
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploration of potential-limited protocols to prevent inefficiencies in Li–O2 batteries during charge†
Zoé
Lacour‡
ab,
Youngjin
Ham
 ad,
Laurence
Brazel
a,
Clare P.
Grey
*ab and
Israel
Temprano
*abc
ad,
Laurence
Brazel
a,
Clare P.
Grey
*ab and
Israel
Temprano
*abc
aYusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB21EW, UK
bThe Faraday Institution, Didcot OX110RA, UK
cCICA - Interdisciplinary Center for Chemistry and Biology, University of A Coruña, 15071, A Coruña, Spain. E-mail: i.temprano@udc.es
dCambridge Graphene Centre, University of Cambridge, Cambridge CB30FA, UK
First published on 7th January 2025
Abstract
Metal–air batteries are promising energy storage systems with high specific energy density and low dependence on critical materials. However, their development is hindered by slow kinetics, low roundtrip efficiency, deficient capacity recovery, and limited lifetime. This work explores the effect of cycling protocols on the lifetime of Li–O2 cells, and the interplay between electrolyte composition and the upper cut-off voltage during charge. Our results suggest that constant-current-constant-voltage (CCCV) protocols accommodate the slower kinetics at the end of charge in Li–O2 cells better than the constant-current (CC) protocols majorly used in the field. These results suggest that CCCV protocols should be standardised to assess performance improvements in Li–O2 cells.
Diversification of energy storage strategies is an increasingly recognised way to accelerate the energy transition, lowering costs and increasing capacity by reducing the burden on critical materials.1 From that perspective, secondary Li–air batteries (LABs) offer an interesting alternative to lithium-ion batteries (LIBs) as they rely solely on lithium and carbonaceous materials for their electrode construction, in contrast with the LIBs heavy reliance on transition metals such as cobalt, nickel, manganese, etc.2
The headline-grabbing high theoretical specific energy of LABs (∼3500 W h kg−1) compared to LIBs (∼800 W h kg−1), is often cited in support of their potential use in applications where energy density is the primary priority, such as transport.3,4 However, slow kinetics in both the discharge and charge processes, as well as gas filtration/purification requirements, may prove challenging to overcome for the deployment of LABs in electric vehicles.5 The use of LABs in stationary battery systems to support decentralised renewable energy generation may, however, be a more suitable application given the lower demands on charging rates and energy density at the system level of this sector. The expected growth in demand for residential and industrial decentralised power generation will add an enormous pressure to global battery production, and therefore to many critical materials, even if EV batteries are widely used in their second-life. The widespread use of LABs as small-scale energy storage can therefore alleviate the huge demand for LIBs and their associated critical resources.6
The development of LABs is still, however, at a low technology readiness level (TRL), with multiple fundamental challenges needing to be overcome for their commercialisation.7 LABs store energy via a conversion (rather than intercalation) chemistry, with oxygen as the active cathodic material.8 During discharge, electrons generated by the oxidation of lithium metal at the anode reach the air electrode, where oxygen is reduced and combined with Li-ions to form insoluble products through the oxygen reduction reaction (ORR).9 During the charging process, these insoluble discharge products are broken down, with reduced oxygen species being oxidised to O2 through the oxygen evolution reaction (OER).10
The kinetics of the OER are strongly dependent on factors such as the nature, abundance and morphology of discharge products, as well as the mean electron transfer path.11,12 Therefore, it is expected that these kinetics slow down towards the end of the charging process, as discharge product becomes scarce and the electron transfer path lengthens.13 This is typically experimentally observed as an increase in overpotential at the end of galvanostatic (constant current, CC) charging of Li–O2 cells (schematically represented in Fig. 1a).4
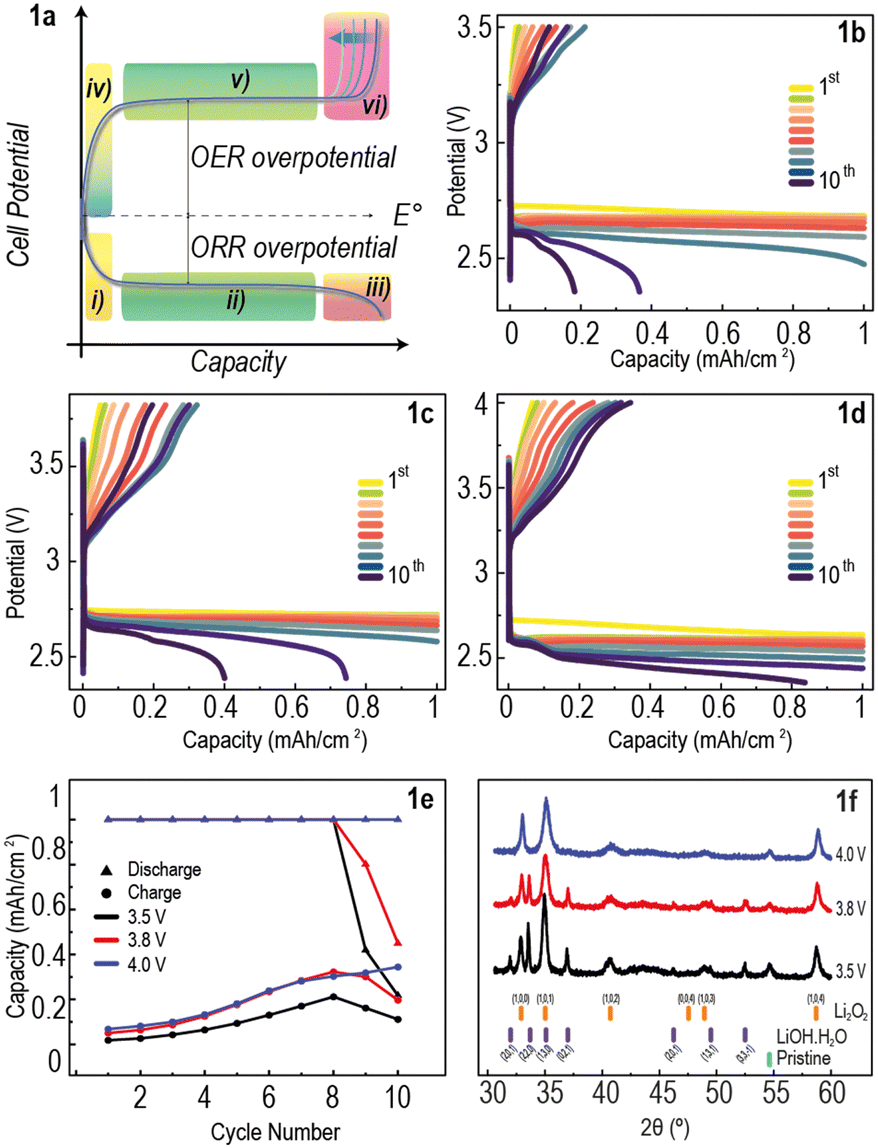 | ||
| Fig. 1 Constant current protocol. (a) Typical galvanostatic cycling (CC) potential curves of Li–O2 cells. (b)–(d) CC potential curves of Li–O2 cells cycled at a range of upper-cut-off voltages (UCVs); (b) 3.5 V, (c) 3.8 V, and (d) 4.0 V. (e) Discharge and charge capacity per cycle comparison. (f) XRD patterns of electrodes taken after the 10th discharge–charge cycles. | ||
Although the theoretical LAB cell potential is high, the reaction kinetics are slow, requiring the application of large overpotentials to achieve sufficiently fast charge rates.14,15 However, these conditions promote undesirable parasitic reactions which rapidly degrade the battery components.16,17 The decomposition of the discharge products and oxidation of reduced oxygen species at the air electrode is a multifaceted process influenced by the properties of the electrode (physical, chemical, and morphological), the electrolyte (viscosity, acceptor number, etc.), and the discharge products themselves.18,19 For instance, slow discharges can produce larger discharge product crystals, which are preferable to the small crystals produced by fast cycles, for high capacity.20 The latter form thin layers that covers the cathode surface, eventually preventing electron transfer and thus shortening the lifetime.21
In this study, we investigate two key and interconnected factors, cycling protocols and electrolyte compositions, both affecting the capacity recovery, overpotentials, and lifetime of Li–O2 cells. We compare CC and CCCV cycling protocols with a range of upper-cut-off voltages (UCVs) to explore the effect of reducing the current at the end of the charge process, and thus limiting cell overpotentials. The evaluation of these cycling protocols has been performed in cells with and without a redox mediator(RM), which reduces charge overpotentials by catalysing the electron transfer reaction.22
A typical plot of voltage vs capacity in galvanostatic (constant current, CC) discharge and charge of a Li–O2 cell is shown in Fig. 1a. The overall reaction at the cathode is 2Li+ + 2e− + O2 ⇄ Li2O2, corresponding to a thermodynamic potential of 2.96 V vs. Li/Li+ (dashed line).23 Even at low rates, the discharge potential is significantly lower than the thermodynamic potential (by ∼0.3 to 0.5 V) and the charging potential is significantly higher (by ∼0.5 to 1 V). These deviations, resulting in high voltage hysteresis (overpotential difference between charge and discharge potential curves), indicate significant energy inefficiency. The potential curves of Li–O2 cells cycled using a CC protocol typically exhibit 6 regions as shown in Fig. 1a: (i) steep drop in cell potential at the beginning of discharge due to nucleation of the discharge product;24 (ii) discharge plateau from growth of discharge product;23 (iii) steep drop in cell potential due to lack of electrode surface and starvation of the ORR;25 (iv) steep increase in potential at beginning of charge;26 (v) charge plateau;18 (vi) steep increase in cell potential at the end of charge due to sluggish kinetics of discharge product decomposition.9 During step (vi), the faradaic efficiency of the OER reaction drops dramatically (observed in electrochemical mass spectrometry experiments),17,19,27,28 while parasitic reactions take over.
Li–O2 cells composed of a lithium metal anode, 1 M LiTFSI in DME electrolyte and a commercial carbon cathode were assembled and cycled using a capacity-limited CC protocol, with upper-cut-off voltages (UCVs) set at 3.5, 3.8, and 4.0 V (Fig. 1b–d). The protocol consisted of a resting period of 8 hours to allow for electrode wetting and oxygen diffusion in the electrolyte, followed by applying a constant current of 100 μA cm−2 until the discharge capacity reached 1 mA h cm−2, or until the potential dropped below 2 V. Subsequently, the same constant current was applied until the charge capacity reached 1 mA h cm−2, or the UCV was reached. The discharge–charge potential curves of these cells over 10 cycles show a steep increase in potential at the beginning of charge, but in all cases the UCV was reached before a charge plateau, and consequently high capacity recovery, could not be achieved (Fig. 1b–d). Fig. 1e shows that, while the discharge capacity limit was reached for most cycles of all cells, very little capacity was recovered upon charge. All cells showed a general increase in charge capacity with cycle number followed by a decrease after cycle 8 for cells with UCV 3.5 V and 3.8 V. The continuous accumulation of discharge product in the air-electrode, as demonstrated by the low charge capacity, is the most likely explanation for this small increase in charging capacity with increasing cycle number.
Postmortem X-ray diffraction (XRD) was performed on the electrodes after the 10 discharge–charge cycles (Fig. 1f). All electrodes cycled using the CC protocol show evidence of residual Li2O2, with toroidal crystals, typical for Li2O2,29 seen in scanning electron microscopy (SEM) images (Fig. S1a–c, ESI†). The cells with UCV 3.5 V and 3.8 V show additional peaks corresponding to LiOH,30 (Fig. 1f) likely formed through electrolyte decomposition. The absence of LiOH peaks in the XRD pattern of the cell cycled at UCV 4.0 V suggests that this byproduct can be decomposed with sufficient charging time at these potentials. This electrolyte decomposition highlights the need for maintaining low overpotentials in order to extend battery lifetime. The poor capacity recovery observed also indicates the need for rethinking the cycling protocols typically used in the field, which should prioritise energy efficiency whilst maintaining the UCV to a range in which parasitic reactions are minimised.
Li–O2 cells, assembled in the same way, were then cycled using a capacity-limited CCCV protocol with UCVs also set at 3.5, 3.8 and 4.0 V. The CCCV protocol was identical to the CC protocol during discharge, but after reaching the UCV during charge the voltage was held at this value until the charge capacity reached 1 mA h cm−2, or until the current dropped below 10% of its initial value. This potential hold appears as a charge plateau, seen in the discharge–charge potential curves of these cells over 10 cycles (Fig. 2a–c). A higher UCV enables both longer constant current (CC) and constant voltage (CV) phases during charging. The former accelerates capacity recovery, as the total current during the CC phase is higher, and the latter increases the capacity recovered before the lower current limit is reached. This is illustrated in Fig. 2d, which shows the first discharge–charge cycle for each cell. As the UCV increases from 3.5 V to 4.0 V, the capacity recovered in the CC phase increases from 1% to 8%, and the total capacity recovered increases from 5% to 74%.
 | ||
| Fig. 2 Constant current constant voltage protocol. (a)–(c) CCCV potential curves of Li–O2 cells cycled at a range of upper-cut-off voltages (UCVs); (a) 3.5 V, (b) 3.8 V and (c) 4.0 V. (d) CCCV potential curves of the first discharge–charge cycle of Li–O2 cells showing capacity recovered during the CC portion of charge and subsequent capacity recovered during the CV portion of charge. (e) Discharge and charge capacity per cycle comparison. (f) XRD patterns of electrodes taken after the 10th discharge–charge cycles. | ||
Fig. 2e, similarly to Fig. 1e, shows a general increase in charge capacity with cycle number for all cells, which again is likely attributed to the continuous accumulation of discharge product in the air-electrode, especially in the low UCV cells. An improvement over the CC protocol is evident, particularly for the cells at UCVs 3.8 V and 4.0 V, which demonstrate increases in capacity recovery (CR) for the 10th cycle from 51 to 63% and 34 to 72%, respectively. Fig. 2e also shows this trend, where cells with a higher UCV exhibited greater capacity recovery in most cycles.
Postmortem XRD was performed on the electrodes after the 10 discharge–charge cycles (Fig. 2f). There is no evidence of LiOH, suggesting that any byproduct initially generated by electrolyte decomposition was then fully decomposed due to the increased charging times compared to the CC protocol. The intensity of Li2O2 peaks is significantly lower in the cell cycled with UCV 4.0 V, which corresponds to the higher charge capacity seen in Fig. 2e, confirming the removal of more Li2O2 from this electrode.This is also observed in the SEM images (Fig. S2a–c, ESI†), where significantly less Li2O2 can be seen on the surface of the UCV 4.0 V electrode. These results indicate that CCCV protocols, with reduced currents at the end of charge to accommodate slower OER kinetics,31–33 enable notably higher CR upon charging than CC protocols, albeit by extending charging times considerably (Fig. S4, ESI†).
Subsequently, we studied the use of the redox mediator LiI in conjunction with CCCV protocols. LiI is oxidised to I3− at the positive electrode, I3− then oxidising the Li2O2 and evolving O2 gas,22 reducing charge overpotential and increasing capacity recovery.34–36 Li–O2 cells with 1 M LiTFSI and 0.1 M LiI in DME were assembled and cycled using the same capacity-limited CCCV protocol with UCVs set at 3.4, 3.5 and 3.8 V. Lower UCVs than in cells without LiI were investigated, to limit the oxidation of I− to I2 above 3.55 V, a highly reactive species that degrades battery components and in turn shortens battery lifetime. The discharge–charge potential curves of these cells (Fig. 3a–c) display a lower charge overpotential than the analogous cells without LiI, and both cells with UCV 3.5 V and 3.8 V show 100% CR upon charge for all 10 cycles (Fig. 3d). A sharp decrease in charge capacity with cycle number for the UCV 3.4 V cell shows that the UCV is too low and therefore reached too quickly to allow full charging on each cycle. Conversely, despite achieving 100% CR in each cycle, the UCV 3.8 V cell forms the degrading I2 species, which occurs at the third plateau of its charge profile, above 3.55 V.17,37 Therefore, the cell with UCV 3.5 V is the best-performing in terms of CR at potentials at which OER-competing processes are not likely to happen. The first plateau in this cell's charge profile corresponds to the oxidation of I− to I3−, which subsequently oxidises Li2O2 to Li metal and O2 gas, shown by the second plateau (Fig. 3b).
 | ||
| Fig. 3 Constant current constant voltage protocol with redox mediator. (a)–(c) CCCV potential curves of Li–O2 cells containing LiI as a redox mediator in the electrolyte cycled at a range of upper-cut-off voltages (UCVs); (a) 3.4 V, (b) 3.5 V and (c) 3.8 V. (d) Discharge and charge capacity per cycle comparison. (e) XRD patterns of electrodes taken after the 10th discharge–charge cycles. | ||
Postmortem XRD was performed on the electrodes after the 10 discharge–charge cycles (Fig. 3e). The cells with UCV 3.4 V and 3.5 V show low intensity peaks, whereas there is a total absence of peaks in the XRD pattern of the cell cycled at UCV 3.8 V. This correlates well with the high charge capacity seen in Fig. 3d for UCV 3.5 V and 3.8 V, suggesting the discharge product was mostly decomposed upon charging, even without reaching the potentiostatic stage of the CCCV protocol. This is also observed in the SEM images of these electrodes (Fig. S3a–c, ESI†), where some film-like Li2O2 can be seen in the UCV 3.4 V cell, whereas in the UCV 3.5 V and 3.8 V cells there is very little to no discharge product visible. This suggests that cycling Li–O2 cells using a CCCV protocol with a UCV of 3.5 V, combined with a RM, can achieve both higher capacity recovery and longer battery lifetime.
This work illustrates the interplay of cycling protocols and electrolyte composition, and their impact on Li–O2 cell lifetime. Our results show that allowing for high UCVs to match the discharge capacity in strictly galvanostatic charging results in a shorter lifetime of Li–O2 cells than adding a potentiostatic stage at the end of charge with lower UCVs. We demonstrate that capacity-limited CCCV charging protocols offer a pronounced improvement over CC protocols in terms of both CR and battery lifetime. While lowering the UCV is key for extending cell lifetime, achieving this without compromising CR remains difficult. Therefore, the challenge of keeping the UCV as low as possible to manage parasitic reactions while still recovering 100% capacity at a reasonable charging rate should be the focus of much more attention in the field.
This work was supported by the Faraday Institution (grant number FITG-FUSE-078). I. T. acknowledges support from a Beatriz Galindo senior fellowship (BG22/00148) from the Spanish Ministry of Science and Innovation. L. B. acknowledges support from the EPSRC through the NanoDTC (grant no. EP/S022953/1), as well as from Cambridge Display Technology Ltd.
Data availability
Data for this article is available at symplectic elements at https://elements.admin.cam.ac.uk/viewobject.html?cid=1&id=1656199.Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Arbabzadeh, R. Sioshansi, J. X. Johnson and G. A. Keoleian, Nat. Commun., 2019, 10, 3413 CrossRef PubMed.
- C. Grey and J. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef.
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS PubMed.
- Z. Gao, I. Temprano, J. Lei, L. Tang, J. Li, C. P. Grey and T. Liu, Adv. Mater., 2023, 35, 2201384 CrossRef CAS.
- L. A. Archer, P. G. Bruce, E. J. Calvo, D. Dewar, J. H. J. Ellison, S. A. Freunberger, X. Gao, L. J. Hardwick, G. Horwitz, J. Janek, L. R. Johnson, J. W. Jordan, S. Matsuda, S. Menkin, S. Mondal, Q. Qiu, T. Samarakoon, I. Temprano, K. Uosaki, G. Vailaya, E. D. Wachsman, Y. Wu and S. Ye, Faraday Discuss., 2024, 248, 392 RSC.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun and A. A. Frimer, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- G. A. Attard, E. J. Calvo, L. A. Curtiss, D. Dewar, J. H. J. Ellison, X. Gao, C. P. Grey, L. J. Hardwick, G. Horwitz, J. Janek, L. R. Johnson, J. W. Jordan, S. Matsuda, S. Mondal, A. R. Neale, N. Ortiz-Vitoriano, I. Temprano, G. Vailaya, E. D. Wachsman, H.-H. Wang, Y. Wu and S. Ye, Faraday Discuss., 2024, 248, 75 Search PubMed.
- L. Luo, B. Liu, S. Song, W. Xu, J.-G. Zhang and C. Wang, Nat. Nanotechnol., 2017, 12, 535–539 CrossRef CAS PubMed.
- Y.-C. Lu, B. M. Gallant, D. G. Kwabi, J. R. Harding, R. R. Mitchell, M. S. Whittingham and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 750–768 RSC.
- H.-D. Lim, B. Lee, Y. Bae, H. Park, Y. Ko, H. Kim, J. Kim and K. Kang, Chem. Soc. Rev., 2017, 46, 2873–2888 RSC.
- L. Liu, Y. Liu, C. Wang, X. Peng, W. Fang, Y. Hou, J. Wang, J. Ye and Y. Wu, Small Methods, 2022, 6, 2101280 CrossRef CAS.
- G. A. Attard, P. G. Bruce, E. J. Calvo, Y. Chen, L. A. Curtiss, D. Dewar, J. H. J. Ellison, J. Fernández-Vidal, S. A. Freunberger, X. Gao, C. P. Grey, L. J. Hardwick, G. Horwitz, J. Janek, L. R. Johnson, E. Jónsson, S. Karunarathne, S. Matsuda, S. Menkin, S. Mondal, S. Nakanishi, N. Ortiz-Vitoriano, Z. Peng, J. P. Rivera, I. Temprano, K. Uosaki, E. D. Wachsman, Y. Wu and S. Ye, Faraday Discuss., 2024, 248, 210 RSC.
- N. B. Aetukuri, B. D. McCloskey, J. M. García, L. E. Krupp, V. Viswanathan and A. C. Luntz, Nat. Chem., 2015, 7, 50–56 CrossRef CAS.
- R. Gao, X. Liang, P. Yin, J. Wang, Y. L. Lee, Z. Hu and X. Liu, Nano Energy, 2017, 41, 535–542 CrossRef CAS.
- C. Xia, M. Waletzko, L. Chen, K. Peppler and P. J. Klar, and J. R. Janek, ACS Appl. Mater. Interfaces, 2014, 6, 12083–12092 CrossRef CAS.
- P. Zhang, M. Ding, X. Li, C. Li, Z. Li and L. Yin, Adv. Energy Mater., 2020, 10, 2001789 CrossRef CAS.
- A. Nakanishi, M. L. Thomas, H.-M. Kwon, Y. Kobayashi, R. Tatara, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2018, 122, 1522–1534 CrossRef CAS.
- L. Johnson, C. Li, Z. Liu, Y. Chen, S. A. Freunberger, P. C. Ashok, B. B. Praveen, K. Dholakia, J.-M. Tarascon and P. G. Bruce, Nat. Chem., 2014, 6, 1091–1099 CrossRef CAS PubMed.
- I. Temprano, T. Liu, E. Petrucco, J. H. Ellison, G. Kim, E. Jónsson and C. P. Grey, Joule, 2020, 4, 2501–2520 CrossRef CAS.
- B. M. Gallant, D. G. Kwabi, R. R. Mitchell, J. Zhou, C. V. Thompson and Y. Shao-Horn, Energy Environ. Sci., 2013, 6, 2518–2528 RSC.
- B. D. Adams, C. Radtke, R. Black, M. L. Trudeau, K. Zaghib and L. F. Nazar, Energy Environ. Sci., 2013, 6, 1772–1778 RSC.
- J. B. Park, S. H. Lee, H. G. Jung, D. Aurbach and Y. K. Sun, Adv. Mater., 2018, 30, 1704162 CrossRef.
- Y.-C. Lu, H. A. Gasteiger, M. C. Parent, V. Chiloyan and Y. Shao-Horn, Electrochem. Solid-State Lett., 2010, 13, A69 CrossRef CAS.
- S. Lau and L. A. Archer, Nano Lett., 2015, 15, 5995–6002 CrossRef CAS.
- J. Højberg, B. D. McCloskey, J. Hjelm, T. Vegge, K. Johansen, P. Norby and A. C. Luntz, ACS Appl. Mater. Interfaces, 2015, 7, 4039–4047 CrossRef PubMed.
- J. Chen, E. Quattrocchi, F. Ciucci and Y. Chen, Chem, 2023, 9, 2267–2281 CAS.
- B. D. McCloskey, D. Bethune, R. Shelby, T. Mori, R. Scheffler, A. Speidel, M. Sherwood and A. Luntz, J. Phys. Chem. Lett., 2012, 3, 3043–3047 CrossRef CAS PubMed.
- S. Das, J. Højberg, K. B. Knudsen, R. Younesi, P. Johansson, P. Norby and T. Vegge, J. Phys. Chem. C, 2015, 119, 18084–18090 CrossRef CAS.
- Z. Z. Shen, C. Zhou, R. Wen and L. J. Wan, Chin. J. Chem., 2021, 39, 2668–2672 CrossRef CAS.
- N. Alcock, Acta Crystallogr., 1971, 27, 1682–1683 CrossRef CAS.
- Z. Su, I. Temprano, N. Folastre, V. Vanpeene, J. Villanova, G. Gachot, E. V. Shevchenko, C. P. Grey, A. A. Franco and A. Demortière, Small Methods, 2024, 8, 2300452 CrossRef CAS.
- K. Ku, S.-B. Son, J. Gim, J. Park, Y. Liang, A. Stark, E. Lee and J. Libera, J. Mater. Chem. A, 2022, 10, 288–295 RSC.
- E. A. Kedzie, J. E. Nichols and B. D. McCloskey, J. Mater. Res., 2022, 37, 3227–3236 CrossRef CAS.
- H.-D. Lim, B. Lee, Y. Zheng, J. Hong, J. Kim, H. Gwon, Y. Ko, M. Lee, K. Cho and K. Kang, Nat. Energy, 2016, 1, 1–9 Search PubMed.
- T. Liu, J. T. Frith, G. Kim, R. N. Kerber, N. Dubouis, Y. Shao, Z. Liu, P. C. Magusin, M. T. Casford and N. Garcia-Araez, J. Am. Chem. Soc., 2018, 140, 1428–1437 CrossRef CAS PubMed.
- E. Jónsson, A. H. Berge, C. P. Grey and I. Temprano, Faraday Discuss., 2024, 248, 145 Search PubMed.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, J. Phys. Chem. C, 2015, 119, 22392–22403 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05801a |
| ‡ Current address: Department of Chemistry, University of Oxford, Oxford OX13TA, UK. |
| This journal is © The Royal Society of Chemistry 2025 |