
DMSO–K2S2O8 mediated iodine-free conversion of glycal C-3 ether to 3-enopyranones: synthesis of furo[3,2-c] pyrans†
Bisma
Rasool
ab,
Sanchari
Kundu
c,
Irshad Ahmad
Zargar
ab and
Debaraj
Mukherjee
 *bc
*bc
aNatural Products and Medicinal Chemistry Division, Indian Institute of Integrative Medicine (CSIR-IIIM), Jammu-180001, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
cDepartment of Chemical Sciences, Bose Institute Kolkata, EN 80, Sector V, Bidhan Nagar, Kolkata-700091, WB, India. E-mail: debaraj@jcbose.ac.in
First published on 2nd December 2024
Abstract
A straightforward, highly efficient, and regioselective method for directly converting 3-O-benzylated and silylated glycals into their corresponding enones has been developed using a DMSO–K2S2O8 reagent system. This reaction is scalable to gram quantities under mild conditions, achieving up to 80% yields. The resulting enones are valuable intermediates for the synthesis of furo[3,2-c] pyrans, which are integral components of various biologically significant scaffolds.
Pyranones, pyranose sugars having α,β-unsaturated enones, are a privileged scaffold commonly found in a wide range of bioactive natural compounds. For example, ascopyrone P, a natural pyranone isolated from fungi of the order Pezizales, has demonstrated antioxidant and antibacterial properties. Anthracycline antibiotics vineomycins A1 and B2 contain a deoxydisaccharide linkage that connects the terminal hex-2-enosyl hexose to the aglycone via a C-glycosidic bond (Fig. 1a). The pyranone moiety is responsible for a variety of biological and pharmacological activities, enhancing the therapeutic potential of these above molecules, thus underscoring their importance in medicinal chemistry and drug discovery.1,2
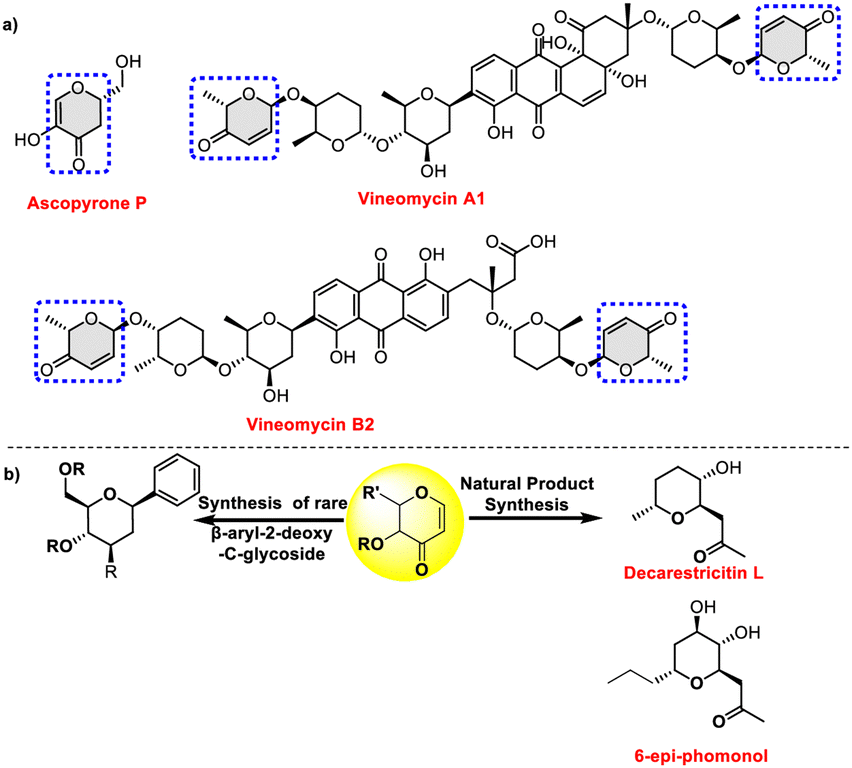 | ||
| Fig. 1 (a) Examples of sugar enones as substructures found in microbial natural products. (b) Synthetic applications of 3-oxo glycals. | ||
Owing to the presence of a highly reactive α,β-unsaturated ketone structure at the C1, C2, and C3 positions,3 pyranone is extensively used in the synthesis of various scaffolds such as 2-deoxy-C- and O-glycosides, including 2-phosphono-α-C-glycosides.4 Numerous studies have documented the synthesis of aryl glycosides from enopyranones.5 Recently, our group successfully synthesized β-aryl glycosides, which are typically challenging to produce, using aryl enopyranones.6 Furthermore, the keto group in these compounds can be readily reduced to produce rare sugars such as D-allal and its derivatives.7
Consequently, the development of methodologies for synthesizing compounds containing 2,3-dihydro-4H-pyran-4-ones is a significant target in synthetic organic chemistry.
There are two approaches to access selectively protected 3-oxo glycals via allylic ether oxidation avoiding protecting group manipulations. Typically, this oxidation is achieved from C-3 vinyl ethers through C-2 C–H activation by an electrophile which undergoes elimination with adjacent C-3 C–H via an ionic pathway [Fig. 2(a), pathway a]. This approach requires either multiple steps like electrophilic iodination at C2, HI elimination by a base to 2,3-enol ether and TfOH-mediated hydrolysis in subsequent steps (Fig. 2(b))8a or a cocktail of expensive iodinating agents like [bis(trifluoroacetoxy)iodo]benzene (PIFA) or diacetoxyiodo-benzene (BAIB) in the presence of additional co-oxidants like TEMPO, which require sulfate reagent to get rid of excess iodine after reaction (Fig. 2(c)).8b,c We considered the possibility of utilizing a radical pathway instead, which could proceed via C-3 using SET thereby avoiding the involvement of external electrophiles [Fig. 2(a), pathway b]. Recently, we have successfully utilized K2S2O8 as a radical initiator for C-2 C–H activation of glycals. In the present communication, we present a novel efficient cost-effective approach for generating 3-oxo-glycals from glycals using a potassium persulfate/DMSO system (Fig. 2(d)) and thereafter application in the synthesis of a chiral furo-pyran scaffold with an exo double bond.
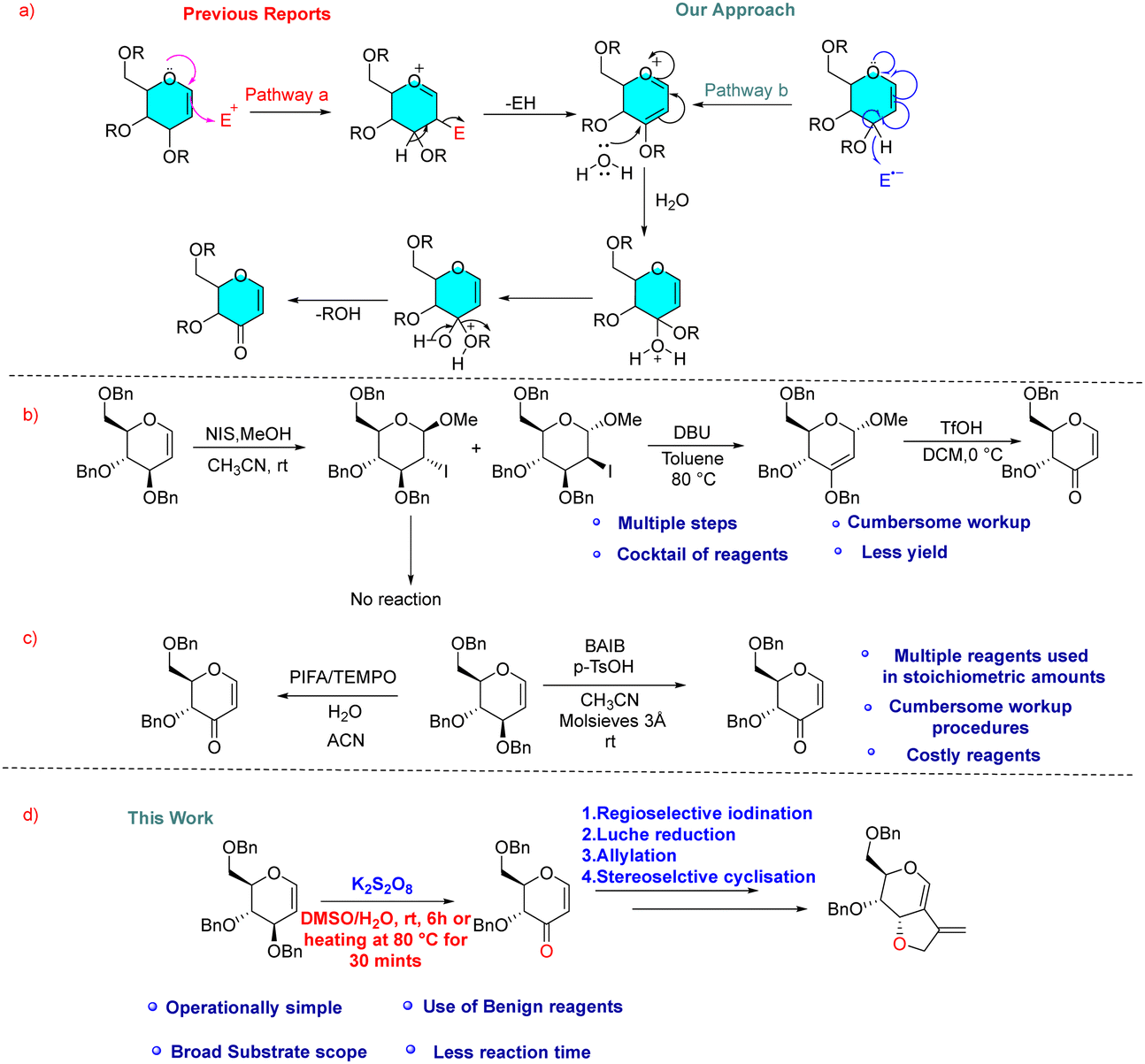 | ||
| Fig. 2 (a) Previous & current approaches for the allylic ether oxidation of glycals. (b) and (c) Literature reports for the synthesis of 3-oxo-glycals. (d) Present work. | ||
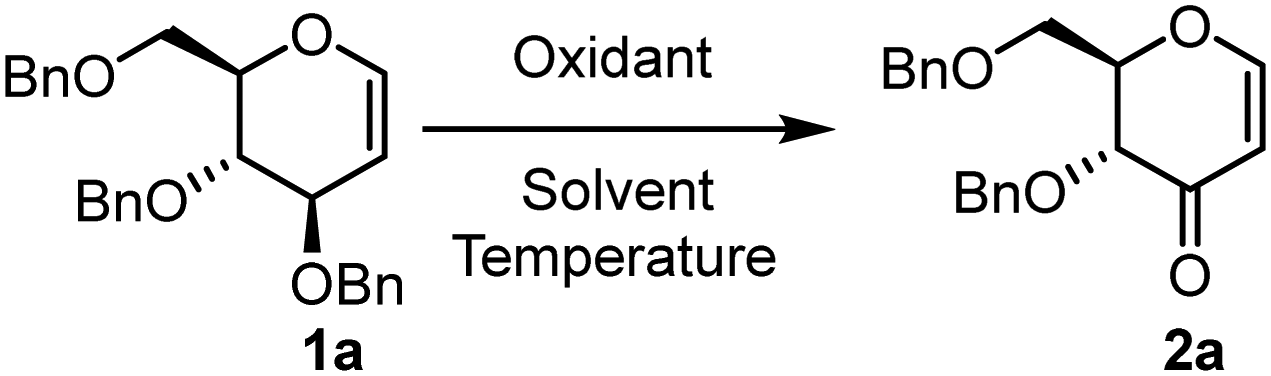
For the initial optimization studies, benzyl-protected glucal (1a) was selected as the model substrate and tested with various oxidants, excluding iodinating agents (Table 1). Given that K2S2O8-mediated radical reactions are effective in organic solvent–water mixtures, we began our optimization using combinations of organic solvents and water. The initial attempt to conduct the reaction in ACN/H2O with oxone as the oxidant at room temperature showed no progress (Table 1, entry 1). Even after raising the reaction temperature to 80 °C, no product formation was observed (Table 1, entry 2). We then tested different oxidants such as Cu(OAc)2, TEMPO, and K2S2O8, but none yielded any product (Table 1, entries 3–6). Consequently, we explored different mixtures of solvents like DMF/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and DMSO/H2O (3:1). Interestingly, the reaction succeeded with good yield when performed in DMSO/H2O (3:1) using 1 equivalent of K2S2O8, both at room temperature and at 80 °C. However, at room temperature, the product formation took 6 hours, while at 80 °C, it occurred within just 30 minutes (Table 1, entries 7 and 8). The product was characterized as di-O-benzyl-2,3-dihydro-4H-pyran-4-one. Furthermore, adjusting the oxidant stoichiometry revealed that increasing the amount of oxidant to 2 equivalents improved the yield, but further increases in the oxidant did not enhance the yield (Table 1, entries 9 and 10). Altering the DMSO:H2O ratio to 2:1 resulted in a decreased product yield (Table 1, entry 11). Notably, using dry DMSO as the solvent reduced the product yield (Table 1, entry 12), and using water alone did not produce the desired product (Table 1, entry 13). Thus, two optimal reaction conditions were identified: 1a (1 equiv.) with K2S2O8 (2 equiv.) in DMSO/H2O (3:1) at room temperature for 6 hours (Table 1, entry 9), and at 80 °C for 30 minutes (Table 1, entry 10). It is pertinent to mention that the reaction goes well even while using rack DMSO without the addition of any external water.
:1) and DMSO/H2O (3:1). Interestingly, the reaction succeeded with good yield when performed in DMSO/H2O (3:1) using 1 equivalent of K2S2O8, both at room temperature and at 80 °C. However, at room temperature, the product formation took 6 hours, while at 80 °C, it occurred within just 30 minutes (Table 1, entries 7 and 8). The product was characterized as di-O-benzyl-2,3-dihydro-4H-pyran-4-one. Furthermore, adjusting the oxidant stoichiometry revealed that increasing the amount of oxidant to 2 equivalents improved the yield, but further increases in the oxidant did not enhance the yield (Table 1, entries 9 and 10). Altering the DMSO:H2O ratio to 2:1 resulted in a decreased product yield (Table 1, entry 11). Notably, using dry DMSO as the solvent reduced the product yield (Table 1, entry 12), and using water alone did not produce the desired product (Table 1, entry 13). Thus, two optimal reaction conditions were identified: 1a (1 equiv.) with K2S2O8 (2 equiv.) in DMSO/H2O (3:1) at room temperature for 6 hours (Table 1, entry 9), and at 80 °C for 30 minutes (Table 1, entry 10). It is pertinent to mention that the reaction goes well even while using rack DMSO without the addition of any external water.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Oxidant (equiv.) | Solvent | Temp. (°C) | Yieldb (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Reactions conditions: 1a (1 equiv.), oxidant, solvent (5 ml). b Isolated yields after column chromatography. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | Oxone (1) | ACN/H2O (3:1) |
rt | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Oxone (1) | ACN/H2O (3:1) |
80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | Cu(OAc)2 | ACN/H2O (3:1) |
80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | TEMPO | ACN/H2O (3:1) |
80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | K2S2O8 (1) | ACN/H2O (3:1) |
80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | K2S2O8 (1) | DMF/H2O (3:1) |
80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | K2S2O8 (1) | DMSO/H2O (3:1) |
rt | 50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | K2S2O8 (1) | DMSO/H2O (3:1) |
80 | 65 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | K 2 S 2 O 8 (2) |
DMSO/H
2
O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
rt | 75 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | K 2 S 2 O 8 (2) |
DMSO/H
2
O (3:1)
|
80 | 77 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | K2S2O8 (2) | DMSO/H2O (2:1) |
80 | 55 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | K2S2O8 (2) | DMSO (dry) | 80 | 47 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | K2S2O8 (2) | H2O | 80 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
After establishing the optimized reaction conditions, we proceeded to assess the substrate scope, as detailed in Scheme 1. Our study encompassed both ester- and ether-protected glycals. Attempts to convert acetyl-protected glycals were unsuccessful. However, under the optimized reaction conditions, the conversion of ether-protected glycals proceeded efficiently with better yields. It is pertinent to mention that ether-protected glycals in both D- and L-configurations exhibited enhanced yields. Importantly, glycals featuring acid-labile protecting groups, such as those having silicon-based protecting groups, were easily tolerated under our optimized conditions, emphasizing the gentle nature of the experimental parameters utilized.
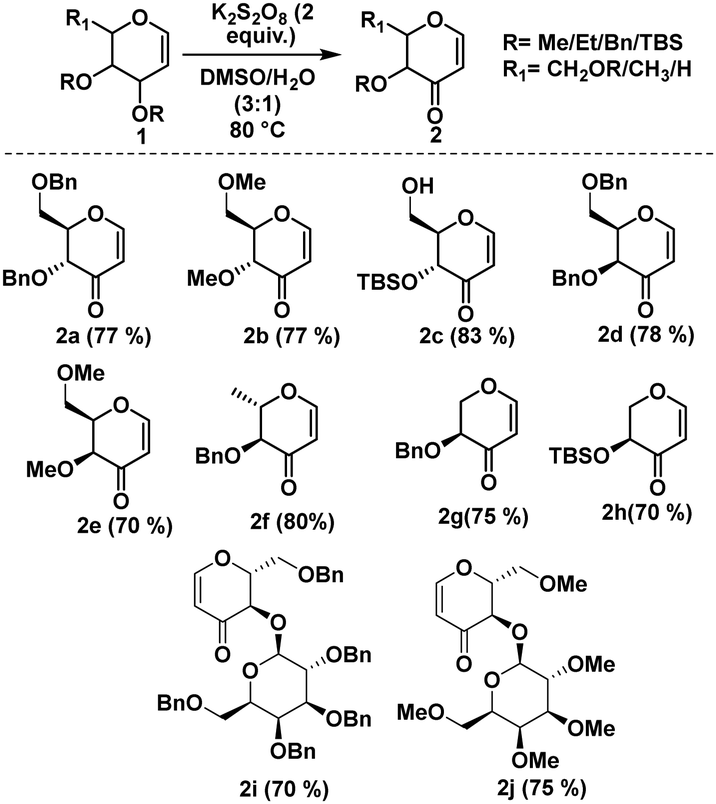 | ||
| Scheme 1 Substrate scope with respect to differently protected glycals. | ||
After successfully synthesizing the 3-oxo glycals, we became interested in the synthesis of bicyclic compounds containing fused pyro-furan scaffolds, which possess biological importance as these types of scaffolds are present in numerous biologically active compounds, as illustrated in Fig. 3.9,10 Towards the synthesis of bicyclic pyran-fused furanoses from 3-oxo glycals, initial C-2 iodination was followed by stereoselective reduction under Luche conditions to obtain C-3 OH, which upon allylation and Pd-catalysed 5-endo dig type cyclization resulted in the target furo[3,2-c] pyrans in good overall yield (Scheme 2).
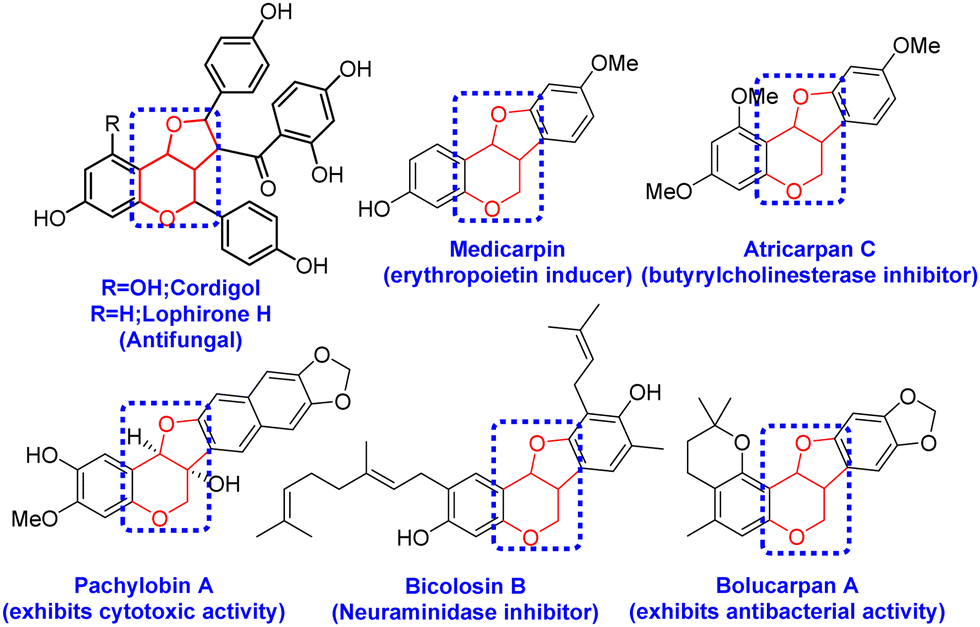 | ||
| Fig. 3 Examples of biologically active compounds containing furo[3,2-c] pyrans as core structures. | ||
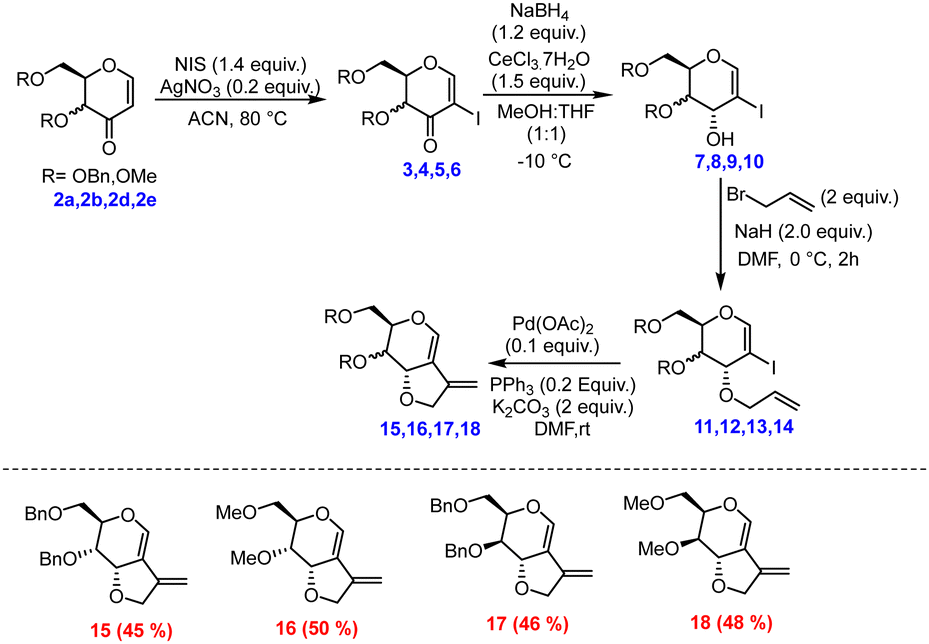 | ||
| Scheme 2 Synthesis of furo[3,2-c] pyrans from hex-3-enuloses. | ||
To shed light on the reaction mechanism, we carried out a series of control experiments. Initially, the reaction of compound 1a was carried out in the presence of K2S2O8 (1 equiv.) and TEMPO (3 equiv.) in DMSO/H2O (3:1) as the solvent. The absence of product formation under these conditions suggests that the reaction proceeds via a radical mechanism (Scheme 3(i)).
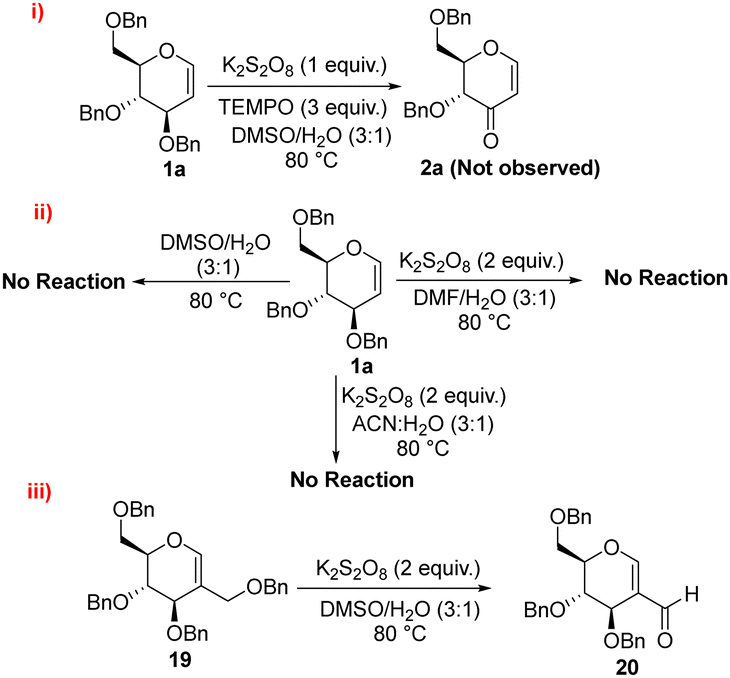 | ||
| Scheme 3 Control experiments: (i) Radical quenching, (ii) influence of solvent and oxidant, (iii) selectivity between primary and secondary allylic oxidation. | ||
Furthermore, the adduct formed by quenching of the allylic radical was confirmed via LCMS (see ESI†). To further investigate the role of DMSO in the reaction, we tested various alternative solvents like DMF/H2O (3:1), and ACN/H2O (3:1). In these experiments, either no product formation was observed, with the starting material being recovered was unchanged, or degradation of the starting material occurred. Additionally, no product formation was detected when the reaction was performed in neat DMSO. These results indicate that potassium persulfate is essential for the conversion, highlighting its critical role in the reaction mechanism (Scheme 3(ii)). In a control experiment using substrate 4, it was observed that under the optimized reaction conditions, compound 5 having C-2 aldehyde was formed with the elimination of the primary benzylic ether group. This outcome demonstrates that the reagent system selectively oxidizes the allylic position; in this case primary getting more preference over the secondary allylic radical (Scheme 3(iii)).
A plausible reaction mechanism is proposed based on the control experiments and the literature findings (Fig. 4). The process initiated with the decomposition of persulfate (S2O82−) into sulfate radical anions (SO4˙−), which are captured by a resonance-stabilized allylic radical at the allylic position of glycal. A second sulfate radical anion then performs a single electron transfer (SET) with this allylic radical, forming an oxocarbenium ion. Attack of DMSO at electron deficient C-3 along with eliminating benzyl alcohol (further oxidized to benzaldehyde by K2S2O8) (see ESI†) in the presence of water provides the target enopyranone.
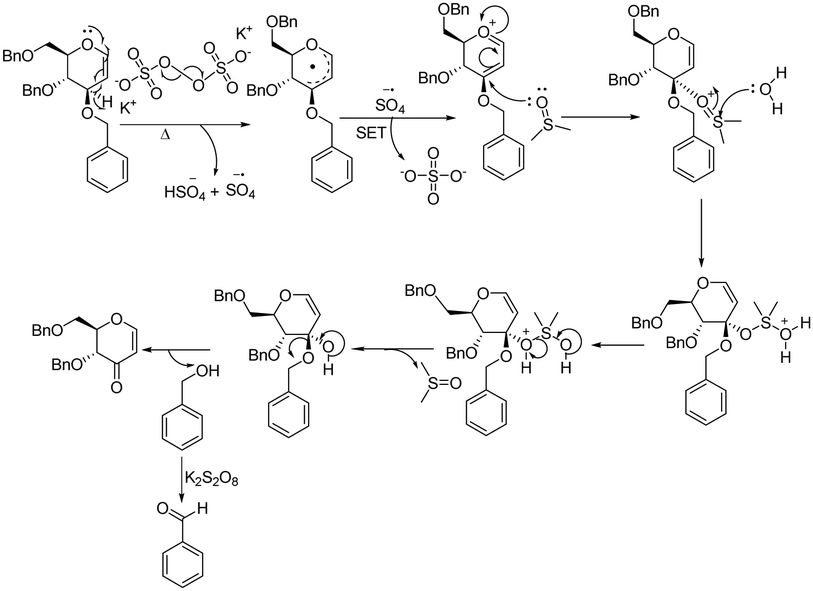 | ||
| Fig. 4 Plausible reaction mechanism of the formation of 3-oxo enopyranone. | ||
In conclusion, we were able to selectively synthesize 3-keto glycals from benzyl and silyl-ether protected glycals using a benign reagent system DMSO/K2S2O8. Under the ideal reaction conditions, a wide variety of ether-protected glycals, such as D-glucal, D-galactal, and L-rhamnal, were found to be compatible.
Furthermore, we were able to synthesize the medicinally important furo[3,2-c] pyrans in good to excellent yields which demonstrates the importance of the current protocol.
The authors are thankful to DST-SERB CRG2021004142 for funding. BR and IAZ are thankful to UGC for the SRF, AcSIR for PhD registration & director CSIR-IIIM Jammu for providing the lab space & facilities. IIIM Publication No. CSIR-IIIM/IPR/00786. DM is thankful to the director Bose Institute for providing lab space and facilities.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
-
(a) S. Amslinger, ChemMedChem, 2010, 5, 351–356 CrossRef CAS PubMed
; (b) H. Ikeda, E. Kaneko, S. Okuzawa, D. Takahashi and K. Toshima, Org. Biomol. Chem., 2014, 12, 8832–8835 RSC
-
(a) T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed
- N. L. Holder, Chem. Rev., 1982, 82, 287–332 CrossRef CAS
-
(a) M. Andreassen and I. Lundt, Carbohydr. Res., 2006, 341, 1692–1696 CrossRef CAS PubMed
-
(a) S. Tang, Q. Zheng, D. C. Xiong, S. Jiang, Q. Li and X. S. Ye, Org. Lett., 2018, 24, 3079–3082 CrossRef PubMed
- I. A. Zargar, B. Rasool, S. K. Bappa and D. Mukherjee, Org. Biomol. Chem., 2024, 22, 6941–6945 RSC
- T. Fujiwara and M. Hayashi, J. Org. Chem., 2008, 73, 9161–9163 CrossRef CAS
-
(a) P. R. Sridhar, I. Ali and M. K. Lakshmi, J. Org. Chem., 2022, 87, 8939–8955 CrossRef CAS PubMed
- M. Bakthadoss and V. Agarwal, J. Org. Chem., 2020, 85, 15221–15231 CrossRef CAS
-
(a) N. Sakander, A. Ahmed, I. A. Zargar and D. Mukherjee, J. Org. Chem., 2023, 88, 8300–8309 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04278f |
| This journal is © The Royal Society of Chemistry 2025 |