
Deamidation analysis of therapeutic drugs using matrix-assisted laser desorption ionization mass spectrometry and a novel algorithm QuanDA†
Han
Zhang‡
a,
Yinran
Xiong‡
bc,
Xiaonan
Shi
a,
Lijia
Zhu
a,
Qiong
Wu
a,
Ting
Wu
 *a and
Yiping
Du
a
*a and
Yiping
Du
a
aSchool of Chemistry and Molecular Engineering and Research Centre of Analysis and Test, East China University of Science and Technology, Shanghai, 200237, P. R. China. E-mail: wu_ting@ecust.edu.cn
bIntegrative Science Center of Germplasm Creation in Western China (CHONGQING) Science City, Bio-logical Science Research Center, Southwest University, Chongqing, 400715, China
cChongqing Municipal Key Laboratory of Scientific Utilization of Tobacco Resources, Chongqing, 400060, China
First published on 7th November 2024
Abstract
A robust deamidation quantification method, called QuanDA, was developed to quantify the spontaneous nonenzymatic deamidation of peptides based on the isotopic distribution change of peptides in matrix-assisted laser desorption ionization (MALDI) mass spectra and non-negative least squares calculation. The predictive model of QuanDA using theoretical spectra of pure un-deamidated and deamidated peptides for a series of simulated partial deamidated peptides is satisfying, with a coefficient of determination (R2) and root mean squared error (RMSE) of 0.9914 and 0.03356, respectively. It was applicable in cases where there is a lack of reference standards of un-deamidated and deamidated peptides. The only requirements were the chemical formulae of un-deamidated and deamidated peptides for isotopic pattern calculation. QuanDA provided a rapid, low-cost and easily accessible method for deamidation analysis in therapeutic drugs.
1. Introduction
Nonenzymatic deamidation of asparagine (Asn) and glutamine (Gln) is one of the best known and studied posttranslational modifications in proteins and peptides.1–3 But the deamidation of Asn has been paid more attention than Gln,4,5 as the half-life of deamidation at the Gln site (100 ∼ 5000 days at 37 °C) is much slower than that at Asn (1 ∼ 500 days at 37 °C).1,6,7 For therapeutic protein/peptide drugs, the degree of deamidation often controls the biological activity and functions of proteins8–10 and influences the metabolic kinetics of proteins. Some regulations have stipulated the limit of deamidation for therapeutical drugs, for example, maximum 5% and 6.5% of deamidated forms are limited for somatropin concentrated solution and somatropin for injection in European Pharmacopoeia 7.0.11 Quantitative analysis of deamidation is needed to monitor the quality attributes of drugs and understand how deamidation contributes to the loss of protein activity during the production and storage of biopharmaceuticals.The deamidation mechanism of Asn undergoes two paths. Under basic conditions, nonenzymatic deamidation of Asn is a two-step reaction process of forming a five-membered ring aminosuccinimidyl (Asu), and then hydrolyzing the cyclic amido bond. The hydrolyzation of the cyclic amido bond can occur at any end of the Asu and form aspartic acid (Asp) or β-Asp (iso-Asp) residues in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio. Under acidic conditions, deamidation is catalyzed by acid, and Asp is directly formed (Fig. 1).10,12 The deamidation rate of Asn is affected by the amino acid sequence and structure of protein, temperature, pH, time and so on. Deamidation conditions are particularly favored in sequences where Asn precedes glycine (Gly), serine (Ser) and threonine (Thr).13 It is associated with the flexibility in the residue connected C-terminal of Asn.14 High temperature and basic pH always promoted the progress of deamidation of peptides.15
:3 ratio. Under acidic conditions, deamidation is catalyzed by acid, and Asp is directly formed (Fig. 1).10,12 The deamidation rate of Asn is affected by the amino acid sequence and structure of protein, temperature, pH, time and so on. Deamidation conditions are particularly favored in sequences where Asn precedes glycine (Gly), serine (Ser) and threonine (Thr).13 It is associated with the flexibility in the residue connected C-terminal of Asn.14 High temperature and basic pH always promoted the progress of deamidation of peptides.15
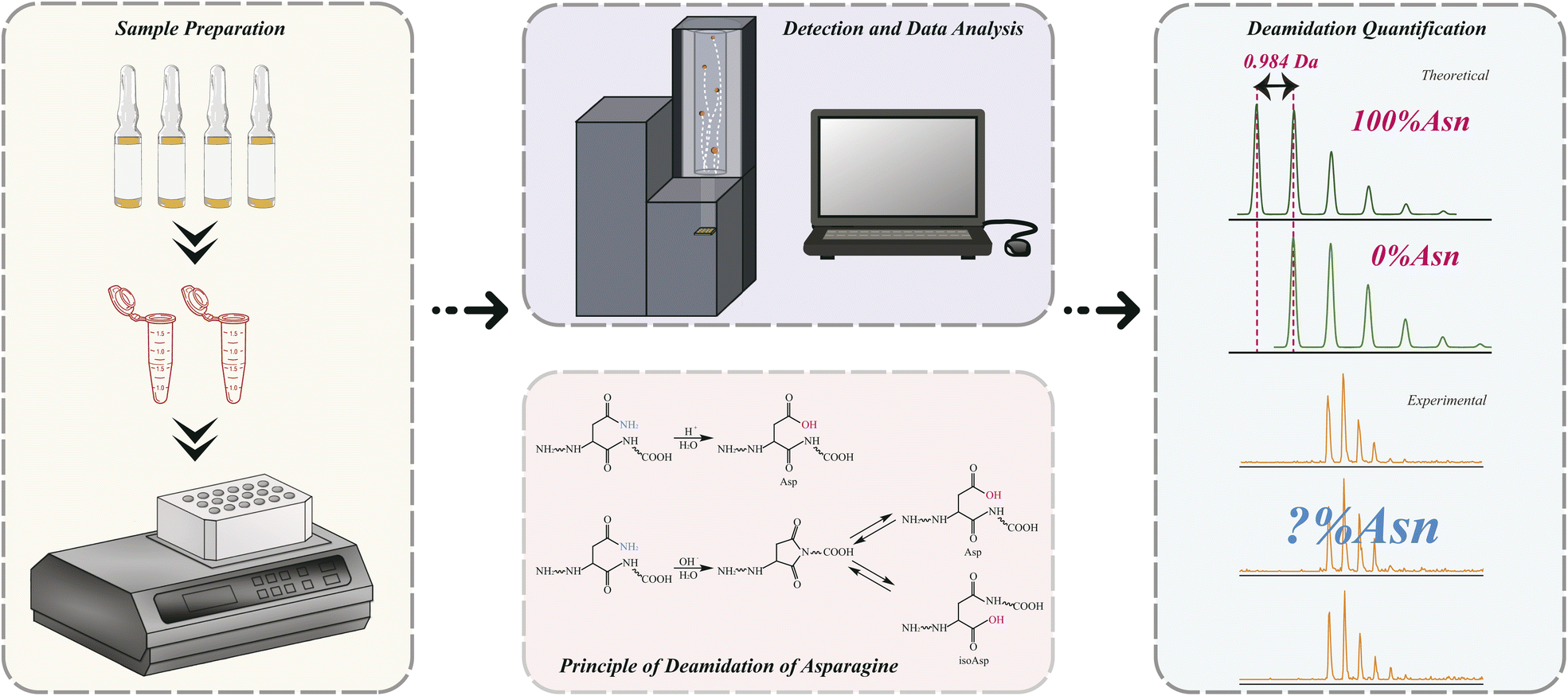 | ||
| Fig. 1 Workflow of QuanDA for deamidation quantification of a given peptide. | ||
Many methods are available to detect and quantify deamidation of Asn residues.13 Capillary electrophoresis (CE) techniques, including capillary zone electrophoresis (CZE)16 and isoelectric focusing (IEF), separate deamidation products according to the charge heterogeneity and isoelectric point.17 High-performance liquid chromatography (HPLC) stands as a highly effective technique for assessing protein purity and precise separation of proteins and their deamidated degradation products.18,19 However, the limitations of conventional chromatographic methods are low sensitivity, specificity, and time-consuming procedures. Sara Carillo et al. employed CE-ESI-MS to analyze numerous monoclonal antibodies, reliably identifying protein variants due to deamidation.20 Liquid chromatography-mass spectrometry (LC-MS), as a powerful analytical method, has become the main analytical method for in-depth characterization and analysis of amino acid sequences, and also the modification site and proportion of deamidation.21,22 Despite the high specificity of chromatography-mass spectrometry, deamidation quantification relies heavily on the chromatographic resolution of the deamidated species for quantitation.22 Now, the analysis of deamidation based on matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) has been attracting more attention due to its high sensitivity and specificity, high-throughput, and ease of operation,23–25 and has garnered significant attention in the deamidation analysis of proteins and peptides. Deamidation introduces a 0.984 Da increase in peptide mass and causes an overlapping isotopic envelope of the un-deamidated and deamidated peptides in MALDI MS spectra.26 Some cases to quantify deamidation were reported based on the centroid mass shift in MALDI spectra.7,27 Another strategy for the quantification of deamidation was based on a shift of the isotope distribution in MALDI spectra caused by deamidation, e.g. q2e is an R package that uses a genetic algorithm to determine the extent of deamidation.28,29 On the other hand, the isotope internal standard method, combining enzyme digestion by endoproteinase Asp-N and the usage of digests of 15N-labeled protein as an internal standard, was proposed for the quantification of deamidation.30 In this study, we described an algorithm called QuanDA based on the isotopic distribution shift in MALDI MS and non-negative least squares calculation to measure the deamidation of therapeutic drugs. QuanDA was much simpler than the existing methods without extra parameters and isotope internal standards, except the monoisotopic masses of un-deamidated and deamidated peptides. It will be applied for both MALDI-TOF and MALDI-FTICR mass spectrometry analysis and helpful in the quality control and characterization of critical quality attributes of therapeutic drugs.
2. Experimental
2.1. Chemicals and reagents
Custom synthesized peptides YTHQGLSSPVTKSFNRGE (Pep-N) and YTHQGLSSPVTKSFDRGE (Pep-D) were provided by Bankpeptide Biological technology Co., Ltd. (Hefei, China). Somatostatin (AGCKNFFWKTFTSC) was purchased from Beyotime (Shanghai, China). Bevacizumab used in this paper was purchased from Qilu pharmaceutical Co., Ltd., and LC-MS grade ammonium acetate and acetonitrile (ACN) were purchased from Fisher Chemical (Fair Lawn, USA). Endoprotease Glu-C was obtained from Shanghai Personal bio Technology Co., Ltd. (Shanghai, China). α-Cyano-4-hydroxycinnamic acid (CHCA) was obtained from Sigma-Aldrich (St. Louis, USA). All other chemicals were of analytical grade and commercially available. Ultrapure water was purified by a Milli-Q advantage A10 water purification system (18.2 MΩ·cm, Millipore, USA).2.2. Simulated deamidated solution preparation
Pep-N and Pep-D were dissolved in CH3COONH4 (pH = 4.5, 50 mM) to get stock solutions of 1 mg mL−1. Then, a series of simulated deamidated samples were obtained by mixing Pep-N and Pep-D at volume ratios of 20:0, 19:1, 18:2, 17:3, 16:4, 15:5, 14:6, 13:7, 12:8, 11:9, 10:10, 9:11, 8:12, 7:13, 6:14, 5:15, 4:16, 3:17, 2:18, 1:19, and 0:20, respectively, for the following MALDI-TOF MS analysis. Additionally, mixtures of Pep-N and Pep-D at 18:2 and 2:18 ratios were analyzed by LC-DAD to assess the outcomes obtained from MALDI MS and QuanDA methodology.
2.3. Aging process for deamidation
Pure Pep-N solutions (1 mg mL−1) were dissolved in different pH solutions (50 mM acetic acid/ammonium acetate at pH = 4 and 5; 50 mM NH4HCO3/NH3·H2O at pH = 7 and 8), and incubated at 37 °C for a total time of 96 h. Each solution was sampled at an interval time of 24 h (0, 24 h, 48 h, 72 h, 96 h) and analyzed by MALDI-TOF MS.Pure somatostatin solution (1 mg mL−1) was dissolved in different pH solutions: 50 mM acetic acid/ammonium acetate at pH 4 and 50 mM NH4HCO3 at pH 7.4, respectively. Subsequently, somatostatin samples in pH 4 and pH 7.4 solutions were stored at 4 °C and 37 °C, respectively, for 13 days. The degree of deamidation of each solution was monitored on the 0, 1, 3, 5, 7, 9, 11, and 13th days using MALDI-TOF MS.
2.4. Bevacizumab digestion
Bevacizumab (4 μL, 1 mg mL−1) was denaturized with 8 M urea (96 μL) and reduced with 20 mM Tris(2-carboxyethyl)phosphine (TCEP, 10 μL) at 56 °C for 45 min. After cooling down to room temperature, it was alkylated with 20 mM iodoacetamide (IAA) in the dark at room temperature for 30 min. The solution was replaced by different digestion buffer solutions using ultrafiltration three times (50 mM NH4HCO3 at pH 8; 50 mM CH3COONH4 at pH 5; 3 times, 10 kDa molecular weight cutoff). Then bevacizumab was digested with endoprotease Glu-C at 37 °C overnight (enzyme:protein = 1:50). Finally, the reaction was stopped by adding formic acid to the final concentration of 1%.
2.5. MALDI-TOF MS analysis
MALDI MS spectra were acquired on a 4800 plus MALDI-TOF mass spectrometer (AB Sciex, USA) equipped with a 355 nm Nd:YAG laser in positive reflector mode. Each spectrum was acquired by averaging spectra over 200 laser shots, and the delay time was set to 450 ns. In order to avoid the sweet spot effect caused by the molecular inhomogeneity of the matrix and the analyte, each sample was collected ten times.Samples were prepared by a layer-by-layer method on a stainless-steel plate. 1 μL sample solution was spotted first, followed by 1 μL α-cyano-4-hydroxycinnamic acid (CHCA), which was utilized as a matrix for peptide ionization.
Theoretical isotope patterns of peptides were calculated by Data Explorer (AB Sciex, USA).
2.6. LC-DAD analysis
An Ultimate 3000 UHPLC system combined with a diode array detector (DAD) was used to evaluate the quantitative results of deamidation of two simulated deamidated Pep-N samples. The separation was performed on a Waters ACQUITY UPLC BEH C18 column (2.1 mm × 50 mm, 1.7 μm). The mobile phase A was 0.1% formic acid in water, B was acetonitrile, and the flow rate was 0.25 mL min−1. The temperature of the column was kept at 45 °C, and the ultraviolet detection wavelength was set at 214 nm. The injection volume was 25 μL. The total analysis time was 10 min. The elution gradient program was started at 0.1% B for 1 min, then increased to 40% B within 8 min, and then increased to 90% within 0.5 min, remained for 0.5 min, then quickly decreased to 0.1% within 0.1 min, and kept for 1 min. The injected volume was 10 μL for a 0.1 mg mL−1 peptide mixture.2.7. LC-MS analysis
An Ultimate 3000 UHPLC system combined with a Q Exactive Plus mass spectrometer was used for deamidation site analysis. The separation was performed on a Welch Xtimate UHPLC C18 column (2.1 mm × 100 mm, 1.8 μm). The mobile phase A was 0.1% formic acid in water, B was 0.1% formic acid in acetonitrile, and the flow rate was 0.25 mL min−1. The temperature of the column was kept at 60 °C, and the injection volume was 10 μL for bevacizumab digestion (0.1 mg mL−1). The total analysis time was 19 min in ESI.† The elution gradient program was started at 0.1% B for 1 min, then increased to 90% B within 16 min, remained for 1 min, then decreased to the initial 0.1% B within 0.1 min, and kept for 1 min.The full-scan MS/data-dependent MS/MS (Full MS/ddMS2) was adopted at a resolution of 70000 for MS acquisition, and the maximum injection time was set as 200 ms. The mass range was from 500 to 3000. In MS/MS mode, a resolution was set at 17500, and the maximum injection time was set as 100 ms. The 10 most intense ions from the full scan were selected for high energy collision dissociation (HCD). The normalized collision energy (NCE) was set as 30 eV.
2.8. Deamidation site analysis
The examination of the deamidation site was performed by Proteome Discoverer 2.2. The peptide confidence at least is set as high. The mass tolerances of precursor and fragment ions were set as 10 ppm and 0.01 Da, respectively. Cysteine carbamidomethylation (C) was set as a fixed modification, oxidation on methionine (M) and deamidation (N) were set as the variable modifications. Up to two missing cleavages were allowed. A false discovery rate (FDR) of 1% was set for peptide identification. Moreover, the localization of the deamidation site was further determined by a mass increase of 0.984 Da on Asn.2.9. Calculation of deamidation by QuanDA
The whole calculation process assumes that Asn and Asp have a proximate signal response at unit concentration in MALDI MS.7,27 Therefore, the math model for the total concentration of the peptide mixture containing Asn (un-deamidated peptide) and Asp (deamidated peptide) in a sample was shown as follows:| cAsnkAsn + cAspkAsp = αx |
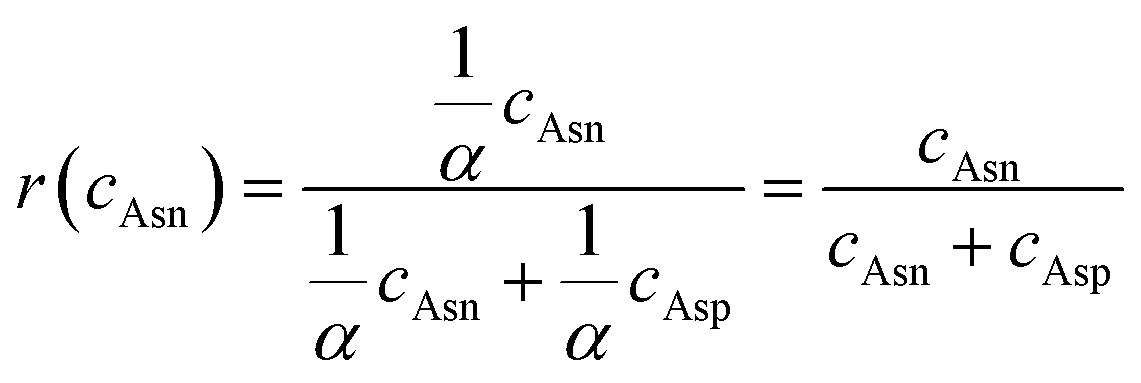
| r(cAsp) = 1 − r(cAsn) |
The quantification of deamidation in peptides was realized based on the above calculation and a corresponding algorithm called QuanDA was built under a Python environment. Ten MALDI MS spectra of each sample were acquired and treated simultaneously in QuanDA. Only the theoretical MALDI MS spectra of the un-deamidated peptide before deamidation and its corresponding deamidated peptide were required to calculate the deamidation quantitation in real samples. The python code can be downloaded at https://github.com/speakinside/QuanDA. The relevant data could be accessible at https://zenodo.org/records/13903594.
3. Results and discussion
3.1. Performance of QuanDA
The principle of QuanDA for the deamidation of asparagine is demonstrated in Fig. 1. Nonenzymatic deamidation during storage or proteolytic digestion affects the isotopic pattern in MALDI MS spectra, as m/z values for the products of this reaction are shifted by 0.984 Da compared to the original peptide. Two theoretical mass spectra of un-deamidated and deamidated peptides could be obtained from open-source or commercial programs. The overlapping isotopic pattern of the mixture of un-deamidated and deamidated peptides with different ratios could be simulated by non-negative least squares, depending on the equal mass spectrometric responses for both derivatives. Each experimental isotopic pattern with deamidation could be calculated using QuanDA.First, in order to test whether QuanDA could fulfil the requirement of deamidation calculation, a pair of synthetic peptides (Pep-N: YTHQGLSSPVTKSFNRGE and Pep-D: YTHQGLSSPVTKSFDRGE) were mixed in different ratios. The isotopic pattern observed by MALDI MS was calculated and compared with their theoretical ratios. The detailed results of each mixture are shown in Fig. 2A. The obtained correlation plot showed an excellent correlation between the theoretical deamidation and real deamidation ratios (Fig. 2B). The correlation coefficient, coefficient of determination (R2) and root mean squared error (RMSE) were 0.9956, 0.9913, and 0.04159, respectively. Considering the purity of synthesized pure Pep-N and Pep-D which represented 0% and 100% deamidation, the calculated theoretical isotopic patterns of Pep-N and Pep-D could be applied to replace the spectra of synthesized pure Pep-N and Pep-D. The results in Fig. 2C showed that the correlation coefficient, determination coefficient (R2) and root mean squared error (RMSE) were 0.9957, 0.9914, and 0.03356, respectively. Obviously, the predictive model of QuanDA is more accurate when using theoretical pure Pep-N and Pep-D spectra with a better coefficient of determination (R2) and root mean squared error (RMSE) on prediction. Moreover, the standard deviations for all predicted deamidation quantitative results from theoretical results were less than 5%. Hence, QuanDA was applicable for any deamidation analysis in cases where there is a lack of reference standards of un-deamidated and deamidated peptides. The only requirements were the chemical formulae of un-deamidated and deamidated peptides for isotopic pattern calculation. It was easily accessible for therapeutic drugs. The input files for deamidation calculation in QuanDA were .txt files of calculated theoretical MALDI spectra of the un-deamidated peptide and deamidated peptide, and experimental MALDI spectra of real samples. Ten replicated acquisitions for each peptide were recommended. Two MALDI spectra with the ratios of Pep-N:Pep-D = 18:2 and Pep-N:Pep-D = 2:18 are demonstrated in Fig. 2D, which demonstrated a distinct right shift of its isotopic distribution caused by deamidation. Meanwhile, Pep-N and Pep-D mixed at 18:2 and 2:18 were analyzed by LC-DAD to evaluate the results from MALDI MS and QuanDA. The LC-DAD spectra of Pep-N and Pep-D mixed at 18:2 and 2:18 are shown in Fig. 2E. The comparison results between LC-DAD and MALDI MS/QuanDA are shown in Table 1. The RSD between the two methods was within 5% which was satisfying for deamidation quantification.
 | ||
| Fig. 2 Predicted results of simulated deamination of Pep-N using QuanDA. (A) The comparison of the calculated percentage of Pep-N from experimental and theoretical un-deamidated Pep-N and Pep-D spectra. (B) The prediction curve of simulated deamination peptides from experimental pure Pep-N and Pep-D spectra. (C) The prediction curve of simulated deamination peptides from theoretical pure Pep-N and Pep-D spectra. (D) The MALDI spectra of Pep-N and Pep-D mixed at 18:2 and 2:18, respectively. (E) LC-DAD spectra of Pep-N and Pep-D mixed at 18:2 and 2:18, respectively. | ||
| Ratio | Contents of un-deamidated Asn (%) | RSD (%) | |
|---|---|---|---|
| MALDI MS | LC-DAD | ||
| Pep-N:Pep-D = 18:2 |
89.37 | 89.88 | 0.40 |
| Pep-N:Pep-D = 2:18 |
13.72 | 11.25 | 2.00 |
3.2. Deamidation quantification in the aging process by QuanDA
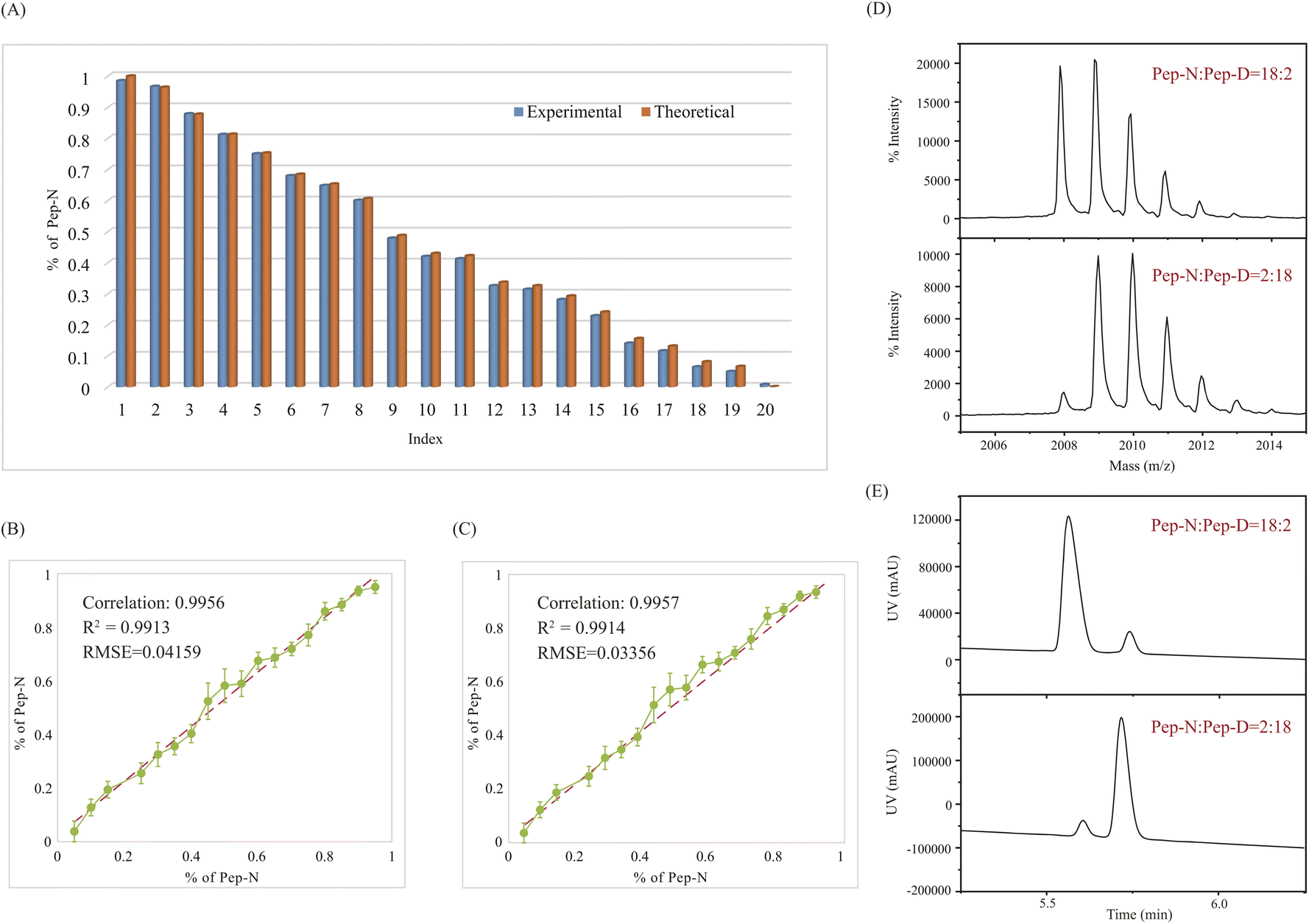
The application of QuanDA was tested in the aging process of Pep-N along with different pHs and aging times. The effect of pH and time on deamidation is particularly important to select appropriate storage conditions and reduce protein activity loss in the biopharmaceutical industry.10,17,31 Five pH conditions (pH = 4, 5, 6, 7, and 8) were tested during 4 days at room temperature. MALDI MS spectra of Pep-N aged at different pHs on the third day in Fig. 3A demonstrated the change of the isotopic pattern at different pHs. The decrease of the monoisotopic peak of Pep-N was apparently observed along with the increase of the pH value. The results shown in Fig. 3B depicted that the deamidation rate was faster under basic conditions than under acidic conditions with an ascending order of pH values. The deamidation percentage at pH 4 was less than 1% on the first day. It increased to 8.5% when aging at pH 8 for one day. The effect of time is also observed in Fig. 3B. After being aged for four days, the deamidation percentage of Pep-N increased to 3.8% at pH 4, 19.1% at pH 8. This suggested that the deamidation catalyzed by acid was much slower than the hydrolyzation of Asu under basic conditions and the storage of therapeutic drugs should be under acidic conditions for a short period of time. | ||
| Fig. 3 The deamidation of Pep-N after aging at different pHs and aging days. (A) MALDI MS spectra of Pep-N after aging at different pHs on the third day. (B) Deamidation change along with different pHs and time. | ||
3.3. Deamidation quantification in real therapeutic drugs
In addition to the synthetic peptides studied above, deamidation in real therapeutic drugs was studied under physiological conditions (pH 7.4, 37 °C) and storage conditions (pH 4, 4 °C). Somatostatin is a naturally occurring peptide hormone that regulates the endocrine system, which has one asparagine site. Somatostatin underwent deamidation following the first-order kinetic model, as shown in eqn (1):| −ln(Ct × C0−1) = k × t | (1) |
| T50 = ln2 × k−1 | (2) |
The calculated deamidation of commercial Somatostatin was 1.7%. Following the deamidation of asparagine residues in somatostatin over time, we found that deamidation has very little likelihood for 13 days at 4 °C. The RSD of the calculated concentration of somatostatin was only 3.07%. But under physiological conditions (at pH 7.4, 37 °C), deamidation started right away. The concentration of somatostatin decreased to be 68.7% on the 13th day (Fig. 4A). The rate constant at 37 °C was calculated to be 0.0296 day−1 according to the fitted kinetic model in Fig. 4B, and half-life was 23.42 days.
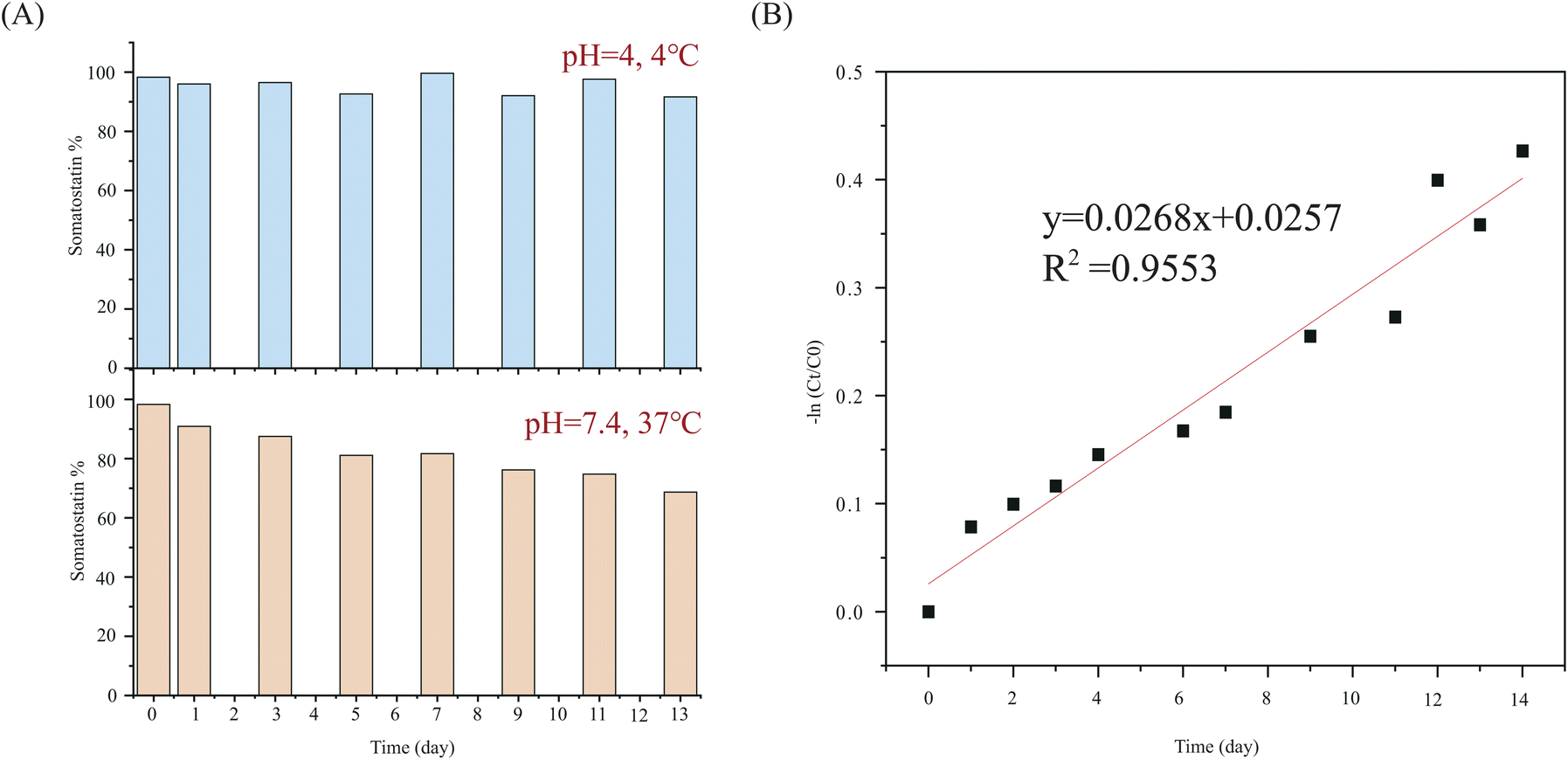 | ||
| Fig. 4 Deamidation of somatostatin under different conditions. (Based on the percentage of intact somatostatin). (A) The percentage of intact somatostatin stored at 4 °C and the percentage of intact somatostatin after deamidation under physiological conditions. (B) The degradation kinetic model of somatostatin at 37 °C. | ||
Bevacizumab is a recombinant monoclonal antibody against the human vascular endothelial growth factor (VEGF), which can be used for treatment of colorectal cancer.32,33 As a complex protein drug, monoclonal antibodies may undergo many posttranslational modifications, among which deamidation is a common posttranslational modification. The sequence of bevacizumab is known (Fig. S1†), and the molecular weight is about 149 kDa. Bevacizumab consists of 214 amino acids and contains 26 asparagine sites. The deamidation sites were analyzed using liquid chromatography-mass spectrometry (LC-MS) after digestion by Glu-C at 37 °C under pH 5 and pH 8. The deamidation results of digestions at pH 5 and pH 8 are shown in Fig. S2.† Table S1† demonstrates the peptides and sites which LC-MS has detected. Similar to the aging process of Pep-N, the deamidation of bevacizumab was intensified under basic conditions as more deamidation peptides were found at pH 8 than pH 5. The generated peptides exhibit distinct specificity based on the ionization methods employed.34 MALDI detected much fewer peptides than LC-MS because of the lack of separation. Considering the intensity and signal-to-noise ratio, we selected the light-chain peptide 196–214 of bevacizumab (VTHQGLSSPVTKSFNRGEC, Pep-N-B, [M + H]+ = 2104.0 Da with carbamidomethyl) for deamidation experiments. The corresponding deamidated peptide was VTHQGLSSPVTKSFDRGEC (Pep-D-B, [M + H]+ = 2105.0 Da). Their theoretical MALDI spectra were calculated by an isotope calculator of data explorer and are shown in Fig. 5A. The full MALDI spectra of bevacizumab after enzymolysis with Glu-C under different pH conditions (pH = 5, 8) have been shown in Fig. 5B. Actually, deamidation has occurred during the digestion under basic conditions, both the un-deamidated peptide and deamidated peptide could be found at pH 8. Compared with the original spectrum of Pep-N-B of Fig. 5A, its isotope pattern at pH 8 was unquestionably changed with a lower intensity of the monoisotopic peak, as the deamidation product (Pep-D-B) which provided a +0.984 Da increase overlapped with the 13C isotope of Pep-N-B in the MALDI-TOF MS spectrum. The peptide was partially deamidated and the deamidation increased dramatically under basic conditions (43.83%, pH = 8), whereas deamidation was not detected at pH 5. We also applied the QuanDA algorithm to analyze LC-MS data from bevacizumab digestion, however, we observed variability in the modified versus unmodified Asn ratio with retention time, preventing the establishment of a consistent isotopic pattern for accurate deamidation quantification. Additionally, the charge state distribution (CSD) of peptides influenced quantitative results, leading to discrepancies between the observed and theoretical isotopic distributions for different charge states. Consequently, the straightforward application of QuanDA for deamidation quantification in LC-MS data proved unsuitable. In contrast, QuanDA effectively processes MALDI MS data featuring singly charged ions and merged isotopic patterns of both modified and unmodified Asn. The deamidation difference of Pep-N-B between pH 5 and pH 8 was pretty evident, as it might serve as a marker peptide for assessing bevacizumab's deamidation behavior using QuanDA and MALDI MS. Enzymatic hydrolysis under basic conditions was an important cause of deamidation. The digestion under acidic conditions might provide real deamidation information of protein drugs.
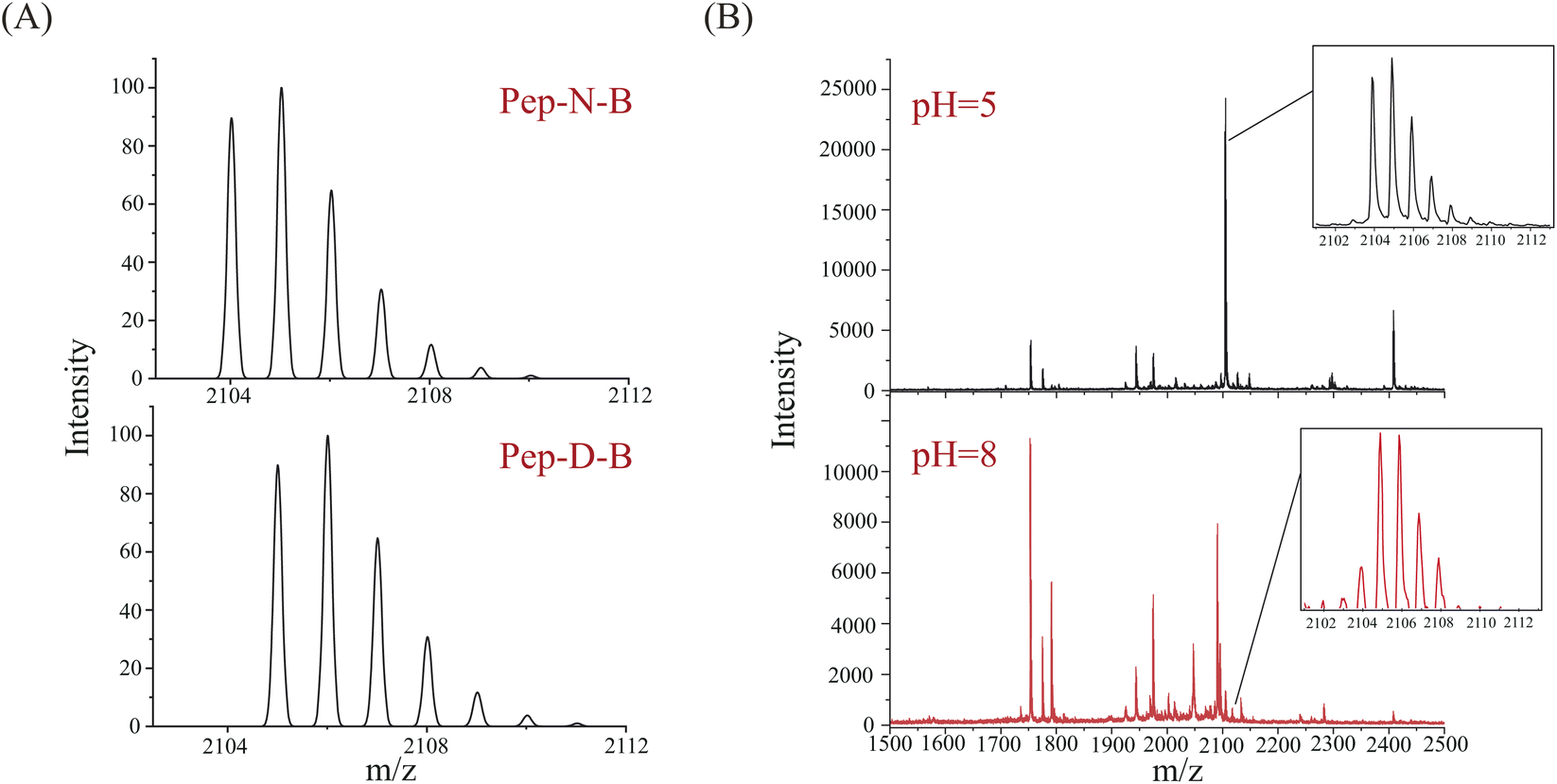 | ||
| Fig. 5 Deamidation analysis of Pep-N-B. (A) Theoretical MALDI spectra of Pep-N-B and Pep-D-B. (B) Full experimental MALDI spectra of bevacizumab digestion at pH 5 and pH 8, zoomed spectra were for Pep-N-B. | ||
4. Conclusions
MALDI MS combined with the new developed algorithm QuanDA is simple, feasible and credible in the deamidation analysis of therapeutic drugs. In this study, a computational model of asparagine deamidation was first validated using QuanDA, demonstrating a good correlation and accuracy by comparing theoretical isotope patterns of synthetic peptide mixtures with patterns observed by actual mass spectrometry. Experimental results show that in a mixture of synthetic peptides Pep-N and Pep-D, QuanDA can accurately calculate the deamidation ratio, which is highly consistent with the pattern of actual spectral observations. The correlation coefficient (R2) and root mean squared error (RMSE) were 0.9914 and 0.03356, respectively. The QuanDA model has also shown applicability in the absence of un-deamidated and deamidated peptide reference standards, all you need are only a pair of theoretical MALDI spectra of un-deamidated and deamidated peptides and experimental spectra of samples. Compared with other algorithms, QuanDA could provide you with the deamidation percentage of the sample directly with less parameter settings, which facilitates deamidation analysis. In contrast to the utilization of CE and HPLC, the QuanDA method, relying on mass spectrometry technology, exhibits a pronounced advantage in terms of heightened sensitivity. The deamidation reaction induces a mass increment of approximately +1 Da, a change readily discernible through the application of sensitive mass spectrometric techniques, and there is no disadvantage like that CE-MS may have protein adsorbed on the capillary wall. Meanwhile, the application of QuanDA was applied for two real therapeutic drugs (somatostatin and bevacizumab) under different conditions. The results were evaluated by LC-DAD with RSD less than 5%. The findings indicate that MALDI MS combined with QuanDA is an effective and accurate method for deamidation quantification in the biopharmaceutical industry.Abbreviations
| QuanDA | Quantification deamidation |
Data availability
All data included in this study are available upon request by contact with the corresponding author.Author contributions
Han Zhang: investigation and writing – original draft. Yinran Xiong: conceptualization, methodology. Xiaonan Shi: investigation, resources. Lijia Zhu: investigation, resources. Qiong Wu: investigation, resources. Ting Wu: resources, supervision, writing – original draft, writing – review & editing. Yiping Du: resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Qi Zhang of Suzhou Mild Bio-Tec. Co., Ltd. for the discussion of the deamidation of therapeutic drugs.References
- Y. Jin, Y. Yi and B. Yeung, Methods, 2022, 200, 58–66 CrossRef.
- G. Leo, I. Bonaduce, A. Andreotti, G. Marino, P. Pucci, M. P. Colombini and L. Birolo, Anal. Chem., 2011, 83, 2056–2064 CrossRef PubMed.
- H. Tonie Wright and D. W. Urry, Crit. Rev. Biochem. Mol. Biol., 1991, 26, 1–52 CrossRef PubMed.
- X. Li, Mechanisms of electron capture dissociation and the application to protein deamidation, Doctor thesis, Boston University, 2010.
- N. E. Robinson and A. B. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 944–949 CrossRef.
- A. B. Robinson, J. W. Scotchler and J. H. McKerrow, J. Am. Chem. Soc., 1973, 95, 8156–8159 CrossRef PubMed.
- N. E. Robinson and A. B. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12409–12413 CrossRef.
- D. Gervais, J. Chem. Technol. Biotechnol., 2015, 91, 569–575 CrossRef.
- E. B. Dunkelberger, L. E. Buchanan, P. Marek, P. Cao, D. P. Raleigh and M. T. Zanni, J. Am. Chem. Soc., 2012, 134, 12658–12667 CrossRef PubMed.
- D. Chelius, D. S. Rehder and P. V. Bondarenko, Anal. Chem., 2005, 77, 6004–6011 CrossRef PubMed.
- E. P. Commission, European Directorate for the Quality of Medicines & HealthCare, European Pharmacopoeia, Council of Europe, 2010 Search PubMed.
- D. W. Aswad, M. V. Paranandi and B. T. Schurter, J. Pharm. Biomed. Anal., 2000, 21, 1129–1136 CrossRef.
- L. Huang, J. Lu, V. J. Wroblewski, J. M. Beals and R. M. Riggin, Anal. Chem., 2005, 77, 1432–1439 CrossRef PubMed.
- Y. T. Zhang, J. Hu, A. L. Pace, R. Wong, Y. J. Wang and Y. H. Kao, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 965, 65–71 CrossRef.
- Q. Yan, M. Huang, M. J. Lewis and P. Hu, mAbs, 2018, 10, 901–912 CrossRef PubMed.
- L. G. Stock, S. Wildner, C. Regl, G. Gadermaier, C. G. Huber and H. Stutz, Anal. Chem., 2018, 90, 11933–11940 CrossRef.
- S. Gupta, W. Jiskoot, C. Schouroneich and A. S. Rathore, J. Pharm. Sci., 2022, 111, 903–918 CrossRef PubMed.
- N. AbuHeshmeh, A. Sallam, Y. Mater and M. Alawi, Int J Res Pharm Biomed Sci., 2013, 4, 428–435 Search PubMed.
- A. Najjar, M. Alawi, N. AbuHeshmeh and A. Sallam, Int. J. Adv. Pharm., 2014, 2014, 1–6 Search PubMed.
- S. Carillo, C. Jakes and J. Bones, J. Pharm. Biomed. Anal., 2020, 185, 113218 CrossRef PubMed.
- A. D. Melnikov, Y. P. Tsentalovich and V. V. Yanshole, Anal. Chem., 2020, 92, 588–592 CrossRef PubMed.
- M. Mackie, P. Rüther, D. Samodova, F. Di Gianvincenzo, C. Granzotto, D. Lyon, D. A. Peggie, H. Howard, L. Harrison, L. J. Jensen, J. V. Olsen and E. Cappellini, Angew. Chem., Int. Ed., 2018, 57, 7369–7374 CrossRef PubMed.
- R. A. Everley, T. M. Mott, S. A. Wyatt, D. M. Toney and T. R. Croley, J. Am. Soc. Mass Spectrom., 2008, 19, 1621–1628 CrossRef.
- N. Singhal, M. Kumar, P. K. Kanaujia and J. S. Virdi, Front. Microbiol., 2015, 6, 791 Search PubMed.
- D. Li, J. Yi, G. Han and L. Qiao, ACS Meas. Sci. Au, 2022, 2, 385–404 CrossRef.
- A. Boudier-Lemosquet, A. Mahler, C. Bobo, M. Dufossee and M. Priault, Methods, 2022, 200, 3–14 CrossRef.
- S. Dorum, S. W. Qiao, L. M. Sollid and B. Fleckenstein, J. Proteome Res., 2009, 8, 1748–1755 CrossRef PubMed.
- J. Wilson, N. L. van Doorn and M. J. Collins, Anal. Chem., 2012, 84, 9041–9048 CrossRef.
- F. Bray, I. Fabrizi, S. Flament, J.-L. Locht, P. Antoine, P. Auguste and C. Rolando, Anal. Chem., 2023, 95, 7422–7432 CrossRef PubMed.
- D. Kameoka, T. Ueda and T. Imoto, J. Biochem., 2003, 134, 129–135 CrossRef PubMed.
- S. Liu, K. R. Moulton, J. R. Auclair and Z. S. Zhou, Amino Acids, 2016, 48, 1059–1067 CrossRef.
- C. Emmanouilides, M. Pegram, R. Robinson, R. Hecht, F. Kabbinavar and W. Isacoff, Tech Coloproctol, 2004, 8(1), s50–s52 CrossRef PubMed.
- G. V. Koukourakis and A. Sotiropoulou-Lontou, Clin. Transl. Oncol., 2011, 13, 710–714 CrossRef PubMed.
- W. M. Nadler, D. Waidelich, A. Kerner, S. Hanke, R. Berg, A. Trumpp and C. Rosli, J. Proteome Res., 2017, 16, 1207–1215 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01595a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |