
Metalloporphyrinic metal–organic frameworks for enhanced photocatalytic degradation of a mustard gas simulant†
Alisa S.
Quon‡
 a,
Doroteo
Manriquez‡
a,
Anna
Nguyen§
a,
Edgar K.
Papazyan§
a,
Pavithra
Wijeratne
a,
Lun
An
b,
Long
Qi
b,
Matthew J.
Tang
a,
Austin D.
Ready
c,
Omar K.
Farha
*d and
Yangyang
Liu
*a
a,
Doroteo
Manriquez‡
a,
Anna
Nguyen§
a,
Edgar K.
Papazyan§
a,
Pavithra
Wijeratne
a,
Lun
An
b,
Long
Qi
b,
Matthew J.
Tang
a,
Austin D.
Ready
c,
Omar K.
Farha
*d and
Yangyang
Liu
*a
aDepartment of Chemistry and Biochemistry, California State University, Los Angeles, 5151 State University Drive, Los Angeles, California 90032, USA. E-mail: yliu114@calstatela.edu
bU.S. DOE Ames National Laboratory, Iowa State University, Ames, Iowa 50011, USA
cDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095, USA
dDepartment of Chemistry and International Institute for Nanotechnology (IIN) and Department of Chemical & Biological Engineering, Northwestern University, Evanston, Illinois 60208, USA. E-mail: o-farha@northwestern.edu
First published on 28th October 2024
Abstract
Four metalloporphyrinic metal–organic frameworks (MOFs) were successfully synthesized and exhibited enhanced activities for the photooxidation of a sulfur mustard simulant, 2-chloroethyl ethyl sulfide (CEES). Among them, a Sn-porphyrin functionalized 2D MOF, namely CSLA-21-NH2(Sn), showed a half-life of 1.5 min for CEES oxidation under blue LED, featuring as one of the fastest photocatalysts for CEES degradation.
Sulfur mustard, also known as HD or mustard gas, is a blistering agent that can cause severe irritation to the skin and mucous membranes. It can cause chronic damage to the nervous, cardiac, and respiratory systems, among other organ systems, sometimes resulting in death.1–3 Since its mass production as a chemical warfare agent (CWA) during World War I, stockpiles of sulfur mustard remain and need to be safely degraded. Dehydrohalogenation and hydrolysis are two widely reported methods to degrade sulfur mustard.4 However, they are typically resource-intensive and ineffective; both reactions occur too slowly for degradation,5 and hydrolysis can result in incomplete degradation.6 A more promising method is to selectively oxidize sulfur mustard into nontoxic sulfoxide products while avoiding overoxidation into the toxic sulfone product (Scheme 1).
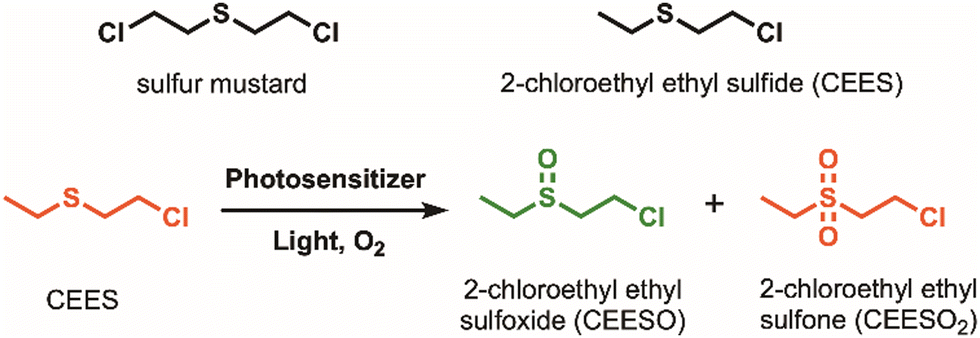 | ||
| Scheme 1 Structural comparison of sulfur mustard and its simulant, 2-chloroethyl ethyl sulfide (CEES), along with the oxidation reaction of CEES into nontoxic sulfoxide (green) and toxic sulfone (red) products. | ||
Materials such as porphyrins,4,7 metal oxides,8–10 covalent-organic frameworks (COFs),11,12 and metal–organic frameworks (MOFs)4,5,13–16 have been used to degrade sulfur mustard and its simulant, 2-chloroethyl ethyl sulfide (CEES) (Scheme 1). Among these materials, MOFs have shown considerable promise as reusable catalysts due to their tunable structures, high porosity and surface areas, crystallinity, and heterogeneous nature. Porphyrinic zirconium MOFs, in particular, have been used in several photooxidation studies due to their exceptional stability and singlet oxygen production.17–19 The high chemical and thermal stability of Zr-porphyrin MOFs originates from their highly oxophilic ZrIV nodes. The rigid frameworks of these MOFs prevent photobleaching and aggregation of porphyrin molecules, thus increasing the amount of singlet oxygen that can be generated compared to free porphyrin molecules in solution.20
PCN-222/MOF-545 is one such MOF, consisting of 8-connected Zr6 clusters and tetrakis(4-carboxyphenyl)porphyrin (TCPP) linkers, which can selectively oxidize CEES into CEESO with a half-life of 9.0 min in methanol.4 Our previous study also found that the CEES oxidation rate was positively correlated to the surface area of some MOF catalysts; MOF-525, a 12-connected porphyrinic zirconium-based MOF with a higher surface area than PCN-222, was tested for the same reaction and achieved a faster half-life of 6.2 min in methanol.4 In addition, Zhao et al. found that CEES photooxidation could be further accelerated by using porphyrin-functionalized 2D metal–organic layers, which exhibited a half-life of only 1.2 min due to the more accessible porphyrin units and enhanced substrate transport on the 2D material.21
While studies have shown how additional photosensitizers, topologies, pore shapes and sizes, and metal node connectivities of MOF catalysts may impact the CEES oxidation rate, fewer studies have illustrated the effect of incorporating metal ions into the linkers of these MOFs. One study found that using Zn–TCPP instead of H2TCPP in a silver-chalcogenide cluster-based MOF produced a 2D framework and enhanced CEES oxidation by 1 min, with complete conversion after 6 min.22 In another study, a lanthanide MOF with Fe(II)–TCPP ligands, Fe–TCPP–La, catalyzed the oxidation of CEES to completion within 5 min, which was 2 min faster than its free base counterpart MOF, TCPP–La.23 More recently, Li et al. reported that Sn–TCPP and PCN-222(Sn) showed superior superoxide dismutase (SOD) catalytic activity compared to their free-base counterparts and other commonly used SOD nanozymes.24 We expected that the coordination of certain metal ions at the centers of porphyrinic ligands in Zr-MOFs would also accelerate the photooxidation of CEES, compared to free base porphyrin MOFs. In this study, we first prepared two metalated porphyrin ligands, In–TCPP and Sn–TCPP, and compared the two metalloporphyrinic MOFs, PCN-222(In) and PCN-222(Sn), with free base PCN-222 for the photooxidation of CEES (Fig. 1a).
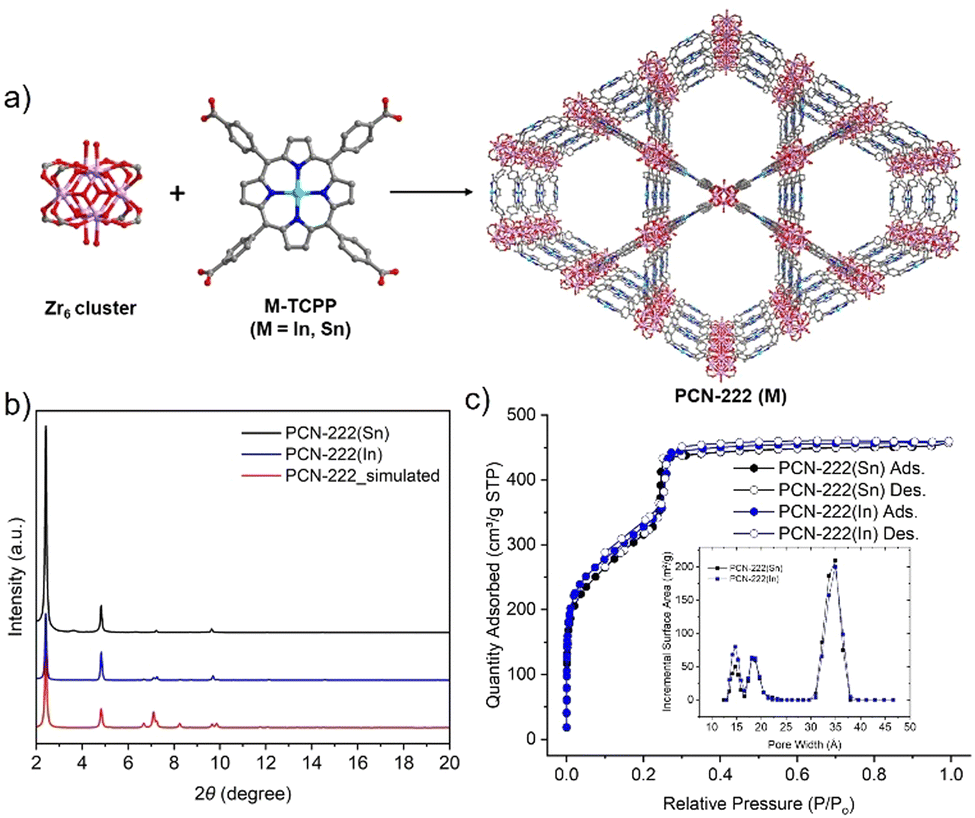 | ||
| Fig. 1 (a) The structures of Zr6-oxo cluster, metalated tetrakis(4-carboxyphenyl)porphyrin (TCPP) with indium (In) and tin (Sn), and metalated PCN-222. Chlorines attached to the center atoms are omitted for clarity. (b) Powder X-ray diffraction (PXRD) patterns of PCN-222(Sn) and PCN-222(In) compared to the simulated pattern of PCN-222. (c) Nitrogen sorption at 77 K and DFT pore size distributions (inset) for PCN-222(In) and PCN-222(Sn). | ||
In- and Sn–TCPP were synthesized following procedures adapted from literature (see ESI,† for more details).25,26 The UV-vis absorption spectra of the two metalated porphyrins exhibited bathochromically shifted Soret bands and Q bands relative to the UV-vis spectrum of free base TCPP, confirming the incorporation of metal ions at the center of the porphyrin (Fig. S2, ESI†). PCN-222(M), M = Sn(IV) or In(III), were synthesized following previously reported procedures for PCN-222(Fe) (see ESI,† for more details).25 The powder X-ray diffraction (PXRD) patterns of the activated PCN-222(M) were consistent with the simulated pattern of PCN-222 (Fig. 1b), confirming the phase purity of PCN-222(M). The permanent porosity of PCN-222(M) was studied using nitrogen sorption experiments at 77 K (Fig. 1c). PCN-222(Sn) and PCN-222(In) both showed type IV reversible nitrogen isotherms with similar nitrogen uptakes and Brunauer–Emmett–Teller (BET) surface areas of 1140 m2 g−1 and 1194 m2 g−1, respectively. The lower gravimetric surface areas of PCN-222(M) compared to that of PCN-222 can be attributed to the added mass of the metalated porphyrin ligands. The pore size distributions of PCN-222(M) are similar to that of PCN-222, with slightly smaller pores due to the chlorines on In/Sn pointing to the MOF open channels. Scanning electron microscope (SEM) images of PCN-222(Sn) and PCN-222(In) showed hexagonal rod-shaped crystals with similar crystal sizes between the two MOFs (Fig. S4a and b, ESI†). SEM images also revealed that PCN-222(Sn) has smooth crystal surfaces, while PCN-222(In) has cracks in the crystals. The metal content analysis of the digested PCN-222(M) using inductively coupled plasma optical emission spectroscopy (ICP-OES) further confirmed the incorporation of In and Sn in these MOFs (Table S2, ESI†).
As expected, the UV-vis spectra of PCN-222(M) showed intense Soret bands at 430 nm followed by smaller Q bands in the 500–700 nm range, indicating high absorptive ability for blue light (Fig. S3, ESI†). Upon this observation, we implemented a blue LED light source and performed photooxidation experiments at room temperature. Preliminary experiments using 0.5 mol% metalated porphyrin ligands Sn–TCPP and In–TCPP in methanol (homogeneous) yielded faster oxidation than 1.0 mol% loading (Fig. S5, ESI†). The slower oxidation observed with higher porphyrin loading could be attributed to porphyrin aggregation at higher concentrations, blocking potential catalytic sites. For all remaining reactions, we used 0.5 mol% catalyst loading.
We then compared the reaction rates of the activated PCN-222(M) in methanol to that of free base PCN-222. PCN-222(Sn) achieved the fastest CEES half-life of 3.3 min, followed by free base PCN-222 (t1/2 = 9 min), and then PCN-222(In) (t1/2 = 10 min) (Fig. 2a). PCN-222(Sn) exhibited a significantly shorter half-life and higher turnover frequency (TOF) than free base PCN-222 (Table S1, ESI†), suggesting that incorporating Sn into the porphyrin linkers increased the efficiency of photooxidation. Consistent with previous reports, the homogeneous metalloporphyrin solutions exhibited slightly faster CEES half-lives (2.5 min with In–TCPP and 2.6 min with Sn–TCPP) than their MOF counterparts.7,21,27 However, compared to homogeneous porphyrin solutions, heterogeneous MOF catalysts are easier to recycle and have superior long-term stability.271H NMR spectra confirmed that the only oxidation products were nontoxic sulfoxides, as shown in Fig. 2b and Fig. S7–S13 (ESI†). Remarkably, all CEES were converted to nontoxic products after 12 min with PCN-222(Sn), whereas the same reaction catalyzed by free base PCN-222 required 25 min to reach 100% conversion.4
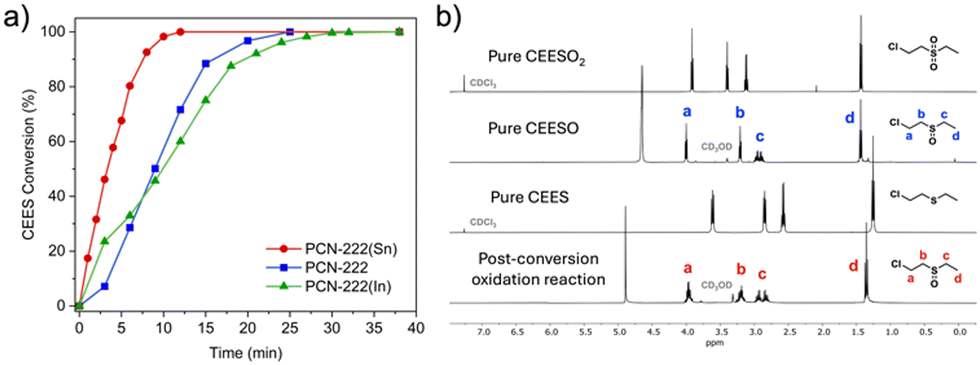 | ||
| Fig. 2 (a) Kinetics of CEES photooxidation with 0.5 mol% loading of PCN-222(Sn), and PCN-222(In) compared to 1.0 mol% loading of free base PCN-222 in MeOH. (b) 1H NMR spectra of pure CEESO2 in CDCl3, pure CEESO in CD3OD, and pure CEES in CDCl3, along with 1H NMR spectrum taken in CD3OD after complete CEES oxidation with PCN-222(Sn), indicating that the only product was CEESO. | ||
We used trapping experiments to identify the reactive oxygen species (ROS) generated by the MOFs. In addition to singlet oxygen (1O2), we also observed free radicals (˙O2−/˙OH) and peroxides (H2O2) (Fig. S17–S19, ESI†). PCN-222(Sn) generated significantly more singlet oxygen than free base PCN-222 (Fig. S17, ESI†), which could explain PCN-222(Sn)'s faster CEES oxidation rate. This difference can be attributed to the higher rate of intersystem crossing (ISC) of Sn–TCPP than that of free base TCPP. The faster CEES oxidation in the presence of PCN-222(Sn) compared to PCN-222(In) could also be explained by more efficient ISC in PCN-222(Sn) due to the slightly higher atomic number of Sn over In. Using heavy atoms is a common strategy to enhance ISC because spin–orbit coupling increases with atomic number.28,29 Interestingly, the reaction catalyzed by PCN-222(In) started faster than that with PCN-222 but ended up taking longer to reach 100% conversion than PCN-222. The slower reaction rate with PCN-222(In) could be attributed to the slightly less accessible pores in PCN-222(In) compared to PCN-222, which could slow down the substrate diffusion. The much faster singlet oxygen production by PCN-222(Sn) offset this slight decrease in pore size.
Encouraged by the high efficiency of the reaction catalyzed by PCN-222(Sn), we grafted the Sn–TCPP on 2D MOFs to further enhance the photooxidation efficiency. We hypothesized that Sn–TCPP (active centers) grafted on 2D MOFs would be more accessible by substrates than those in 3D MOFs, leading to faster CEES oxidation. To test this hypothesis, Sn–TCPP was incorporated into CAU-26 and CAU-26-NH2 using solvent-assisted linker incorporation (SALI)30 in DMF at 65 °C for 18 h (Fig. 3a). CAU-26 is a 2D MOF with Zr6-oxo clusters and benzene dicarboxylic acids (BDC) linkers, previously reported by Leubner et al.31 CAU-26-NH2 is a new 2D MOF we synthesized using an amine-functionalized BDC under similar synthetic conditions to CAU-26. After SALI, the dark purple precipitates were washed with DMF/acetone and dried under a vacuum.
 | ||
| Fig. 3 (a) The schematic structure of CSLA-21-NH2(Sn). (b) Powder X-ray diffraction (PXRD) patterns of simulated CAU-26 (black), and experimental CAU-26 (red), CAU-26-NH2 (blue), CSLA-21(Sn) (green), and CSLA-21-NH2(Sn) (purple). (c) Nitrogen sorption isotherms at 77 K for CAU-26, CAU-26-NH2, and Sn–TCPP modified MOFs. (d) The reaction kinetics of CEES oxidation in the presence of 0.5 mol% of CSLA-21(Sn) and CSLA-21-NH2(Sn) under blue LED irradiation. | ||
The modified MOFs, named CSLA-21(Sn) and CSLA-21-NH2(Sn), were characterized by PXRD, confirming the retention of the 2D MOF structures and phase purity (Fig. 3b). SEM images of the two modified MOFs showed layered structures with small sheets, indicating the retention of the 2D MOF structures after modification (Fig. S4c and d, ESI†). The BET surface areas of the 2D MOFs and Sn–TCPP-modified MOFs were determined using nitrogen sorption experiments at 77 K (Fig. 3c). As expected, the Sn–TCPP functionalized MOFs, CSLA-21(Sn) and CSLA-21-NH2(Sn), exhibited lower surface areas (326 m2 g−1 and 194 m2 g−1, respectively) than their parent MOFs (766 m2 g−1 and 590 m2 g−1 for CAU-26 and CAU-26-NH2, respectively) due to the occupation of the free space by Sn–TCPP. The successful incorporation of Sn–TCPP was further confirmed using ICP-OES analysis of the digested CSLA-21(Sn) and CSLA-21-NH2(Sn) (Table S2, ESI†).
The photooxidation of CEES was carried out using CSLA-21(Sn) and CSLA-21-NH2(Sn) under blue LED, and the reaction kinetics are shown in Fig. 3d. With a 0.5 mol% catalyst loading (calculated based on Sn–TCPP), the CEES degradation showed half-lives of 2.4 and 1.5 min for CSLA-21(Sn) and CSLA-21-NH2(Sn), respectively (Fig. 3d). Remarkably, in the presence of CSLA-21-NH2(Sn), all the CEES were oxidized to CEESO within 5.5 min, making it among the fastest MOFs studied for this reaction to date (Fig. S6 and Table S1, ESI†). The relatively short half-lives and high TOFs of the Sn–TCPP functionalized 2D MOF confirmed our hypothesis of their more accessible active sites and, thus, faster catalysis. Furthermore, the faster oxidation with CSLA-21-NH2(Sn) compared to CSLA-21(Sn) could be attributed to the hydrogen-bond donating properties of the amine functional groups, which was found to favor the formation of an intermediate in CEES oxidation in one of our previous studies.4 PXRD patterns of the MOFs taken after photooxidation experiments confirmed their structural stability as heterogeneous catalysts (Fig. S14 and S15, ESI†). Recyclability experiments also showed that all four MOFs can be used for at least four cycles of photooxidation without significant loss in catalytic activity (Fig. S16, ESI†). However, post-catalysis ICP experiments showed a loss in Sn and In, which was more significant for the 2D MOFs than the 3D MOFs (Table S2, ESI†). The ICP results suggested that the 2D MOFs are less robust than the 3D MOFs.
In this work, we aimed to improve the efficiency of CEES photooxidation using the metalloporphyrinic MOFs, PCN-222(Sn) and PCN-222(In), and the Sn-porphyrin functionalized 2D MOFs, CSLA-21(Sn) and CSLA-21-NH2(Sn). These four zirconium-based MOFs were synthesized, characterized, and tested in the presence of O2 under blue LED irradiation. PCN-222(Sn) achieved a significantly faster CEES half-life than free-base PCN-222, which suggests that incorporating certain metals such as tin into porphyrinic MOFs can enhance singlet oxygen production and, thus, CEES oxidation. In addition, incorporating the Sn-porphyrin linkers into 2D MOFs led to two new MOFs that exhibited even faster CEES oxidation with half-lives of 2.4 and 1.5 min due to more accessible active sites. Compared to currently used materials, CSLA-21-NH2(Sn) yields one of the fastest CEES half-lives and shortest completion times while maintaining high selectivity for nontoxic sulfoxides in methanol. This method of incorporating metal ions into porphyrins and MOF structures may be used in other material syntheses and catalytic reactions to significantly increase their photocatalytic efficiency. Future work may focus on increasing the robustness of 2D MOFs to improve their reusability.
A. S. Q. and D. M. carried out the experiments and data analysis. A. S. Q. wrote the initial draft of the manuscript. O. K. F. and Y. L. conceptualized the study, designed experiments, and revised the manuscript. A. N., E. K. P., P. W., and M. J. T. assisted with experiments and data analysis. L. A. and L. Q. assisted with SEM data collection, analysis, and manuscript editing. A. D. R. assisted with ICP analysis. All authors provided feedback for the manuscript.
This research was sponsored by the Army Research Office (W911NF-19-1-0001). Y. L. also acknowledges the NSF CREST program (HRD-2112554). P. W. was supported by the NIH Bridge to the Doctorate grant (5T32GM146700). L. Q. was supported by the U. S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, Catalysis Science program. The Ames National Laboratory is operated for the U. S. DOE by Iowa State University under Contract No. DE-AC02-07CH11358. We thank Omar Santana and Daniel Castaneda for assisting with some samples.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. C. G. Dacre, Pharmacol. Rev., 1996, 48(2), 289–326 CAS.
- A. Emad and G. R. Rezaian, Chest, 1997, 112(3), 734–738 CrossRef CAS PubMed.
- A. Jiang and H. Maibach, J. Appl. Toxicol., 2018, 38(1), 108–112 CrossRef CAS PubMed.
- Y. Hao, E. K. Papazyan, Y. Ba and Y. Liu, ACS Catal., 2022, 12(1), 363–371 CrossRef CAS.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54(31), 9001–9005 CrossRef CAS PubMed.
- N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. King and V. Hauschild, Environ. Health Perspect., 1999, 107(12), 933–974 CrossRef CAS PubMed.
- M. S. Lee, S. J. Garibay, A. M. Ploskonka and J. B. Decoste, MRS Commun., 2019, 9(2), 464–473 CrossRef CAS.
- T. H. Mahato, B. Singh, A. K. Srivastava, G. K. Prasad, A. R. Srivastava, K. Ganesan and R. Vijayaraghavan, J. Hazard. Mater., 2011, 192(3), 1890–1895 CrossRef CAS PubMed.
- V. Štengl, T. M. Grygar, F. Opluštil and T. Němec, J. Hazard. Mater., 2011, 192(3), 1491–1504 CrossRef PubMed.
- M. T. Naseri, M. Sarabadani, D. Ashrafi, H. Saeidian and M. Babri, Environ. Sci. Pollut. Res., 2013, 20(2), 907–916 CrossRef CAS.
- L. Zhang, C. Sun, S.-J. Xiao, Q.-G. Tan, G.-P. Yang, J.-Q. Fan, Y.-T. Luo, R.-P. Liang and J.-D. Qiu, ACS Appl. Nano Mater., 2023, 6(18), 17083–17091 CrossRef CAS.
- Q. Y. Wang, J. Liu, M. Cao, J. H. Hu, R. Pang, S. Wang, M. Asad, Y. L. Wei and S. Q. Zang, Angew. Chem., Int. Ed., 2022, 61(32), e202207130 CrossRef CAS PubMed.
- C. F. Pereira, Y. Liu, A. Howarth, F. Figueira, J. Rocha, J. T. Hupp, O. K. Farha, J. P. C. Tomé and F. A. Almeida Paz, ACS Appl. Nano Mater., 2019, 2(1), 465–469 CrossRef CAS.
- Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. Decoste, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4(36), 13809–13813 RSC.
- C. T. Buru, M. B. Majewski, A. J. Howarth, R. H. Lavroff, C.-W. Kung, A. W. Peters, S. Goswami and O. K. Farha, ACS Appl. Mater. Interfaces, 2018, 10(28), 23802–23806 CrossRef CAS PubMed.
- D. T. Lee, J. D. Jamir, G. W. Peterson and G. N. Parsons, Matter, 2020, 2(2), 404–415 CrossRef CAS.
- M. C. Oliver and L. Huang, Nanomaterials, 2023, 13(15), 2178 CrossRef CAS PubMed.
- P. Kalita, R. Paul, A. Boruah, G. Q. Dao, A. Bhaumik and J. Mondal, Green Chem., 2023, 25(15), 5789–5812 RSC.
- K. Yu, D.-I. Won, W. I. Lee and W.-S. Ahn, Korean J. Chem. Eng., 2021, 38(4), 653–673 CrossRef CAS.
- A. G. Griesbeck, T. T. El-Idreesy and A. Bartoschek, Adv. Synth. Catal., 2004, 346(2–3), 245–251 CrossRef CAS.
- H. Zhao, C.-A. Tao, S. Zhao, X. Zou, F. Wang and J. Wang, ACS Appl. Mater. Interfaces, 2023, 15(2), 3297–3306 CrossRef CAS PubMed.
- C.-H. Gong, Z.-B. Sun, M. Cao, X.-M. Luo, J. Wu, Q.-Y. Wang, S.-Q. Zang and T. C. W. Mak, Chem. Commun., 2022, 58(70), 9806–9809 RSC.
- Z.-H. Long, D. Luo, K. Wu, Z.-Y. Chen, M.-M. Wu, X.-P. Zhou and D. Li, ACS Appl. Mater. Interfaces, 2021, 13(31), 37102–37110 CrossRef CAS PubMed.
- L. Li, H. Li, L. Shi, L. Shi and T. Li, Langmuir, 2022, 38(23), 7272–7279 CrossRef CAS PubMed.
- D. Feng, Z. Y. Gu, J. R. Li, H. L. Jiang, Z. Wei and H. C. Zhou, Angew. Chem., Int. Ed., 2012, 51(41), 10307–10310 CrossRef CAS PubMed.
- A.-M. Manke, K. Geisel, A. Fetzer and P. Kurz, Phys. Chem. Chem. Phys., 2014, 16(24), 12029–12042 RSC.
- Y. Hao, B. M. Liu, T. F. Bennett, C. G. Monsour, M. Selke and Y. Liu, J. Phys. Chem. C, 2021, 125(13), 7392–7400 CrossRef CAS.
- A. Garci, J. A. Weber, R. M. Young, M. Kazem-Rostami, M. Ovalle, Y. Beldjoudi, A. Atilgan, Y. J. Bae, W. Liu, L. O. Jones, C. L. Stern, G. C. Schatz, O. K. Farha, M. R. Wasielewski and J. Fraser Stoddart, Nat. Catal., 2022, 5(6), 524–533 CrossRef CAS.
- Z. W. Jiang, Y. C. Zou, T. T. Zhao, S. J. Zhen, Y. F. Li and C. Z. Huang, Angew. Chem., Int. Ed., 2020, 59(8), 3300–3306 CrossRef CAS PubMed.
- P. Deria, J. E. Mondloch, E. Tylianakis, P. Ghosh, W. Bury, R. Q. Snurr, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2013, 135(45), 16801–16804 CrossRef CAS PubMed.
- S. Leubner, H. Zhao, N. Van Velthoven, M. Henrion, H. Reinsch, D. E. De Vos, U. Kolb and N. Stock, Angew. Chem., Int. Ed., 2019, 58(32), 10995–11000 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03890h |
| ‡ A. S. Q and D. M. contributed equally. |
| § Current address: California University of Science and Medicine, Colton, California 92324, United States. |
| This journal is © The Royal Society of Chemistry 2025 |