
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe temperature dependence of Mössbauer quadrupole splitting values: a quantum chemical analysis†
Niklas
von Rhein
 and
Vera
Krewald
*
and
Vera
Krewald
*
Department of Chemistry, Quantum Chemistry, TU Darmstadt, Peter-Grünberg-Str. 4, 64287 Darmstadt, Germany. E-mail: vera.krewald@tu-darmstadt.de
First published on 15th January 2025
Abstract
The two key parameters extracted from Mössbauer spectroscopy, isomer shift and quadrupole splitting, have well-known temperature dependencies. While the behavior of the values following a temperature change has long been known, its microscopic origins are less clear. For quantum chemical calculations – formally representing the situation at 0 K – significant discrepancies with the experiment can arise, especially at elevated temperatures. Herein, we introduce an ensemble-based approach for capturing the temperature dependence of the quadrupole splitting. Our method is exemplified with [Fe(TPP)(2-MeHIm)], an iron(II) high spin system. We rationalise the temperature dependence by changes in the shape of the charge distribution due to vibrational distortion. By using a normal mode fitting approach, we isolated collective nuclear movements associated with the change in the quadrupole splitting.
Mössbauer spectroscopy is a key method for investigating structural and electronic properties of iron nuclei in molecular environments,1e.g. bioinorganic systems like hemoglobin, P450, and nitrogenase,2–7 molecular complexes and catalysts,8–14 or single atom catalysts15–18 such as FeNC catalysts for the oxygen reduction reaction.19–26 Mössbauer spectroscopy is often used to elucidate structures and electronic structures, usually in combination with density functional theory calculations on known or plausible systems.27–35 While Mössbauer spectroscopy—like many other spectroscopies—shows a temperature dependence,11,12,36–38 quantum chemical calculations are usually restricted to a rigid equilibrium geometry at absolute zero.39 For Mössbauer spectroscopy, this limits the direct comparison of experiment and theory to experimental temperatures below ca. 80 K, and makes the assignment of signals observed under in situ or operando conditions at higher temperatures significantly less reliable.
The two key Mössbauer parameters, isomer shift and quadrupole splitting (see Fig. 1), are altered by several temperature-dependent effects,1 such as the second-order Mössbauer effect on the isomer shift.1,40,41 An effect that may influence both isomer shift and quadrupole splitting is that different electronic states may be thermally accessible at a given temperature due to the energetic proximity of different configurations or multiplicities, or due to even smaller energy gaps introduced by spin–orbit coupling. These effects are exploited in spin crossover complexes.42–49
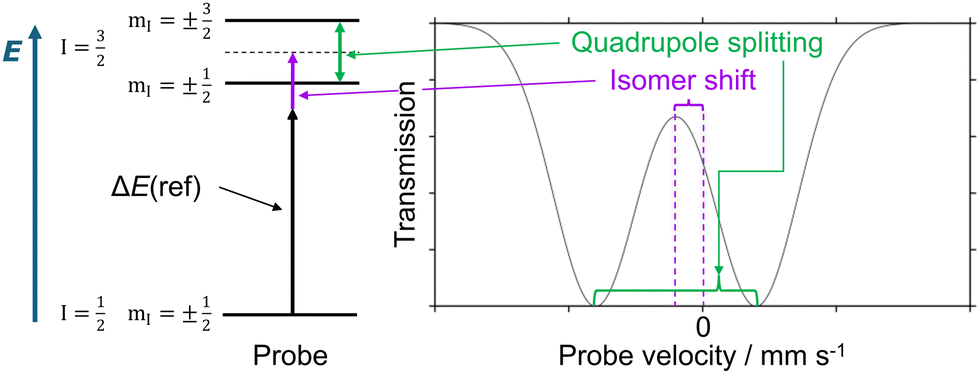 | ||
| Fig. 1 Right: Exemplary Mössbauer doublet signal plotted as transmission vs. probe velocity. The isomer shift (violet) refers to the center of the doublet with respect to a reference (typically, α-iron). The quadrupole splitting (green) measures the separation of the doublet minima. Left: Diagram with the nuclear energy levels; labels refer to the nuclear spin quantum number I and the nuclear magnetic quantum number mI. | ||
Herein, we focus on an effect that mainly influences the quadrupole splitting: the influence of vibrational movements of a molecular complex that are inherently temperature-sensitive.
The quadrupole splitting and its sign depend on the shape of the charge distribution around the iron nucleus. The electric field gradient tensor (EFG) has diagonal components |Vzz| ≥ |Vyy| ≥ |Vxx|. If |Vzz| ≫ |Vyy|, a positive sign of the quadrupole splitting indicates an oblate charge distribution, while a negative sign corresponds to a prolate one. Even from basic crystal field theory considerations it is clear that the EFG will be influenced by the ligand field splitting.1 Vibrational movements will influence the charge distribution as well as the ligand field splitting and consequently the magnitude and shape of the EFG. Since the macroscopic measurement probes the superposition of all vibrationally accessible structures, the observed quadrupole splitting is expected to be sensitive to the temperature.
To the best of our knowledge, the influence of vibrational distortions on Mössbauer parameters has not yet been evaluated in a quantum chemical study. Our approach is to generate an ensemble representative for a given temperature using Wigner sampling.50–52 This allows us to study the temperature-dependence of the quadrupole splitting explicitly for the first time. Moreover, our analysis provides possible explanations for the experimental observations that are rooted in an electronic structure and normal mode analysis of the ensemble.
Our approach requires three steps as described briefly in the following; details are given in the ESI.† All quantum chemical calculations were performed using the ORCA 5.0 program suite (see ESI,† Method S1).53–55 (1) A DFT geometry optimization and frequency calculation is carried out; here, TPSS/def2-TZVP:def2-SVP (heteroatoms: carbon, hydrogen) is chosen as the theory level.56–58 (2) Using Wigner sampling50,51 as implemented in the SHARC program,59,60 an ensemble of displaced structures is created that is representative for a given temperature (see ESI,† Method S2); here, 600 structures are generated per temperature of interest (see ESI,† Fig. S1, for convergence behaviour). In contrast to MD-based approaches, Wigner sampling uses the harmonic approximation and takes into account zero-point energies.61 (3) For all structures in the ensemble, the Mössbauer parameters are calculated using DFT; here, in accordance with a previous calibration study,27 B3LYP/CP(PPP):def2-TZVP (iron:all other atoms) is chosen as the theory level.58,62,63 This theory level has uncertainties of ±0.13 mm s−1 and ±0.22 mm s−1 for the isomer shift and the quadrupole splitting, respectively, as determined by an in-house calibration study following the work of Gallenkamp et al.27 using ORCA 5.0 instead of ORCA 4.2 (see ESI,† Method S1).
To test our approach, we chose the Fe(II) high spin complex [Fe(TPP)(2-MeHIm)], see Fig. 2. Its experimental Mössbauer parameters are taken from ref. 11, see Table 1.11,12 Fe(II) high spin complexes are advantageous as they have exceptionally high isomer shifts which are easily discernible from other spin and oxidation states.1 While iron is notorious for its complicated electronic structure, which often renders it challenging for DFT as a single-determinant method,64–67 the large number of single point calculations per ensemble required DFT as an efficient computational method. The high spin state of [Fe(TPP)(2-MeHIm)] was identified as the ground state (see ESI,† Table S1, for spin state energies). The Mössbauer parameters for the equilibrium structure are δ = 0.94 mm s−1 and ΔEQ = 2.84 mm s−1, in good to reasonable agreement with the experiment. We note that the quadrupole splitting lies outside of the error bars from the calibration study, which may be due to zero-point vibrational effects as discussed in the following.
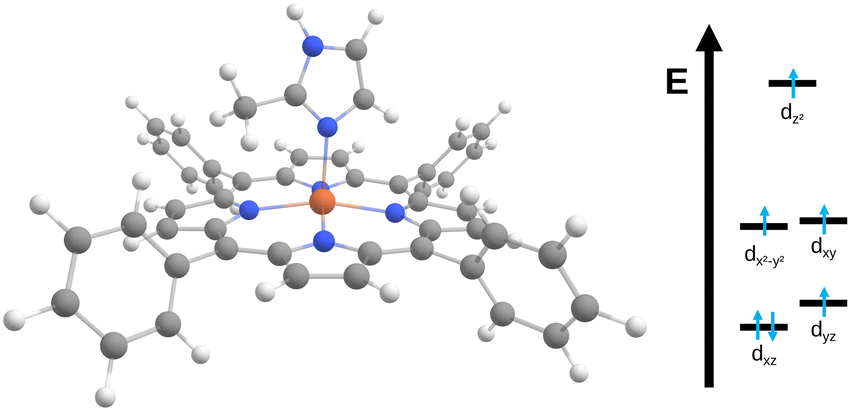 | ||
| Fig. 2 Structure of the [Fe(TPP)(2-MeHIm)] complex11 studied here alongside a qualitative molecular orbital diagram. | ||
| Experiment | Theory (all) | Theory (filtered) | ||||
|---|---|---|---|---|---|---|
| T/K | δ/mm s−1 | ΔEQ/mm s−1 | δ/mm s−1 | ΔEQ/mm s−1 | δ/mm s−1 | ΔEQ/mm s−1 |
| 77 | 0.92 | 2.26 | 0.94 | 2.09 | 0.94 | 2.00 |
| 195 | 0.87 | 1.97 | 0.94 | 1.69 | 0.94 | 1.59 |
| 300 | 0.82 | 1.74 | 0.94 | 1.65 | 0.94 | 1.47 |
The Mössbauer parameters of all structures in the generated ensembles at 77 K, 195 K and 300 K are shown in Fig. 3. For the isomer shift, no temperature dependence is observed in agreement with experiment. Strikingly, for the quadrupole splitting we not only observe the expected scatter around the positive value of the equilibrium structure, but in addition that some members of the ensemble have negative quadrupole splitting values. Moreover, the number of species with negative quadrupole splitting values increases with increasing temperature. We thus conclude that a key contributor to the temperature dependence of the quadrupole splitting are vibrational distortions that—crucially—lead to a swap in the sign of the quadrupole splitting. Note that experimentally, only the absolute value of the quadrupole splitting is routinely measured. Indeed, averaging the quadrupole splitting value over the ensemble, see Table 1 and Fig. 4, qualitatively reproduces the experimental trend of a decreasing quadrupole splitting with higher temperature. We also note that the agreement with the experimental quadrupole splitting value at low temperatures improves.
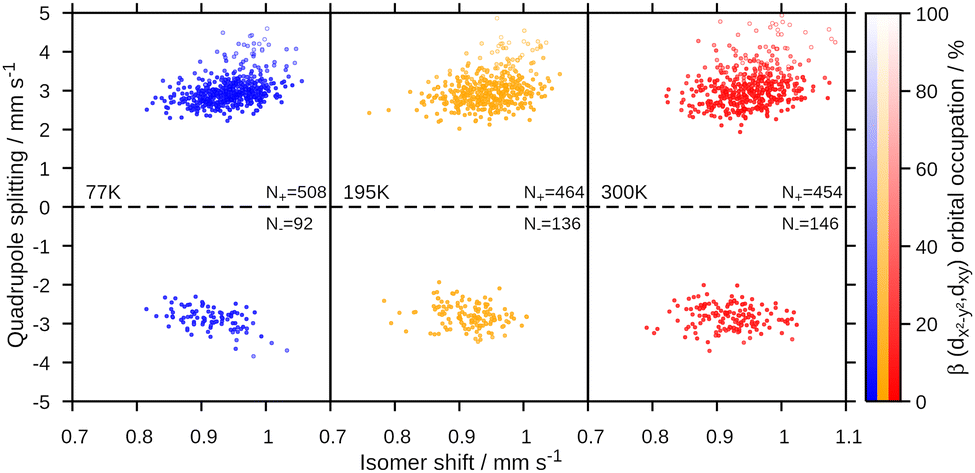 | ||
| Fig. 3 Quadrupole splitting vs. isomer shift for all structures of the created ensembles at different temperatures. Labels indicate the temperature (left) and the number of structures with a positive/negative quadrupole splitting (right). The brightness of the points is defined by the sum of the dx2−y2(β) and dxy(β) orbital occupation. | ||
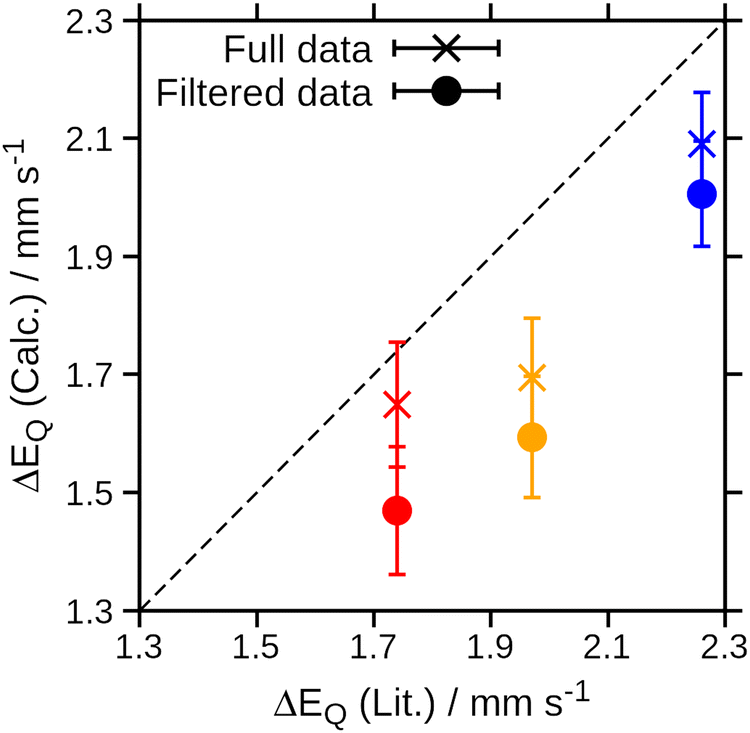 | ||
| Fig. 4 Correlation of the experimental quadrupole splitting versus the computed quadrupole splitting averaged after Wigner sampling. Data points shown as crosses refer to the full data set; those shown as circles refer to the filtered data set. Error bars denote the statistical error obtained for the respective ensemble. The dashed line shows the ideal behaviour with a slope of 1. | ||
The observed changes in experimental and computed quadrupole splitting values can be traced back to the interlinked electronic and molecular structures. For an iron(II) high spin complex, one d orbital is doubly occupied. In the equilibrium structure, this is dxz, see Fig. 2. When the structure is distorted along the normal coordinates, different orbitals can be stabilized, leading to different electronic configurations, hence different charge distributions and thus ultimately different quadrupole splitting values. For [Fe(TPP)(2-MeHIm)], two relevant electronic configurations were identified. In one configuration, three electrons are distributed between the dxz and dyz orbitals, with the exact distribution depending on the deformation of the molecule and the mixing of those two orbitals. In the other configuration, the electrons are distributed between the dx2–y2 and dxy orbitals. This is represented in Fig. 3 by the brightness of the data points, which is scaled by the sum of the dx2−y2(β) and dxy(β) orbital occupation. The configuration with (dx2−y2, dxy)3 has an overall larger quadrupole splitting than that with the (dxz, dyz)3 configuration.
We note that filtering out the (dxz, dyz)3 configuration better reproduces the trend in the temperature dependence of the quadrupole splitting (see Table 1 and Fig. 4), albeit with a more pronounced discrepancy with the experimental values. This may indicate that on one hand, the vibrational component of the temperature effect is adequately captured for a single spin state. On the other hand, it may be the case that different spin states contribute in the experiment, either due to structural distortions caused by zero-point vibrations or electronic (near-)degeneracies, the ratio of which is likely not adequately represented by the single-determinant DFT calculations used here.
With the ensemble of vibrationally distorted structures in hand, the changes in Mössbauer parameters can be correlated to displacements along the normal coordinates of the [Fe(TPP)(2-MeHIm)] complex. Correlating these changes to individual normal coordinates does not deliver satisfactory results, see ESI,† supplemental file “Ensemble_Data.xlsx”. Therefore, we turned to a multicomponent fitting procedure, in which the normal coordinates are used as basis functions to obtain collective motions that represent the observed changes. Methodological details are given in the ESI,† Methods S3 and S4.
For the isomer shift, a motion with a high correlation coefficient (R2 > 0.9) was obtained. The fit parameters and nuclear movements are similar at all temperatures (see ESI,† Fig. S2).
For the quadrupole splitting, the R2 value is lower (0.6183 to 0.6758) and the coefficients for each normal coordinate vary more strongly for the different temperatures, see Fig. S3 (ESI†). The resulting movement is similar at all temperatures. It comprises a strong out-of-plane movement of the iron ion combined with substantial stretching of the axial iron-nitrogen bond and weaker stretching of the in-plane iron-nitrogen bonds, see Fig. 5. This fits the expectations: the charge distribution in the out-of-plane direction changes strongly with this movement, thus converting its overall shape from oblate to prolate and vice versa, which hence results in the observed sign swap of the quadrupole splitting. The differences in the calculated nuclear movements for different temperatures lie mostly in the motion of the in-plane nitrogen atoms. Their motion is more difficult to capture due to the asymmetry around the iron nucleus introduced by the 2-MeHIm ligand and its CH3 group close to the FeN4 centre combined with the strong symmetry dependence of the quadrupole splitting.
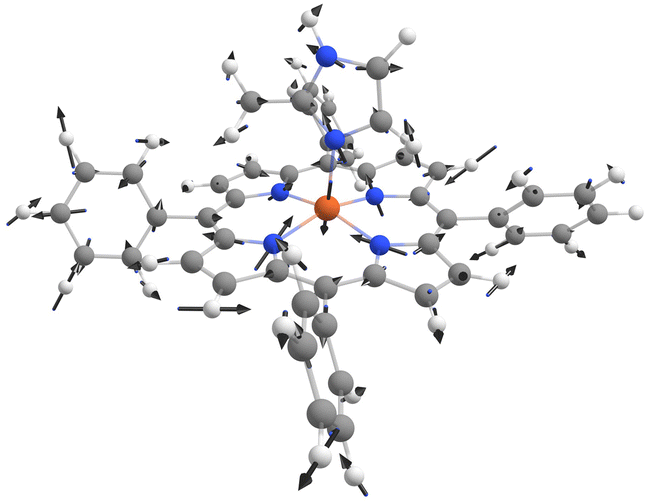 | ||
| Fig. 5 Nuclear movement of the atoms in a [Fe(TPP)(2-MeHIm)] complex correlated to the quadrupole splitting calculated by a linear combination of normal modes at 77 K. The correlation coefficient is R2 = 0.6183. | ||
To summarise, we have shown that a Wigner sampling approach generating a representative ensemble at different temperatures is indeed able to reproduce the vibrational temperature dependence of the quadrupole splitting. We identified as the root cause of the vibrational component of its temperature dependence that distortions lead to a swap in sign. A quantitative comparison with experimental data is likely hampered by different electronic spin ground states upon distortion incorrectly represented with DFT. Using a linear combination of normal modes, we have shown which collective nuclear movements are responsible for the observed changes. This provides a deeper understanding of the quadrupole splitting and its temperature dependence in general: the calculated movement explains how the charge distribution is converted from an oblate to a prolate shape and hence rationalises the sign swap for the quadrupole splitting.
N. V. R. gratefully acknowledges funding by the Deutsche Bundesstiftung Umweld (DBU). N. V. R. and V. K. gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – CRC 1487, “Iron, upgraded!” – project number 443703006. Computing time on the high-performance computer Lichtenberg II at NHR4CES at TU Darmstadt is gratefully acknowledged. This paper is dedicated to the memory of Dr Eckhard Bill.
Data availability
The data supporting this article have been included as part of the ESI.† The computational codes used to generate the primary data are available free of charge for academic users.Conflicts of interest
There are no conflicts to declare.Notes and references
-
P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011 Search PubMed
.
-
E. Bill, Practical Approaches to Biological Inorganic Chemistry, Elsevier, 2020, pp. 201–228 Search PubMed
-
Bioinorganic Chemistry, ed. M. J. Clarke, Springer, Berlin and Heidelberg, 1991, vol. 78 Search PubMed
- F. Neese, Curr. Opin. Chem. Biol., 2003, 7, 125–135 CrossRef CAS PubMed
- M. D. Pluth and Z. J. Tonzetich, Chem. Soc. Rev., 2020, 49, 4070–4134 RSC
- H. T. Dong, C. J. White, B. Zhang, C. Krebs and N. Lehnert, J. Am. Chem. Soc., 2018, 140, 13429–13440 CrossRef CAS PubMed
- D. Nam, J.-P. Bacik, R. L. Khade, M. C. Aguilera, Y. Wei, J. D. Villada, M. L. Neidig, Y. Zhang, N. Ando and R. Fasan, Nat. Commun., 2023, 14, 7985 CrossRef CAS PubMed
- B. Boso, G. Lang and C. A. Reed, J. Chem. Phys., 1983, 78, 2561–2567 CrossRef CAS
- C. Hu, J. An, B. C. Noll, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2006, 45, 4177–4185 CrossRef CAS PubMed
- C. Hu, B. C. Noll, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2018, 57, 793–803 CrossRef CAS PubMed
- J. P. Collman, J. L. Hoard, N. Kim, G. Lang and C. A. Reed, J. Am. Chem. Soc., 1975, 97, 2676–2681 CrossRef CAS PubMed
- D. Dolphin, J. R. Sams, T. B. Tsin and K. L. Wong, J. Am. Chem. Soc., 1976, 98, 6970–6975 CrossRef CAS PubMed
- M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischauer, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2019, 52, 140–150 CrossRef CAS PubMed
-
V. Schünemann, in Modern Mössbauer Spectroscopy, ed. Y. Yoshida and G. Langouche, Springer Singapore, Singapore, 2021, vol. 137 of Topics in Applied Physics, pp. 173–219 Search PubMed
- C. Gallenkamp, U. I. Kramm and V. Krewald, Chem. Commun., 2021, 57, 859–862 RSC
- X. Li, K. Zhu, J. Pang, M. Tian, J. Liu, A. I. Rykov, M. Zheng, X. Wang, X. Zhu, Y. Huang, B. Liu, J. Wang, W. Yang and T. Zhang, Appl. Catal., B, 2018, 224, 518–532 CrossRef CAS
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Adv. Mater., 2019, 31, e1808193 CrossRef PubMed
- Y. Zeng, X. Li, J. Wang, M. T. Sougrati, Y. Huang, T. Zhang and B. Liu, Chem. Catal., 2021, 1, 1215–1233 CrossRef CAS
- J. S. Bates, J. J. Martinez, M. N. Hall, A. A. Al-Omari, E. Murphy, Y. Zeng, F. Luo, M. Primbs, D. Menga, N. Bibent, M. T. Sougrati, F. E. Wagner, P. Atanassov, G. Wu, P. Strasser, T.-P. Fellinger, F. Jaouen, T. W. Root and S. S. Stahl, J. Am. Chem. Soc., 2023, 145, 26222–26237 CrossRef CAS PubMed
- U. I. Koslowski, I. Abs-Wurmbach, S. Fiechter and P. Bogdanoff, J. Phys. Chem. C, 2008, 112, 15356–15366 CrossRef CAS
- U. I. Kramm, I. Herrmann-Geppert, S. Fiechter, G. Zehl, I. Zizak, I. Dorbandt, D. Schmeißer and P. Bogdanoff, J. Mater. Chem. A, 2014, 2, 2663–2670 RSC
- U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J.-P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS PubMed
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef CAS PubMed
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, e1805623 CrossRef PubMed
- J. Li, S. Ghoshal, W. Liang, M.-T. Sougrati, F. Jaouen, B. Halevi, S. McKinney, G. McCool, C. Ma, X. Yuan, Z.-F. Ma, S. Mukerjee and Q. Jia, Energy Environ. Sci., 2016, 9, 2418–2432 RSC
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed
- C. Gallenkamp, U. I. Kramm, J. Proppe and V. Krewald, Int. J. Quantum Chem., 2021, 121, e26394 CrossRef CAS
- C. Gallenkamp, U. I. Kramm and V. Krewald, JACS Au, 2024, 4, 940–950 CrossRef CAS PubMed
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chemistry, 2020, 6, 3440–3454 CrossRef CAS
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS
- D. Menga, A. Guilherme Buzanich, F. Wagner and T.-P. Fellinger, Angew. Chem., 2022, 61, e202207089 CrossRef CAS PubMed
- L. Ni, C. Gallenkamp, S. Paul, M. Kübler, P. Theis, S. Chabbra, K. Hofmann, E. Bill, A. Schnegg, B. Albert, V. Krewald and U. I. Kramm, Adv. Energy Sustainability Res., 2021, 2, 2000064 CrossRef CAS
- L. Ni, C. Gallenkamp, S. Wagner, E. Bill, V. Krewald and U. I. Kramm, J. Am. Chem. Soc., 2022, 144, 16827–16840 CrossRef CAS PubMed
- M. Pápai and G. Vankó, J. Chem. Theory Comput., 2013, 9, 5004–5020 CrossRef PubMed
- U. N. Morzan, D. J. Alonso de Armiño, N. O. Foglia, F. Ramírez, M. C. González Lebrero, D. A. Scherlis and D. A. Estrin, Chem. Rev., 2018, 118, 4071–4113 CrossRef CAS PubMed
- P. R. Edwards, C. E. Johnson and R. J. P. Williams, J. Chem. Phys., 1967, 47, 2074–2082 CrossRef CAS
- D. E. Freedman, W. H. Harman, T. D. Harris, G. J. Long, C. J. Chang and J. R. Long, J. Am. Chem. Soc., 2010, 132, 1224–1225 CrossRef CAS PubMed
- M. T. Sougrati, V. Goellner, A. K. Schuppert, L. Stievano and F. Jaouen, Catal. Today, 2016, 262, 110–120 CrossRef CAS
-
F. Jensen, Introduction to Computational Chemistry, John Wiley & Sons Incorporated, Newark, 1st edn, 2017 Search PubMed
- A. L. Kholmetskii, T. Yarman, O. V. Missevitch and B. I. Rogozev, Phys. Scr., 2009, 79, 065007 CrossRef
- R. F. Wallis and D. C. Gazis, Phys. Rev., 1962, 128, 106–110 CrossRef CAS
- C. M. Grunert, S. Reiman, H. Spiering, J. A. Kitchen, S. Brooker and P. Gütlich, Angew. Chem., 2008, 120, 3039–3041 CrossRef
-
P. Gütlich and H. A. Goodwin, in Spin Crossover in Transition Metal Compounds I, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004, vol. 233 of Topics in Current Chemistry, pp. 1–47 Search PubMed
- P. Gütlich, Eur. J. Inorg. Chem., 2013, 581–591 CrossRef
-
Spin Crossover in Transition Metal Compounds I, ed. P. Gütlich and H. A. Goodwin, Springer Berlin Heidelberg, Berlin, Heidelberg, 2004 Search PubMed
- P. Gütlich and A. Hauser, Coord. Chem. Rev., 1990, 97, 1–22 CrossRef
- F.-L. Yang, B. Li, T. Hanajima, Y. Einaga, R.-B. Huang, L.-S. Zheng and J. Tao, Dalton Trans., 2010, 39, 2288–2292 RSC
- B. Weber, Coord. Chem. Rev., 2009, 253, 2432–2449 CrossRef CAS
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303–7338 RSC
- E. Wigner, Phys. Rev., 1932, 40, 749–759 CrossRef CAS
- L. Sun and W. L. Hase, J. Chem. Phys., 2010, 133, 044313 CrossRef PubMed
- A. Šrut, B. J. Lear and V. Krewald, Chem. Sci., 2023, 14, 9213–9225 RSC
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS
- F. Neese, F. Wennmohs, D. Aravena, M. Atanasov, A. Auer, A. Alexander, U. Becker, G. Bistoni and D. Bykov, et al., ORCA – An ab initio, DFT and semiempirical SCF-MO package – Version 5.0.3: Manual.
- F. Neese, WIREs Comput. Mol. Sci., 2022, 12, 12753 Search PubMed
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- S. Mai, P. Marquetand and L. Gonzalez, WIREs Comput. Mol. Sci., 2018, 8, e1370 CrossRef PubMed
- S. Mai, D. Avagliano, M. Heindl, P. Marquetand, M. F. S. J. Menger, M. Oppel, F. Plasser, S. Polonius, M. Ruckenbauer, Y. Shu, D. G. Truhlar, L. Zhang, P. Zobel and L. Gonzalez, SHARC3.0: Surface Hopping Including Arbitrary Couplings—Program Package for Non-Adiabatic Dynamics, https://sharc-md.org/, 2023 Search PubMed.
- J. P. Zobel, O. S. Bokareva, P. Zimmer, C. Wölper, M. Bauer and L. González, Inorg. Chem., 2020, 59, 14666–14678 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef
-
M. Radoń, in Computational Chemistry, ed. R. van Eldik and R. Puchta, Elsevier, Amsterdam, 2019, vol. 73 of Advances in Inorganic Chemistry, pp. 221–264 Search PubMed
- M. Radoń, J. Chem. Theory Comput., 2014, 10, 2306–2321 CrossRef PubMed
- M. Swart, J. Chem. Theory Comput., 2008, 4, 2057–2066 CrossRef CAS PubMed
- G. Li Manni, D. Kats, D. P. Tew and A. Alavi, J. Chem. Theory Comput., 2019, 15, 1492–1497 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc03943b |
| This journal is © The Royal Society of Chemistry 2025 |