
Recent advances in photochemical strain-release reactions of bicyclo[1.1.0]butanes
Xiang
Zhou†
,
Ye
Hu†
,
Yao
Huang†
and
Yang
Xiong
 *
*
Chongqing Key Laboratory of Natural Product Synthesis and Drug Research, Innovative Drug Research Center, School of Pharmaceutical Sciences, Chongqing University, Chongqing 401331, P. R. China
First published on 11th November 2024
Abstract
Bicyclo[1.1.0]butanes (BCBs) are attractive compounds for their beautiful “butterfly” conformations, distinctive properties, and novel reactivities. As soon as the first example had been synthesized, a wide range of strain-release reactions were explored for the preparation of cyclobutanes and bicyclic systems in the ground state or excited state. In particular, with the demand for the construction of rigid three-dimensional aliphatic skeletons to “escape from flatland” in drug discovery programs, numerous efforts have been devoted in this area to expanding the boundaries of their reactivities and broadening the chemical space of their attractive bioisosteric products. In recent years, with the great resurgence and dramatic evolution of photochemistry, photochemical strain-release reactions generally relying on single electron transfer (SET) or energy transfer (EnT) strategies can provide much more opportunities and capability for innovative transformations of BCBs. In this review, we summarize and highlight the recent advances (year > 2016) of this topic and hope that it will inspire much more wonderful chemistry of BCBs.
 Xiang Zhou | Xiang Zhou received his bachelor degree at Chongqing University of Arts and Sciences in 2020. Then, he did master's study as a joint student at Jiangxi Science and Technology Normal University and Sun Yat-Sen University under the supervision of Prof. Jiang Weng and Dr. Yanshi Xiong and obtained his master's degree in 2023. Later, he continued PhD study at the Innovative Drug Research Center, School of Pharmaceutical Sciences, Chongqing University under the supervision of Prof. Yang Xiong. His current research interests focus on the development of enantioselective diradical reactions. |
 Ye Hu | Ye Hu received her bachelor's degree in pharmacy at Chongqing University in 2022. She then studied for her master's degree at Innovative Drug Research Center, School of Pharmaceutical Sciences, Chongqing University under the supervision of Prof. Yang Xiong. Her current research interests focus on diradical-mediated hydrogen atom transfer reactions of C(sp3)–H bonds. |
 Yao Huang | Yao Huang received her bachelor's degree in pharmacy at Guizhou Medical University in 2023. She then studied for her master's degree at Innovative Drug Research Center, School of Pharmaceutical Sciences, Chongqing University under the supervision of Prof. Yang Xiong. Her current research interests focus on diradical-mediated C(sp3)–H bond functionalizations. |
 Yang Xiong | Yang Xiong received his bachelor's degree at Southwest University in 2014 and obtained a PhD in 2019 at the Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences (UCAS), Chinese Academy of Sciences (CAS). After a 3 months’ research stay in Prof. Can Li's group at Dalian Institute of Chemical Physics, UCAS, CAS, he moved to Prof. Thorsten Bach's group, Technical University of Munich, as an Alexander von Humboldt postdoctoral fellow. In 2022, he began his independent research career at School of Pharmaceutical Sciences, Chongqing University. His research focuses on the discovery, development and application of diradical reactions. |
1. Introduction
Since the proposal of strain theory by Adolf von Baeyer in 1885,1 the explorations of the properties and reactivity of strained cyclic systems have been greatly expanded and widely used in organic chemistry. The concept of strain theory is mainly discussed in terms of bond length, bond angle distortions, torsional terms and nonbonded interactions. Meanwhile, strain energies are also valuable quantities to examine unusual geometries or intramolecular interactions of organic compounds.2 These strain energy factors provide a convenient way to understand the driving force for organic reactions. Based on strain theory, strain-release processes have been widely applied to generate novel highly rigid aliphatic skeletons. It is noted that the distortion of bond geometry in strained ring systems is inherently destabilizing, especially 3 and 4 membered rings.3 Among these ring systems, bicyclo[1.1.0]butane (BCB) with a beautiful “butterfly” conformation is one of the fascinating building-blocks in organic synthesis and medicinal chemistry (Fig. 1).4 | ||
| Fig. 1 “Butterfly” conformation and its strain-release reactions. | ||
It was widely known that BCBs were firstly introduced in 1959.5 However, the real renaissance of strain-release reactions of these “spring-loaded” molecules wasn’t until 2016.6 Since that time, a large variety of strain-release reactions have been realized to access rigid three-dimensional aliphatic skeletons to “escape from flatland” in drug discovery programs.7 In fact, BCBs have two cyclopropane rings distorting from a planar conformation by 120–125° and all C–C bonds have almost the same length of 1.50 Å.8 Meanwhile, the von Baeyer strain energy of BCB is approximately 66 kcal mol−1.9 Moreover, the bridging bent σ bond is more than 90% formed of two p orbitals, which can display its π-like reactivity.10 Furthermore, the different substituents in these unique structures feature some distinctive properties and novel reactivities, which stimulate the exploitation of a wide range of strain-release reactions. In particular, with the great development of photocatalysis,11 numerous efforts have been devoted in this area to expanding the boundaries of their photochemical reactivities and broadening the chemical space of their attractive bioisosteric products by utilizing radical or diradical species. In this review, we summarize and highlight the recent advances (year > 2016) of photochemical strain-release reactions mainly relying on single electron transfer (SET) or energy transfer (EnT) strategies. The topic of their thermal transformations involving 2e, which have been included in other articles, will not be discussed in this review.12
2. Photochemical strain-release reactions via SET
With its great resurgence and dramatic evolution, visible light photoredox catalysis has emerged as a powerful tool to provide considerable generality, synthetic capability and various opportunities for organic chemistry by utilizing radical intermediates with single electron transfer (SET) processes.13 As such, in 2019, Cintrat and coworkers report an unprecedented formal Giese-type addition of C(sp3)-centered radicals from a diverse range of α-amino and α-oxy carboxylic acids into highly strained BCBs to access valuable alkylated cyclobutanes by visible light-mediated photoredox catalysis.14 It is noted that when the reaction was carried out with the cesium salt in the presence of deuterium oxide, the desired product [2H]-3a was formed with 72% d-incorporation to the phenyl sulfonyl group, thus confirming the formation of the transient sulfonyl anion. Based on the kinetic studies and DFT calculations, the proposed mechanistic details are outlined in Scheme 1. Initially, the photoexcited IrIII catalyst undergoes SET with the in situ formed cesium carboxylate 1A to generate a radical 1B, and extrusion of CO2 to form the carbon-centered radical 1C, which undergoes a Giese radical addition to BCB 2a to produce sulfonyl radical 1D. Then the radical intermediate 1D was reduced by the IrII photocatalyst, followed by a protonation to deliver the “cyclobutylated” sulfone 3a. | ||
| Scheme 1 Giese-type addition to BCBs. | ||
In 2020, the Gryko group presented a polarity-reversal strategy to generate nucleophilic radicals through strain release via light-driven cobalt catalysis including Giese-type addition to electrophilic alkenes and Co/Ni catalyzed cross-coupling with aryl iodides (Scheme 2).15 Based on a series of electrochemical, spectroscopic, and kinetic experiments as well as X-ray structural analysis of the intermediate alkylcobalt(III) complex 2B and DFT calculations, it is anticipated that in the presence of a proton source the nucleophilic Co(I) formed and then underwent conjugate addition to the π-like central C–C bond of BCB, furnishing Co(III)-alkyl complex 2A. The resulting intermediate 2B can generate alkyl radical 2C and Co(II) upon homolytic cleavage initiated by light irradiation. The radical 2C is able to engage in addition to the SOMOphile or interception by a transition metal for further transformations.
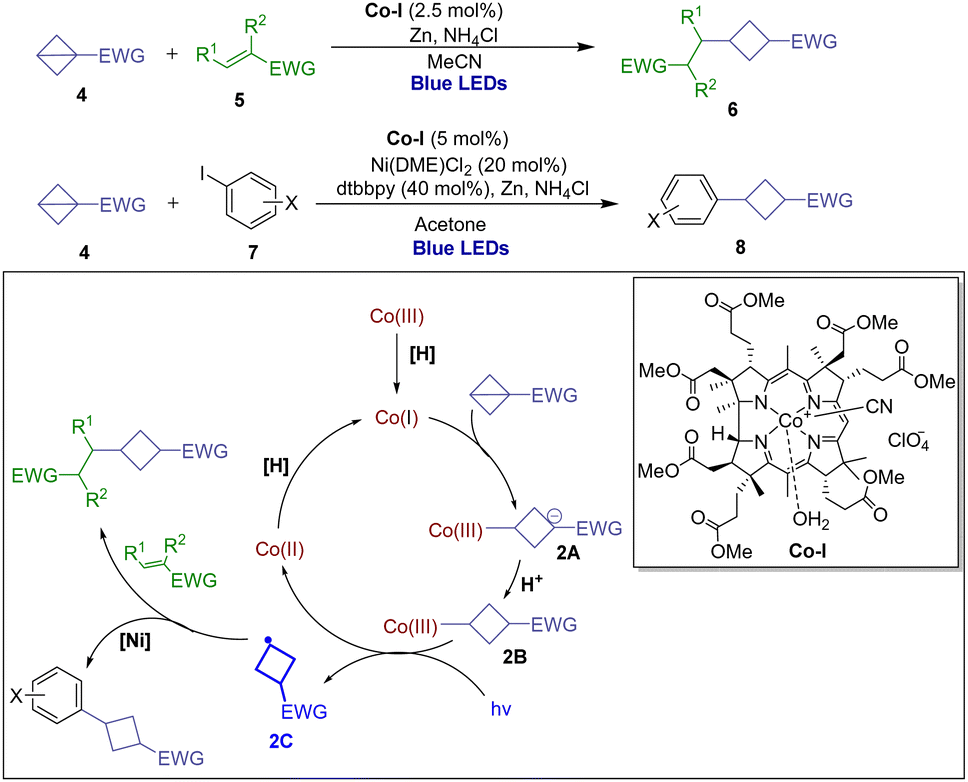 | ||
| Scheme 2 Light-driven cobalt catalysis in strain-release reactions of BCBs. | ||
In 2022, the Molander group disclosed the first photoinduced [3σ+2σ] cycloaddition for the synthesis of trisubstituted bicyclo[3.1.1]heptanes using BCBs and cyclopropylamines, which can provide unique meta-substituted arene bioisosteres for chemical space explorations (Scheme 3).16 The authors proposed that the photoexcited state (IrIII*) goes through SET with the cyclopropylaniline 10a to form the radical cation species 3A which is followed by ring opening via β-scission to generate radical cation 3B. The reactive intermediate 3B adds to BCB 9a to form a stabilized benzylic radical cation 3C. Subsequently, 3C undergoes cyclization to form the radical cation 3D, which is reduced by the IrII species generated in the reductive quenching photoredox cycle or can be reduced by the presence of the cyclopropylaniline 10avia an enthalpically favourable reaction.
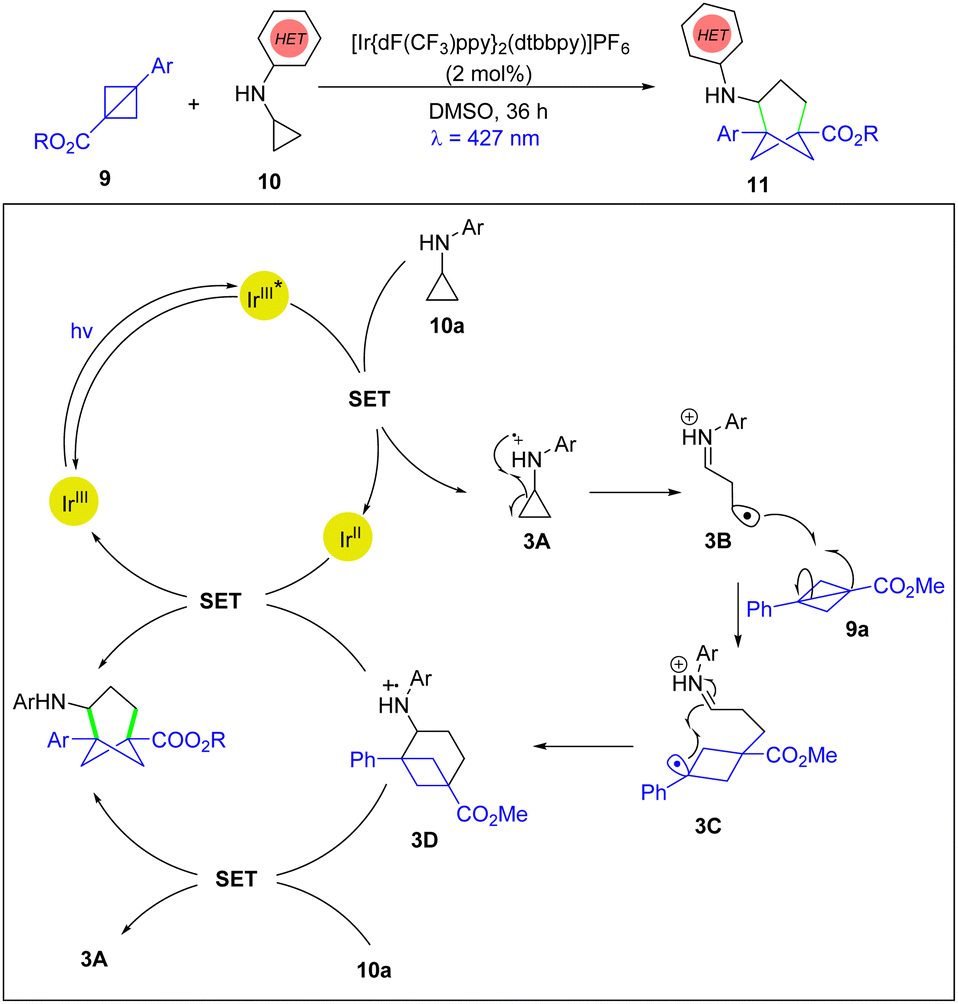 | ||
| Scheme 3 Photoinduced [3σ+2σ] cycloaddition. | ||
In the same year, as a continuation of their research on photoinduced palladium chemistry,17 the Gevorgyan group reported the first visible light-induced palladium hydride enabled hydroalkenylation of BCBs, delivering a diverse array of highly valuable alkenylated cyclobutanes (Scheme 4).18 This transformation demonstrates broad functional group tolerance and is amenable to late-stage functionalization of complex molecules. Based on mechanistic studies (deuterium labelling and controlled experiments), it is suggested that the catalytically active H–Pd(II)–X species (X = Br− or I−) is formed via an oxidative addition of Pd(0) into the HX precursor. Then, a migratory insertion of PdH into the π-like central C–C bond of BCBs provides complex 4A, which can undergo homolysis by light irradiation to generate 4B. Next, 4B adds to the alkene to produce a benzylic radical intermediate 4C. Finally, 4C undergoes a β-H-elimination to deliver hydroalkenylation product 14 and regenerate the H–Pd(II)–X complex.
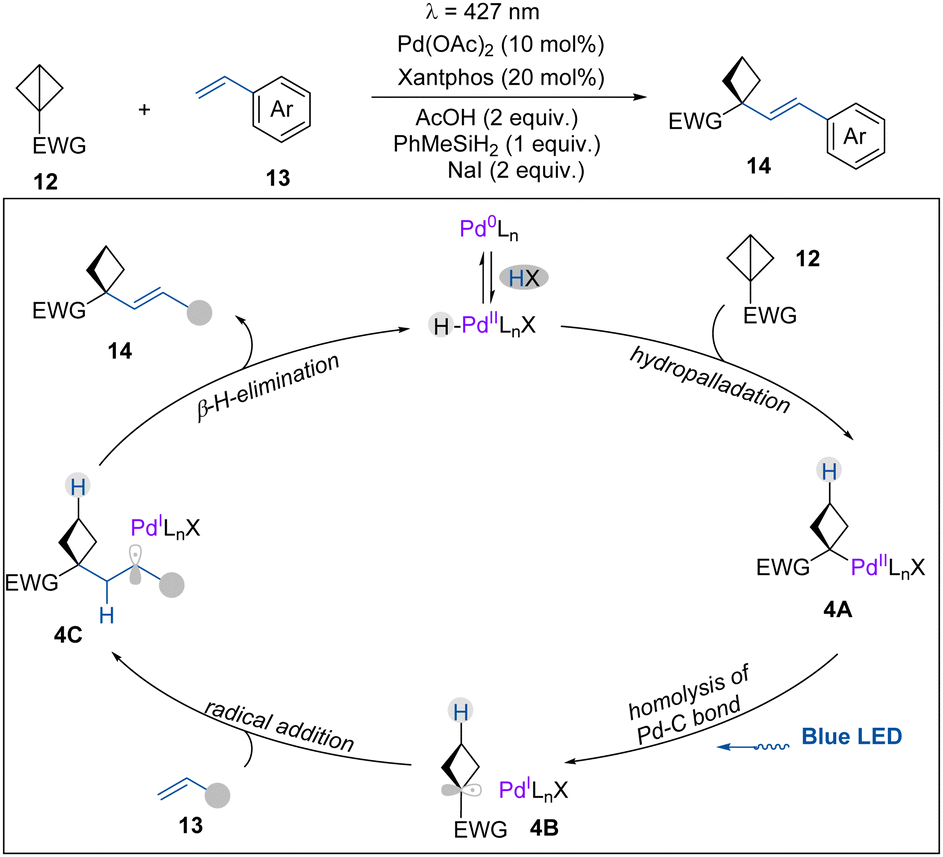 | ||
| Scheme 4 Palladium hydride enabled hydroalkenylation of BCBs. | ||
In 2023, Glorius and co-workers have disclosed an elegant photoinduced dearomative ring enlargement of thiophenes by insertion of BCBs to access eight-membered bicyclic rings (Scheme 5).19 This novel method demonstrates high synthetic value, broad functional-group compatibility, and excellent selectivity. The cyclic voltammetry analysis suggests that 15a not 16a was oxidized by the PC*. Moreover, no desired product was observed in the presence of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy, which suggests a radical pathway. Based on further mechanistic studies such as Stern–Volmer luminescence quenching analysis, and DFT calculations, the authors proposed that [Acr-Mes2]+BF4− undergoes SET with (benzo)thiophenes to produce a radical 5A, which can attack onto BCBs to release the ring strain and form the more stable radical cation 5B. Then, 5B can proceed a further cyclization to generate the delocalized radical cation 5C, which can be reduced to form 5D. Finally, the product can be effectively attained with a ring opening of 5D and facile C–S fragmentation of 5E. For benzothiophenes, the mechanism follows an analogous pathway. However, the initial S-radical attacks the BCBs and affords the opposite regioisomer.
 | ||
| Scheme 5 Photoinduced dearomative ring enlargement of thiophenes by insertion of BCBs. | ||
Continuing their research interests in this area, the Glorius group has reported a photoredox strategy for the difunctionalization of BCBs with thioalkynes, thioalkenes, and allylthioethers as bifunctional reagents to access 1,1,3,3-tetrasubstituted and 1,1,3-trisubstituted cyclobutane products (Scheme 6).20 This protocol features an excellent atom economy, mild reaction conditions, and good functional group tolerance. Based on the mechanistic studies, especially cyclic voltammetry analysis, the authors proposed a mechanism for the thio-alkynylation of BCBs, as shown in Scheme 6. Firstly, the excited state PC* undergoes a SET with 20a to generate radical cation 6A, which adds to BCB to produce radical cationic intermediate 6B. Then, the subsequent addition of 6B to the alkyne forms 6C, which can undergo a homolytic scission of the weakened C–S σ-bond to give a radical cationic 6D. Next, 6D is reduced by reduced PC or oxidation of 20a to form radical cation chain propagation. For the thio-alkenylation and thio-allylation, these processes realize the difunctionalizations likely through cyclic radical cationic intermediates 6E and 6F, respectively.
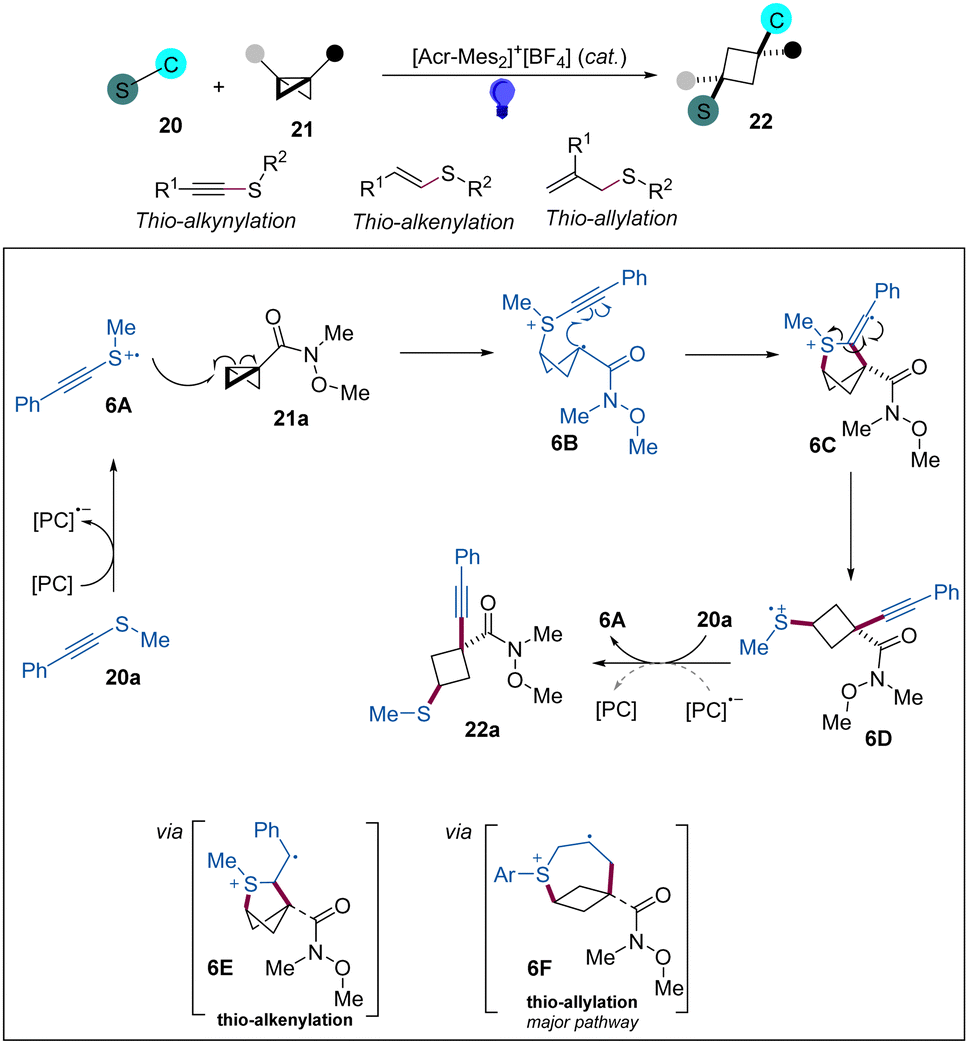 | ||
| Scheme 6 Difunctionalization of BCBs with thioalkynes, thioalkenes, and allylthioethers. | ||
In 2024, the Glorius group reported an intermolecular approach for the dearomative photocycloaddition of phenols with BCBs.21 This novel methodology paves a new pathway to overcome the typical ground-state resonance energy of the phenol unit, giving access to a bicyclo[2.1.1]hexane framework (Scheme 7). According to mechanistic analysis, Stern–Volmer quenching unveiled both phenol 23a and BCB 24a as potential quenchers of the photoexcited catalyst. It is anticipated that the excited photocatalyst can oxidize both BCB and phenol via SET to lead to delocalized radical cations 7A and 7B, respectively. Thus, the common intermediate 7C can be formed by two pathways: (i) a Friedel–Crafts type alkylation from phenol 23a with 7A; (ii) a strain-release event of BCB 24a with 7B. Then, the benzylic radical 7C undergoes a 5-exo-trig cyclization to form α-carbonyl radical 7D, which is reduced to generate the desired [2π+2σ] cycloadduct.
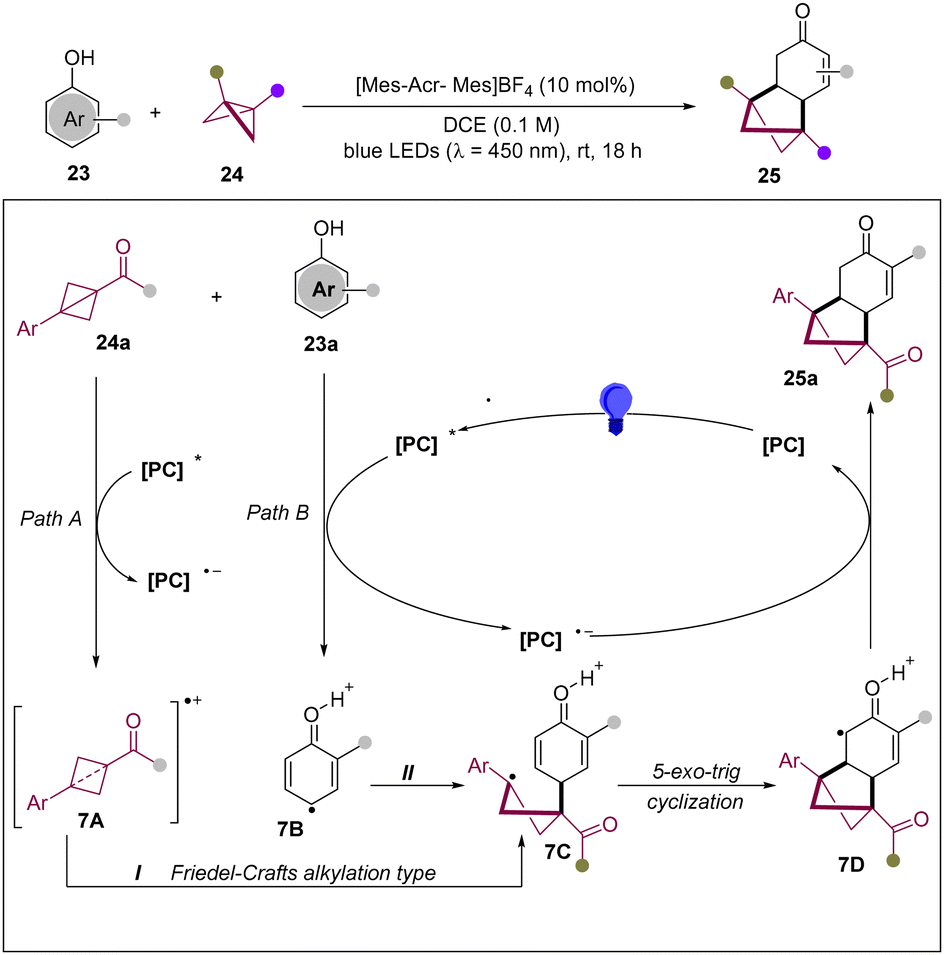 | ||
| Scheme 7 Dearomative photocycloaddition of phenols with BCBs. | ||
Recently, the Glorius group has realized a photoredox-catalyzed highly regio- and chemoselective insertion of amidyl radicals to BCBs, providing direct access to 2-oxa-4-azabicyclo[3.1.1]hept-3-enes.22 This elegant work provides an effective platform to access important C(sp3)-rich heterobicyclic motifs with pyridine and pyrimidine derivatives. The mechanistic experiments strongly support the photoredox pathway. Based on DFT calculations, the proposed mechanism is shown in Scheme 8. Firstly, the excited-state Ir(III) photocatalyst undergoes SET with N-amidocollidinium tetrafluoroborate 27a to lead to an amidyl radical 8A, which subsequently adds to BCB 26a to afford the cyclobutyl radical 8B. Next, 8B is oxidized by Ir(IV) to generate the stabilized benzylic carbocation 8C and regenerate the Ir(III) photocatalyst. Finally, via base-mediated nucleophilic attack from the amidyl oxygen atom, 8C undergoes a cyclization to afford the observed product 28a.
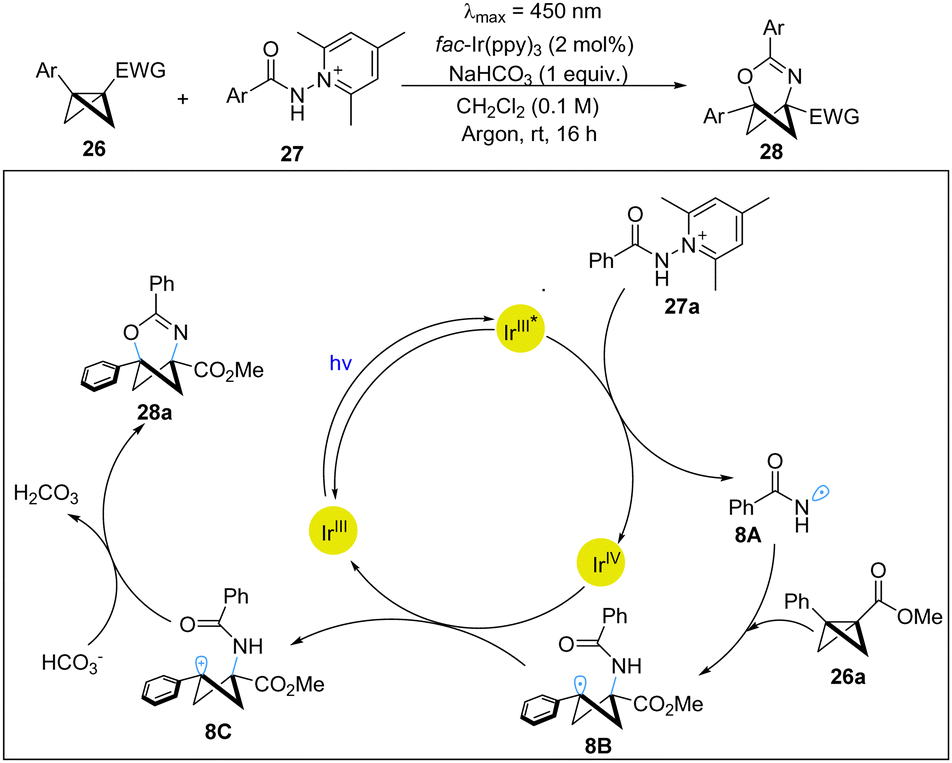 | ||
| Scheme 8 Photoredox-catalyzed insertion of amidyl radicals to BCBs. | ||
In 2023, the Shi group reported a photoexcited HE* promoted [2π+2σ] cycloaddition of BCBs with alkenes for the synthesis of bicyclo[2.1.1]-hexanes (Scheme 9).23 This effective method exhibits a wide scope of application of substrate and functional group compatibility and provides a cost-effective and convenient method for the synthesis of various bicyclo[2.1.1]hexanes. The results of mechanistic experiments fully demonstrated the presence of radical intermediates in the reaction. The authors proposed that by visible light-induction, the direct hydrogen atom transfer (HAT) proceeded from the C4–H of HE to BCB 29a generating 9E and ketyl radical 9A, which undergoes a ring opening to produce radical 9B or 9B′. Then, 9B or 9B′ adds to alkene 30 to form more stable radical 9C and 9C′, which subsequently undertakes a cyclization to lead to a radical intermediate 9D. Finally, product 31 and HE were formed via HAT by the radical species of 9E.
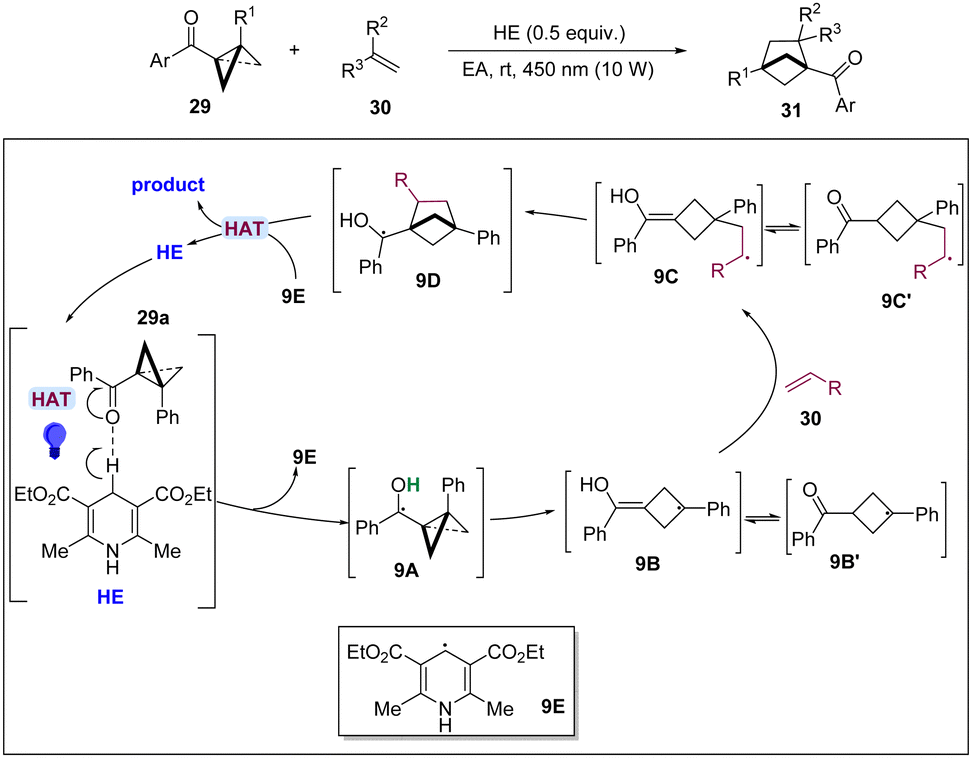 | ||
| Scheme 9 [2π+2σ] photocycloadditions of BCBs with alkenes. | ||
In 2024, Li and co-workers have developed a visible-light-induced radical alkylarylation of N-aryl bicyclobutyl amides with α-carbonyl alkyl bromides to access functionalized 3-spirocyclobutyl oxindoles (Scheme 10).24 Based on the radical clock experiments, it is proposed that the alkyl bromide 33a is firstly reduced by the excited photoredox catalyst PC* to form alkyl radical 10A, which adds to the BCB 32a giving 10B. Then, 10B was intercepted by the aryl moiety of BCB in an intramolecular pathway to afford a delocalized radical species 10C. Next, undergoing a SET oxidation of 10C with PC delivers carbocation 10D. Finally, the siprocyclobutyl-oxindole 34a was obtained by deprotonation of 10D in the presence of a base.
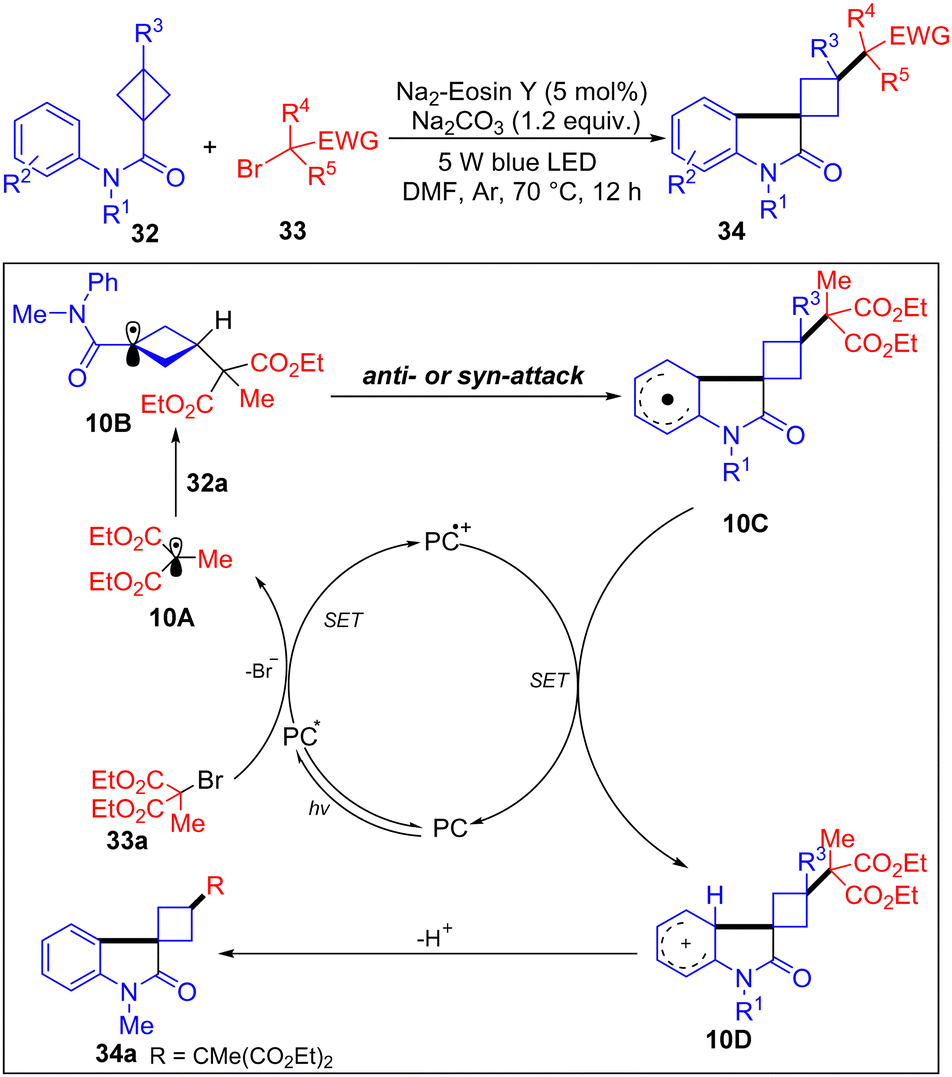 | ||
| Scheme 10 Visible-light-induced radical alkylarylation of N-aryl bicyclobutyl amides. | ||
3. Photochemical strain-release reactions via EnT
Triplet energy transfer is an enormously valuable tool to enable complementary reactivity modes in the framework of organic synthesis and to achieve high selectivity control in photochemical excited reactions via diradical intermediates.25 Among these reactions, the [2+2]-photocycloaddition is one of the most widely used by synthetic chemists to access various cyclobutanes for more than one century. Despite the enormous development of such reactions, however, explorations have been focused on [2π+2π]-systems, in which two π-bonds are converted into two new σ-bonds. In 2022, the Glorius group reported a visible-light-mediated intermolecular strain-release-driven [2π+2σ] photo-cycloaddition that uses BCBs as 2σ-electron reactants, extending the system to σ-bonds (Scheme 11).26 Notably, coumarins, flavones and indoles, and even imines can act as π-bond coupling partners with a variety of BCBs to access three-dimensional bioisosteres. The Stern–Volmer analysis revealed that coumarin quenches the photoexcited photocatalyst, whereas BCB 36 shows no quenching. Based on mechanistic studies and DFT calculations, the authors proposed that visible light excited TXT sensitizes coumarin by EnT and the resulting triplet state coumarin approaches BCB to form an exciplex 11A which gives rise to the first C–C bond forming event, determining the regioselectivity. The observed regioisomer was favourable by the α-carbonyl-site of coumarin engaging BCB in the 3-position (11B). After intersystem crossing of the triplet 1,5-diradical 11C, the cis-selective diradical recombination of 11D occurred as the diastereoselectivity determining step to deliver the cycloaddition products.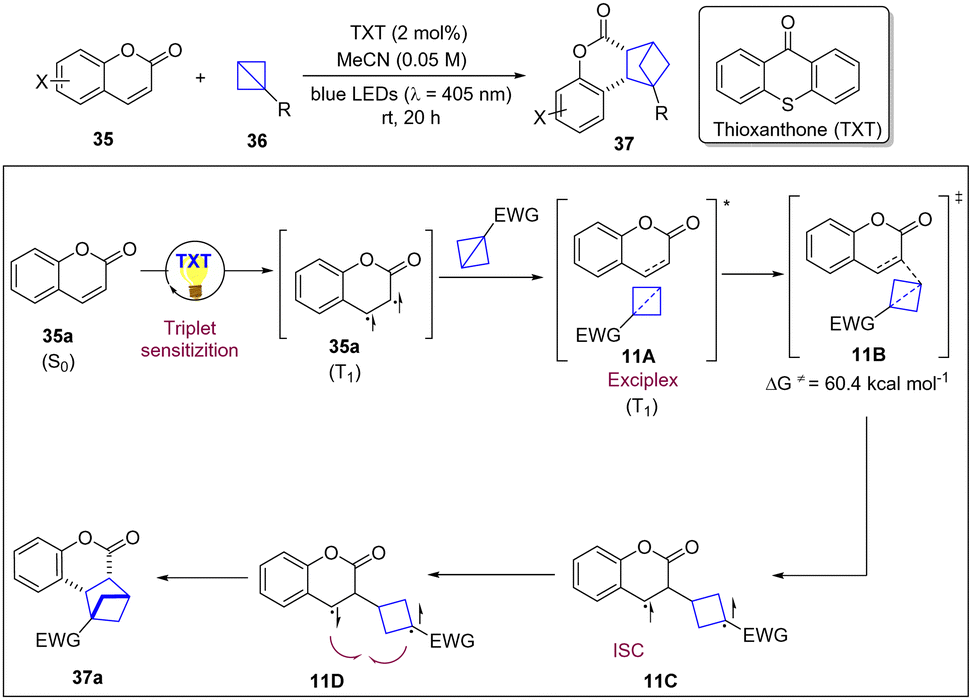 | ||
| Scheme 11 Intermolecular strain-release-driven [2π+2σ] photocycloaddition of BCBs. | ||
Meanwhile, the Brown group also reported an elegant work of strain-release-driven excited BCB with alkenes for the synthesis of bicyclo[2.1.1]hexanes.27 It was noted that the reaction proceeded in 60% yield upon direct irradiation with 365 nm LEDs. Meanwhile, UV/vis measurements showed that the absorption spectrum of BCB 39 overlaps with the emission spectrum of the 365 nm LEDs. It is proposed that the reaction proceeds via energy transfer of 40 or direct excitation followed by rapid ISC to generate the T1 (π–π*) state of 39 (Scheme 12). Then, it undergoes a strain-release induced bond cleavage to generate triplet diradical 12A, which can be captured by the alkene to lead to 12B. Finally, undergoing an ISC and bond formation of the diradical delivers the desired product. This new approach unlocks a new strategy for strain-release-driven transformations of excited BCBs.
 | ||
| Scheme 12 Strain-release-driven excited BCB with alkenes. | ||
In 2023, the Bach group firstly achieved the asymmetric photocycloaddition of 2(1H)-quinolones 42 with 1-substituted BCB upon irradiation (λ = 366 nm) in the presence of a chiral complexing agent 44 (Scheme 13).28 It is found that a two-point hydrogen bond between the quinolone and the template is responsible for the excellent stereocontrol. This asymmetric reaction opens a new window for the enantioselective photochemical transformations of BCBs.
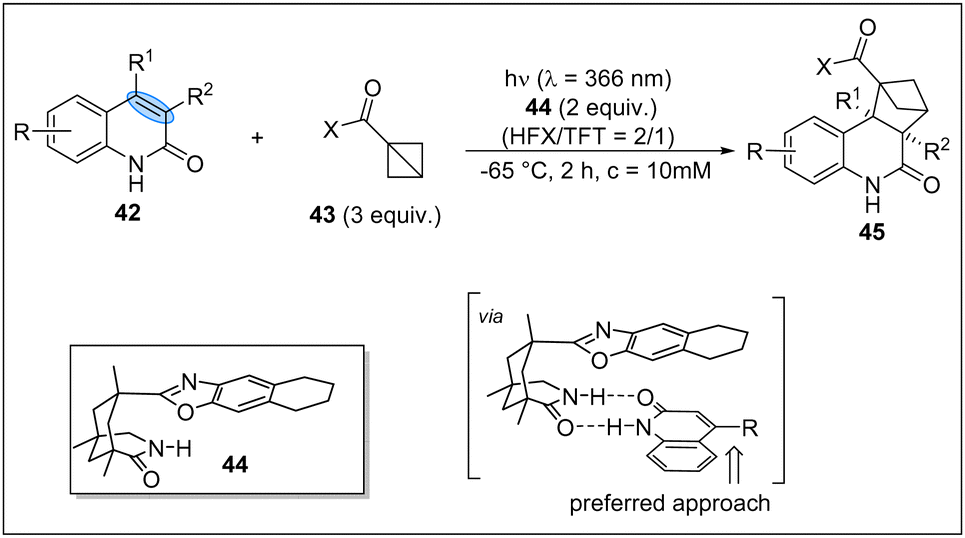 | ||
| Scheme 13 The asymmetric photocycloaddition of 2(1H)-quinolones with BCBs. | ||
Later, the Jiang group developed a highly enantioselective catalytic [2π+2σ] photo-cycloaddition of BCBs with vinylazaarenes (Scheme 14).29 Notably, the diverse 1-ketocarbonyl-3-substituted BCBs, α/β-substituted vinylazaarenes can effectively realize successful assembly of an all-carbon quaternary stereocenter or two adjacent tertiary stereocenters on the product with high yields and ee's. Based on DFT calculations and control experiments, it is proposed that the bifunctional chiral catalyst is crucial for the success of such novel transformations via a H-bond in the proton coupled electron transfer (PCET) process to avoid the elusive racemic background reaction and the control of the enantioselectivity cyclization of the diradical intermediate. This new activation mode of PAH containing chiral Brønsted acid catalysts will encourage the pursuit of devising more types of photochemical transformations.
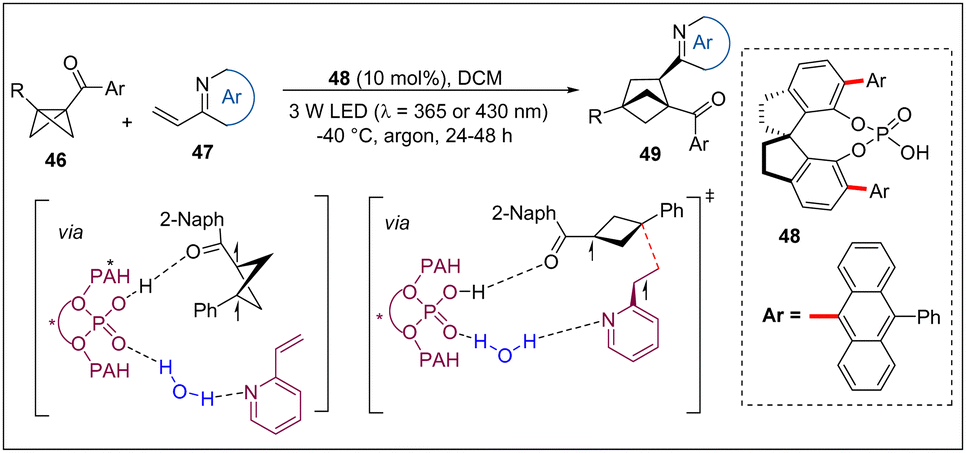 | ||
| Scheme 14 Enantioselective catalytic [2π+2σ] photocycloaddition of BCBs with vinylazaarenes. | ||
In 2022, the Glorius group discovered a new strategy to access polysubstituted 2-oxabicyclo[2.1.1]hexanes in a single operation from readily accessible benzoylformate esters and BCBs via visible-light-induced triplet energy transfer catalysis.30 The process is proposed to involve a formal [2π+2σ] photocycloaddition/backbone C–H abstraction/aryl group migration sequence. Importantly, Stern–Volmer quenching experiments exhibited that the two substrates can quench the excited-state photocatalyst at similar rates. Meanwhile, cyclic voltammetry experiments ruled out a single electron transfer process. These results support an energy transfer mechanism and suggest that both substrates can be sensitized into their triplet states. Based on mechanistic studies and their previous report,26 a feasible mechanism is proposed in Scheme 15. Initially, the diradical species 15A was generated via energy transfer from the excited-state photocatalyst [IrIII]* with the benzoylformate esters. Then, 15A adds to the BCB to form the more stabilized diradical species 15B. Undergoing an intersystem crossing (ISC) and radical–radical coupling leads to 15C, which undergoes a HAT by another excited species 15A to generate radical 15D. Next, 15D undergoes aryl group migration via intermediate 15E to afford the more stabilized radical species 15F. Finally, as a result of the steric effect, the hydrogen-donor can access species 15F from a less sterically hindered position to deliver the final product 52a.
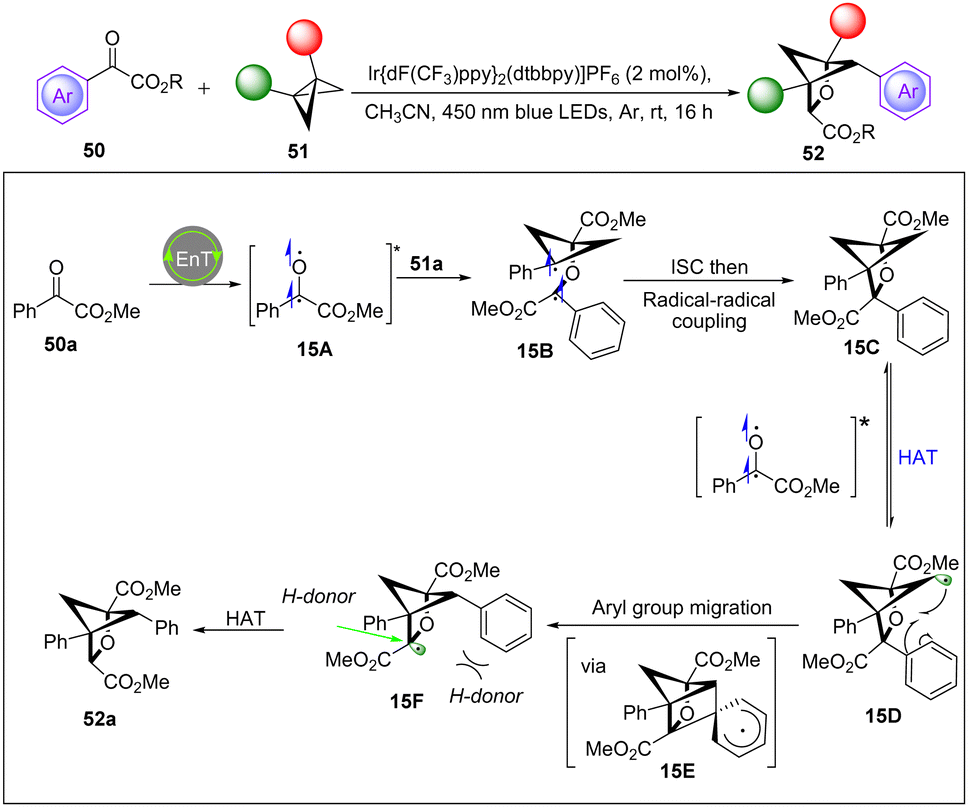 | ||
| Scheme 15 Visible-light-induced reactions of benzoylformate esters with BCBs. | ||
Later, the same group reported an ortho-selective intermolecular dearomative [2π+2σ] photocycloaddition of bicyclic aza-arenes including (iso)quinolines, quinazolines, and quinoxalines with BCBs for the construction of C(sp3)-rich bicyclo[2.1.1]hexanes.31 This novel methodology displayed a broad functional group tolerance and excellent compatibility of various bicyclic aza-arenes to give access to highly decorated bicyclo[2.1.1]hexane frameworks. The UV/vis studies revealed that quinoline 53a and BCB 54a do not absorb near the irradiated wavelength (λ = 450 nm). However, the addition of Sc(OTf)3 resulted in a bathochromic shift for both substrates. Moreover, Stern–Volmer studies displayed that the excited photocatalyst was quenched by 53a, whereas BCB 54a did not show quenching. Based on these experiments and DFT calculations, a possible mechanism is proposed to explain the chain reaction (Scheme 16). Firstly, the diradical species 16B is generated from 16A by EnT with photo-excited Ir* or direct excitation. However, there are two possible ways to afford the final product. The 16B can selectively add to BCBs to form cyclobutane diradical 16F which undergoes an ISC and radical–radical coupling to lead to 55. On the other hand, 16D can also be generated by oxidation of diradical 16F or a SET and subsequently radical addition from 16B. Then, a cyclization of 16D forms BCH radical cation 16E, which undergoes a SET with neutral quinoline 16A by liberating the product 55 and 16C. It is noted that preliminary DFT calculations further suggest that the activation of 16E by acid renders the SET event thermodynamically favourable.
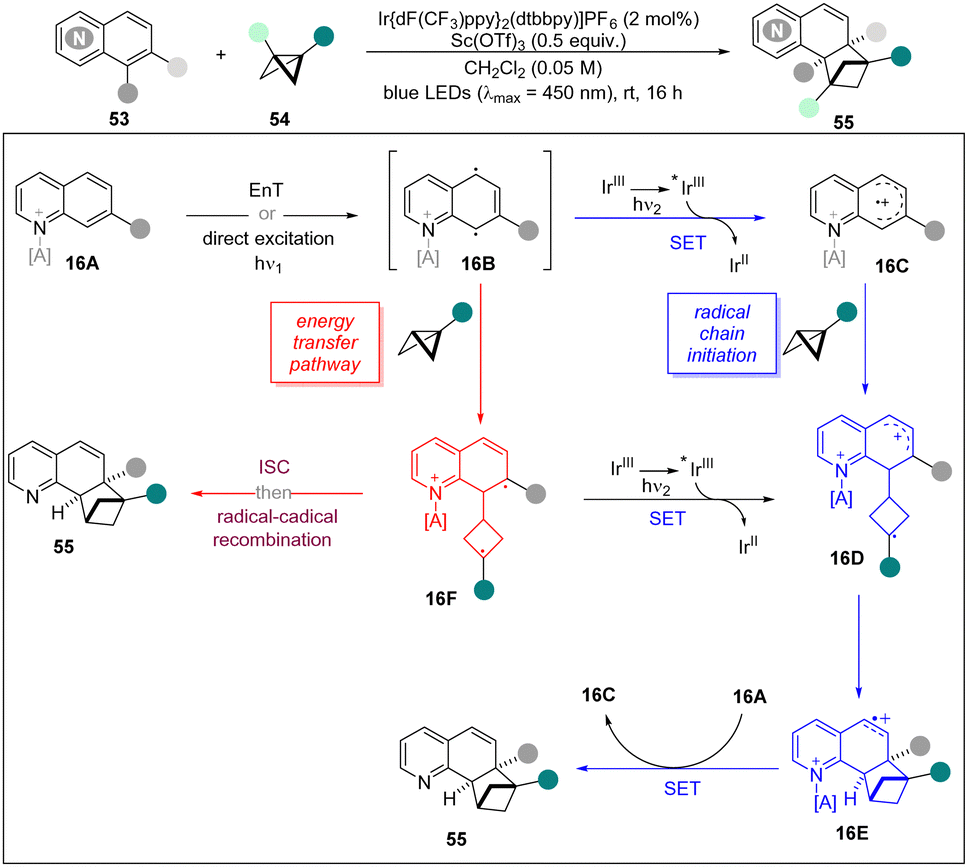 | ||
| Scheme 16 ortho-Selective intermolecular dearomative [2π+2σ] photocycloaddition. | ||
In 2024, Glorius and co-workers have achieved a double strain-release driven [2π+2σ]-photocycloaddition of cyclobutenone as a strained precursor with BCBs to access diverse hetero-bicyclo[2.1.1]hexane units under catalyst-free conditions (Scheme 17).32 The broad functional group tolerance and the bicyclic core modification demonstrate the high synthetic utility of this methodology. It was noted that only cyclobutenone 56a absorbs in the visible region of the UV-visible spectrum, suggesting that direct excitation of cyclobutanone 56a forms the ketene intermediate via a retro 4π-electrocyclic ring opening reaction. Based on DFT calculations, the authors proposed that the excited ketene intermediate 17A undergoes an ISC to form the diradical intermediate 17B, in which the spin densities are located on the dichloro-substituted alkene (π to π* excitation). Then, 1,3-disubstituted BCB 57a and mono-substituted BCB 57b can insert into the carbonyl radical in 17Bvia17C and 17E, respectively. However, the origin of C–C/C–O selectivity is decided in the radical–radical recombination step from 17D and 17G, which requires further investigation.
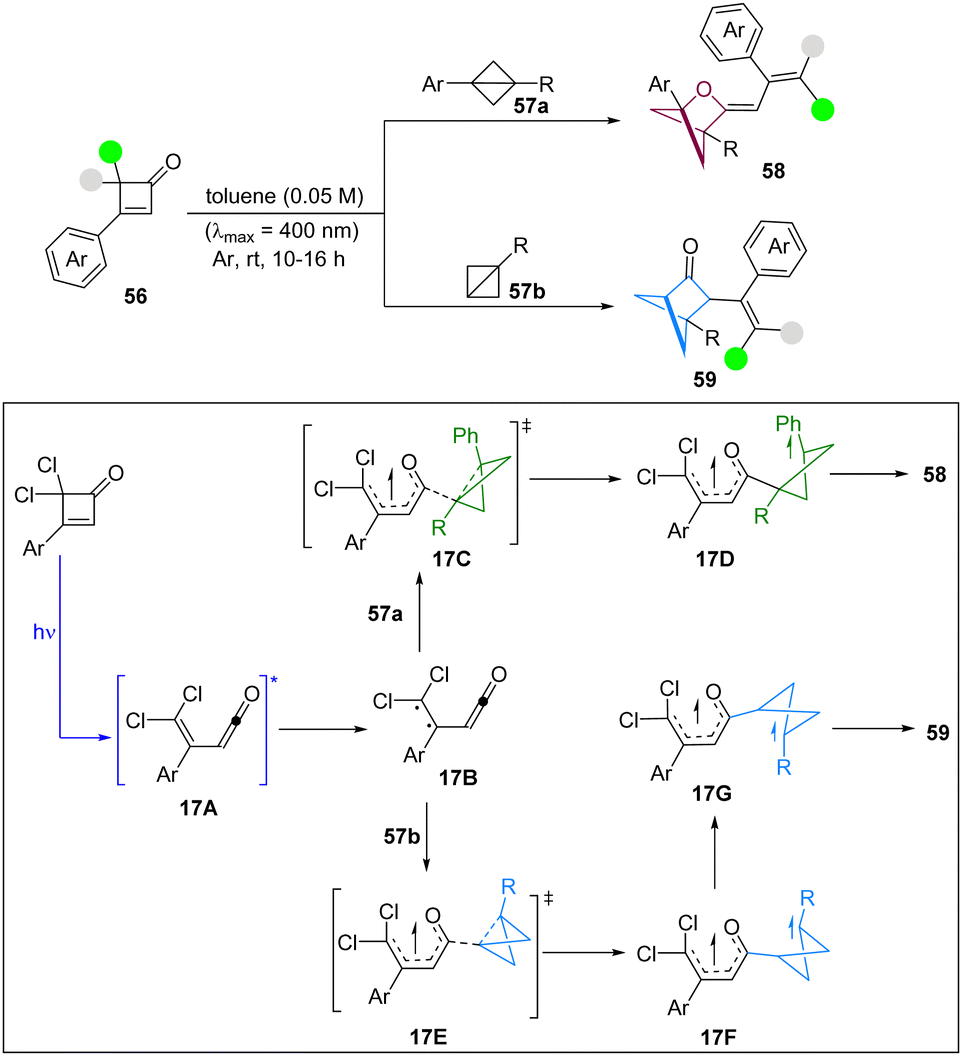 | ||
| Scheme 17 [2π+2σ]-photocycloaddition of cyclobutenone with BCBs. | ||
In 2023, the Waser group reported a novel photochemical [2σ+2π] annulation of carbonyl cyclopropanes with BCBs for the synthesis of bicyclo[3.1.1]heptanes (Scheme 18).33 Mechanistic studies, including DFT computations and a trapping experiment with TEMPO and DMPO, support a 1,3-biradical 18B generated from homolytic ring-opening of cyclopropane as a key intermediate. They proposed that 18B can proceed a radical addition process with BCBs to form a new biradical 18C. Finally, 18c undergoes an intersystem crossing and radical recombination to lead to the products 62.
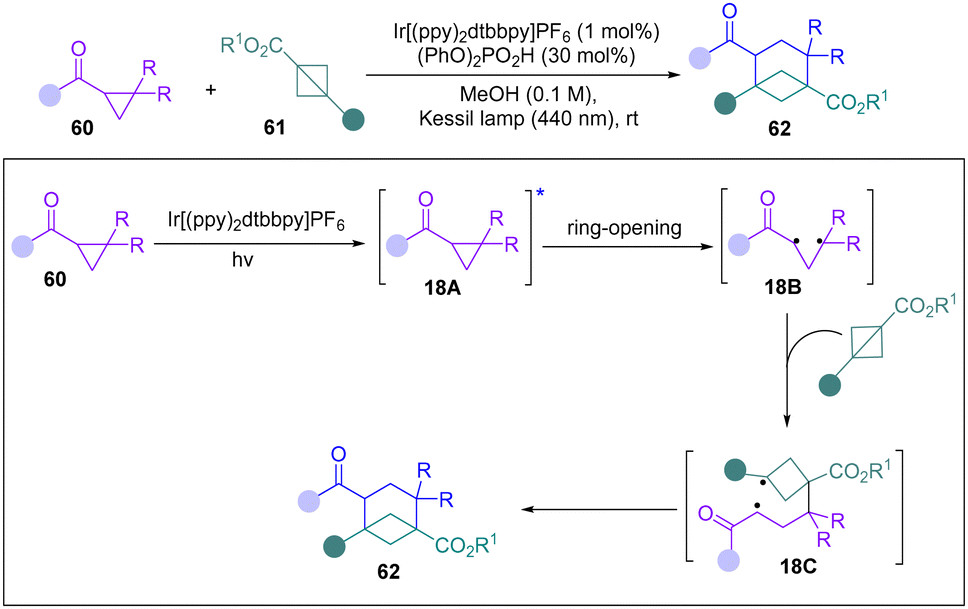 | ||
| Scheme 18 Photochemical [2σ+2π] annulation of carbonyl cyclopropanes with BCBs. | ||
4. Conclusions and outlook
In the past few years, photochemical strain-release reactions of BCBs have been rapidly developing and applied in a series of constructions of rigid three-dimensional aliphatic skeletons to “escape from flatland” in drug discovery programs, due to their unique photochemical properties. Indeed, the distinctive characteristics of the bridging bent σ bond display its π-like reactivity which enables a flexible strategy for the discovery and development of various interesting organic transformations. It is likely that the design of the photochemical reactions mainly relies on SET or EnT strategies by using radical and diradical intermediates to push the boundary of their reactivities. But of course, on the basis of the creativities and efforts of chemists, the fascination of photochemical strain-release reactions of BCBs is awaiting further investigations, especially in asymmetric synthesis with transition metals, enzyme catalysis, and electrocatalysis. Meanwhile, using photochemical C–H bond functionalization reactions to realize strain-release reactions of BCBs is still rare.34 Moreover, it is also a big challenge to use some electron-rich substituted BCBs or aza-BCBs for photochemical strain-release reactions due to their reactivities.35 Furthermore, new concepts and modes in such reactions are also required for improving the understanding of the mechanisms. Lastly, it is likely a dream to achieve selective transformations of any σ bonds in BCBs besides the bridging bent σ bond. However, stimulated by achievements of past years and driven by the attractions of these beautiful architectures, it is believed that much more wonderful chemistry of this topic will be unlocked in the future.Author contributions
Y. Xiong conceptualized the topic, and wrote the manuscript with contributions from all authors.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as a part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (Grant No: 22301026), the Fundamental Research Funds for the Central Universities (Project No. 2024CDJXY-002, No. 2024IAIS-QN001), the Natural Science Foundation of Chongqing (CSTB2024NSCQ-MSX0206), and the Hongshen Young Scholars Program from Chongqing University (02470011080002) are gratefully acknowledged for financial support. We thank Dr Thomas Rigotti (Universidad Autónoma of Madrid) for proofreading the manuscript.Notes and references
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1885, 18, 2269–2281 CrossRef
.
- K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef
- J. Turkowska, J. Durka and D. Gryko, Chem. Commun., 2020, 56, 5718–5734 RSC
- C. B. Kelly, J. A. Milligan, L. J. Tilley and T. M. Sodano, Chem. Sci., 2022, 13, 11721–11737 RSC
- K. B. Wiberg and R. P. Ciula, J. Am. Chem. Soc., 1959, 81, 5261–5262 CrossRef CAS
- R. Gianatassio, J. M. Lopchuk, J. Wang, C. M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed
-
(a) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046–14128 CrossRef CAS PubMed
-
(a) S. Meiboom and L. C. Snyder, Acc. Chem. Res., 1971, 4, 81–87 CrossRef CAS
-
(a) P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103–8112 CrossRef CAS
-
(a) J. M. Schulman and G. J. Fisanick, J. Am. Chem. Soc., 1970, 92, 6653–6654 CrossRef CAS
-
(a) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS
-
(a) J. A. Milligan and P. Wipf, Nat. Chem., 2016, 8, 296–297 CrossRef CAS PubMed
- For selected reviews see:
(a) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS
- G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J. C. Cintrat, Angew. Chem., Int. Ed., 2020, 59, 2618–2622 CrossRef CAS PubMed
- M. Ociepa, Aleksandra J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355–5361 CrossRef CAS PubMed
- Y. Zheng, W. Huang, R. K. Dhungana, A. Granados, S. Keess, M. Makvandi and G. A. Molander, J. Am. Chem. Soc., 2022, 144, 23685–23690 CrossRef CAS PubMed
-
(a) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Angew. Chem., Int. Ed., 2024, 63, e202311972 CrossRef CAS PubMed
- Z. Zhang and V. Gevorgyan, J. Am. Chem. Soc., 2022, 144, 20875–20883 CrossRef CAS PubMed
- H. Wang, H. Shao, A. Das, S. Dutta, H. T. Chan, C. Daniliuc, K. N. Houk and F. Glorius, Science, 2023, 381, 75–81 CrossRef CAS
- H. Wang, J. E. Erchinger, M. Lenz, S. Dutta, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 23771–23780 CrossRef CAS PubMed
- S. Dutta, Y. Lu, J. E. Erchinger, H. Shao, E. Studer, F. Schäfer, H. Wang, D. Rana, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2024, 146, 5232–5241 CrossRef CAS PubMed
- C. C. Chintawar, R. Laskar, D. Rana, F. Schäfer, N. V. Wyngaerden, S. Dutta, C. G. Daniliuc and F. Glorius, Nat. Catal., 2024 DOI:10.1038/s41929-024-01239-9
- H. Yan, Y. Liu, X. Feng and L. Shi, Org. Lett., 2023, 25, 8116–8120 CrossRef CAS PubMed
- J. Fan, J. Yuan, P. Xia, J. Zhou, L. Zhong, P. Huang, Y. Liu, K. Tang and J. Li, Org. Lett., 2024, 26, 2073–2078 CrossRef CAS PubMed
- For selected reviews see:
(a) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS
- R. Kleinmans, T. Pinkert, S. Dutta, T. O. Paulisch, H. Keum, C. G. Daniliuc and F. Glorius, Nature, 2022, 605, 477–482 CrossRef CAS PubMed
- R. Guo, Y. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed
- M. de Robichon, T. Kratz, F. Beyer, J. Zuber, C. Merten and T. Bach, J. Am. Chem. Soc., 2023, 145, 24466–24470 CAS
- Q. Fu, S. Cao, J. Wang, X. Lv, H. Wang, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2024, 146, 8372–8380 CrossRef CAS PubMed
- Y. Liang, R. Kleinmans, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2022, 144, 20207–20213 CrossRef CAS PubMed
- R. Kleinmans, S. Dutta, K. Ozols, H. Shao, F. Schäfer, R. E. Thielemann, H. T. Chan, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2023, 145, 12324–12332 CrossRef CAS PubMed
- S. Dutta, Y. Lu, J. E. Erchinger, H. Shao, E. Studer, F. Schäfer, H. Wang, D. Rana, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2024, 146, 5232–5241 CrossRef CAS PubMed
- T. V. T. Nguyen, A. Bossonnet, M. D. Wodrich and J. Waser, J. Am. Chem. Soc., 2023, 145, 25411–25421 CrossRef CAS PubMed
- S. Cuadros, J. Paut, E. Anselmi, G. Dagousset, E. Magnier and L. Dell’Amico, Angew. Chem., Int. Ed., 2024, 63, e202317333 CrossRef CAS PubMed
-
(a) A. Fawcett, A. Murtaza, C. H. U. Gregson and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 4573–4578 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |