
Recent developments in the photoredox catalyzed Minisci-type reactions under continuous flow
Serena
Pillitteri
 a,
Erik V.
Van der Eycken
ab and
Upendra K.
Sharma
*c
a,
Erik V.
Van der Eycken
ab and
Upendra K.
Sharma
*c
aLaboratory for Organic & Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, University of Leuven (KU Leuven), Celestijnenlaan 200F, B-3001, Leuven, Belgium
bPeoples’ Friendship University of Russia (RUDN University), Miklukho-Maklaya street 6, 117198 Moscow, Russia
cDepartment of Chemistry and Biochemistry, University of Missouri-St. Louis, One University Boulevard, St. Louis, MO 63121, USA. E-mail: sharmauk@umsl.edu
First published on 1st November 2024
Abstract
The Minisci reaction constitutes a straightforward and convenient strategy to achieve the direct C–H functionalization of heterocyclic molecules. This radical-based reaction platform has received increasing attention due to the predictability of its outcome according to the nature of the radicals and heterocycles involved. Considering the importance of these heterocyclic scaffolds in the development and production of drug molecules, it is inevitable that scaling up this reaction manifold is of utmost importance. This review will present recent strategies to achieve the goal, which mostly involve implementing the reaction conditions under continuous flow.
 Serena Pillitteri | Serena Pillitteri received her PhD degree from KU Leuven in Chemistry in 2024 with Prof. Erik Van der Eycken and Dr Upendra K. Sharma at KU Leuven (Belgium) as a FWO researcher. During her PhD, she received an FWO travel grant to work with Prof. Burkhard König (University of Regensburg, Germany). Currently, she is working as a postdoctoral researcher in the same research group. Her research focuses on the application of visible light photoredox catalysis in continuous flow and on the development of novel synthetic methodologies for the generation of C-based radicals, with a focus on organoboron compounds. |
 Erik V. Van der Eycken | Erik V. Van der Eycken is a full Professor of Organic Chemistry and Head of the Division of Molecular Design & Synthesis at KU Leuven, Belgium. He received his PhD-degree (1987) in organic chemistry from the University of Ghent, Belgium. He spent time as visiting scientist at the University of Graz (2002) with Prof. C. O. Kappe, at The Scripps Research Institute (La Jolla, USA) (2003) in the group of K. B. Sharpless, and at Uppsala University (2004) with Prof. M. Larhed and Prof. A. Hallberg. The main focus of his research is the development of new synthetic methodologies in combination with enabling techniques. |
 Upendra K. Sharma | Upendra K. Sharma received his PhD (2011) from CSIR-Institute of Himalayan Bioresource Technology, Palampur, India. He followed this with a lecturer position at the National Institute of Technology, Jalandhar, India. He was a postdoctoral fellow at KU Leuven, the University of Cambridge, the University of Eindhoven, and the Shanghai Institute of Organic Chemistry. He has also worked as a visiting research scientist at the Massachusetts Institute of Technology (MIT). In 2020, he joined KU Leuven as a research expert (Jr. group leader) starting his independent research career before joining the University of Missouri - St. Louis faculty in the fall of 2024. His research interests include exploring enabling technologies to address challenges in organic synthesis. |
Introduction
The direct C–H functionalization of heterocyclic scaffolds represents a straight way to synthesize differently functionalized molecular architectures.1,2 Considering the relevance of N-containing heterocyclic cores in drug molecules,3 the scientific community has directed great endeavors in developing synthetic methodologies for the purpose.Where the well-established chemistry of cross-coupling reactions might be complicated by the complexing nature of these scaffolds, radical chemistry comes into help. In the late 1960s, it was discovered that radical addition on basic heterocycles like pyridines and quinolines could be selectively steered to the C2 and C4 positions upon the addition of a strong acid to the reaction mixture.4–6 What would later be known as the Minisci reaction, exploited the LUMO-lowering effect of the acid additive to direct the addition of a nucleophilic radical, overall obtaining a formal substitution reaction.7,8 The mechanism proceeds with the following steps: upon radical addition on the protonated heteroarene, the resulting radical cation can undergo a first deprotonation step followed by oxidation (these two steps could also be reversed) and rearomatization. Alternatively, according to the reaction conditions, a hydrogen atom transfer (HAT) step can happen, to obtain the same final product (Fig. 1).9
 | ||
| Fig. 1 General mechanisms of the Minisci reaction and scheme of the modules of a continuous flow setup. | ||
The group of Minisci first employed alkyl carboxylic acids as a radical source and a combination of a silver catalyst and persulfate as the oxidant under thermal conditions.6 This pioneering work was nevertheless limited by the low yield of primary alkyl carboxylic acids and could not be expanded to the use of aryl radicals.
In recent years, the advent of photoredox catalysis has introduced the possibility to generate radicals in a mild and selective way, increasing the scope of applicability of radical reactions in general.10,11 Considering the utility of Minisci-type reactions, a growing number of applications for this reaction manifold is constantly appearing, enabling in many cases also the late-stage functionalization of differently substituted drug molecules or naturally occurring scaffolds.9 The range of radical precursors employed for the purpose has been drastically expanded. With reference to photoredox catalyzed procedures, together with carboxylic acids, their derivatives (oxalates and especially N-acyloxyphthalimides, also known as redox-active esters, RAEs) represent convenient radical sources. In addition to those, organoboron compounds, sulfur compounds, alkyl halides, and, in many cases, also simple alkyl chains have been introduced as radical sources.9 Reactivity studies have also been directed towards the functionalization of electron-rich heterocyclic cores (pyrroles, as an example), leveraging in this case the affinity of electrophilic radicals for electron-rich scaffolds.8 These kinds of reactions will be further referred to in the discussion as Minisci-type reactions. The importance of this reaction manifold and its applications have been highlighted in recent reviews, that unfold different aspects of the developed procedures so far available, focusing on the radical source, on the energy input or on methods to perform the reaction enantioselectively.9,12–16
The rise of photoredox-catalyzed synthetic procedures can be rationalized with the several benefits that photoredox catalysis holds. The reaction conditions are mild, given that visible light irradiation, mostly at room temperature, is utilized.17,18 In addition, safer and cost-effective procedures and reagents have been introduced, making photoredox catalysis an attractive field, also from an industrial point of view.19 The overall increased mildness of the reaction conditions leads to improved tolerability and applicability. Considering the complex and functionalized nature of drug molecules and their intermediates, this feature is highly desirable to achieve the late-stage functionalization of these molecules.20 If also considering the need to increase the saturation of molecules to expand the chemical space, photoredox catalysis comes as the ideal platform, given the possibility to generate C-centered radicals from a plethora of saturated radical sources used for alkylation reactions.21 All these elements led industrial chemists to look at photoredox catalysis with increasing interest. Recent reviews have pointed out how photoredox catalysis is becoming routinely employed in the development of new methods in both the pharmaceutical and agrochemical industries.19,22
Together with the development of new methods, the issue of scalability of photochemical processes becomes of fundamental importance. In this regard, flow chemistry applied to photoredox-catalyzed processes figures as the obvious solution to the problem.23 The implementation of batch photochemical reactions in continuous flow offers several benefits. First, light penetration is drastically increased. According to the Bouguer–Lambert–Beer law, the light intensity decreases towards the center of a batch reactor, as a result of the presence of high absorbing molecules like the photocatalysts. In a flow tube, the small diameter maximizes light distribution and therefore enhances the reaction rate.24,25
Due to the small dimensions of the tubes, heat and mass transfer also become better. This allows to perform reactions at higher temperatures and, in many cases, avoids the formation of side products. The possibility to finely tune the reaction conditions in terms of reactor volume, temperature, light intensity, and residence time, also allows for higher reproducibility of the reaction outcome, and predictability when considering the scale-up of the developed flow protocols.24,26 In a more practical sense, the modularity that flow setups offer becomes a major benefit, especially when considering not only reproducibility, but also automation, standardization and analysis of a reaction. As it is evident from Fig. 1, a flow reactor can be coupled with different modules, that can aid the purification step or, alternatively, allow on-line analysis, rendering the process as a whole more efficient.27
In light of the benefits deriving from the implementation of photoredox processes in continuous flow, it comes as no surprise that many efforts have been directed to translating batch photochemistry into flow chemistry.19 Recent reviews have appeared discussing strategies to scale-up photochemical reactions performed in flow, highlighting the benefits but also the challenges that still need to be tackled.28–30
Considering the importance of the Minisci and Minisci-type reaction manifolds, we will describe in this highlight recent methodologies employing flow chemistry in the photoredox-catalyzed functionalization of heterocyclic scaffolds. The discussion will be divided in two sections: in the first section, the functionalization of basic heterocycles through a Minisci mechanism will be described, while in the second section, the functionalization of electron-rich heterocycles will be addressed.
Implementation of the Minisci reaction under continuous photo-flow conditions
In 2021, Graham, Noonan and co-workers disclosed an interesting route for synthesizing an intermediate of Ceralasertib, a compound currently being evaluated as an anticancer drug. In this work, the authors drastically shortened its synthetic route by introducing the Minisci conditions to form the desired functionalized dichloropyrimidine (Scheme 1). At first, 1-methylsulfanylcyclopropanecarboxylic acid was mixed with N-hydroxyphthalimide to generate in situ a redox-active ester, that was later mixed with the dichloropyrimidine in the presence of 4CzIPN and camphorsulfonic acid. The desired product was obtained in 25% yield. Extensive optimization led the authors to find the best conditions to ensure a high reaction rate and an increased yield. The in-depth studies of the reaction rate and mechanism were dictated by the aim to find suitable conditions for an efficient scale-up using a continuous-flow setup (aiming at 5 kg day−1 of the functionalized pyrimidine). Interestingly, the authors noticed that the addition of acid was not required because of the matching nucleophilicity of the alkyl radical and the electrophilicity of the dichloropyrimidine, and they found out that an increment in the reaction rate could be achieved with more reducing catalysts of the family of 4CzIPN. In particular, 3DPA2FBN was the photocatalyst of choice. For the flow process, the authors first employed a Vapourtec UV-150 setup. Due to the limited dimensions of the reactor, the process was further optimized in tubes of a diameter of 6 mm (4 mm internal diameter) and then in tubes with 8 mm internal diameter. With a residence time of 33 min, the reaction yield was in both cases higher than 60%. Considering that a pilot plant would be equipped with longer tubing (total length: 204 m), the authors reasoned that, if run continuously for 24 h, this process would in theory generate 6.6 kg day−1 of the desired drug intermediate.31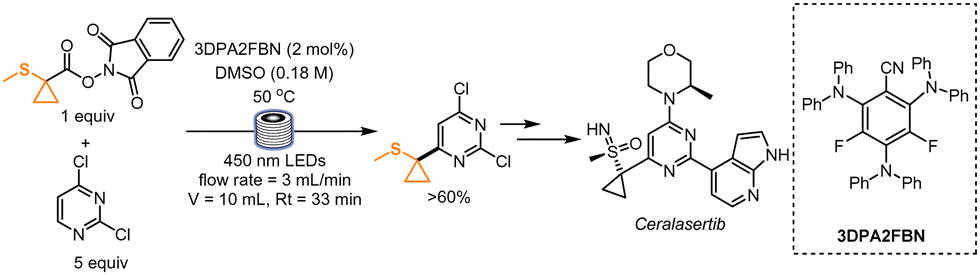 | ||
| Scheme 1 Additive-free Minisci reaction for the synthesis of an intermediate of Ceralasertib. | ||
In the same year, a similar radical generation strategy was developed by Mousseau and co-workers. In their work, 3-(methylester)bicyclo[1,1,1]pentane-1-carboxylic acid (BCP) was first converted into a RAE and then used in a photochemical process to functionalize a wide variety of heterocyclic scaffolds, and therefore obtain a wide array of 1,3-substituted BCPs. The starting material was chosen with the aim to build bioisosteres of benzoates. To determine the best reaction conditions, the authors employed a high-throughput continuous flow reactor coupled with high-performance chromatography where nanomolar scale reactions could be performed. For this reaction, good yields were obtained within a residence time of 90 s. The reactions were run in duplicates and triplicates to confirm the results, ensuring reproducibility. The work revealed that 475 reactions could be performed in 12 h of instrument time. Higher scale (0.25 mmol scale) batch reactions were performed for the scope studies: differently substituted pyridines were utilized, together with pyridones and fused heterocycles like quinolines, pyrazines, ribonucleosides, and 5,6-membered ring systems. In addition, differently functionalized BCPs were formed as well (Scheme 2).32
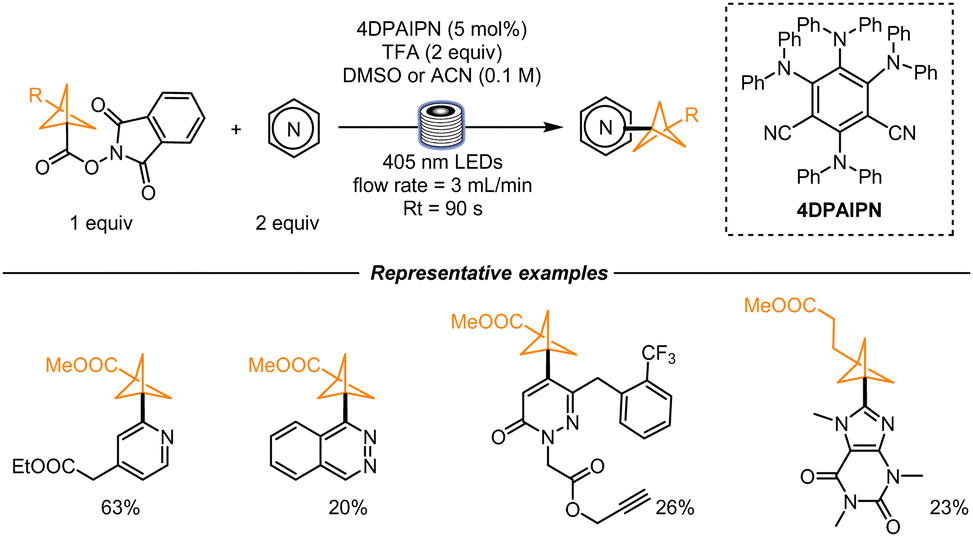 | ||
| Scheme 2 Nanomole-scale reaction screening using BCPs as benzoate bioisosteres. | ||
In 2022, our group reported a photoredox/cobalt-catalyzed Minisci reaction using boronic acids and derivatives (boronic esters and trifluoroborates) as alkyl radical sources. The substitution of stoichiometric amounts of (inorganic) oxidants with a cobalt catalyst resulted in being particularly convenient for the translation of the batch conditions to continuous flow. Throughout the duration of the reaction, no precipitate formation was observed, avoiding clogging issues in the thin tubes of a flow setup (Scheme 3). Mechanistically, upon oxidation of the boron compound by the excited photocatalyst, an alkyl radical is formed. At this stage, a classical Minisci mechanism, as delineated before, takes place. After radical addition on the protonated heterocyclic core, further oxidation and proton loss happens, delivering the final product. A Co(II) species is involved in this last formal HAT step. The Co(II) is generated upon reduction of the Co(III) co-catalyst by the reduced form of the photocatalyst, which is regenerated at the same time. The Co(III)–H species, formed after the formal hydrogen abstraction, then leads in the acidic medium to the formation of H2 with the concomitant Co(III) catalyst regeneration.
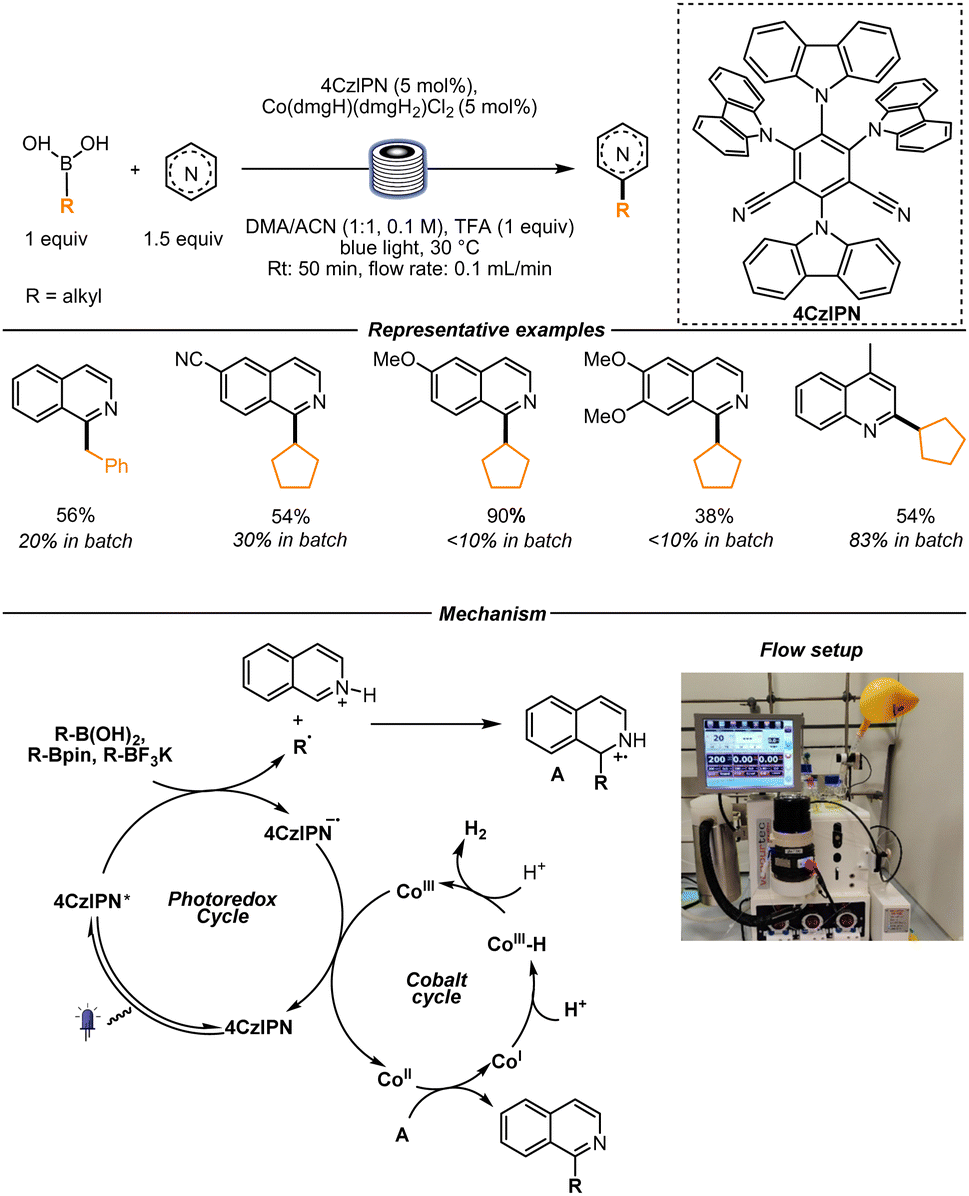 | ||
| Scheme 3 Dual photoredox/cobalt catalyzed Minisci reaction with boronic acids and derivatives as radical source. Reproduced from ref. 33 with permission from the Chinese Chemical Society (CCS), Shanghai Institute of Organic Chemistry (SIOC), and the Royal Society of Chemistry, copyright 2022. | ||
A small scope of substrates was tested in flow. It was observed that, in some cases, reactions that were sluggish in batch worked better in flow. In these cases, cleaner reaction mixtures were obtained. In addition, the yield of some of the substrates was higher than the one obtained in a batch setup, and the reduced reaction time was of particular importance. The flow setup utilized in this case was a Vapourtec UV-150, and an average residence time of 50 min in a 10 mL volume reactor (with exceptions in some cases) was set.33
An innovative platform to perform consecutive photochemical reactions through a telescoped continuous-flow process was presented by Baumann and co-workers. In this report, a dual coil photoreactor was effectively utilized for the formation of quinolines, that were subsequently functionalized in a Minisci fashion using diacetyl as the photocatalyst. Under 365 nm irradiation, the photocatalyst reacts with a wide variety of ethers, which undergo a hydrogen atom transfer (HAT) step, generating a C-centered radical that further adds on differently substituted quinolines (Scheme 4). The authors described the setup used to perform the sequential reactions. A Vapourtec E-series with its UV150 photomodule was utilized. Upon quinoline formation in the first coil, the stream was pumped in the second coil and mixed through a T-mixer with a solution containing the photocatalyst, tert-butyl hydroperoxide (TBHP), and trifluoroacetic acid (TFA), dissolved in the desired ether. Both reactions were irradiated with a 365 nm lamp, with a residence time of 10 min and 5 min respectively (volume of the reactors: 5 mL). The authors also evaluated the best wavelength combination for the two telescoped reactions: while quinoline formation was not affected by the wavelength and performed well under blue light irradiation as well, the Minisci step necessitated a more energetic wavelength, that led the authors to choose a 365 nm irradiation for the dual-coil photoreactor setup, without the need for two separated flow reactors equipped with two different light sources.34
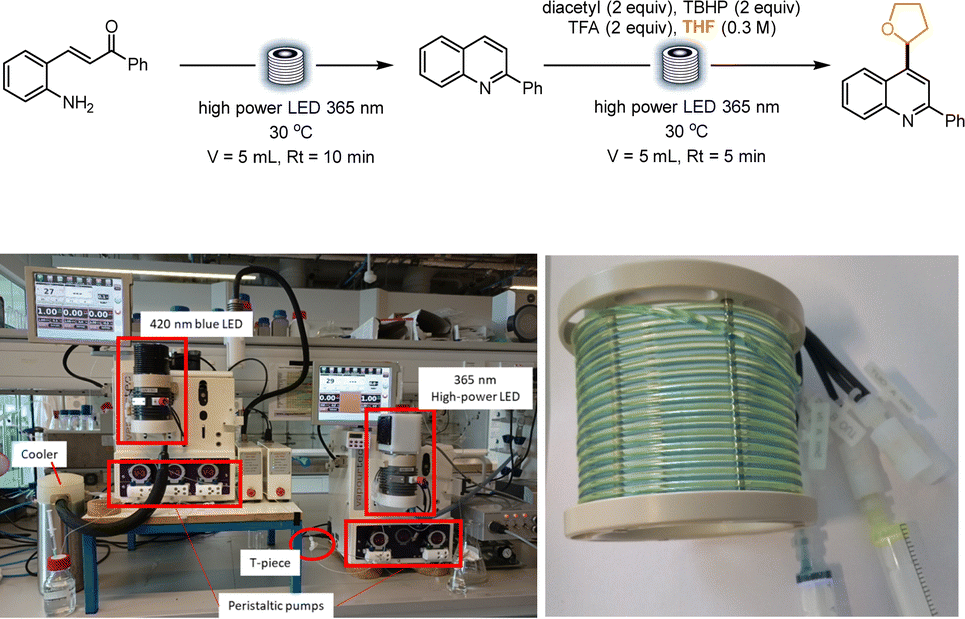 | ||
| Scheme 4 Consecutive photochemical reactions for the synthesis and functionalization of quinolines. In the pictures, the reported setup for the dual photochemical reactor and the dual flow coil. Reproduced from ref. 34 with permission from the Royal Society of Chemistry, copyright 2022. | ||
A different strategy to functionalize basic heterocycles was utilized by Li, Zhu and co-workers. In this case, C-radical generation was obtained using alcohols as radical precursors. In the presence of phenyliodine bis(trifluoroacetate) (PIFA) under blue-light irradiation, an alkoxy radical, together with TFA, are generated, overcoming the challenge of the high O–H bond dissociation energy. At this stage, two different pathways can be devised, according to the structure of the alcohol: either a 1,5-HAT or a β-scission can happen, forming the desired C-centered radical (Scheme 5). The reaction was optimized in a stop-flow regime, and the authors underlined that the corresponding reaction in batch, under the same reaction conditions, resulted in a lower yield. This result is due to better light penetration in the microtubes. Operationally, the reaction was pumped into PFA tubes with an internal diameter of 760 μm and was irradiated for 12 h with blue light. When performed on a gram scale, a longer irradiation time (48 h) was required. The scope of the reaction proved to be quite broad, both in terms of heterocyclic molecules and alcohols. Quinolines bearing electron-withdrawing and electron-donating groups were functionalized, together with phenanthridines, pyridines, bipyridines and quinazolines as examples. The alcohol counterpart was also versatile. Primary, secondary, and tertiary alcohols generated the corresponding C-centered radicals upon PIFA activation. Similarly, a diverse array of alcohols underwent β-scission to afford moderate to good yields of functionalized heterocycles.35
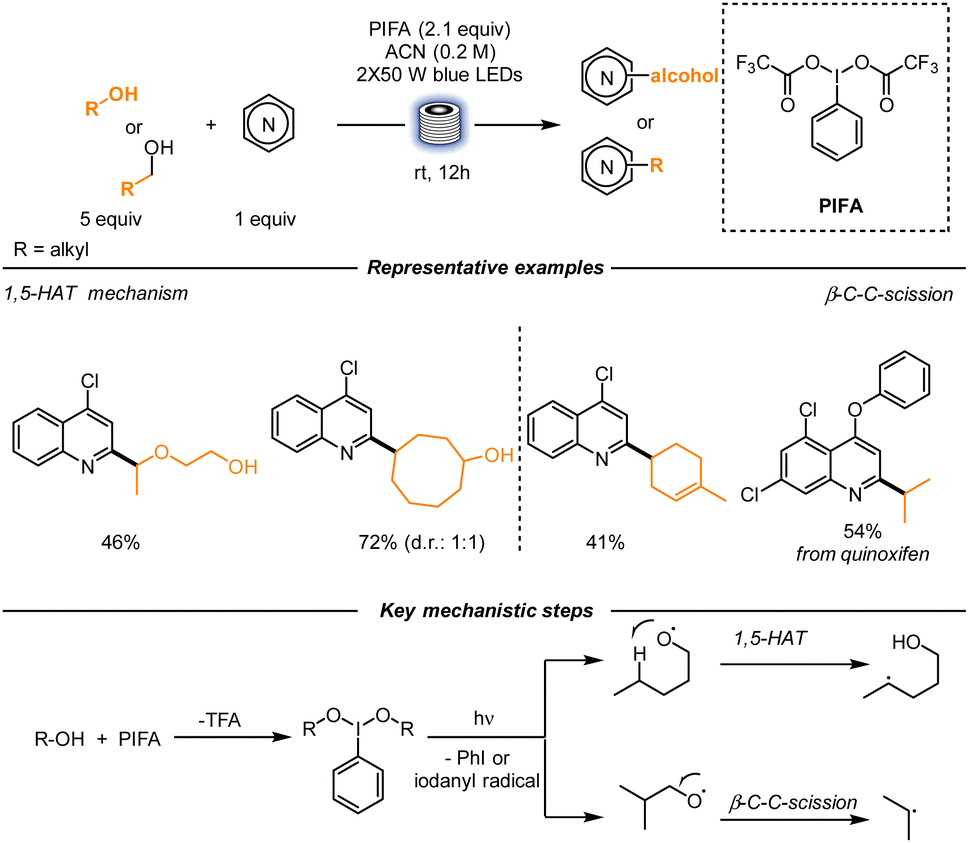 | ||
| Scheme 5 Heteroarylation of aliphatic alcohols using stop-flow conditions. | ||
Considering the importance of diazines in drug molecules, Jubault, Poisson and co-workers devised a convenient method to achieve the alkylation of these heterocyclic cores using carboxylic acids as alkyl radical source and a combination of benzoyl peroxide and 4CzIPN to enable radical formation. Though mostly developed in batch, the reaction was also implemented in a flow setup. Upon white LED irradiation with a flow rate of 0.5 mL min−1 in a 10 mL volume reactor, 72% yield of the product was formed, with an improved productivity (3.24 mmol h−1 against 0.85 mmol h−1 in batch, Scheme 6).36
 | ||
| Scheme 6 Flow implementation for the alkylation of diazines using carboxylic acids as radical source. | ||
Development of the Minisci-type reaction using electron-rich heterocycles under continuous-flow
Complementarily to basic heterocycles, the functionalization of electron-rich heterocyclic scaffolds can be achieved using Minisci-type conditions as well. In this case, the nucleophilicity of the heterocycle has to be matched with the electrophilicity of the radical species generated. For this subclass of transformations, flow conditions were implemented as well over the years.Stephenson and co-workers demonstrated in 2015 that trifluoroacetic anhydride (TFAA), under the right reaction conditions, can be an effective source of CF3 radicals, that can further react with (hetero)aryl scaffolds, generating interesting trifluoromethylated compounds. In light of the unattainable oxidation potential of the TFA anion, they reasoned that its redox activation could become feasible in the presence of a sacrificial redox auxiliary. They identified pyridine-N-oxide as an inexpensive and convenient mediator. Upon the generation of the TFAA/N-oxide adduct, the reduction potential becomes considerably lower (−1.10 V), and a photocatalyst-mediated single electron transfer leads to the cleavage of the newly formed N–O bond and the subsequent extrusion of pyridine and CO2, together with the desired CF3 radical (Scheme 7). A few heterocyclic cores were trifluoromethylated, including indoles, pyrroles, and oxazoles. In light of the poor reactivity of pyridines (due to the polar mismatch between the CF3 radical and the heterocycle), N-methylpyridone was utilized instead. After the trifluoromethylation reaction, that afforded the product in 54% yield, the pyridone was converted into a pyridine of high importance for drug development. The authors then tried to scale up the reaction. While batch scale-up was found to be feasible, but requiring consistently longer reaction time, the implementation of the reaction under flow conditions constituted an obvious solution to improve light penetration. With a residence time of 10 min in a 10 mL volume reactor, a 20 g scale reaction was performed, with a promising 71% yield.37
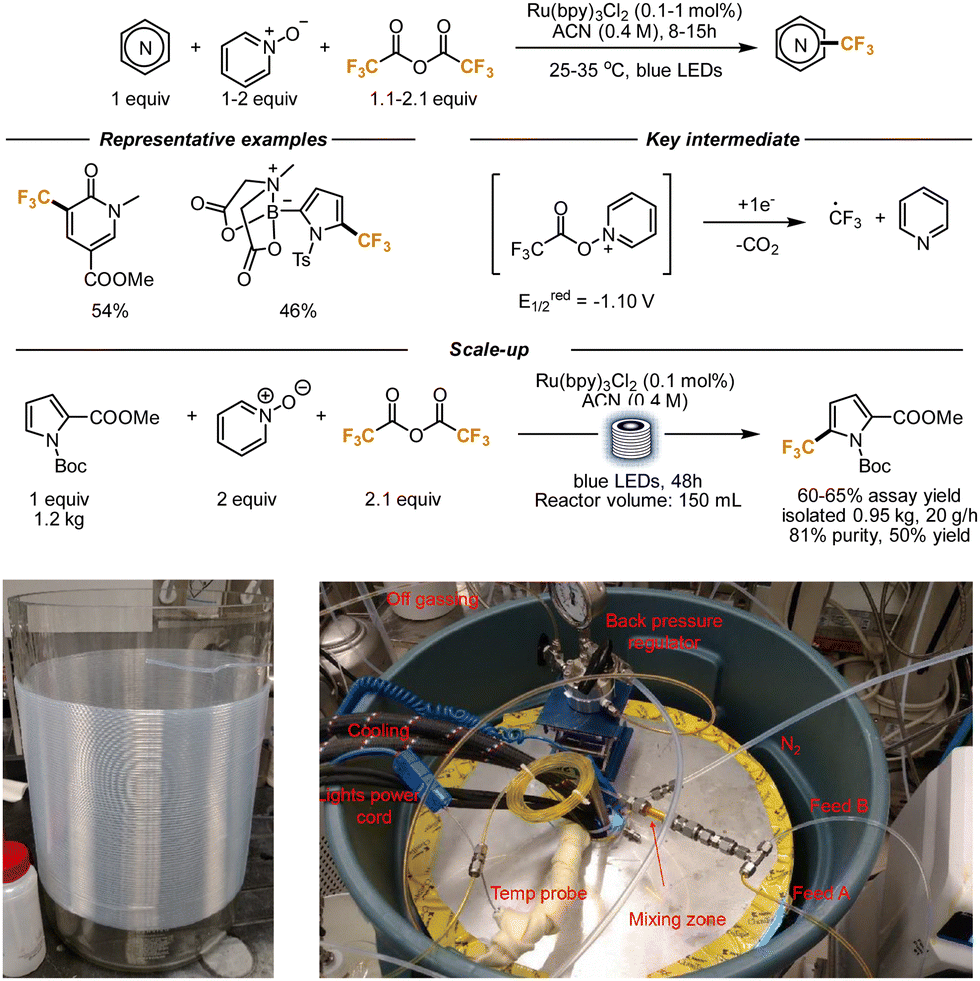 | ||
| Scheme 7 TFAA as trifluoromethylating reagent. Application to the kg-scale synthesis of a trifluoromethylated pyrrole. In the pictures, details of the glass reactor and tubing (on the left) and overview of the whole setup (on the right); reproduced from ref. 38, with permission from Elsevier, copyright 2016. | ||
Aware of the potential that this reaction manifold held, the same authors also reported a reaction variant that was scaled up to 1 kg. The authors realized that the electronics of the pyridine-N-oxides play a role in the reaction outcome. Upon extensive optimization, they found out that the best results could be obtained by introducing 4-phenyl pyridine-N-oxide as a redox mediator. Its addition lowered the reduction potential of the TFAA/N-oxide adduct to −0.91 V. 0.1 Mol % of the photocatalyst was also sufficient to obtain good results. The newly developed conditions proved to be broadly applicable to a wide range of electron-rich heterocycles, and the reactivity was further extended to the perfluoroethylation of similar scaffolds. Interestingly, N-Boc protecting groups and MIDA boronates remained untouched during the reaction. To further scale up the reaction, an in-house flow reactor was developed. The blue LEDs were installed in a glass beaker, that was wrapped with PFA tubing (1.6 mm internal diameter) from the outside, for a total volume of 150 mL. The setup was placed into a steel casing and cooled down with water (see pictures underneath Scheme 7). A peristaltic pump was employed to pump the reaction in the flow setup. The reaction was performed on a pyrrole moiety, using in this case pyridine-N-oxide for its cheaper price and 0.1 mol% of a ruthenium photocatalyst. Assays of the reaction revealed that an average yield of 60–65% was constantly obtained. After 48 h of irradiation and purification, 0.95 kg of the final product was obtained. Considering the dependence of the reaction outcome on the light intensity, the authors hypothesize that a light source with a higher photon flux would allow for the use of wider tubes, with a consequent further increment in productivity.38
Aware of the convenience of using TFAA as a trifluoromethylating reagent, a different strategy leveraging the efficiency of an EDA complex was developed by the same group. Previous studies revealed that N-(trifluoroacetoxy)-pyridinium (see the structure in the box Key intermediate, Scheme 7) can form an EDA complex in the presence of electron-rich heterocycles. To effectively exploit this possibility, the authors introduced a catalytic amount of 2-methoxynaphthalene to act as the electron-donor moiety. They hypothesized that, upon irradiation, the EDA complex between the N-(trifluoromethylacetoxy)-pyridinium and 2-methoxynaphthalene would form a 2-methoxynaphthalene radical cation and the desired CF3 radical (Scheme 8). The CF3 radical would then undergo the addition on electron-rich heterocycles. The arising radical intermediate would then be finally oxidized by the 2-methoxynaphthalene radical cation, therefore forming the final product and regenerating 2-methoxynaphthalene for a new catalytic cycle. The reaction platform proved to be amenable to a range of functionalized pyrroles, benzopyrroles and azaindoles. The authors noticed higher yields in the presence of a Boc-protection rather than different protecting groups. To prove the scalability of the process, the reaction was performed in a flow setup. With a throughput of 0.12 mmol min−1, the gram scale reaction afforded the final product in moderate yield (46% yield, 1.19 g).39
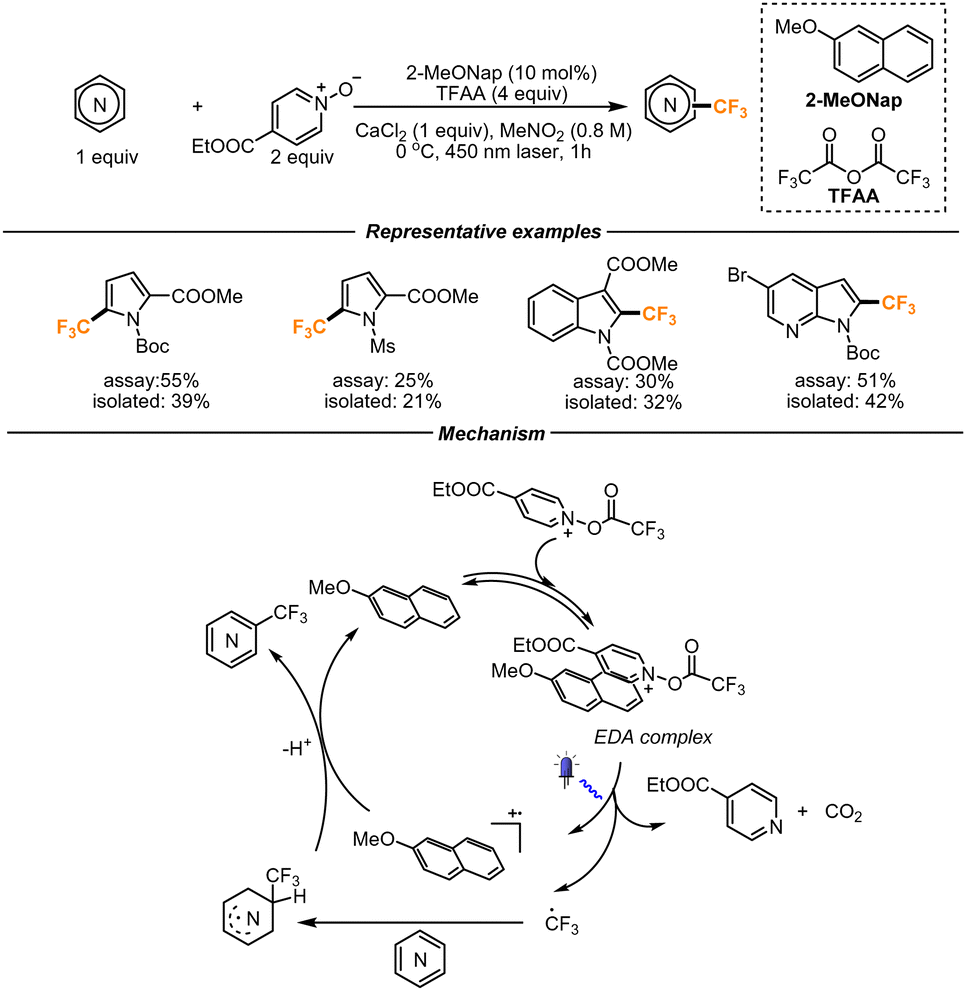 | ||
| Scheme 8 EDA-mediated trifluoromethylation of heterocyclic scaffolds. | ||
In 2017, Guttman, Kappe, and co-workers were able to achieve the incorporation of fluorinated moieties in pyrroles and indoles as well. No visible-light irradiation was required for this transformation, which, following previous studies, involved the use of H2O2, a Fe(II) catalyst, and DMSO for the generation of radicals from fluorinated alkyl iodides. These conditions were also implemented under continuous flow, demonstrating that the improved heat and mass transfer can substantially impact the reaction rate. With a residence time of a few seconds in a microflow reactor, the process showed potential for further industrial applications (Scheme 9).40
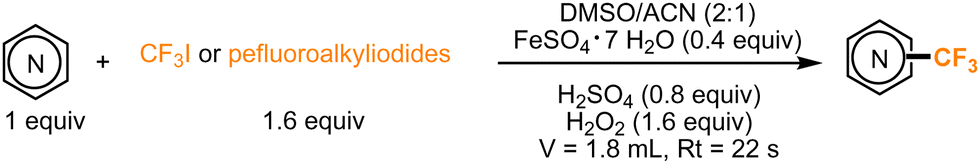 | ||
| Scheme 9 Continuous-flow perfluoroalkylation using Minisci-type conditions. | ||
Following these first approaches, photoredox-catalyzed Minisci-type reactions performed under continuous flow started to appear, as a result of the convenience in radical generation and in terms of selectivity.
In 2018, Fagnoni and co-workers developed a photocatalyzed strategy for the trifluoromethylation of electron-rich cores, including heterocyclic scaffolds. In this work, the authors employed N-aryltrifluoromethanesulfonimide as a CF3 radical precursor. This compound, upon irradiation with 310 nm light, undergoes a N-S homolytic break, generating radicals I and II. Radical II undergoes SO2 loss, forming a CF3 radical, that further attacks the heterocyclic molecule (Scheme 10). Radical I might instead be involved in the final HAT step that forms the final product, and the resulting sulfonamide can be the source of another equivalent of CF3 radicals. Alternatively, the CF3 radical itself can promote the HAT step from the intermediate. The authors proposed this step considering that CF3H was observed as a side product of the reaction. For the flow setup, coils of UV-transparent FEP tubing were wrapped around a water-cooled 500 W medium-pressure mercury lamp. The reaction was pumped at a flow rate of 10 mL h−1 in a 50 mL volume reactor. A small scope of reactions was attempted, and the reactions performed better than the corresponding batch variant, with yield increments up to 30% and shorter reaction times (5 h versus 12 h).41
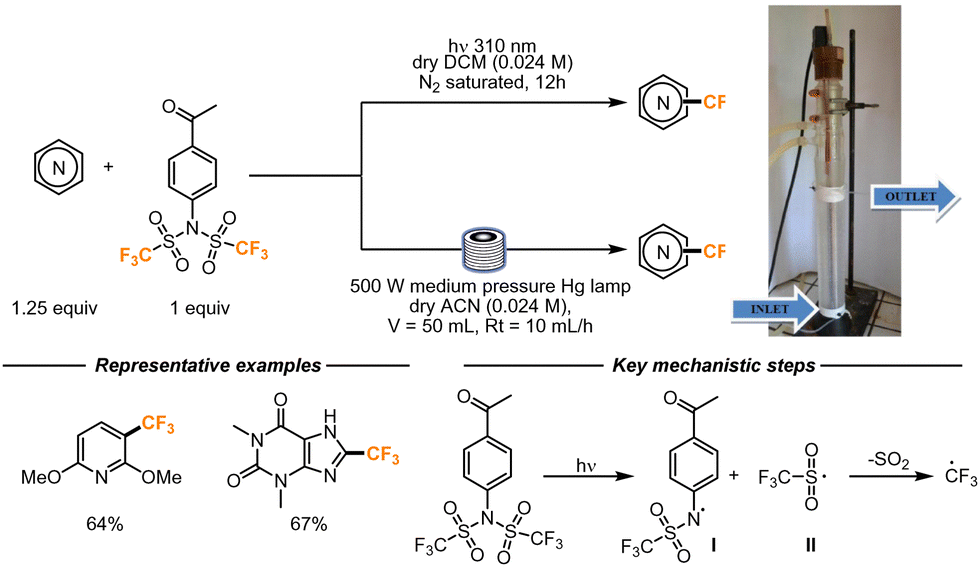 | ||
| Scheme 10 N-Aryltrifluoromethanesulfonimide employed as trifluoromethylating reagent employing Minisci-type conditions. Reproduced from ref. 41 with permission from the Royal Society of Chemistry, copyright 2018. | ||
The trifluoromethylation of functionalized heterocycles was also studied by Noël and co-workers. The Langlois reagent (CF3SO2Na) was chosen to be used as a precursor of the CF3 radical, in the presence of (Ir[dF(CF3)ppy]2(dtbpy))PF6 as a photocatalyst, that was proved to be the best catalyst through fluorescence quenching experiments (Scheme 11). The flow setup used for the transformation consisted of a Vapourtec UV-150 photoreactor, equipped with fluoropolymer tubing with an internal diameter of 1.3 mm and a total volume of 10 mL. A residence time of 30 min was found to be optimal to obtain the final products. First, caffeine was trifluoromethylated in a moderate yield, and then the scope was further expanded to indoles, benzimidazoles, pyridones, and pyrimidones. The reaction was also scaled up: the trifluoromethylation of 5-bromopyridone was accomplished. Within 3.5 h, 545 mg (56%) of the trifluoromethylated scaffold was obtained.42
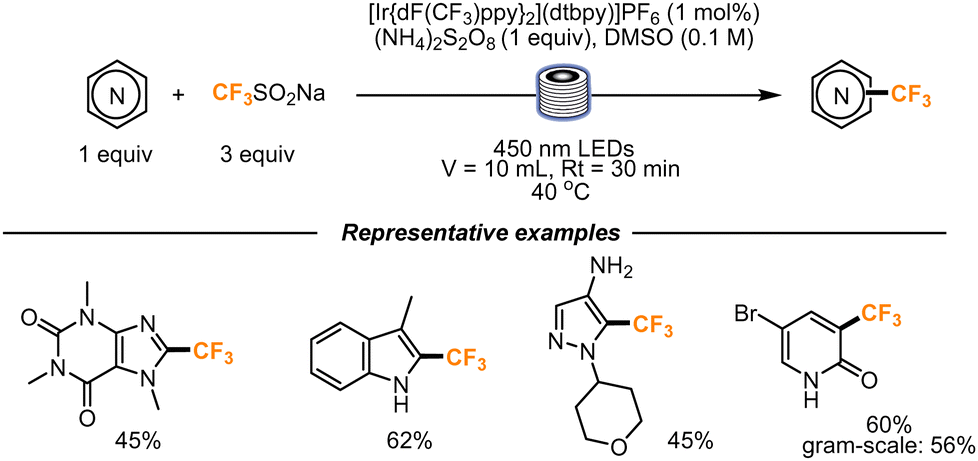 | ||
| Scheme 11 Trifluoromethylation of highly functionalized heterocyclic scaffolds using the Langlois reagent. | ||
With a similar aim, the trifluoromethylation of comparable scaffolds using the Langlois reagent was also developed employing 3DPA2FBN as a photocatalyst and implemented in a flow setup as well.43
The functionalization of electron-rich heterocycles with bromomalonate or trifluoromethylating and perfluoroalkylating reagents was then further studied by the same group soon after. In this work, a novel class of organic photoredox catalysts was utilized (2,2′-bithiophenebased derivatives), that proved to be as effective as metal-based photocatalysts. For these transformations, the implementation under continuous flow became particularly important to drastically reduce the reaction time to few minutes (Scheme 12).
 | ||
| Scheme 12 Functionalization of heteroarenes under photo-flow conditions. | ||
When using bromomalonate, triphenylamine was used as a sacrificial electron donor and irradiation with 400 nm light was found to give better results considering the higher match with the absorption maximum of the chosen photocatalyst. For the flow setup, microtubes with an internal diameter of 760 μm and an overall volume of 2.5 mL were used. For the trifluoromethylation and difluoroalkylation reactions, the corresponding perfluorinated iodides were employed. In this case, 450 nm light was utilized and a Vapourtec Photoreactor (tubing internal diameter: 1.3 mm, reactor volume 5 mL) was employed, with a residence time of 20 min. In the case of the gaseous trifluoromethyliodide, a slug flow reaction regime was conveniently used, providing the desired product in good yield.44
The Langlois reagent was also found to be an efficient trifluoromethylating agent of non-functionalized heterocyclic cores by Ackermann and co-workers. In their work, they demonstrated the viability of a photo-electrochemical transformation, which also effectively translated in a continuous-flow setup (Scheme 13). Both an acridinium-based photocatalyst (Mes-Acr+ClO4) and Ru(bpy)3(PF6)2 could be employed, together with a graphitic felt anode and a platinum cathode. The trifluoromethylated reaction proved to be feasible also on natural product-derived compounds, showing high tolerability. Mechanistically, the authors hypothesize that, upon excitation, the photocatalyst can oxidize the Langlois reagent, generating in turn a CF3 radical. The photocatalyst is then regenerated by anodic oxidation. The CF3 radical simultaneously attacks the heterocycle, forming a radical intermediate that can be then oxidized to generate a cation. Proton loss then determines the formation of the final product, while proton reduction happens at the cathode, with the development of H2.45
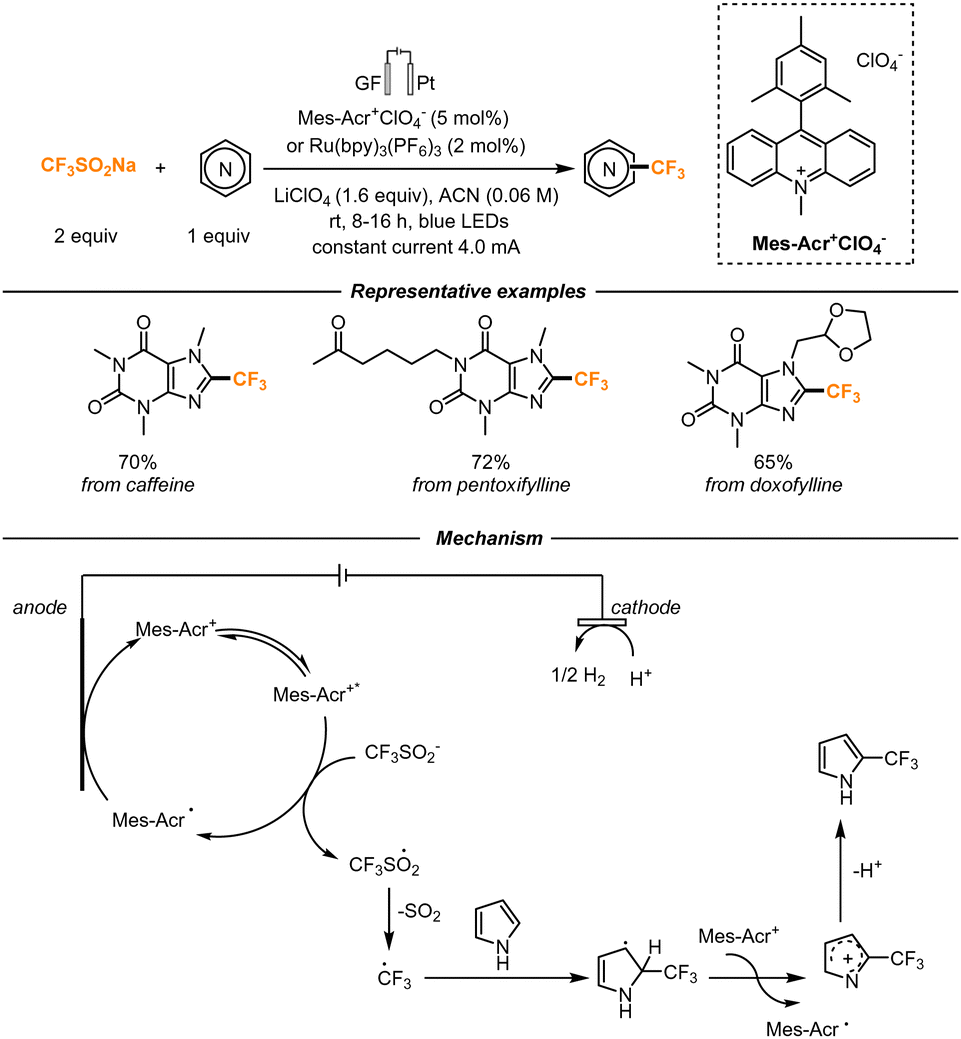 | ||
| Scheme 13 Photo-electrochemical trifluoromethylation of heterocyclic scaffolds. | ||
Last year, Vilé and co-workers further explored the Minisci-type reaction using a heterogenous catalyst in a flow setup (Scheme 14). Mesoporous graphitic carbon nitride (mpgC3N4) represents a convenient catalyst with high surface area and porosity. The catalyst was therefore packed with glass beads (2.5 wt%) in a transparent FEP tube (length = 500 mm, internal diameter = 2.1 mm). The presence of glass beads ensured a uniform catalyst separation and dilution. A residence time of 20 min was found to be optimal for the formation of the desired product. Longer residence times determined instead the formation of di-trifluoromethylated products. The catalyst stability was also evaluated: when the reaction was continuously streamed in the flow setup for 5 h, no catalyst decomposition was observed, and no decrease in product formation could be detected over time. This result, if performing the reaction in batch, would mean that the catalyst could be reutilized for 15 cycles without any product and catalyst loss. The choice of the base was another important parameter. In the presence of a strong base like K2HPO4, the yield increased, as a result of the improved re-aromatization rate (following CF3 radical addition). Under the described conditions, pyrroles, indoles and indazoles could be functionalized, though the latter heterocyclic core suffered from multiple CF3 additions.46
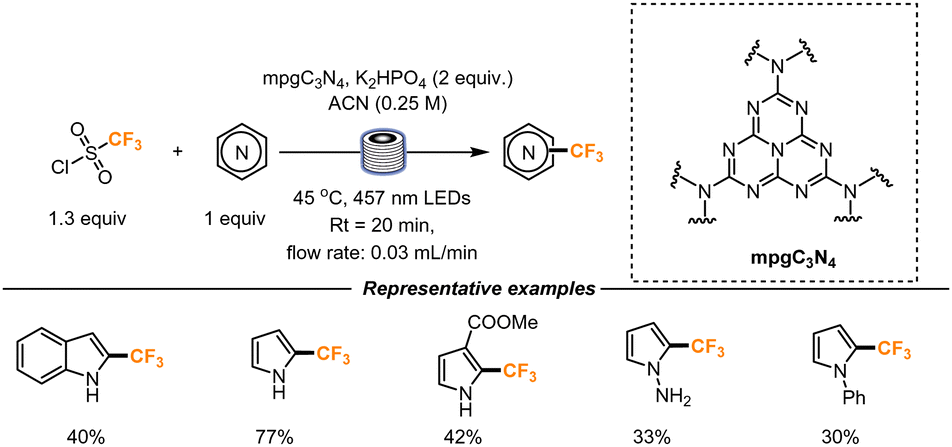 | ||
| Scheme 14 Trifluoromethylation of heterocycles catalyzed by nanostructured carbon nitride in a packed bad flow reactor. | ||
Conclusions and future perspectives
In light of the examples reported in this perspective, it is evident that the Minisci reaction has become an indispensable transformation, whose scope has undergone a tremendous expansion since the surge of photoredox-catalyzed processes. At the same time, the need for new protocols has to be accompanied by technological advances. In this regard, especially when considering photoredox-catalyzed reactions, flow chemistry has seen widespread application. When specifically analyzing the Minisci reaction, it is evident that considerable efforts have been devoted to the purpose of scaling up the reaction. For Minisci-type reactions, often involving trifluoromethylation of electron-rich substrates, several examples involving flow implementation are available, including a kilogram scale reaction. For basic heterocycles, on the other hand, fewer examples can be found, probably as a result of the need for insoluble reaction partners (like inorganic oxidants), which might undermine the development of flow procedures due to the risk of incurring in clogging issues. Despite some recent example avoiding the need for stoichiometric amounts of acids or oxidants, further alternatives to be applicable to a broader variety of substrates are desirable. This will also meet the need of tolerant reaction conditions to be applied for the late stage modifications of drug molecules. Some examples have been presented here, but they are still to some extent limited.The examples here described also show the variability of flow setups that can be utilized. While in many of the studies here presented commercially available setups were used (e.g. Vapourtec Photoreactors), in-house built flow reactors also proved to work efficiently. This underscores the versatility and modularity of flow chemistry, which does not require specialized equipment, and becomes particularly effective when scaling up photoreactions, thanks to the improved light penetration, and better control over the reaction parameters.
Though most of the examples here presented are a result of academic efforts, it can be anticipated that photoredox catalysis in combination with flow chemistry will play a central role in future industrial implementation. To reach the goal, it can be foreseen the need for standardized processes and setups. Considering the high variability of flow setups that have been described in this Highlight, it is evident the necessity to standardize the setups, or to provide a detailed description of the necessary properties to ensure reproducibility, that will lead to an easier assessment of the requirements for further scale-up. To this end, many of the examples here presented have shown the optimization of a small scale reaction, with limited examples showing the scalability of the reactions to a (multi)kilogram scale. Nevertheless, close collaboration between synthetic organic chemistry and chemical engineering is desirable to develop not only sustainable and efficient reactions, but also a reaction design that meets the needs of the industry, allowing the development of effective and more widely applicable scale-up strategies employing flow chemistry.
Author contributions
S. P. wrote the manuscript. E. V. V. d. E. and U. K. S. revised the manuscript. All authors approved the submitted version.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. P. (writing) is thankful to the FWO for obtaining a PhD scholarship (S. P. grant no. 11H0121N). The publication has been prepared with the support of the “RUDN University Strategic Academic Leadership Program” (recipient E. V. V. d. E.; supervision). U. K. S. is thankful to the University of Missouri-St. Louis (USA) for the start-up funds. This article is dedicated to honoring the remarkable career of Dr Rajender (Raj) S. Varma, of the United States Environmental Protection Agency (US EPA).References
-
A. Fiorati, C. Gambarotti, L. Melone, N. Pastori, C. Punta, G. Raffaini and A. Truscello, in Green Synthetic Approaches for Biologically Relevant Heterocycles, Elsevier, 2021, pp. 189–206 Search PubMed
.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC
- F. Minisci, R. Galli, M. Cecere, V. Malatesta and T. Caronna, Tetrahedron Lett., 1968, 9, 5609–5612 CrossRef
- F. Minisci, R. Galli, V. Malatesta and T. Caronna, Tetrahedron, 1970, 26, 4083–4091 CrossRef CAS
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS
- F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489–519 CrossRef CAS
- F. O’Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC
- X. Zhang, S. Li, F. Qiu, H. T. Ang, J. Wu and P. Jia, Green Chem., 2024, 26, 3595–3626 RSC
- M. Martos, I. Bosque and J. C. Gonzalez-Gomez, Synthesis, 2024, 1655253, DOI:10.1055/s-0043-1775387
- P. D. Bacoş, A. S. K. Lahdenperä and R. J. Phipps, Acc. Chem. Res., 2023, 56, 2037–2049 CrossRef
- J. Predygier, J. Szczepanik and M. Giedyk, Synlett, 2022, 103–108 CrossRef CAS
- M. Kim, Y. Koo and S. Hong, Acc. Chem. Res., 2022, 55, 3043–3056 CrossRef CAS PubMed
- J. P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed
- L. Candish, K. D. Collins, G. C. Cook, J. J. Douglas, A. Gómez-Suárez, A. Jolit and S. Keess, Chem. Rev., 2022, 122, 2907–2980 CrossRef CAS
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Synthesis, 2021, 3963–3976 CrossRef CAS
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 9790–9833 CrossRef CAS
- E. G. Moschetta, G. C. Cook, L. J. Edwards, M. A. Ischay, Z. Lei, F. Buono, F. Lévesque, J. A. O. Garber, M. MacTaggart, M. Sezen-Edmonds, K. P. Cole, M. G. Beaver, J. Doerfler, S. M. Opalka, W. Liang, P. D. Morse and N. Miyake, Org. Process Res. Dev., 2024, 28, 831–846 CrossRef CAS
- C. Sambiagio and T. Noël, Trends Chem., 2020, 2, 92–106 CrossRef CAS
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef
- L. Capaldo, Z. Wen and T. Noël, Chem. Sci., 2023, 14, 4230–4247 RSC
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS
- L. Y. Vázquez-Amaya, G. A. Coppola, E. V. Van der Eycken and U. K. Sharma, J. Flow Chem., 2024, 14, 257–279 CrossRef
- S. D. A. Zondag, D. Mazzarella and T. Noël, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 283–300 CrossRef CAS PubMed
- K. Donnelly and M. Baumann, J. Flow Chem., 2021, 11, 223–241 CrossRef CAS
- M. Zhang and P. Roth, Curr. Opin. Chem. Eng., 2023, 39, 100897 CrossRef
- M. A. Graham, G. Noonan, J. H. Cherryman, J. J. Douglas, M. Gonzalez, L. V. Jackson, K. Leslie, Z. Liu, D. McKinney, R. H. Munday, C. D. Parsons, D. T. E. Whittaker, E. Zhang and J. Zhang, Org. Process Res. Dev., 2021, 25, 57–67 CrossRef CAS
- J. J. Mousseau, M. A. Perry, M. W. Bundesmann, G. M. Chinigo, C. Choi, G. Gallego, R. W. Hicklin, S. Hoy, D. C. Limburg, N. W. Sach and Y. Zhang, ACS Catal., 2022, 12, 600–606 CrossRef CAS
- S. Pillitteri, P. Ranjan, G. M. Ojeda-Carralero, L. Y. Vázquez Amaya, J. E. Alfonso-Ramos, E. V. Van der Eycken and U. K. Sharma, Org. Chem. Front., 2022, 9, 6958–6967 RSC
- R. Crawford, M. Di Filippo, D. Guthrie and M. Baumann, Chem. Commun., 2022, 58, 13274–13277 RSC
- Z. Cao, M. Ji, X. Wang, X. Wu, Y. Li and C. Zhu, Green Chem., 2022, 24, 4498–4503 RSC
- M. Auvray, M. Jeanty, P. Jubault and T. Poisson, Chem. – Eur. J., 2023, 43, e202301417 CrossRef
- J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed
- J. W. Beatty, J. J. Douglas, R. Miller, R. C. McAtee, K. P. Cole and C. R. J. Stephenson, Chem, 2016, 1, 456–472 CAS
- E. J. McClain, T. M. Monos, M. Mori, J. W. Beatty and C. R. J. Stephenson, ACS Catal., 2020, 10, 12636–12641 CrossRef CAS
- J. L. Monteiro, P. F. Carneiro, P. Elsner, D. M. Roberge, P. G. M. Wuts, K. C. Kurjan, B. Gutmann and C. O. Kappe, Chem. – Eur. J., 2017, 23, 176–186 CrossRef CAS
- E. Torti, S. Protti and M. Fagnoni, Chem. Commun., 2018, 54, 4144–4147 RSC
- I. Abdiaj, C. Bottecchia, J. Alcazar and T. Noël, Synthesis, 2017, 4978–4985 CAS
- J. Yang, S. A. Kawale, X. Yang and D. Kim, Eur. J. Org. Chem., 2023, e202201287 CrossRef CAS
- C. Bottecchia, R. Martín, I. Abdiaj, E. Crovini, J. Alcazar, J. Orduna, M. J. Blesa, J. R. Carrillo, P. Prieto and T. Noël, Adv. Synth. Catal., 2019, 361, 945–950 CrossRef CAS
- Y. Qiu, A. Scheremetjew, L. H. Finger and L. Ackermann, Chem. – Eur. J., 2020, 26, 3241–3246 CrossRef CAS PubMed
- A. Sivo, V. Ruta, V. Granata, O. Savateev, M. A. Bajada and G. Vilé, ACS Sustainable Chem. Eng., 2023, 11, 5284–5292 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |