
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational engineering of an antimalarial peptide with enhanced proteolytic stability and preserved parasite invasion inhibitory activity†
Abhisek
Kar
 ,
Akash
Narayan
,
Vishal
Malik
and
Kalyaneswar
Mandal
*
,
Akash
Narayan
,
Vishal
Malik
and
Kalyaneswar
Mandal
*
Tata Institute of Fundamental Research Hyderabad, 36/p Gopanpally, Hyderabad, Telangana – 500046, India. E-mail: kmandal@tifrh.res.in
First published on 14th November 2024
Abstract
We describe rational chemical engineering to enhance the proteolytic stability of a chimeric peptide using a combination of unique strategies that involve the incorporation of a series of D-amino acids into the parent L-peptide sequence and restricting the conformational freedom of the peptide by covalent stitching. We hypothesize that replacing a stretch of sequence of an unstructured peptide motif with D-amino acids would increase its proteolytic stability without significantly affecting its affinity to the target protein. Also, considering the Cβ–Cβ distances, replacing an appropriate pair of residues with cysteine to form an additional disulfide bond in the molecule would provide additional stability to the engineered peptide. To verify this hypothesis, we have implemented these strategies to a previously reported peptidic inhibitor RR, against P. falciparum invasion into red blood cells (RBCs) and designed two novel heterochiral chimeric peptides, RR-I and RR-II. We have demonstrated that these peptides exhibit remarkable inhibitory activity with dramatically enhanced proteolytic stability. Finally, we have designed a cyclic analog, RR-III, to enhance the stability of the peptide against endopeptidases. The RR-III peptide exhibits the same inhibitory activity as RR-II while demonstrating impressive resistance to enzymatic degradation and prolonged stability in human plasma. These developments hold promise for a new generation of peptide-based therapeutics, showcasing the potential of residue selection for tailored modifications, as demonstrated in this work.
Introduction
Peptides have several advantageous properties that make them perfect candidates for the design of novel therapeutics.1 In contrast to a small molecule drug, a peptide drug has the potential to exhibit very high affinity and specificity to its targets. In addition, peptides are usually less toxic.1 Moreover, the sequence diversity of a peptide allows it to have access to a wide range of disease targets. Despite these advantages, naturally occurring peptides are not always suitable for therapeutic applications as they rapidly get degraded in vivo by proteases.1,2 As a consequence, the less bioavailable natural peptide drug candidates often struggle to cross the preclinical experimental stages. To overcome this issue, several chemical strategies have recently been developed to increase the stability of peptides in serum, which include peptide head-to-tail cyclization,3a–c restriction of conformational freedom by the formation of additional covalent bonds,3b,d,e or incorporation of D-amino acids in the native sequence.3d,fIn our laboratory, we are particularly interested in developing novel peptides or small protein-based inhibitors that would interfere with the malaria parasite invasion into red blood cells (RBCs). Plasmodium falciparum (P. falciparum) is responsible for the most severe form of human malaria amongst all malaria parasites. The only mainstay treatment for P. falciparum malaria, which has resulted in a substantial decline in the figure of malaria-related deaths, is artemisinin-based combination therapies (ACTs). However, the decline in death rate is now threatened by the recent emergence of drug-resistant P. falciparum strains.4 Invasion of P. falciparum merozoites into new red blood cells (RBCs) is a critical pinch point in the parasite life cycle. In the first step, merozoite attaches to the RBC surface through RBC ligand–parasite receptor interactions. Next, reorientation of the merozoite brings the apical end close to the RBC surface.5,6 Subsequently, the formation of a tight moving junction between two parasitic proteins, apical membrane antigen-1 (AMA1) and rhoptry neck protein-2 (RON2), triggers the invasion process.7,8
There are only a few peptide inhibitors known to interfere with PfAMA1–PfRON2 interactions to prevent parasite entry into RBCs. The native extracellular domain of PfRON2 (PfRON22021–2059) is an efficient inhibitor for parasite growth.9 Also, a mutant peptide with twenty-nine residues of the PfRON2 ectodomain (PfRON22027–2055, F2038W, Q2046M) was reported to be effective against the P. falciparum 3D7 strain with an IC50 of 0.16 μM. There are only a few non-PfRON2 sequence-based peptide inhibitors known against PfAMA1.10,11 Out of which, a 20-residue peptide obtained by Harris et al.11 from a random peptide phage display library screen has an IC50 (3D7 strain) of ∼4 μM.
In a recent work, we demonstrated the design of a hybrid peptide originating from the native ligand PfRON22027–2055 and R1 peptide obtained from a phage-display screen to generate a highly potent chimeric peptide inhibitor of PfAMA1 (Fig. 1A).12 The hybrid peptide (RR) exhibited a hundred-fold better potency compared to any of its parent peptides. However, despite remarkable inhibitory activity, it showed extremely poor proteolytic stability and a short plasma half-life (see Fig. 1B). Therefore, like most potential therapeutic peptides, this one also suffered from proteolytic instability, making it a less bioavailable therapeutic candidate. The remarkable inhibitory activity and yet very poor bioavailability compelled us to rationally design novel peptides, based on the RR sequence, that would be resistant to proteolysis without compromising the potency of the peptide molecule.
 | ||
Fig. 1 (A) The sequence of the parent chimeric peptide RR derived from RON22027–2055 (blue) and R1 (green) peptide fusion. (B) Stability of the RR peptide against enzyme (pepsin, chymotrypsin, and pronase) and human plasma treatment. For the enzyme degradation assay, the enzyme-to-peptide ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (w/w). For the plasma digestion assay, the peptide was incubated in 20% diluted human plasma in 20 mM phosphate buffer of pH 7.4 to a final concentration of 0.5 mg mL−1 and incubated at 37 °C. :100 (w/w). For the plasma digestion assay, the peptide was incubated in 20% diluted human plasma in 20 mM phosphate buffer of pH 7.4 to a final concentration of 0.5 mg mL−1 and incubated at 37 °C. | ||
Herein, we systematically study the stability of the chimeric peptide (RR) using a combination of unique strategies, which include incorporation of a series of D-amino acids into a specific location of the parent peptide sequence13 and restriction of conformational freedom of the peptide by covalent stitching. Replacing even a single L-amino acid in a peptide secondary structure with a D-amino acid usually compromises the biological activity of the peptide, as the sidechain orientations of the peptide with respect to the target protein get inverted. We hypothesize that replacing a stretch of sequence with D-amino acids of an unstructured peptide motif will increase its serum stability without significantly compromising its affinity to target proteins. To verify our hypothesis, we applied our strategies to the RR peptide and designed three novel heterochiral chimeric peptides, RR-I, RR-II and RR-III, and showed that these peptides exhibit remarkable inhibitory activity against P. falciparum invasion into RBCs with enhanced proteolytic stability.
Results and discussion
The main objective of this study was to rationally engineer synthetic peptides that exhibit increased resistance to proteolytic degradation while maintaining their high affinity for specific targets. Our starting point was the reported chimeric peptide molecule RR (VFAEFLPLFS10 CFTTRMSPPQ20 QICLN25).12 First, we designed the 25-residue peptide RR-I (vfaeflPlfs10 CFTTRMSPPQ20 QICLN25). In this design, we replaced the first ten N-terminal residues of RR with D-amino acids, except for the proline (highlighted in bold in the sequence above), originated from the R1 peptide sequence (Fig. 2A). The configuration of the proline was not changed as it can induce a turn in a peptide structure. We hypothesized that replacing the unstructured peptide motif of RR, derived from the R1 peptide, with D-amino acids (except proline) would result in a peptide that retains activity while providing increased resistance to proteolysis. In our second design iteration (Fig. 2A), we introduced an additional disulfide bond into the loop region of the RR-I peptide, which is a part of the PfRON22027–2055 ectodomain loop, forming the peptide RR-II (vfaeflPlfs10 CFCTRMSPPQ20 CICLN25). Since the loop region of the RR-I peptide was composed entirely of L-amino acids, we hypothesized that the incorporation of an additional disulfide bond into the loop could be achieved without disturbing the interaction interface. We expected that this modification would significantly improve the in vivo stability of the peptide. To achieve this, we performed Cβ–Cβ distance calculations between pairs of residues in the loop region (12Phe-22Ile) of the RR-I peptide. Our analysis revealed that residues 13Thr and 21Gln were spatially close (Cβ–Cβ distance = 4.4 Å) to each other, making them suitable candidates for disulfide bond formation when replaced by cysteine.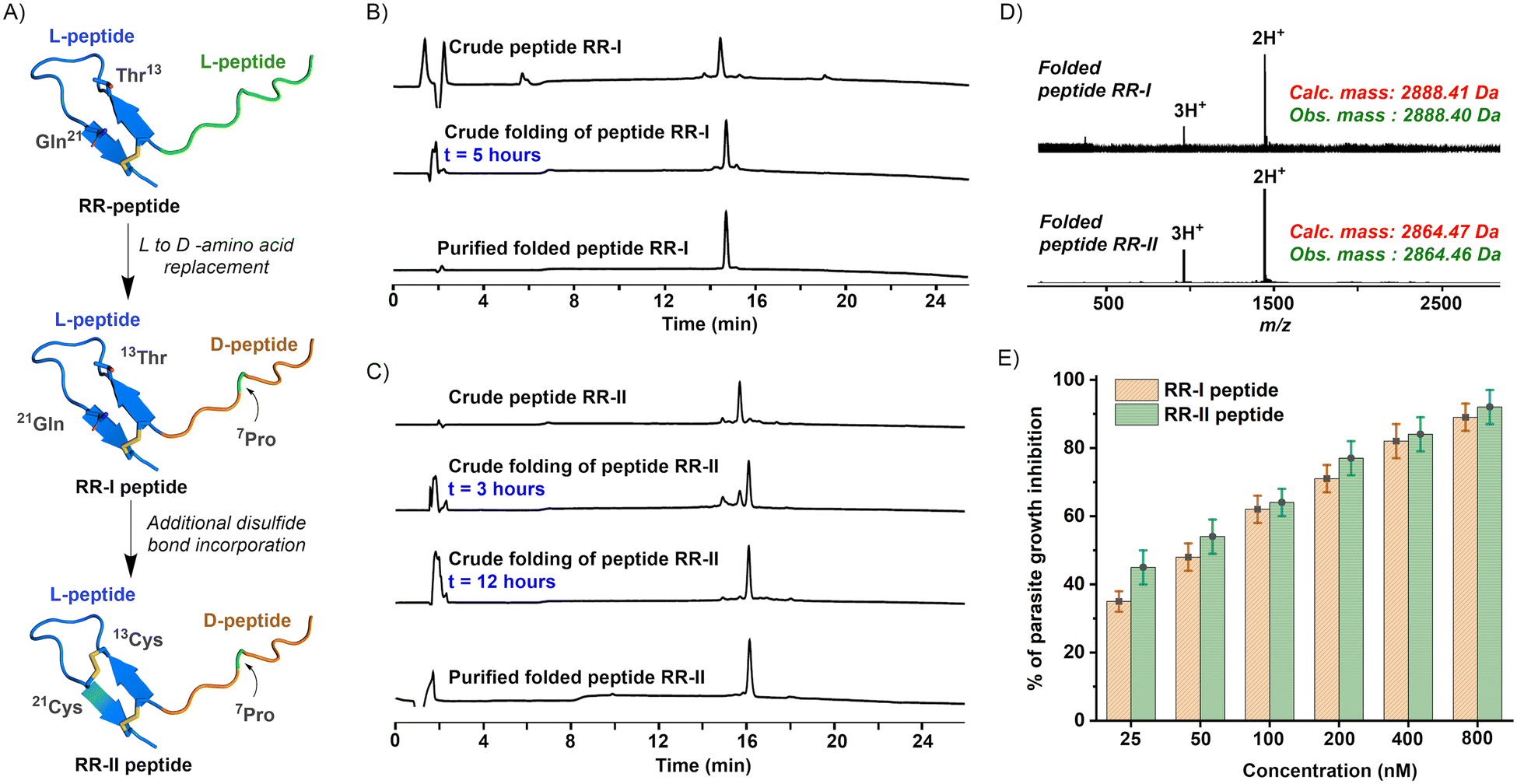 | ||
| Fig. 2 (A) Schematic depiction of the designed peptides. The RR-I peptide was designed by replacing all but not the 7Pro residue of the peptide sequence derived from the R1 peptide (in orange) in the RR peptide with D-amino acids. Furthermore, the RR-II peptide was designed by replacing 13Thr and 21Gln of RR-I with two cysteine residues to form an additional disulfide bond. (B) Analytical HPLC profile (λ = 214 nm) of the folded peptide: crude, crude folded, and purified folded peptide RR-I. (C) Analytical HPLC profile (λ = 214 nm) of the folded peptide: crude, crude folded, and purified folded peptide RR-II. (D) ESI-MS data (most abundant isotopologue mass with an uncertainty of ± 0.02 Da) of the folded peptide RR-I and RR-II. (E) Malaria parasite (3D7 strain) growth inhibition assay. A dose-dependent response of peptides RR-I (orange) and RR-II (green) was observed in the GIA assay. Synchronized schizonts were treated with inhibitors with varying concentrations (25–800 nM). | ||
The designed peptides RR-I and RR-II were synthesized via solid phase peptide synthesis (SPPS),14 as outlined in Section S1.4 (ESI†). The amino acid composition of the synthesized peptides was confirmed through ESI-MS analysis. Given the satisfactory quality of the crude peptide profiles, as evidenced by analytical HPLC and ESI-MS, we proceeded to fold the peptides directly, which involved the formation of disulfide bonds without further purification. The folding of the crude peptides was accomplished through air oxidation, as detailed in Section S1.5 (ESI†). We confirmed the correct number of disulfide bonds formed using ESI-MS analysis, as shown in Fig. 2D. The TFA counter-anions of the purified peptides were then exchanged with HCl for a parasite growth inhibition assay (GIA) by lyophilization of the peptides three times after dissolving in acetonitrile in water (50% v/v) containing 0.1 N HCl.
To assess the biological activity of the designed peptides, we conducted a parasite growth inhibition assay (GIA) using the frequently observed P. falciparum strain 3D7, detailed in Section S1.7 (ESI†). This functional assay quantifies the inhibitory effect of peptides present in a blood sample on the growth of specific malaria parasites. It is worth noting that the IC50 value of the RR chimeric peptide in the inhibition assay was previously determined to be ∼30 nM as reported in our earlier work.12 In contrast, the designed heterochiral chimeric peptides, RR-I and RR-II, exhibited IC50 values of 56 nM and 45 nM, respectively. While the activity of these engineered heterochiral peptides was modestly reduced compared to the RR peptide, it is important to highlight that they still demonstrated superior inhibitory activity when compared to the parent peptide R1 (IC50 = 4.0 μM) and native 29-residue PfRON22027–2055 ectodomain (IC50 = 0.2 μM).9,11 Despite low nanomolar IC50 values for both peptides, we could not achieve 100% inhibition with these peptides even at higher concentrations. This could be due to the potential aggregation of peptides at higher concentrations in the culture media during the assay period, given their hydrophobic nature.
We next addressed the proteolytic stability (see Section S1.8, ESI†) of the engineered heterochiral peptides RR-I and RR-II along with their parent peptides using a series of enzymes: pepsin, chymotrypsin, and pronase (Fig. 3). Pepsin, known for its enzymatic activity to cleave the peptide bond at the C-terminus of phenylalanine, leucine, tyrosine, and tryptophan residues, resulted in complete degradation of R1 and RR within just 15 minutes, and the PfRON22027–2055 peptide experienced complete degradation within 1.5 hours. Interestingly, both the heterochiral peptides, RR-I and RR-II, showed remarkable resistance and remained completely intact in the pepsin digestion assay for a prolonged period of up to 20 hours (Fig. 3A). Chymotrypsin preferentially cleaves at the carboxy terminus of aromatic amino acids, such as tyrosine, phenylalanine, and tryptophan, and occasionally leucine or methionine. As a result, the L-peptides R1, RR, and PfRON22027–2055 are broken down within 15 minutes. In contrast, 90% of the RR-I peptide remained intact in the first 15 minutes but, completely cleaved within 4 hours. Impressively, 95% of the RR-II peptide remained intact without any degradation even after 20 hours in the chymotrypsin digestion assay (Fig. 3B). In the pronase (a mixture of non-specific bacterial proteases) digestion assay, the peptides PfRON22027–2055, R1, and RR were completely degraded within just 15 minutes. In contrast, the RR-I peptide showed a slower degradation rate, requiring 1.5 hours for complete degradation, while 55% of the RR-II peptide remained intact even after 20 hours (Fig. 3C). Pepsin and chymotrypsin mainly target residues located at the N-terminus of the peptides, particularly those derived from the R1 peptide. Replacing these residues with their corresponding D-amino acids resulted in a significant improvement in enzymatic stability as seen in the pepsin and chymotrypsin digestion assays. Although both RR-I and RR-II peptides contain the same number of D-amino acid residues in their sequences, the presence of an additional disulfide bond in the RR-II peptide likely contributed to its superior enzymatic stability as demonstrated by the chymotrypsin and pronase digestion tests.
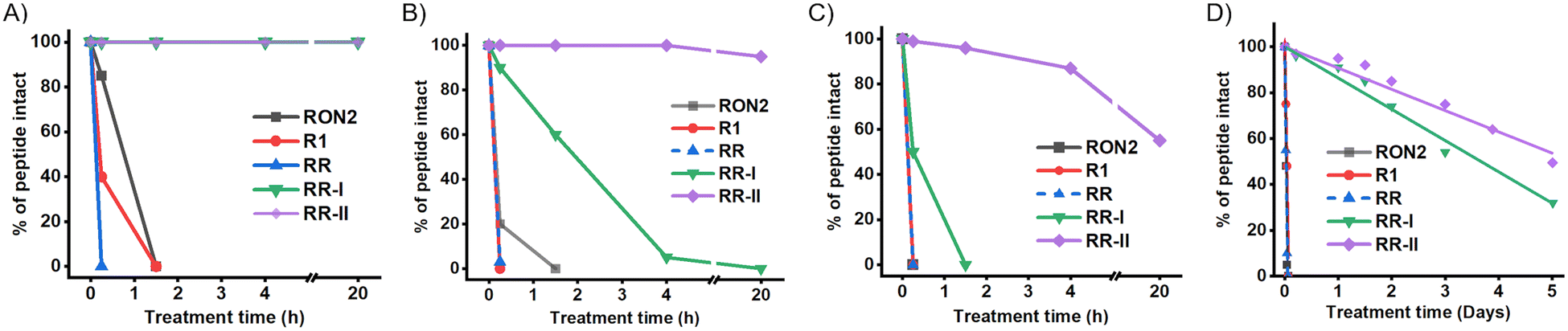 | ||
| Fig. 3 In vitro enzyme digestion (pepsin (A), chymotrypsin (B), and pronase (C)) of the designed RR-I and RR-II peptides along with the parent peptides R1, PfRON22027–2055, and RR. Data are represented as the percentage of peptides intact with an average of two repeats. The intact peptide amount was calculated from the area under the respective UV absorbance peak measured at 214 nm. Solid lines represent the plot over connecting experimental data points. (D) The plasma digestion assay of the designed peptides along with the parent peptides. The same concentration of each peptide was treated with diluted plasma (20% v/v) in a 20 mM phosphate buffer of pH 7.4. The percentage of the intact peptides was quantified by measuring the intensity of the peak detected at 214 nm wavelength in analytical RP-HPLC. Solid lines represent the fitted plot over the experimental data points. | ||
Our subsequent investigation involved assessing the stability of the three peptides, RR, RR-I, and RR-II, in 20% human plasma (Fig. 3D). These peptides were subjected to human plasma treatment following the protocol detailed in Section S1.9 (ESI†). Notably, the peptides R1, PfRON22027–2055, and RR experienced rapid decomposition within the initial hour of the plasma treatment. However, to our surprise, both RR-I and RR-II peptides displayed remarkable stability in the human plasma used in the experiment. Approximately 50% of RR-I remained intact for up to 3 days, while RR-II exhibited an even more impressive resilience, with 50% of the peptide remaining intact for up to 5 days.
Our findings suggest that substituting the unstructured peptide motif in the RR peptide, originally derived from the R1 peptide, with D-amino acids (excluding 7Pro) significantly enhances resistance to proteolytic degradation in diluted human plasma. Furthermore, the incorporation of an additional disulfide bond into the loop region in RR-II, composed entirely of L-amino acids, further enhanced its stability when compared to RR-I.
Subsequently, to investigate the impact of the chirality of the junction residue (10Ser) within the fused peptide, which serves as the first residue followed by the β-hairpin loop derived from the PfRON22027–2055 peptide, we introduced a chemical mutation by replacing the D-Ser in the RR-I peptide with L-Ser (see Fig. 4A). The resultant peptide, named RR-Ia, was synthesized and folded as detailed in Section S1.4 and S1.5 (ESI†). When subjected to a parasite growth inhibition assay (GIA) at a fixed concentration of 50 nM, RR-Ia exhibited a notable reduction in inhibitory activity, as illustrated in Fig. 4B, in comparison to the original parent peptide RR-I. This result unequivocally highlights the preference for D-chirality in the 10Ser residue at the fusion point.
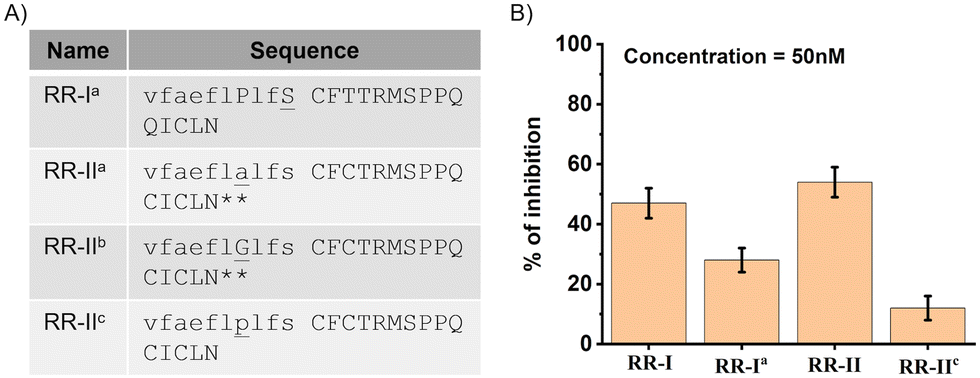 | ||
| Fig. 4 (A) The sequence of the chemically mutated peptides. ** Indicates the peptides were not soluble in growth inhibition assay media. (B) Malaria parasite growth inhibition assay with the 3D7 strain. Synchronized schizonts were treated with inhibitors with a fixed concentration of 50 nM peptide. | ||
To assess the impact of chemically mutating the original 7Pro residue, we substituted it with D-Ala (RR-IIa) and Gly (RR-IIb) in the RR-II peptide. Unexpectedly, we encountered challenges with the solubility of these chemically mutated peptides in various buffer conditions while performing the parasite growth inhibition assay. The issue was particularly evident when dissolved in the complete media utilized throughout the parasite growth inhibition assay, even during the screening at the lowest concentration (25 nM) of the peptide. Furthermore, de novo secondary structure predictions, generated using the alpha-fold15 (predicted model structures are shown in Fig. S4, ESI†), revealed an intriguing secondary structural change in the previously unstructured region of the fused peptide derived from the R1 peptide when the 7Pro was chemically mutated to 7D-Ala and 7Gly. Specifically, this alteration induced an α-helical structure. This observation led us to infer that the presence of the 7Pro residue serves as a helix-breaking element within the sequence, emphasizing its crucial role in maintaining an unstructured conformation essential for biological activity. Next, we changed the chirality of the residue Pro from 7L-Pro to 7D-Pro (RR-IIc, Fig. 4A) of the peptide RR-II and evaluated the activity by parasite growth inhibition assay. Again, the parasite growth inhibition assay at a fixed concentration of 50 nM exhibited a three-fourth reduction in inhibitory activity in comparison to the original parent peptide, as illustrated in Fig. 4B. This outcome certainly proved our hypothesis that the requirement of 7L-Pro is crucial for maintaining the spatial orientation of other residues during the interaction.
Natural cyclic peptides are well-known for their inherent resistance to in vivo enzymatic degradation.16 By incorporating D-amino acids into our designed peptide, we achieved significant improvement in stability while preserving its activity. However, analysis using ESI-MS during the enzymatic digestion assay revealed continued susceptibility to digestion possibly by endopeptidases, primarily from the C-terminal end of the peptides. To enhance the molecule's stability against enzyme degradation further, we designed another version of the peptide through peptide main chain-to-side chain cyclization. In this new design, we substituted the 10D-Ser residue with 10D-Lys, where the lysine residue features a side chain amine linked to cysteine, effectively replacing 25Asn in the original RR-II peptide. Subsequent native chemical ligation (NCL)17 of the cysteine residue, linked to 10D-Lys, with the thioester at the C-terminal residue, 24Leu, resulted in the formation of a macrolactam ring through the backbone-to-side chain cyclization, as illustrated in Fig. 5A. The thioester peptide was prepared as peptide o-aminoanilide followed by NaNO2 oxidation and exchange with MESNa.18 As we wanted to eliminate an odd cysteine, we subjected the cysteine that underwent ligation to desulfurization,19 converting it to an alanine after the NCL reaction. For orthogonal desulfurization, we had to keep the other four cysteine residues protected with the acetamido-methyl (Acm) group. Once the ligation and desulfurization were completed, we removed the Acm groups and proceeded with the folding to get the final desired peptide, RR-III (Fig. 5A). The progress of the synthesis, as monitored by LC-MS, is shown in Fig. 5B. The details of the synthesis have been discussed in Section S1.6 (ESI†). The folded macrolactam peptide had a mass of 2843.34 ± 0.02 Da (calculated mass 2843.34 Da, most abundant isotopologue), as determined by ESI-MS (Fig. 5C).
 | ||
| Fig. 5 (A) A scheme for the synthesis of RR-III (5). (B) Analytical HPLC profile (λ= 214 nm) for the synthesis of the peptide RR-III: (a) crude peptide 1, (b) crude reaction mixture after the cyclization via NCL reaction, (c) purified cyclized peptide 2, (d) desulfurization of the product 2 to derive peptide 3, (e) purified desulfurized peptide 3, (f) reaction mixture after Acm removal to get peptide 4, (g) purified peptide 4, (h) reaction mixture after folding to get the final desired peptide RR-III (5), and (i) the purified peptide RR-III. ** denoting MPAA peak. (C) ESI-MS data of the peptide 4 and folded peptide 5 (RR-III). Four Dalton mass decrease indicates two disulfide bond formation. (D) Malaria parasite (3D7 strain) growth inhibition assay. A dose-dependent response of peptides RR-III was observed in the GIA assay. Synchronized schizonts were treated with inhibitors with varying concentrations (25–800 nM). (E) In vitro enzyme digestion (pepsin, chymotrypsin, and pronase) of the designed RR-III. Solid lines represent the plot over connecting experimental data points. (F) The plasma digestion assay of the RR-III peptide. The peptide was treated with diluted plasma (20% v/v) in a 20 mM phosphate buffer of pH 7.4. The percentage of the intact peptides was quantified by measuring the intensity of the peak detected at 214 nm wavelength in analytical RP-HPLC. Solid lines represent the fitted plot over experimental data points. The data for RR-I and RR-II are the same as in Fig. 3D. | ||
The subsequent experiment aimed at assessing the GIA of the engineered peptide RR-III against the same P. falciparum strain 3D7. The parasite growth inhibition assay involving RR-III revealed a dose-dependent response (see Fig. 5D), demonstrating an IC50 value of approximately 52 nM, consistent with the IC50 value observed for the RR-II peptide. This consistent pattern in activity strongly reinforces the notion that there has been no compromise in the structural integrity of the designed peptide, thus affirming its capacity to exert its intended biological activity.
In our final phase of experimentation, we evaluated the stability of the RR-III peptide when exposed to in vitro enzyme treatments and human plasma. Specifically, RR-III was subjected to pepsin and chymotrypsin digestion assays, where the peptide exhibited remarkable resistance to degradation for an extended period of up to 48 hours. Conversely, in the pronase digestion assay, we observed that 85% of the RR-III peptide remained intact even after 48 hours of incubation (see Fig. 5E). Furthermore, upon exposure to 20% human plasma, only 25% of the peptide underwent degradation (see Fig. 5F) within five days. These compelling findings unequivocally demonstrate that the main chain-to-backbone cyclization employed in the design of the RR-III peptide contributed significantly to its enhanced enzymatic stability compared to RR-II.
Finally, surface plasmon resonance (SPR) experiments were performed to evaluate the binding interactions between the manipulated peptides and the target protein P. falciparum 3D7 AMA-1104–438. The AMA-1 protein was expressed and folded (see Section S1.11, ESI†) according to the protocol described in a previous publication.20 Briefly, the expression was performed in Escherichia coli BL21(DE3) cells using a pET-based expression vector. After induction with IPTG, cells were harvested and lysed, and AMA-1 protein was purified from inclusion bodies. Protein refolding was achieved by a rapid dilution method in which the denatured protein was gradually diluted in a refolding buffer with appropriate redox conditions. Folded protein was purified by size exclusion chromatography and used for subsequent studies. Using a BI-4500AP SPR Instrument, the AMA-1 protein was immobilized on a Ni-NTA CM5 sensor chip, and a range of concentrations of the target peptides were injected to monitor binding in real-time. The resulting sensograms provided kinetic data that revealed the association and dissociation rates of the interactions. The equilibrium dissociation constant (KD) was calculated to quantify the binding affinity of each peptide. Kinetic analysis showed that the designed peptides RR-I, RR-II, and RR-III exhibit very fast association and dissociation rates as observed for parent peptide RR. This means that the modifications, including the incorporation of D-amino acids and the formation of disulfide bonds, did not significantly change the association and dissociation rate of the peptides to the target protein while it increased their proteolytic stability. The mutant peptide RR-Ia showed a very weak binding profile consistent with SPR sensograms that cannot be fitted with the one-to-one Langmuir isotherm. On the other hand, the mutated peptide RR-IIc showed no detectable binding signature even at a concentration of up to 1 μM (Fig. 6).
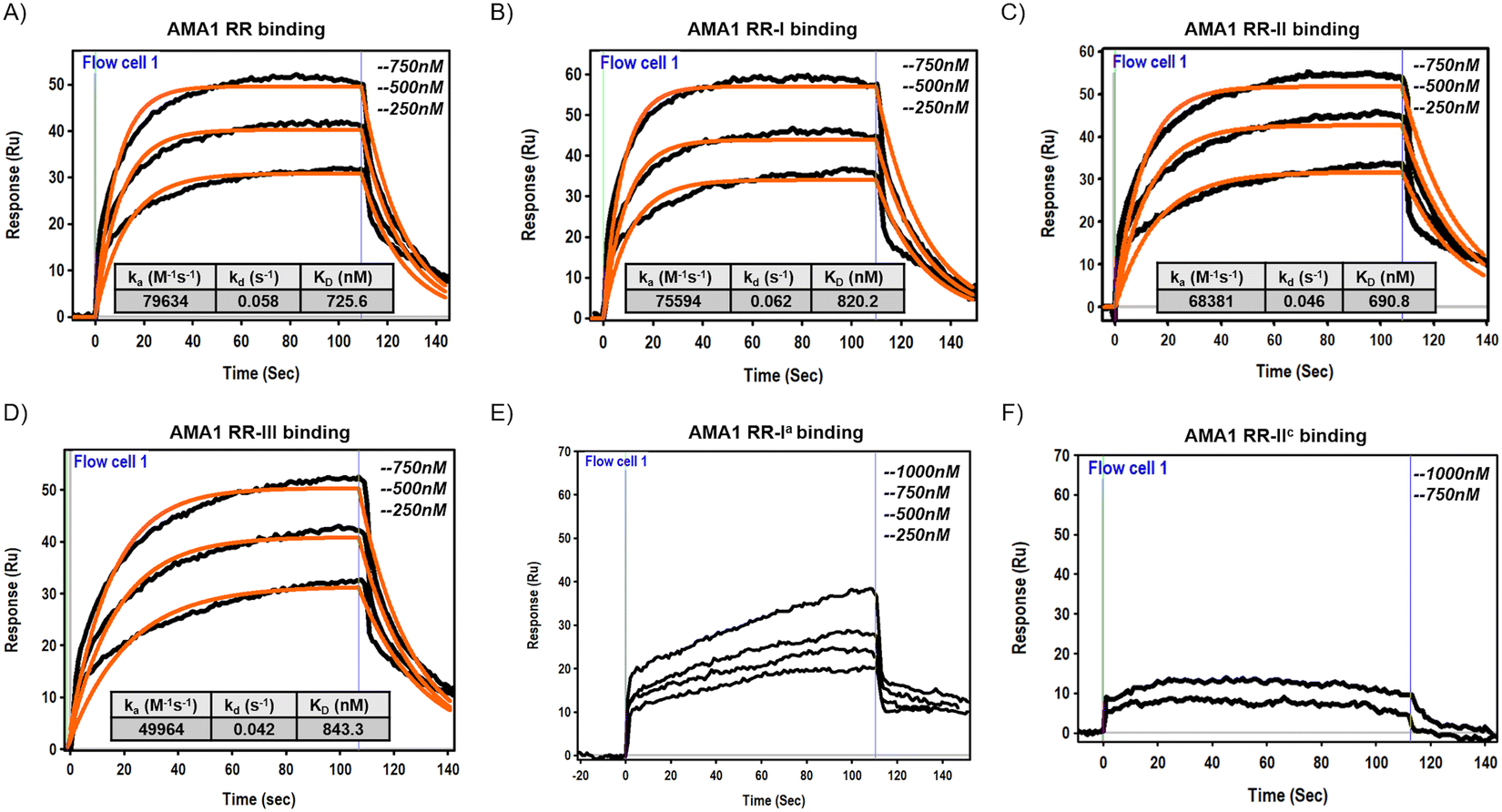 | ||
| Fig. 6 The SPR sensorgrams of the binding of the designed peptides (RR (A), RR-I (B), RR-II (C), RR-III (D), RR-Ia (E), and RR-IIc (F)) with P. falciparum 3D7 AMA-1104–438. The solid line (orange) is the fitted data over experimental data points. | ||
Conclusions
In conclusion, our designed peptide RR-I, which contains ten D-amino acids replacing the N-terminal first ten residues derived from the R1 peptide, demonstrated impressive proteolytic resistance while retaining inhibitory activity in the parasite growth inhibition assay (GIA). Building on this success, RR-II was constructed by introducing an additional disulfide bond into the loop region of RR-I. These modifications significantly improved the in vitro stability and made both RR-I and RR-II highly resistant to enzymatic degradation. Further investigations included assessing the stability of these peptides in diluted human plasma. To our delight, both RR-I and RR-II demonstrated remarkable stability, with RR-II exhibiting a half-life of up to 5 days in a 20% human plasma. These findings contributed significantly to overcoming the common hurdle of proteolytic instability associated with therapeutic peptides. To understand the influence of chirality at specific residues, we chemically introduced mutations into vulnerable locations of the peptide. The results clearly demonstrated the preference for specific chirality in key residues to maintain structural integrity and biological activity. Finally, we introduced a backbone-to-side chain cyclized analog, RR-III, to increase stability against endopeptidases. This modification proved successful as the RR-III peptide exhibited the same inhibitory activity as RR-II while showing quite impressive and the highest (among all) resistance to enzymatic degradation with prolonged stability in human plasma.Judicious replacement of an unstructured L-amino acid segment with D-amino acids, as demonstrated here, offers a versatile approach to enhancing the proteolytic stability of other miniprotein or peptide candidates that contain segments lacking defined secondary structure. Also, various strategies, including peptide cyclization and restricting conformational freedom through additional covalent bonds, have historically been employed to stabilize proteins or peptides across different folds. Hence, our approach with an anti-parallel beta-sheet can similarly be extended to various secondary structures in other peptides or mini-proteins with diverse folding patterns. While a completely rational approach may not always be as effective as ours, prior in silico calculations may be beneficial for effective covalent modifications.
Our study represents a significant advance in the rational chemical engineering of peptides for therapeutic applications. By combining structural insights with innovative chemical modifications, we have created peptides that not only retain potent inhibitory activity but also exhibit robust stability against proteolytic degradation. These advances hold promise for the development of a new generation of peptide-based therapeutics that may expand the repertoire of effective treatment options for various disease areas while exploiting the appropriate selection of residues for modifications, as demonstrated in this work.
Author contributions
Conceptualization: A. K. & K. M.; investigation: A. K., A. N. & V. M.; formal analysis: A. K., A. N., V. M. & K. M.; funding acquisition: K. M.; supervision: K. M.; writing – original draft: A. K. with the input received from all co-authors; writing – review & editing: A. K. & K. M.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the DBT/Wellcome Trust India Alliance grant (grant no. IA/I/15/1/501847) and the intramural funds at TIFR Hyderabad from the Department of Atomic Energy (DAE), Government of India, under Project Identification No. RTI-4007 to K. M.References
- (a) L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Therapeutic Peptides: Current Applications and Future Directions, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS; (b) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in Peptide Drug Discovery, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS.
- B. J. Bruno, G. D. Miller and C. S. Lim, Basics and recent advances in peptide and protein drug delivery, Ther. Delivery, 2013, 11, 1443 CrossRef.
- (a) V. J. Hruby, Designing peptide receptor agonists and antagonists, Nat. Rev. Drug Discovery, 2002, 1, 847–858 CrossRef CAS; (b) L. Gentilucci, R. De Marco and L. Cerisoli, Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization, Curr. Pharm. Des., 2010, 16, 3185–3203 CrossRef CAS PubMed; (c) R. J. Clark, H. Fischer, L. Dempster, N. L. Daly, K. J. Rosengren, S. T. Nevin, F. A. Meunier, D. J. Adams and D. J. Craik, Engineering stable peptide toxins by means of backbone cyclization: Stabilization of the α-conotoxin MII, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13767–13772 CrossRef CAS PubMed; (d) G. L. Verdine and G. Hilinski, Stapled peptides for intracellular drug targets, J. Met. Enzymol., 2012, 503, 3–33 CAS; (e) D. P. Fairlie, G. Abbenante and D. R. March, Macrocyclic peptidomimetics - Forcing peptides into bioactive conformations, Curr. Med. Chem., 1995, 2, 654–686 CrossRef CAS; (f) J. J. Nestor, The medicinal chemistry of peptides, Curr. Med. Chem., 2009, 16, 4399–4418 CrossRef CAS PubMed.
- B. Blasco, D. Leroy and D. A. Fidock, Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic, Nat. Med., 2017, 23, 917–928 CrossRef CAS PubMed.
- J. A. Dvorak, L. H. Miller and W. C. Whitehouse, Invasion of erythrocytes by malaria merozoites, Science, 1975, 87, 748–750 CrossRef PubMed.
- M. Aikawa, L. H. Miller, J. Johnson and J. Rabbege, Erythrocyte entry by malarial parasites. A moving junction between erythrocyte and parasite, J. Cell Biol., 1978, 77, 72–82 CrossRef CAS PubMed.
- J. Baum, D. Richard, J. Healer, M. Rug, Z. Krnajski, T. W. Gilberger, J. L. Green, A. A. Holder and A. F. Cowman, A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites, J. Biol. Chem., 2006, 281, 5197–5208 CrossRef CAS.
- T. J. Jewett and L. D. Sibley, Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites, Mol. Cell, 2003, 11, 885–894 CrossRef PubMed.
- B. Vulliez-Le Normand, M. L. Tonkin, M. H. Lamarque, S. Langer, S. Hoos, M. Roques, F. A. Saul, B. W. Faber, G. A. Bentley, M. J. Boulanger and M. Lebrun, Structural and Functional Insights into the Malaria Parasite Moving Junction Complex, PLoS Pathog., 2012, 8, e1002755 CrossRef.
- F. Li, A. Dluzewski, A. M. Coley, A. Thomas, L. Tilley, R. F. Anders and M. Fole, Phage displayed peptides bind to the malarial protein apical membrane antigen-1 and inhibit the merozoite invasion of host erythrocytes, J. Biol. Chem., 2002, 277, 50303 CrossRef.
- K. S. Harris, J. L. Casey, A. M. Coley, R. Masciantonio, J. K. Sabo, D. W. Keizer, E. F. Lee, A. McMahon, R. S. Norton, R. F. Anders and M. Foley, Binding hot spot for invasion inhibitory molecules on Plasmodium falciparum apical membrane antigen 1, Infect. Immun., 2005, 73, 6981 CrossRef.
- J. Mannuthodikayil, S. Sinha, S. Singh, A. Biswas, I. Ali, P. C. Mashurabad, W. Tabassum, P. Vydyam, M. K. Bhattacharyya and K. Mandal, A Chimeric Peptide Inhibits Red Blood Cell Invasion by Plasmodium falciparum with Hundredfold Increased Efficacy, ChemBioChem, 2023, 24, e202200533 CrossRef PubMed.
- (a) G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. S. R. K. Pulavarti, Q. Kaas, A. Eletsky, P. S. Huang, W. A. Johnsen, P. J. Greisen, G. J. Rocklin, Y. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Accurate de novo design of hyperstable constrained peptides, Nature, 2016, 538, 329–335 CrossRef PubMed; (b) S. Imanishi, T. Katoh, Y. Yin, M. Yamada, M. Kawai and H. Suga, In Vitro Selection of Macrocyclic d/l-Hybrid Peptides against Human EGFR, J. Am. Chem. Soc., 2021, 143, 5680–5684 CrossRef PubMed.
- R. B. Merrifield, Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly Accurate Protein Structure Prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef.
- D. Gang, D. W. Kim and H. S. Park, Cyclic Peptides: Promising Scaffolds for Biopharmaceuticals, Genes, 2018, 9, 557 CrossRef.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Synthesis of proteins by native chemical ligation, Science, 1994, 266(5186), 776 CrossRef.
- (a) J. B. Blanco-Canosa and P. E. Dawson, An efficient Fmoc-SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation, Angew. Chem., Int. Ed., 2008, 47, 6851 CrossRef; (b) J. X. Wang, G. M. Fang, Y. He, D. L. Qu, M. Yu, Z. Y. Hong and L. Liu, Peptide o-Aminoanilides as Crypto-Thioesters for Protein Chemical Synthesis, Angew. Chem., Int. Ed., 2015, 54, 2194–2198 CrossRef; (c) J. Mannuthodikayil, S. Singh, A. Biswas, A. Kar, W. Tabassum, P. Vydyam, M. K. Bhattacharyya and K. Mandal, Benzimidazolinone-Free Peptide o-Aminoanilides for Chemical Protein Synthesis, Org. Lett., 2019, 21, 9040–9044 CrossRef.
- (a) L. Z. Yan and P. E. Dawson, Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization, J. Am. Chem. Soc., 2001, 123, 526–533 CrossRef PubMed; (b) Q. Wan and S. J. Danishefsky, Free-radical-based, specific desulfurization of cysteine: a powerful advance in the synthesis of polypeptides and glycopolypeptides, Angew. Chem., Int. Ed., 2007, 46, 9248 CrossRef PubMed; (c) C. Haase, H. Rohde and O. Seitz, Native Chemical Ligation at Valine, Angew. Chem., Int. Ed., 2008, 47, 6807–6810 CrossRef.
- A. Biswas, S. Raran-Kurussi, A. Narayan, A. Kar, P. Chandra Mashurabad, M. K. Bhattacharyya and K. Mandal, Efficient refolding and functional characterization of PfAMA1(DI + DII) expressed in E. coli, Biochem. Biophys. Rep., 2021, 26, 100950 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00229f |
| This journal is © The Royal Society of Chemistry 2025 |