
Functionalized quinolones and isoquinolones via 1,2-difunctionalization of arynes: synthesis of antagonist agent AS2717638 and floxacin key intermediates†
Sachin D.
Mahale
 ab,
Anamika
Prasad
a and
Santosh B.
Mhaske
*ab
ab,
Anamika
Prasad
a and
Santosh B.
Mhaske
*ab
aDivision of Organic Chemistry, CSIR-National Chemical Laboratory (CSIR-NCL), Pune–411008, India. E-mail: sb.mhaske@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad–201002, India
First published on 22nd November 2024
Abstract
Quinolones and isoquinolones are privileged scaffolds in synthetic/medicinal chemistry and drug discovery due to their unique chemical structures and intrinsic properties. Herein, we reveal a transition-metal-free approach for their synthesis from the reaction of dimethyl-2-((phenylamino)methylene)malonate with aryne precursors under mild conditions. The substrate scope is broad, accommodating a wide range of functional groups. The synthetic utility of the developed protocol has been demonstrated in the total synthesis of the potent antagonist agent AS2717638 and key intermediates of floxacin congeners. The gram-scale experiments illustrate its synthetic potential.
The scrutiny for new small molecules with therapeutic and pharmacological activity often entails evaluating manifold collections of drug-like compounds.1 To achieve these libraries, designing new methods for the expeditious and wide-ranging derivatization of novel molecular scaffolds has been a key goal of synthetic chemists in drug discovery programs.2 Exploring the switchable approach to synthesize valuable products from basic building blocks is appealing because it allows diversity oriented production of complex molecules without redesigning the synthetic pathways.3
Quinolones, isoquinolones and their derivatives manifest diverse biological and pharmacological activities, rendering them valuable for drug discovery and development. They have been widely investigated as privileged structural motifs incorporated in plentiful natural products, APIs, bioactive compounds, and pharmaceuticals.4 Quinolones have a great deal of biological and medicinal uses due to their antimalarial, anti-HIV, anti-inflammatory, antimicrobial, and antitumor activities.4,5 One of the famous synthetic quinolones are the floxacin family of APIs, which contain several quinolone drugs such as ciprofloxacin and sarafloxacin.6 Many natural products such as quinolactacin A and sarcomejine (Fig. 1) also feature the quinolone scaffold.7 Similarly, compounds containing an isoquinolone skeleton are renowned for their anti-tumor, anti-hypertensive, anti-thrombotic, anti-malarial, anti-inflammatory, and analgesic properties.4,8 There are many bioactive agents such as SJ000101247 and AS2717638 (Fig. 1) and natural products like ruprechstyril featuring an isoquinolone core.9 The isoquinolones containing a carboxylic acid or amide group at the 4-position are known to have potential bioactivities.10 Additionally, isoquinolone derivatives are extensively used as key intermediates in organic transformations.11

Due to the widespread presence of the pivotal motifs in numerous biologically significant molecules, several reports on the construction of quinolone and isoquinolone scaffolds have been well-documented.6,12 In recent decades, there has been galloping upgradation in the synthesis of quinolone and isoquinolone architectures using inter- and intramolecular annulation methods involving unsaturated hydrocarbons and nitrogen-containing motifs. The synthesis of quinolones by two classical routes comprising an intramolecular cyclization strategy is reported.13 The recent efforts to synthesize isoquinolones have predominantly employed organometallic chemistry. The most straightforward approach for the synthesis of isoquinolones involves the annulation of benzamide with alkyne, alkene, or allenes using transition metals.14 Few reports demonstrate metal-catalyzed decarbonylative coupling of amides/imides with alkynes to obtain isoquinolones.15
Our group and many others have exemplified the usefulness of aryne reactivity in the synthesis of heterocyclic scaffolds.16 In this context, there are few reports on the synthesis of isoquinolones and quinolones utilizing arynes, as depicted in Scheme 1A and B.17,18 Interestingly, Ramtohul et al. demonstrated the reaction between β-enamino esters and ketones with arynes to form the C-arylated β-enamino esters and ketones, but did not observe quinolones/isoquinolones (Scheme 1C).19 Despite the development of numerous synthetic methods to obtain these important heterocyclic scaffolds, there exist some drawbacks like multistep synthesis, use of transition metals, requirement of directing groups, use of oxidants, tedious preparation, and harsh reaction conditions. Moreover, synthesizing both scaffolds typically requires distinct starting materials and synthetic routes, which can make the process exorbitant. Consequently, devising a novel methodology for the selective construction of N-heteroarenes from common starting materials in a controlled manner is highly enticing. Herein, we introduce a switchable approach for selective synthesis of quinolones and isoquinolones utilizing a novel transition-metal-free protocol via 1,2-difunctionalization of arynes under mild reaction conditions (Scheme 1D).
 | ||
| Scheme 1 Prior aryne-based approaches and the present work. | ||
We envisaged a straightforward single-step synthetic process for the key intermediates of the floxacin class of quinolone alkaloids employing β-enamino diesters and arynes. Accordingly, dimethyl 2-((phenylamino)methylene)malonate (1a) was treated with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate (2a) in the presence of CsF. Surprisingly, we observed the selective formation of isoquinolone 3a along with a small amount of our expected product quinolone 4a (Scheme 2).
 | ||
| Scheme 2 Optimized protocol for isoquinolone synthesis. | ||
Nevertheless, considering the importance of the isoquinolone moiety in pharmaceuticals and other applications, we planned to explore our serendipitous observation further by optimizing the protocol. Several permutations and combinations (see ESI†) provided the best reaction conditions (Scheme 2). We proceeded to generalize the protocol by varying enamines. A series of substituted enamines were used, and the results from these reactions were promising, consistently yielding varyingly substituted isoquinolones 3a–3v in good to excellent yields (Scheme 3). Interestingly, along with nitro-substituted isoquinolone 3i, we also observed the formation of quinolone 4i in 21% yield, which indicated the probability to alter the protocol to obtain quinolone selectively. An important example in this series of compounds is the isoquinolone 3l, as the enamine corresponding to this compound was synthesized from benzocaine, a drug known for its use as a topical local anesthetic. We observed a decrease in the yield with electron withdrawing group substituted and heterocyclic enamines. Interestingly, methyl-furan substituted enamine provided isoquinolone product 3t with cycloaddition between furan and aryne in good yield. Note that isoquinolone 3v is the isomeric form of the key intermediate of the API ciprofloxacin. Subsequently, the scope of aryne precursor 2 was tested with enamine 1a under optimal reaction conditions (Scheme 3). Unsymmetrically substituted halo, alkyl, electron withdrawing and electron donating groups containing aryne precursors provided the corresponding products in good yields as inseparable mixtures of regioisomers (3w, 3x, 3z, 3bb and 3cc). Symmetrically substituted difluoro and dimethyl substituted aryne precursors provided excellent yields (3y and 3aa). Interestingly, the ortho methoxy substituted aryne precursor displayed a good electronic and steric effect to regioselectively furnish single isomer 3dd. The sesamol-based aryne precursor with symmetrical substituents provided the desired isoquinolone 3ee in very good yield. Unsymmetrical and symmetrical polyaromatic arynes generated from their corresponding naphthalene precursors worked well under the optimized reaction conditions. Remarkably, the unsymmetrically substituted naphthalene aryne precursor delivered the corresponding product 3ff as a single isomer.
 | ||
Scheme 3 Substrate scope for isoquinolones. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Standard reaction conditions: 1 (40 mg, 1 equiv.), 2 (3 equiv.), CsF (4 equiv.), Cs2CO3 (2.5 equiv.), ACN (2 mL), 70 °C, 30 min. bApprox. 3–5% of quinolone was observed except in 3i where 21% of quinolone 4i was observed. cReaction required 2.5 h for completion. dReaction completed in 1 h. eReaction took 3 h. Standard reaction conditions: 1 (40 mg, 1 equiv.), 2 (3 equiv.), CsF (4 equiv.), Cs2CO3 (2.5 equiv.), ACN (2 mL), 70 °C, 30 min. bApprox. 3–5% of quinolone was observed except in 3i where 21% of quinolone 4i was observed. cReaction required 2.5 h for completion. dReaction completed in 1 h. eReaction took 3 h. | ||
During the substrate scope studies to selectively obtain isoquinolones (Scheme 3), we observed 3–5% of quinolone formation. Notably, as mentioned earlier, N-Ph-pNO2 substituted enamine produced quinolone 4i in a considerable amount. This result prompted us to investigate it further to optimize the protocol for selective formation of quinolones (Scheme 4).
 | ||
| Scheme 4 Optimized protocol for quinolone synthesis. | ||
Several permutations and combinations (see ESI†) with the unsubstituted aryne precursor 2a and enamine 1i provided the expected product 4i in 66% yield (Scheme 4). After establishing the optimal reaction conditions to selectively obtain quinolones, we investigated the scope of different aryne precursors with enamine 1i. Unsymmetrical chloro aryne generated the product 4b in good yield as a mixture of regioisomers. The symmetrically substituted difluoro- and dimethyl aryne precursors also worked well to provide the corresponding products 4c and 4d, respectively. Gratifyingly, 3,6-dimethyl substituted aryne precursor furnished the corresponding quinolone 4e in 74% yield, along with its isoquinolone analogue 3ei in 20% yield. Arynes containing electron withdrawing groups like CF3 provided the corresponding products 4f as a mixture of inseparable regioisomers in good yield. Furthermore, reactions with symmetrically substituted arynes having electron donating groups yielded the corresponding products 4g and 4h in very good yields. The symmetrically substituted polyaromatic aryne provided the corresponding product 4j in good yields (Scheme 5).
 | ||
Scheme 5 Substrate scope for quinolone. aStandard reaction conditions: 1 (40 mg, 1 equiv.), 2 (3 equiv.), CsF (4 equiv.), Cs2CO3 (2.5 equiv.), PhCN (2 mL), 100 °C, 3 h. b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Approximately 4–7% of isoquinolone was observed except in 4e where 20% of isoquinolone 3ei was observed. Approximately 4–7% of isoquinolone was observed except in 4e where 20% of isoquinolone 3ei was observed. | ||
We observed that the 3,6-dimethyl substituted aryne precursor gives the expected product in very good yield; hence, we explored it with various enamines under the optimized conditions to produce quinolone products. Enamines with aryl and substituted aryl groups on nitrogen containing 4-fluoro, 4-chloro, 4-iodo, and 2,4-difluoro delivered the corresponding quinolone products 4k–4o in good to excellent yields. Likewise, enamines containing aryl with 4-methyl or electron withdrawing groups such as CF3 and CO2Et as well as an electron donating methoxy group worked very well to provide quinolones 4p–4s respectively with minimal electronic effect on the yield. It is interesting to note that the enamine synthesized from the benzocaine API smoothly reacted furnishing the quinolone product 4r in very good yield. N-Naphthyl enamine also worked well to provide quinolone 4t. Interestingly, N-pyridyl enamine which worked well under previous isoquinolone formation protocols (Scheme 3, 3p), did not deliver the desired product 4u under the quinolone formation protocol. Instead it provided pyrido-pyrimidine-carboxylate 4ua (see ESI†) via intramolecular cyclization, which is known as Gould–Jacob's reaction.13b,20 The N-benzyl, alkyl substituted enamines provided the desired quinolone products 4v and 4w, respectively, although in low yield, as most of the starting material remained unreacted. The quinolone product 4w is interesting as it features the core of the API ciprofloxacin.
After the substrate scope studies, we planned to demonstrate the application of our protocol in the synthesis of antagonist agent AS2717638.9a It is a selective piperidine-based LPAR5 antagonist with approximately 3 to 90 times more potency than existing antagonists.21 To date, two total syntheses of AS2717638 and related compounds have been reported, each requiring 10 to 11 steps.9a,22 Our synthetic route began with the synthesis of enamine 1q using diester 5 and isoxazole amine 6q.23 The enamine 1q was then treated with aryne 2k using our protocol yielding isoquinolone 3hh. After hydrolysis of the ester and subsequent acid-amine coupling with piperidine using the coupling reagent HATU and DIPEA the product AS2717638 was produced in 72% yield over two steps (Scheme 6).
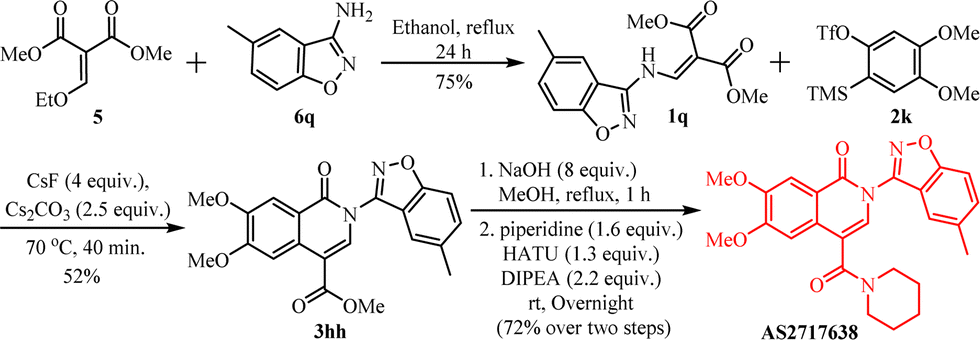 | ||
| Scheme 6 Synthesis of AS2717638. | ||
We have depicted a plausible reaction mechanism in Scheme 7 based on literature precedence and our observations.16–19,24 The aryne intermediate [i] and intermediate [ii] react to form a four-membered cyclic intermediate [iv] via [iii]. It follows two possible pathways. Path a involves opening of the four-membered cyclic intermediate, leading to the insertion of arene into the C–C Sigma bond generating intermediate [v]. Subsequently, it delivers isoquinolone 3via intramolecular cyclization. Alternatively, in path b, intermediate [iv] expels methanol to form intermediate [vi], which undergoes ring-opening and cyclization to yield isoquinolone 3 (Scheme 7A). The proposed mechanism for the formation of quinolones involves simple nucleophilic attack of a nitrogen anion [vii] on the aryne intermediate followed by intramolecular cyclization to furnish quinolone 4 (Scheme 7B). The selective formation of these heterocycles depends on the temperature, solvent, and electronic/steric properties of the substrates. The nucleophilic attack by the nitrogen anion is feasible when the enamine contains an electron-withdrawing group on the nitrogen, likely due to resonance stabilization.
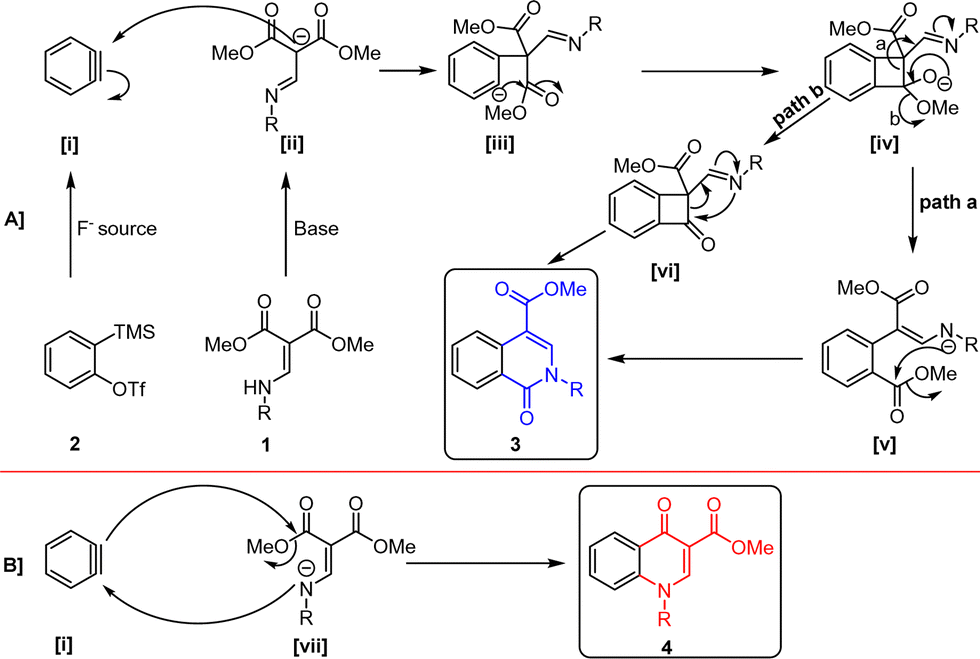 | ||
| Scheme 7 Plausible reaction mechanisms. | ||
In conclusion, we have demonstrated an efficient transition-metal-free protocol for the selective synthesis of pharmaceutically valuable functionalized quinolones and isoquinolones utilizing easily accessible enamine and aryne substrates. The overall process involves the cleavage of the C–C bonds and the formation of C–C and C–N bonds in a single-step to accomplish difunctionalized hetero aromatics of synthetic importance. Additionally, the protocol has been extended to the synthesis of the antagonist agent AS2717638 and floxacin key intermediates. The scalability of the protocol was also demonstrated. It should be feasible to extend this protocol to the total synthesis of the antimalarial agent SJ000101247, related natural products, and the floxacin class of APIs.
S. B. M. acknowledges financial support from SERB, New Delhi, India. S. D. M. thanks CSIR-New Delhi.
Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
A provisional patent has been filed based on this chemistry.Notes and references
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef.
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS.
- X.-Q. Hao, C. Du, X. Zhu, P.-X. Li, J.-H. Zhang, J.-L. Niu and M.-P. Song, Org. Lett., 2016, 18, 3610–3613 CrossRef CAS PubMed.
- (a) A. R. Millanao, A. Y. Mora, N. A. Villagra, S. A. Bucarey and A. A. Hidalgo, Molecules, 2021, 26, 7153–7195 CrossRef CAS; (b) N. T. Dung, D. M. Thanh, N. T. Huong, P. T. Thuy, N. T. Hoan, D. T. M. Thanh, N. Van Trang and N. T. Son, Struct. Chem., 2020, 31, 2435–2450 CrossRef CAS.
- (a) L. A. Mitscher, Chem. Rev., 2005, 105, 559–592 CrossRef CAS; (b) S. C. Kuo, H. Z. Lee, J. P. Juang, Y. T. Lin, T. S. Wu, J. J. Chang, D. Lednicer, K. D. Paull and C. M. Lin, J. Med. Chem., 1993, 36, 1146–1156 CrossRef CAS.
- H. Bai, F. Liu, X. Wang, P. Wang and C. Huang, ACS Omega, 2018, 3, 11233–11251 CrossRef CAS PubMed.
- (a) M. Liu, M. Ohashi and Y. Tang, Org. Lett., 2020, 22, 6637–6641 CrossRef CAS PubMed; (b) N. Fokialakis, P. Magiatis, A.-L. Skaltsounis, F. Tillequin and T. Sévenet, J. Nat. Prod., 2000, 63, 1004–1005 CrossRef CAS PubMed.
- (a) Y. Jang, J. Han, X. Li, H. Shin, W.-J. Cho and M. Kim, Pharmaceutical, 2021, 14, 650 CAS; (b) W. A. Guiguemde, Anang A. Shelat, Jose F. Garcia-Bustos, T. T. Diagana, F.-J. Gamo and R. K. Guy, Chem. Biol., 2012, 19, 116–129 CrossRef CAS PubMed.
- (a) D. Zhang, A. M. Decker, K. Woodhouse, R. Snyder, P. Patel, D. L. Harris, Y.-X. Tao, J.-X. Li and Y. Zhang, Eur. J. Med. Chem., 2022, 243, 114741 CrossRef CAS PubMed; (b) D. M. Floyd, P. Stein, Z. Wang, J. Liu, S. Castro, J. A. Clark, M. Connelly, F. Zhu, G. Holbrook, A. Matheny, M. S. Sigal, J. Min, R. Dhinakaran, S. Krishnan, S. Bashyum, S. Knapp and R. K. Guy, J. Med. Chem., 2016, 59, 7950–7962 CrossRef CAS PubMed; (c) G. R. Pettit, Y. Meng, D. L. Herald, K. A. N. Graham, R. K. Pettit and D. L. Doubek, J. Nat. Prod., 2003, 66, 1065–1069 CrossRef CAS PubMed.
- Q. Wang, K. C. Mgimpatsang, X. Li and A. Dömling, J. Org. Chem., 2021, 86, 9771–9780 CrossRef CAS.
- S. Lee, S. Mah and S. Hong, Org. Lett., 2015, 17, 3864–3867 CrossRef CAS.
- (a) S. Jaime-Figueroa, M. J. Bond, J. I. Vergara, J. C. Swartzel and C. M. Crews, J. Org. Chem., 2021, 86, 8479–8488 CrossRef CAS PubMed; (b) R. Manetsch, D. E. Kyle, R. Neelarapu, J. R. Maignan, C. L. Lichorowic and A. N. LaCrue, US Pat., US10000452B1, 2018 Search PubMed; (c) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS PubMed.
- (a) O. Píša and S. Rádl, Eur. J. Chem., 2016, 2016, 2336–2350 CrossRef; (b) R. G. Gould and W. A. Jacobs, J. Am. Chem. Soc., 1939, 61, 2890–2895 CrossRef CAS.
- S. Wang, L. Yao, J.-S. Wang, J. Ying and X.-F. Wu, Mol. Catal., 2022, 524, 112303 CrossRef CAS.
- X.-T. Min, D.-W. Ji, Y.-Q. Guan, S.-Y. Guo, Y.-C. Hu, B. Wan and Q.-A. Chen, Angew. Chem., Int. Ed., 2021, 60, 1583–1587 CrossRef CAS PubMed.
- (a) V. G. Pandya and S. B. Mhaske, Tetrahedron Lett., 2022, 101, 153901 CrossRef CAS; (b) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892–4044 CrossRef CAS PubMed; (c) M. M. Ahire, M. B. Thoke and S. B. Mhaske, Org. Lett., 2018, 20, 848–851 CrossRef CAS PubMed.
- R. Santhosh Reddy, C. Lagishetti, I. N. C. Kiran, H. You and Y. He, Org. Lett., 2016, 18, 3818–3821 CrossRef CAS PubMed.
- (a) Q. Yan, Z. Zhuang, R. Fan, J. Wang, T. Yao and J. Tan, Org. Lett., 2024, 26, 1840–1844 CrossRef CAS PubMed; (b) R. Fan, S. Liu, Q. Yan, Y. Wei, J. Wang, Y. Lan and J. Tan, Chem. Sci., 2023, 14, 4278–4287 RSC.
- Y. K. Ramtohul and A. Chartrand, Org. Lett., 2007, 9, 1029–1032 CrossRef CAS.
- M. Wernik, P. E. Hartmann, G. Sipos, F. Darvas, A. D. Boese, D. Dallinger and C. O. Kappe, Eur. J. Chem., 2020, 7051–7061 CrossRef CAS.
- I. Plastira, L. Joshi, E. Bernhart, J. Schoene, E. Specker, M. Nazare and W. Sattler, Front. Cell. Neurosci., 2019, 13, 1–12 Search PubMed.
- Y. Kawamoto, R. Seo, N. Murai, H. Hiyama and H. Oka, Bioorg. Med. Chem. Lett., 2018, 26, 257–265 CrossRef CAS.
- (a) B. Hufnagel, W. F. Zhu, H. M. Franz, E. Proschak and V. Hernandez-Olmos, ChemistryOpen, 2022, 11, e202200252 CrossRef CAS PubMed; (b) E. Whiting, M. E. Lanning, J. A. Scheenstra and S. Fletcher, J. Org. Chem., 2015, 80, 1229–1234 CrossRef CAS PubMed.
- (a) M. Asamdi and K. H. Chikhalia, Asian J. Org. Chem., 2017, 6, 1331–1348 CrossRef CAS; (b) C. Wu and F. Shi, Asian J. Org. Chem., 2013, 2, 116–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05671j |
| This journal is © The Royal Society of Chemistry 2025 |