concentrations, and the quantity of FQ reporters. By extending the incubation duration, the chain elongation increases, leading to a higher fluorescence intensity over a longer period of incubation. Using a spectrophotometer, we determined that the fluorescence intensity exhibited a rapid increase within 1 h, followed by a subsequent deceleration (Fig. S3†). The concentrations of H1 and H2 probes were optimized at 500 nM each (Fig. S4†). The optimal concentration of the FQ reporters was determined to be 100 nM based on the findings presented in Fig. S5.†We assessed the sensitivity of the self-priming hairpin-triggered cascade reaction by testing LDL at different concentrations in the multiplex detection system under optimal conditions. To assess the practical diagnostic capability, we measured the LDL concentration in human serum using this method. Initially, we ascertain the concentration of LDL present in the human serum sample by employing an ELISA kit. The sample was considered the LDL standard solution that needed to be diluted to various concentrations. According to the data presented in Fig. 3A, the peak fluorescence intensities rose in direct proportion to the concentration of the target LDL (30–180 mg dL−1). Furthermore, a strong linear correlation was observed when the peak fluorescence intensities were graphed against the logarithm of the corresponding LDL concentration, with an equation of F = 25.29 × CLDL + 178.7 (Fig. 3B). The limit of detection (LOD) was determined to be 12.3 μg dL−1 based on the 3s/slope method, where s is the standard deviation of the blank. The sensitivity of these results is equivalent to or even superior to those obtained from prior isothermal amplification methods for LDL detection. This demonstrates a high level of sensitivity, particularly in the multiplex analytical system.
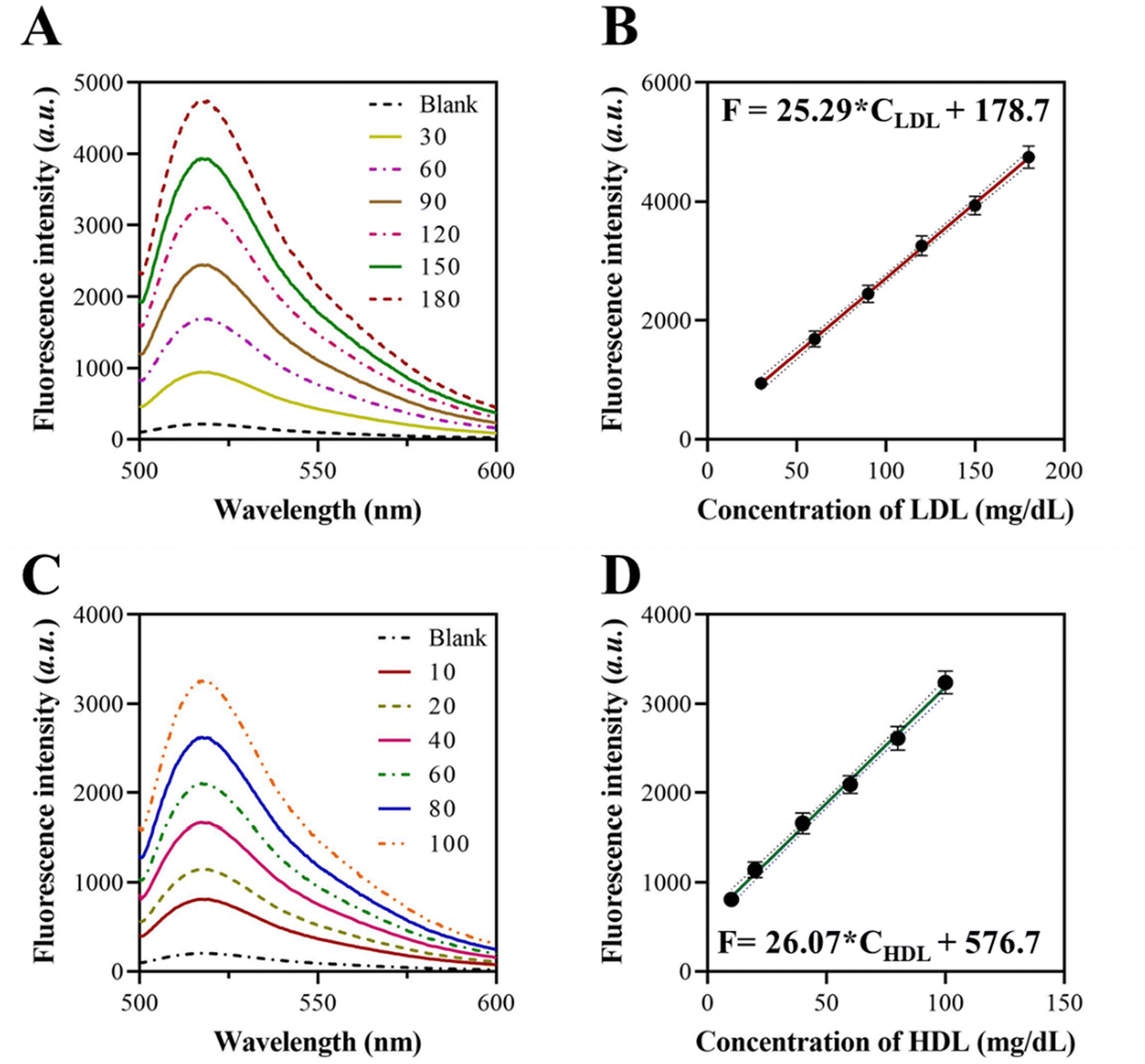 | ||
| Fig. 3 Sensitivity of the approach for LDL and HDL detection. (A) Fluorescence spectrum of the approach when detecting different concentrations of LDL. (B) Linear correlation between the recorded fluorescence intensity and the concentration of LDL. (C) Fluorescence spectrum of the approach when detecting different concentrations of HDL. (D) Linear correlation between the recorded fluorescence intensity and the concentration of HDL. | ||
We also constructed a calibration curve for HDL detection using the proposed approach. The fluorescence spectrum of the approach when detecting different concentrations of HDL was recorded, as shown in Fig. 3C. The results showed a good linear relationship with a linear regression value, R2, of 0.9926 for a concentration range of 10–100 mg dL−1 of HDL (Fig. 3D). The LOD for HDL detection was determined to be 5.4 μg dL−1 using the 3σ/S calculation method, where σ represents the standard deviation and S represents the slope of the linear plot.
3.5 Selectivity analysis
The specificity of the self-priming hairpin-induced cascade reaction was evaluated by examining its response to the target LDL and interfering molecules. When LDL was present, only LDL showed high fluorescence, but there were no fluorescence signals recorded when non-target molecules were present (Fig. 4A). Fig. 4B demonstrates a similar outcome, indicating that the approach yielded substantial signal amplification solely in the presence of HDL in the sensor device. The results demonstrated that the system exhibited a high level of specificity towards its target molecules. This confirms the exceptional specificity of the suggested method.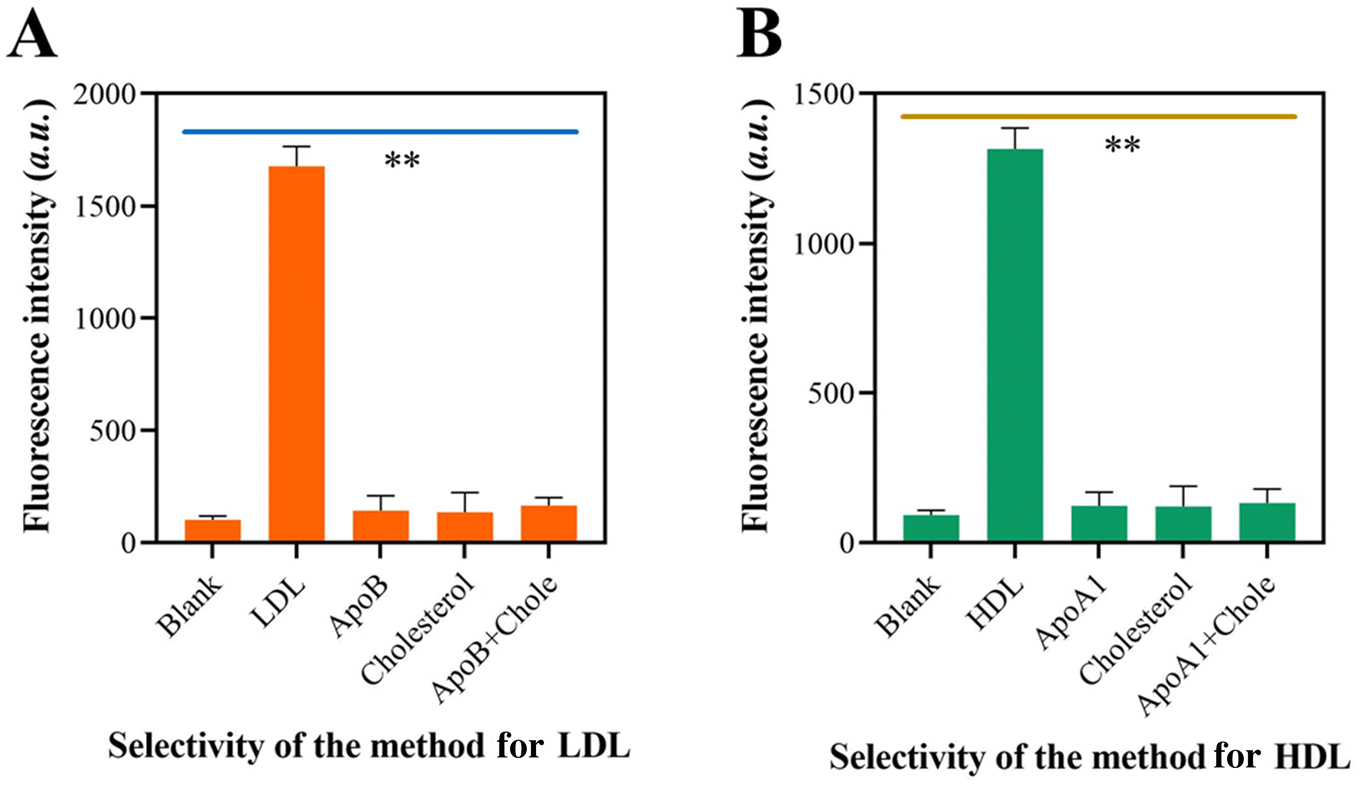 | ||
| Fig. 4 Specificity of the approach for LDL and HDL detection. (A) Fluorescence intensity of the approach when detecting LDL and interfering materials. (B) Fluorescence intensity of the approach when detecting HDL and interfering materials. | ||
3.6 Repeatability and stability of the approach for LDL and HDL detection
The repeatability of the technique for detecting LDL and HDL was assessed by detecting 10 duplicate samples. The outcome depicted in Fig. 5A demonstrates that the method exhibits a coefficient of variation (CV) of 4.21% for LDL detection and a CV of 3.76% for HDL detection. The low CV observed in the strategy for detecting LDL and HDL indicates a good level of repeatability in the method. In addition, the reliability of the method was demonstrated by successfully detecting the target molecule over 5 days. The results depicted in Fig. 5B demonstrated that the CV values for the 5-day detection of LDL and HDL were 4.12% and 3.65%, respectively. These findings indicate the exceptional stability of the technique. Compared with the former proposed LDL/HDL detection methods, the proposed approach exhibited higher specificity and a lower LOD, indicating its promising potential in clinical applications (Table S2†).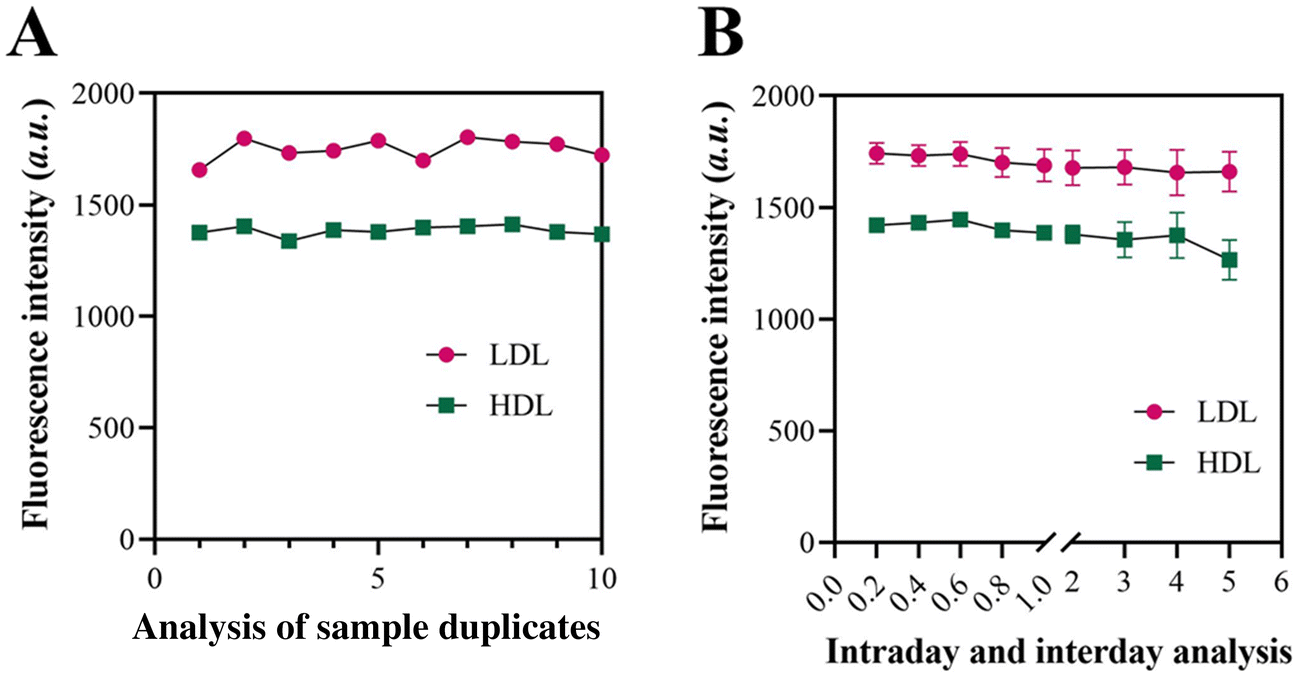 | ||
| Fig. 5 Application potential of the approach for LDL and HDL detection. (A) Fluorescence intensity of the approach when detecting LDL and HDL from 10 sample duplicates. (B) Fluorescence intensity of the approach when detecting LDL and HDL intraday and interday. | ||
4. Conclusion
We have created a precise and sensitive approach for detecting HDL/LDL by combining the immobilization of target molecules and a signaling mechanism based on self-priming hairpin-triggered Cas12a/crRNA. In contrast to current procedures that focus on detecting cholesterol or single marker proteins on HDL or LDL, our work involves the dual recognition of target molecules that target both cholesterol and surface proteins (ApoB for LDL; ApoA1 for HDL). This innovative design allows for the specific detection of the actual HDL and LDL particles. The sensitivity is ensured by utilizing the “a” sequence attached to the cholesterol-binding protein, which triggers a signal amplification process when ApoA1/ApoB and cholesterol are present on the same HDL or LDL particle. This amplification leads to the enhancement of the FQ signal through the trans-cleavage of Cas12a/crRNA. This approach is employed to identify the actual HDL and LDL particles, rather than just one molecule of their components, such as AopA1 and ApoB. With these exceptional characteristics, the platform would offer numerous valuable advantages. The platform would function as an alternative method to the standard lipoprotein detection technique, addressing the issue of low specificity. Furthermore, the platform's isothermal tendency makes it highly suitable for diagnostics in developing countries, since it eliminates the need for complex hardware dependencies. Ultimately, this suggested that uniform tests can improve the identification of lipoprotein particles in clinical settings, which could aid in demonstrating their predictive capabilities in assessing the risk of associated disorders.Author contributions
G. A. and X. Z.: supervision, data curation, funding acquisition, conceptualization, methodology, and reviewing the original draft. X. L., H. P., and L. G.: software, investigation, methodology, and writing the original manuscript. H. Z., C. Z., and W. L.: methodology and formal analysis.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (No. 82202645), the Scientific Research and Innovation Fund of the First Affiliated Hospital of Harbin Medical University (No: 2021B01), and the HeiLongJiang Province Postdoctoral Research Launch Fund (No: LBH-Q21114).References
- A. Garcia-Rios and J. M. Ordovas, Chronodisruption and cardiovascular disease, Clin. Investig. Arterioscler., 2022, 34(Suppl 1), S32–S37 Search PubMed.
- K. Gazzola, L. Reeskamp and B. J. van den Born, Ethnicity, lipids and cardiovascular disease, Curr. Opin. Lipidol., 2017, 28(3), 225–230 CrossRef CAS PubMed.
- M. A. Fischer and T. M. Vondriska, Clinical epigenomics for cardiovascular disease: Diagnostics and therapies, J. Mol. Cell. Cardiol., 2021, 154, 97–105 CrossRef CAS PubMed.
- S. J. Kim, F. C. P. Mesquita and C. Hochman-Mendez, New Biomarkers for Cardiovascular Disease, Tex. Heart Inst. J., 2023, 50(5), e238178 Search PubMed.
- G. H. Dahlen, Lipoprotein(a), atherosclerosis and thrombosis, Prog. Lipid Res., 1991, 30(2–3), 189–194 CrossRef CAS PubMed.
- D. R. Illingworth, Lipoprotein metabolism, Am. J. Kidney Dis., 1993, 22(1), 90–97 CrossRef CAS PubMed.
- A. M. Scanu, R. M. Lawn and K. Berg, Lipoprotein(a) and atherosclerosis, Ann. Intern. Med., 1991, 115(3), 209–218 CrossRef CAS PubMed.
- P. R. Kamstrup, Lipoprotein(a) and Cardiovascular Disease, Clin. Chem., 2021, 67(1), 154–166 CrossRef PubMed.
- R. M. Krauss and S. M. King, Remnant lipoprotein particles and cardiovascular disease risk, Best Pract. Res., Clin. Endocrinol. Metab., 2023, 37(3), 101682 CrossRef CAS PubMed.
- L. Di and A. Maiseyeu, Low-density lipoprotein nanomedicines: mechanisms of targeting, biology, and theranostic potential, Drug Delivery, 2021, 28(1), 408–421 CrossRef CAS PubMed.
- S. M. Marcovina, Lipoprotein(a): a genetically determined risk factor for Cardiovascular disease, Crit. Rev. Clin. Lab. Sci., 2023, 60(8), 560–572 CrossRef CAS PubMed.
- A. Cesaro, A. Schiavo, E. Moscarella, S. Coletta, M. Conte, F. Gragnano, F. Fimiani, E. Monda, M. Caiazza, G. Limongelli, L. D'Erasmo, C. Riccio, M. Arca and P. Calabro, Lipoprotein(a): a genetic marker for cardiovascular disease and target for emerging therapies, J. Cardiovasc. Med., 2021, 22(3), 151–161 CrossRef CAS PubMed.
- F. Gomez-Delgado, N. Katsiki, J. Lopez-Miranda and P. Perez-Martinez, Dietary habits, lipoprotein metabolism and cardiovascular disease: From individual foods to dietary patterns, Crit. Rev. Food Sci. Nutr., 2021, 61(10), 1651–1669 CrossRef PubMed.
- W. Y. Ding, M. B. Protty, I. G. Davies and G. Y. H. Lip, Relationship between lipoproteins, thrombosis, and atrial fibrillation, Cardiovasc. Res., 2022, 118(3), 716–731 CrossRef CAS PubMed.
- P. J. Nestel, High-density lipoprotein turnover, Am. Heart J., 1987, 113(2 Pt 2), 518–521 CrossRef CAS PubMed.
- S. Mundi, M. Massaro, E. Scoditti, M. A. Carluccio, V. W. M. van Hinsbergh, M. L. Iruela-Arispe and R. De Caterina, Endothelial permeability, LDL deposition, and cardiovascular risk factors-a review, Cardiovasc. Res., 2018, 114(1), 35–52 CrossRef CAS PubMed.
- N. Di Pietro, G. Formoso and A. Pandolfi, Physiology and pathophysiology of oxLDL uptake by vascular wall cells in atherosclerosis, Vasc. Pharmacol., 2016, 84, 1–7 CrossRef CAS PubMed.
- C. E. Kosmas, I. Martinez, A. Sourlas, K. V. Bouza, F. N. Campos, V. Torres, P. D. Montan and E. Guzman, High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease, Drugs Context, 2018, 7, 212525 Search PubMed.
- R. Wang, X. Zhao, X. Chen, X. Qiu, G. Qing, H. Zhang, L. Zhang, X. Hu, Z. He, D. Zhong, Y. Wang and Y. Luo, Rolling Circular Amplification (RCA)-Assisted CRISPR/Cas9 Cleavage (RACE) for Highly Specific Detection of Multiple Extracellular Vesicle MicroRNAs, Anal. Chem., 2020, 92(2), 2176–2185 CrossRef CAS PubMed.
- G. Zhang, J. Tong, X. Zhao and J. Ren, CRISPR-Cas12a enhanced rolling circle amplification method for ultrasensitive miRNA detection, Microchem. J., 2020, 158(2020), 105239 CrossRef CAS.
- X. Zhao, L. Zeng, Q. Mei and Y. Luo, Allosteric Probe-Initiated Wash-Free Method for Sensitive Extracellular Vesicle Detection through Dual Cycle-Assisted CRISPR-Cas12a, ACS Sens., 2020, 5(7), 2239–2246 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01167h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |