
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand labilization gates intramolecular electron transfer in molecular photocatalyst†
Louis
Blechschmidt
ab,
Linda
Zedler
ab,
Alexander K.
Mengele
 c,
Sven
Rau
c and
Benjamin
Dietzek-Ivanšić
*ab
c,
Sven
Rau
c and
Benjamin
Dietzek-Ivanšić
*ab
aInstitute of Physical Chemistry, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
bDepartment Functional Interfaces, Leibniz Institute of Photonic Technology (IPHT), Albert-Einstein-Straße 9, 07745 Jena, Germany. E-mail: Benjamin.dietzek@leibniz-ipht.de
cInstitute of Inorganic Chemistry I, Ulm University, Albert-Einstein-Allee 11, 89081 Ulm, Germany
First published on 3rd January 2025
Abstract
Identification of rate determining steps concerning catalyst activation and catalytic turnover is key to optimize molecular photocatalysts. In this contribution, femtosecond transient absorption spectroscopy upon variation of temperature and ionic strength yields new insights into the light-driven reactivity of the benchmark molecular photocatalyst, RutpphzRhCp*.
To understand and optimize molecular photocatalysts it is important to identify rate determining steps – both in catalysis and in the excited-state processes leading to the formation of the catalytically active intermediates. In this context, [(tbbpy)2Ru(tpphz)Rh(Cp*)Cl]Cl(PF6)2 (RutpphzRhCp*; tbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, Cp* = pentamethylcyclopentadienyl)1 has evolved into a well-studied “drosophila” photocatalyst.1–4 It consists of a photoredox active Ru-polypridyl moiety4,5 connected to a catalytically competent RhCp*-center5–13via the well-known tetrapyridophenazine (tpphz)-bridge.14–16 The bridge is fully conjugated and it is structurally rigid – different from other examples capable of light-driven intramolecular redox processes.17–20
In recent years, different tpphz-based architectures have been used to study various mechanistic details of light-driven reductive catalysis in detail, e.g., the role of excitation wavelength in the catalytic activity,21 the dynamics of the charge transfer across the bridging ligand,22–26 the impact of the nature of the catalytically active metal center,27–30 and the ability to actively repair the degraded photocatalyst.31
Zedler et al. characterized the photoinduced activation of RutpphzRhCp* with the help of transient absorption spectroscopy, spectroelectrochemistry, resonance-Raman and quantum-chemical simulations.22 As a result of these investigations the following picture for the light-driven reactivity of the photocatalyst in the presence of substrate and a sacrificial electron donor has emerged: excitation of the Ru-MLCT band (400 nm to 500 nm) populates both, tbbpy- and tpphz-centered 1MLCT states. Intersystem crossing and ultrafast relaxation yields a 3MLCT state with excess electron density localized at the phenanthroline moiety (phen) of tpphz within about 2 ps after light absorption. Intra-ligand charge transfer (ILCT) towards the central phenazine (phz) moiety in tpphz is reflected in a rise of excited state absorption (ESA) at 600 nm (τ2 = 11 ps). This phz-centered state finally transfers the negative charge to the rhodium-center, formally reducing the metal to rhodium(II). Under catalytic conditions a suitable electron donor is used to fill the electron hole at the ruthenium(II)-ion thus enabling a subsequent second light-driven reduction of the rhodium-center, forming rhodium(I) and, by that, photocatalysis.22
Nonetheless, the question remains to be addressed, why the charge-transfer across the rigid and fully conjugated bridge is comparably slow, i.e., associated with a characteristic first-order lifetime of 450 ps (at room temperature in acetonitrile). Both processes, the analogue ligand-to-metal charge transfer (LMCT from the central phz-unit towards the osmium(III)-center), as well as the subsequent charge recombination in closely related photoexcited [(bpy)2RuII(tpphz)OsIII(bpy)2]5+, dissolved in acetonitrile, are faster with time constants of 30 ps and 230 ps, respectively.24 Therefore, structural rearrangements of the bridging ligand, which have been identified to play a key role in structurally flexible molecular photocatalysts,3,17–20 cannot be responsible for the comparably slow electron transfer (ET) in RutpphzRhCp* due to the structural rigidity of the ligand. To study this aspect, the work presented here applies temperature-dependent ultrafast transient absorption spectroscopy in an attempt to identify possible barriers along the excited-state ET path, which might account for the comparably slow charge transfer kinetics. In doing so we recorded transient absorption data in a temperature range between 240 K and 300 K, while focusing on the ultrafast, i.e., sub-ns light-induced dynamics.
The kinetics shown in Fig. 1(b) reflect the transient optical density changes of the sample. The kinetics were obtained by spectrally integrating the differential absorption changes (see Fig. 1(a)) in the wavelength range between 570 and 630 nm. Considering the remaining differential absorption signal after 2 ns, it becomes apparent that the residual ESA signal decreases upon decreasing the temperature from 300 to 240 K. This observation appears counterintuitive, since one would expect that at 300 K a higher kinetic rate is achieved, which would result in less ESA signal 2 ns after the initial excitation, compared to the low temperature experiment at 240 K. However, analysing the rate of the ESA decline by exponential fitting reveals that the decay of the visible ESA band becomes faster (yet less in amplitude) at elevated temperatures.
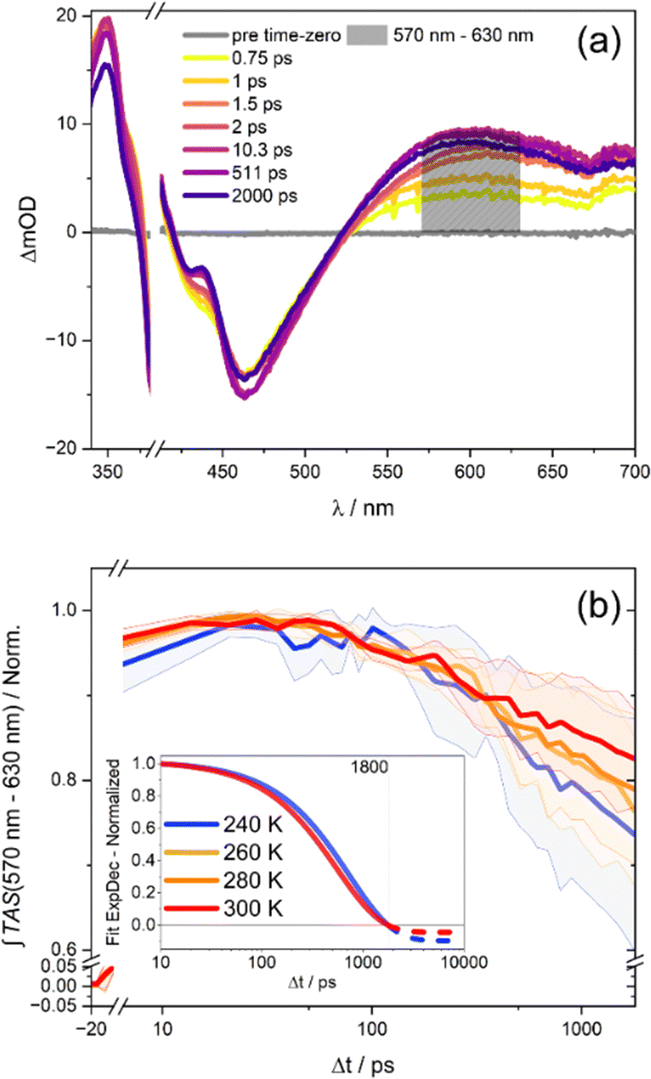 | ||
| Fig. 1 (a) Transient absorption spectra (TAS) of RutpphzRhCp* after excitation at 400 nm and approx. 240 K at different delay times. The changes in the ESA band above 550 nm are assigned to the evolution and consumption of the phz-centered state. (b) The OD values between 570 nm to 630 nm were integrated for TASs at multiple delay-times to estimate the kinetics of the phz-centered state. The inset shows the curves at the highest and lowest temperature after fitting with a monoexponential decay from 10 ps to 1800 ps and [0, 1] normalization within the same borders. Furthermore, the spotted lines in the inset show the extrapolated values of the fits until 10 ns. | ||
The transient absorption data were fitted according to the four-component model (τ1, τ2, τ3, τinf) described above, which resulted in the same decay associated spectra (DAS) at each temperature. Therefore, the assignments of the time constants to their respective transition processes provided by Zedler et al. were adopted.22 A comparison between the fitted time constants over the temperature of the experiment shows, that specifically the characteristic time constant accounting for the ESA decay on a sub-ns timescale, τ3, increases with decreasing temperature (see Fig. 2); the corresponding first-order rate, k3 = 1/τ3 decreases. Fitting also reveals that the long-lived component, which reflects the temperature-dependent long-delay time offset in the transient data (as mentioned before), is altered with temperature. This finding indicates that the phz-centered MLCT state does not fully decay to a state, which involves the rhodium-center. The time-constant likely reflects the formation of an equillibrium of states, which contains phz-centered MLCT and rhodium-centred charge-transfer character. The result of these two temperature-dependent effects, i.e., thermally activated formation of the excited-state equilibrium and temperature-dependent equilibrium, causes the excited-state absorption kinetics as observed.
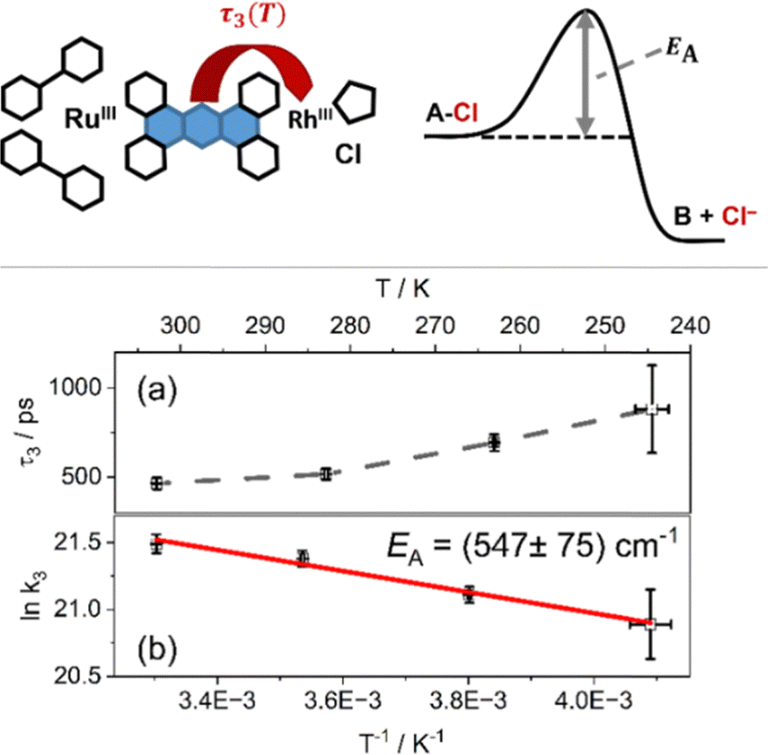 | ||
| Fig. 2 Schematic illustration of the electron transfer in photoexcited RutpphzRhCp* (from the phz-centered state to the rhodium-center) with an energy diagram scheme. (a) Plot of the assigned time constant over the temperature; (b) plot of the logarithm of the correlated kinetic constant over the inverse temperature (Arrhenius-plot). | ||
To quantify the temperature dependence of the transient absorption data, recorded at different temperatures, a global fit using three time-dependent and one infinite component was utilized, a procedure that follows literature precedent.22 The fitting (for a full account of the fitting results, the reader is referred to Tables S2 and S3 in the ESI†) reveals that the first two characteristic time constants, which are associated with vibrational cooling, ISC and LLCT to the phen-like sphere coordinated to ruthenium (τ1) on the one hand and ILCT to the central phz moiety of the tpphz bridge on the other hand (τ2),22 do not significantly vary with temperature. The characteristic time-constant, which relates to the kinetics of charge transfer from the reduced phenazine core of the tpphz ligand to the catalytically active metal-center (τ3),22 however, shows a prominent temperature dependence: the mean value of τ3 varies from 465 ps at 303 K to 882 ps at 246 K.
When plotting the logarithm of the corresponding first order rate constants as a function of the inverse temperature (Fig. 2b) an Arrhenius behavior becomes apparent, which can be associated with an activation energy of 547 ± 75 cm−1 (67.8 meV, 6.54 kJ mol−1).32 This activation energy for the ET process from the reduced phenazine to the rhodium-center is on the same order of magnitude as the energy of the rhodium–chloride stretching vibration for related complexes like [RhIII(phen)2Cl2]+ (347 cm−1 and 352 cm−1)33 and others.33,34 This indicates that thermally driven chloride labilization is the gating process for the reduction of the rhodium(III)-center. This structural rearrangement in the coordination sphere of the rhodium-center, following ET from the bridging ligand to the metal which is exergonic based on CV data,1,22 explains the comparatively slow photoinitiated ET, ca. 450 ps at room temperature, despite the fully-conjugated and rigid nature of the tpphz-bridge.
Following the premise that intramolecular charge transfer is gated by the dissociation of the chloride ligand, we expand our studies to a variation of the chloride concentration in the solution. For mononuclear [(NN)Rh(Cp*)Cl]+ complexes (NN = α-diimine ligand), an equilibrium between the chloride dissociated and chloride associated species was observed even following two-fold electrochemical reduction. The two reduced species, [(NN)RhI(Cp*)Cl]− and [(NN)RhI(Cp*)]0 were distinguishable by their distinct absorption bands between 690–760 nm and 480–530.35
In contrast to this study, we investigate if the equilibrium between chloride dissociated and chloride associated species at the monoreduced rhodium(II) intermediate controls the rate of charge transfer from the reduced phenazine moiety to the rhodium(III)-center. To do so, fs-TA experiments were performed by varying the free chloride concentration in the range from 3 to 300 mM at room temperature by using tetrabutylammonium chloride (TBACl) as additive, while the concentration of RutpphzRhCp* in the solution was fixed at 0.3 mM. Therefore, the excess of salt varied from 10 to 1000 equivalents in the series. As no significant spectral shift were observed, the TA data recorded from these experiments were fitted globally, using the same parameter set (three time-dependent and one infinite component) as before, resulting in DAS with the same spectral features as before22 independent of the amount of added salt. Just like in the temperature-dependent experiments the first two components (representing τ1: vibrational cooling, ISC, ILCT and τ2: LLCT) were not sensitive to the change of the environmental factors (see ESI,† Fig. S17). However, the time constant τ3, associated with the decay of the phz-centered state in question, shows a trend in which its value is rising gradually with increasing TBACl concentration over the whole experimental concentration range, see Fig. 3. With this data alone it cannot be decided if the hindrance in the ET from the phz-centered state to the rhodium center is caused specifically by the presence of excess chloride ions or rather is a more general effect caused by the raise in ionic strength to the solution.36–38 In order to observe the effect of the pure increase of ionic strength under exclusion of chemical reactions or interactions between the added salt and RutpphzRhCp*, tetrabutylammonium hexafluorophosphate (TBAPF6) serving as an additive providing non-coordinating anions was added instead of TBACl in a comprehensive study. The values of τ3 recorded in solutions with TBAPF6 concentrations ranging from 3 mM to 35 mM are either similar or just slightly below the initial value of 400 ps, which was recorded without any external salts added to the solution, see again Fig. 3. This independence of τ3 on the concentration of TBAPF6 indicates that the TBACl-dependence of τ3 does not stem from the increased ionic strength of the solution, but rather suggest that this is specific to the interactions between free chloride and the molecular photocatalyst. This finding supports the idea of a dynamic equilibrium between chloride associated and chloride dissociated species following the decay of the phz-centered state.
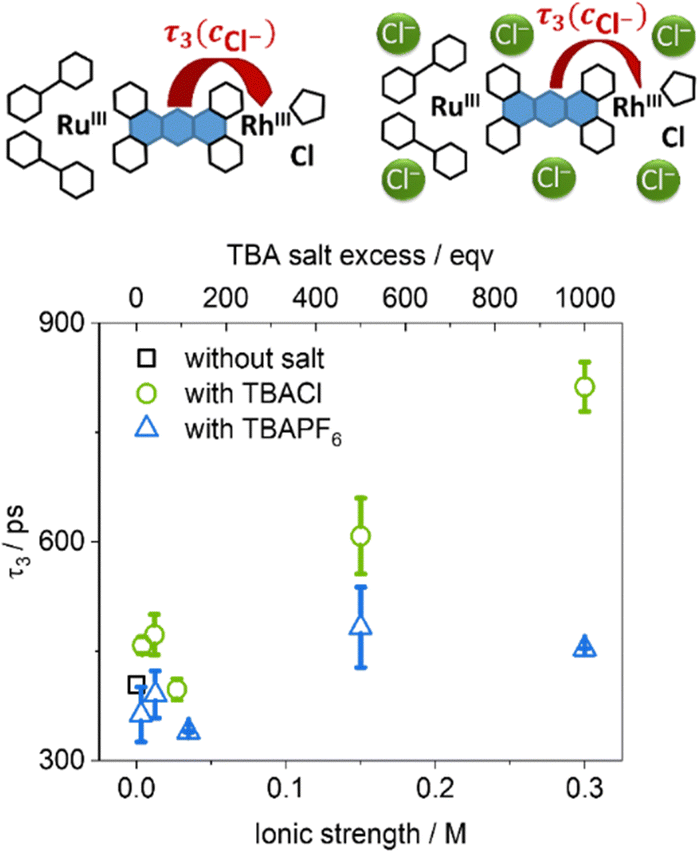 | ||
| Fig. 3 Plot of the time constant τ3, representing the decay the phz-centered state, as function of the ionic strength added to the solution by either TBACl or TBAPF6. | ||
While this prominent effect highlights the reduction-induced cleavage of the rhodium–chloride bond, even at the highest chloride concentration of 0.3 M (1000 equivalents) the kinetic rate of the electron transfer step towards the rhodium(III) center 1/τ3 is with 1.23 GHz still orders of magnitude larger than the maximum turnover frequency (TOF) of the photocatalyst for nicotinamide reduction (4.2 mHz).2 This also explains why large chloride excess had no negative impact on the catalytic performance of RutpphzRhCp*![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 as reaction steps within the catalytic cycle following reductive catalyst activation such as tautomerization processes at the rhodium-center or hydride transfers are associated with energy barriers >60 kJ mol−1 and >43 kJ mol−1, respectively.39,40
2 as reaction steps within the catalytic cycle following reductive catalyst activation such as tautomerization processes at the rhodium-center or hydride transfers are associated with energy barriers >60 kJ mol−1 and >43 kJ mol−1, respectively.39,40
In summary change in temperature and in concentration of free chloride ions significantly impact the rate characterizing the decay of the phz-centered 3MLCT state and the reduction of the rhodium-center in RutpphzRhCp*. The results show that chloride dissociation from the rhodium ion is a key gating process for the light-driven intramolecular electron transfer step. The dissociation of the chloride ligand from the rhodium-center is followed by significant structural reorganization of the complex, where the Cp* ring assumes a position perpendicular to the tpphz–rhodium binding axis. This could kinetically hinder the back-reaction to the Cl-associated species within the lifetime of the reduced species by steric shielding.22
The authors acknowledge funding by the DFG via the CRC TRR 234 CATALIGHT, project A1 and the projects 456209398 and 494988281.
Data availability
Data for this article, including the experimental raw data of the fs-TA experiments, steady-state UV-vis spectroscopic data, data processing and analysing, are available at Zenodo at https://doi.org/10.5281/zenodo.14500795.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. K. Mengele, S. Kaufhold, C. Streb and S. Rau, Dalton Trans., 2016, 45, 6612–6618 RSC.
- A. K. Mengele, G. M. Seibold, B. J. Eikmanns and S. Rau, ChemCatChem, 2017, 9, 4369–4376 CrossRef CAS.
- L. Zedler, P. Wintergerst, A. K. Mengele, C. Müller, C. Li, B. Dietzek-Ivanšić and S. Rau, Nat. Commun., 2022, 13, 2538 CrossRef CAS PubMed.
- G. E. Shillito, S. Rau and S. Kupfer, ChemCatChem, 2023, 15, e202201489 CrossRef CAS.
- E. Steckhan, S. Herrmann, R. Ruppert, E. Dietz, M. Frede and E. Spika, Organometallics, 1991, 10, 1568–1577 CrossRef CAS.
- H. C. Lo, C. Leiva, O. Buriez, J. B. Kerr, M. M. Olmstead and R. H. Fish, Inorg. Chem., 2001, 40, 6705–6716 CrossRef CAS PubMed.
- C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105–9108 RSC.
- U. Kölle and M. Grützel, Angew. Chem., Int. Ed. Engl., 1987, 26, 567–570 CrossRef.
- N. Madern, N. Queyriaux, A. Chevalley, M. Ghasemi, O. Nicolotti, I. Ciofini, G. F. Mangiatordi and M. Salmain, J. Mol. Catal. B: Enzym., 2015, 122, 314–322 CrossRef CAS.
- Y. Peng, M. V. Ramos-Garcés, D. Lionetti and J. D. Blakemore, Inorg. Chem., 2017, 56, 10824–10831 CrossRef CAS PubMed.
- R. Ruppert, S. Herrmann and E. Steckhan, Tetrahedron Lett., 1987, 28, 6583–6586 CrossRef CAS.
- R. Ruppert, S. Herrmann and E. Steckhan, J. Chem. Soc., Chem. Commun., 1988, 1150 RSC.
- M. Lämmle, A. K. Mengele, G. E. Shillito, S. Kupfer and S. Rau, Chem. – Eur. J., 2023, 29, e202202722 CrossRef PubMed.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, J. Chem. Soc., Chem. Commun., 1995, 0, 1799–1800 RSC.
- J. Bolger, A. Gourdon, E. Ishow and J.-P. Launay, Inorg. Chem., 1996, 35, 2937–2944 CrossRef CAS.
- L. Flamigni, S. Encinas, F. Barigelletti, F. M. MacDonnell, K.-J. Kim, F. Puntoriero and S. Campagna, Chem. Commun., 2000, 1185–1186 RSC.
- P. P. Lainé, F. Bedioui, F. Loiseau, C. Chiorboli and S. Campagna, J. Am. Chem. Soc., 2006, 128, 7510–7521 CrossRef PubMed.
- M. E. Walther and O. S. Wenger, ChemPhysChem, 2009, 10, 1203–1206 CrossRef CAS PubMed.
- S. Malzkuhn, X. Guo, D. Häussinger and O. S. Wenger, J. Phys. Chem. A, 2019, 123, 96–102 CrossRef CAS PubMed.
- D. I. Schuster, P. Cheng, P. D. Jarowski, D. M. Guldi, C. Luo, L. Echegoyen, S. Pyo, A. R. Holzwarth, S. E. Braslavsky, R. M. Williams and G. Klihm, J. Am. Chem. Soc., 2004, 126, 7257–7270 CrossRef CAS PubMed.
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. González, M. Schmitt, S. Rau and J. Popp, Angew. Chem., Int. Ed., 2010, 49, 3981–3984 CrossRef CAS PubMed.
- L. Zedler, A. K. Mengele, K. M. Ziems, Y. Zhang, M. Wächtler, S. Gräfe, T. Pascher, S. Rau, S. Kupfer and B. Dietzek, Angew. Chem., Int. Ed., 2019, 58, 13140–13148 CrossRef CAS PubMed.
- C. Chiorboli, C. A. Bignozzi, F. Scandola, E. Ishow, A. Gourdon and J.-P. Launay, Inorg. Chem., 1999, 38, 2402–2410 CrossRef CAS.
- C. Chiorboli, M. A. J. Rodgers and F. Scandola, J. Am. Chem. Soc., 2003, 125, 483–491 CrossRef CAS PubMed.
- S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundström, M. Karnahl, M. Schwalbe, B. Schäfer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15, 7678–7688 CrossRef CAS PubMed.
- M. Staniszewska, S. Kupfer and J. Guthmuller, Chem. – Eur. J., 2018, 24, 11166–11176 CrossRef CAS PubMed.
- M. G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 5044–5048 CrossRef CAS PubMed.
- M. G. Pfeffer, T. Kowacs, M. Wächtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54, 6627–6631 CrossRef CAS PubMed.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45, 6215–6218 CrossRef CAS PubMed.
- M. Staniszewska, S. Kupfer and J. Guthmuller, J. Phys. Chem. C, 2019, 123, 16003–16013 CrossRef CAS.
- M. G. Pfeffer, C. Müller, E. T. E. Kastl, A. K. Mengele, B. Bagemihl, S. S. Fauth, J. Habermehl, L. Petermann, M. Wächtler, M. Schulz, D. Chartrand, F. Laverdière, P. Seeber, S. Kupfer, S. Gräfe, G. S. Hanan, J. G. Vos, B. Dietzek-Ivanšić and S. Rau, Nat. Chem., 2022, 14, 500–506 CrossRef CAS PubMed.
- S. Arrhenius, Z. Phys. Chem., 1889, 4U, 226–248 CrossRef.
- I. I. Bhayat and W. R. McWhinnie, Spectrochim. Acta, Part A, 1972, 28, 743–751 CrossRef CAS.
- K. W. Bowker, E. R. Gardner and J. Burgess, Inorg. Chim. Acta, 1970, 4, 626–628 CrossRef CAS.
- S. Chardon-Noblat, S. Cosnier, A. Deronizer and N. Vlachopoulos, J. Electroanal. Chem., 1993, 352, 213–228 CrossRef CAS.
- H. A. R. Gazi and R. Biswas, J. Chem. Sci., 2011, 123, 265–277 CrossRef.
- E. Waisman, G. Worry and R. A. Marcus, J. Electroanal. Chem. Interfacial. Electrochem., 1977, 82, 9–28 CrossRef CAS.
- A. Willie, M. McLean, R. Q. Liu, S. Hilgen-Willis, A. J. Saunders, G. J. Pielak, S. G. Sligar, B. Durham and F. Millett, Biochemistry, 1993, 32, 7519–7525 CrossRef CAS PubMed.
- T. K. Todorova, T. N. Huan, X. Wang, H. Agarwala and M. Fontecave, Inorg. Chem., 2019, 58, 6893–6903 CrossRef CAS PubMed.
- A. Marrone and R. H. Fish, J. Organomet. Chem., 2021, 943, 121810 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05844e |
| This journal is © The Royal Society of Chemistry 2025 |