
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective binding of rare-earth ions in polymerizable cages†
Jou-Tsen
Ou
 and
Mercedes K.
Taylor
*
and
Mercedes K.
Taylor
*
Department of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742, USA
First published on 12th February 2025
Abstract
We assembled metal–organic cages by combining rare-earth metals with an alkene-terminated ligand (L), allowing for subsequent thiol–ene polymerization of the cages to yield rare-earth-binding polymer networks. Mixed-metal self-assembly experiments with ligand L show that the size ratio of the respective rare-earth metals dictates the metal composition of the cages.
The rapid development of clean-energy technologies, such as electric vehicles and wind turbines, has created an urgent need for rare-earth metals.1,2 Recycling rare-earth metals from electronic waste can offset supply chain vulnerabilities for countries without natural rare-earth deposits, as the major rare-earth deposits are concentrated in Asia.3,4 However, since electronic waste typically contains various rare-earth metals with similar chemical and physical properties, isolating individual rare-earth metals is an ongoing challenge.5
As metal–organic cages (MOCs) have garnered increasing interest due to their porosity and processability,6,7 rare-earth-containing cages have emerged as a supramolecular approach to rare-earth extraction and separation.7–9 Recently, Sun and co-workers reported a highly selective self-sorting process for mixtures of rare-earth metals, using a C3-symmetric ligand with three tridentate binding sites.8 The components were self-assembled into tetrahedral cages of M4L4 geometry in which all four metal binding sites were occupied by the same rare-earth element. Such homo-metallic cages were selectively formed even from mixtures of highly similar rare-earth ions. The authors were able to use this self-sorting behavior in a liquid–liquid extraction to separate lanthanide ions.
In a separation process, the ion-adsorption capacity of cages is limited by the number of binding sites within each cage, and the recyclability is constrained by the disassembly of the cages after metal removal. These challenges could be addressed by linking the rare-earth-binding cages into a solid-state network, as was recently achieved through copolymerization with divinylbenzene.9 However, due to divinylbenzene's ability to self-polymerize, this route limits control over both the distance between the cages and the degree of cross-linking. We aimed to develop a route to polymerizable cages that instead react via step-growth polymerization, which would offer improved control over polymer morphology and composition.
To this end, we developed a novel C3-symmetric ligand (L) with three tridentate arms, featuring three terminal alkenes connected to a rigid core via flexible alkyl chains (Scheme 1). Ligand L assembles with rare-earth ions to form supramolecular cages with M4L4 geometry, where M = Y3+, Eu3+, Tb3+, Dy3+, or Ho3+. (The complete formula for the complexes is M4L4(OTf)12, which we denote M4L4; OTf− = trifluoromethanesulfonate). The Y4L4 cage was then polymerized with various di-thiol co-monomers, and the resulting morphologies were studied using scanning electron microscopy (SEM). Since the cage networks are insoluble in common solvents, the selectivity of the rare-earth binding sites was examined at the molecular level by performing mixed-metal self-assembly experiments with the ligand L. The results show that the formation of homo-metallic versus mixed-metal cages depends on the ionic radius ratio of the two rare-earth metals.
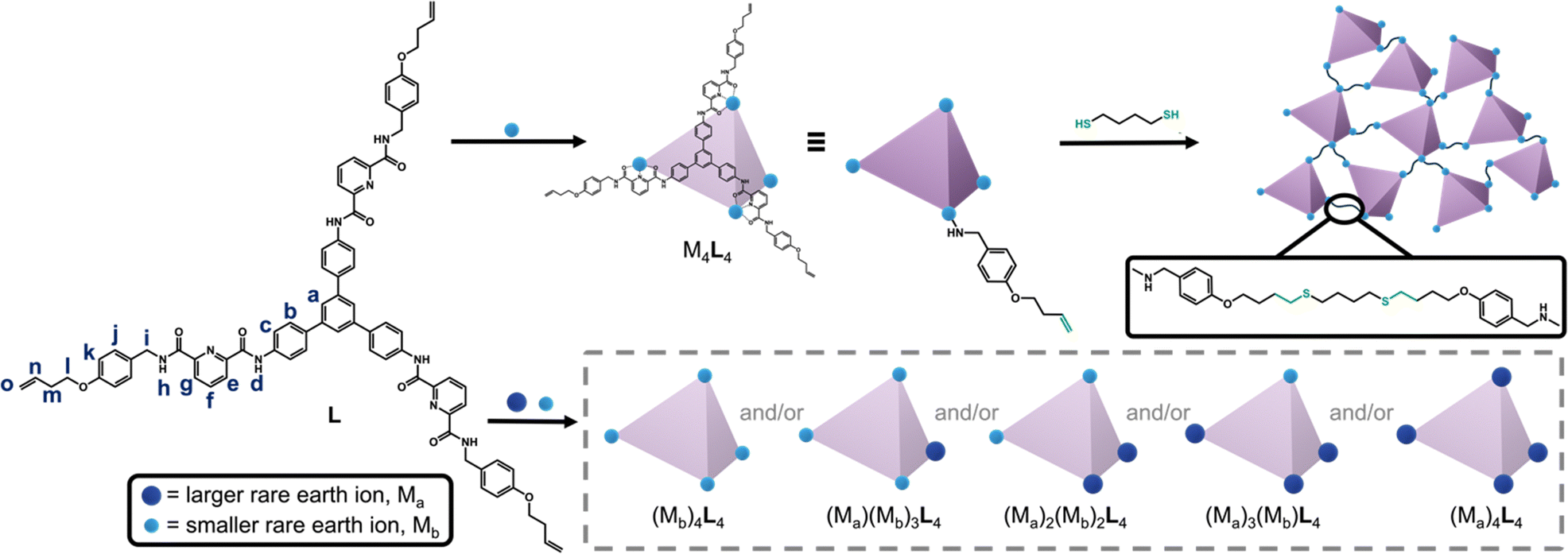 | ||
| Scheme 1 Top: The reaction of ligand L with rare-earth metal ions leads to the formation of metal–organic cages, which can be polymerized through thiol–ene reactions with the terminal vinyl groups on L. Bottom: Mixed-metal self-assembly experiments lead to cages containing either one metal or a mixture of metals, depending on the size difference between the metals. Light blue spheres represent the smaller rare-earth metal ion and dark blue spheres represent the larger rare-earth metal ion in a given mixture. | ||
The ligand L was synthesized by appending three tridentate arms to a rigid, three-fold symmetric core (1,3,5-tris-(4-aminophenyl)benzene, TAPB). The tridentate arm was synthesized through a series of four steps: (1) Williamson ether synthesis,10 (2) reductive amination,10 (3) amide formation,11 and (4) saponification.9 In the first step, 4-bromobut-1-ene was reacted with 4-hydroxybenzaldehyde to form an ether through nucleophilic substitution (SN2). Reductive amination was then carried out to convert the aldehyde to an amine, which was used in amide formation with 6-(methoxycarbonyl)picolinic acid to install the rare-earth binding site. Finally, the ester group was saponified to yield the carboxylic acid-containing tridentate arm. The three-fold symmetric core, TAPB, was synthesized by slightly modifying the well-known synthetic procedure.12 The carboxylic acid-containing tridentate arm was then reacted with TAPB through amide formation,11 yielding the tridentate C3-symmetric ligand L with three terminal alkene groups. The chemical structure of L was confirmed by 1H NMR (Fig. 1a), 13C NMR, HSQC, and COSY spectra; complete synthetic details and characterization for all materials is provided in the (Schemes S1, S2 and Fig. S1–S21, ESI†).
 | ||
| Fig. 1 1H NMR spectra of (a) ligand L (DMSO-d6) and (b) the Y4L4 cage (CD3CN). Different NMR solvents were used due to the differing solubilities of the two species. (c) DOSY spectrum of the Y4L4 cage. (d) ESI-TOF-MS spectrum of the Y4L4 cage with insets showing the observed and simulated isotopic patterns corresponding to [Y4L4(OTf)7]5+. | ||
The ligand L was then reacted with rare-earth metals to form supramolecular metal–organic cages. When 1.0 equivalent of L was treated with 1.1 equivalents of yttrium(III)trifluoromethanesulfonate (Y(OTf)3) at 80 °C for three hours, the turbid suspension gradually turned clear and yellow. A single set of signals with noticeable chemical shifts relative to the free ligand was observed in the 1H NMR spectrum of the Y4L4 complex (Fig. 1b). Through comparison with 1H–1H correlated spectroscopy (COSY) spectra of L and the Y4L4 complex (Fig. S14 and S22, ESI†), all the signals in the 1H NMR spectra were fully assigned. The formation of a single species was further confirmed by 1H diffusion-ordered spectroscopy (DOSY), which showed that all the signals from the protons of the Y4L4 complex have the same diffusion coefficient of 3.44 × 10−10 m2 s−1, corresponding to a dynamic radius of 1.90 nm, as calculated using the Stokes–Einstein equation (Fig. 1c). The composition of Y4L4 was further determined by electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS), and the MS spectrum was analyzed based on the equation [Y4L4(OTf)12-n-m-mH]n+ (Fig. 1d).13 The spectrum exhibits a series of peaks with charge states ranging from 4+ to 8+, resulting from the successive loss of OTf− anions from Y4L4(OTf)12; the MS results show that the Y4L4 complex consists of four Y3+ ions, four L molecules, and 12 OTf− counterions. For example, a peak with m/z equal to 1300.8352 was assigned to the charged molecular formula [Y4L4(OTf)7]5+. Detailed peaks assignment is provided in Fig. S23 (ESI†). The charges of the peaks were verified by the finely resolved isotopic distribution in the MS signal, as shown in the inset of Fig. 1d.
Additional self-assembly experiments were conducted between L and various rare-earth triflate salts, namely La(OTf)3, Eu(OTf)3, Tb(OTf)3, Dy(OTf)3, and Ho(OTf)3. The ESI-TOF-MS spectra (Fig. S23–S28, ESI†) indicate that L forms M4L4 cages with Eu3+, Tb3+, Dy3+, and Ho3+. The M4L4 complexes reported here are analogous to previously reported tetrahedral cages, due the C3-symmetric arrangement of tridentate arms that combine to occupy the nine coordination sites of four rare-earth ions.8,13,14 In contrast, no M4L4 complex was observed to form when L was reacted with La(OTf)3, the largest rare-earth ion tested. This finding suggests that L exhibits selective binding toward smaller rare-earth ions.
Although rare-earth complexation with ligands such as L can accomplish rare-earth separations via liquid–liquid extraction, these solution-state approaches generate large volumes of hazardous waste, making them costly and inefficient.15 In pursuit of a solid-state material that incorporates supramolecular rare-earth binding, we used thiol–ene reactions to polymerize the Y4L4 cages into an extended network. This cage was selected because Y3+ is diamagnetic, allowing the cross-linked cages to be characterized by solid-state NMR (ssNMR; Fig. 2a and Fig. S29, S30, ESI†). The Y4L4 cage was reacted with 1,4-butanedithiol at two different alkene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiol ratios (1:1 and 1:10) at 70 °C under an inert atmosphere. The alkene carbon peaks in the Y4L4 cage were identified at 117 and 135 ppm (Fig. 2a), based on the HSQC NMR spectrum of the Y4L4 cage (Fig. S31, ESI†). Following the thiol–ene polymerizations, the alkene peak at 117 ppm disappeared, and a broad peak appeared at ∼28 ppm, indicating the conversion of alkenes to alkanes. The peak at 135 ppm in the Y4L4 spectrum is comprised of overlapping aromatic and alkene signals, which explains its incomplete disappearance after the thiol–ene reactions. A control experiment in which an alkene was reacted with a 10-fold excess of thiol confirmed that the excess thiol groups do not form disulfide bonds under these polymerization conditions (Fig. S32, ESI†).
:thiol ratios (1:1 and 1:10) at 70 °C under an inert atmosphere. The alkene carbon peaks in the Y4L4 cage were identified at 117 and 135 ppm (Fig. 2a), based on the HSQC NMR spectrum of the Y4L4 cage (Fig. S31, ESI†). Following the thiol–ene polymerizations, the alkene peak at 117 ppm disappeared, and a broad peak appeared at ∼28 ppm, indicating the conversion of alkenes to alkanes. The peak at 135 ppm in the Y4L4 spectrum is comprised of overlapping aromatic and alkene signals, which explains its incomplete disappearance after the thiol–ene reactions. A control experiment in which an alkene was reacted with a 10-fold excess of thiol confirmed that the excess thiol groups do not form disulfide bonds under these polymerization conditions (Fig. S32, ESI†).
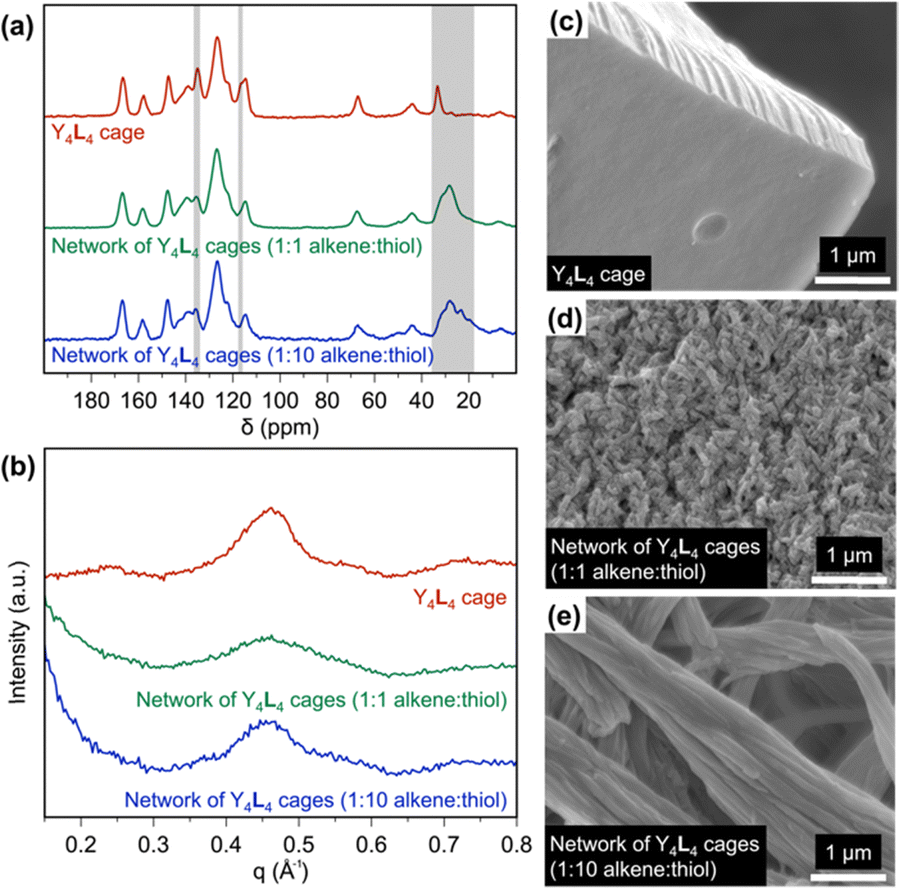 | ||
| Fig. 2 (a) Solid-state 13C NMR spectra and (b) SAXS curves of the Y4L4 cage, the network of Y4L4 cages polymerized with a 1:1 alkene:thiol ratio, and the network of Y4L4 cages with polymerized with a 1:10 alkene:thiol ratio. SEM images of (c) the Y4L4 cage, (d) the network of Y4L4 cages polymerized with a 1:1 alkene:thiol ratio, and (e) the network of Y4L4 cages polymerized with 1:10 alkene:thiol ratio. | ||
Small-angle X-ray scattering (SAXS) was used to further investigate the structure of the materials, as shown in Fig. 2b. The curve for Y4L4 shows a peak centered at 0.45 Å−1, corresponding to a d-spacing of 1.4 nm. This peak is attributed to the scattering of the cages, which should be dominated by the contribution from the relatively massive nucleus of yttrium; this result suggests the average Y–Y distance in the crystal structure is 1.4 nm. The same 0.45 Å−1 peak is present, but broadened, in both cage networks, indicating that the cages are not closely packed after polymerization but maintain an average Y–Y distance of 1.4 nm. The peak in the 1:1 alkene:thiol network is broader than in the 1:10 alkene:thiol network, suggesting the grain size of the 1:1 network is smaller than the grain size of 1:10 network, based on the Scherrer equation.
The morphologies of the cages and cage networks were studied by SEM. The Y4L4 cages appear as aggregates composed of well-defined, smooth blocks at the micrometer scale, but after polymerization, the microscale block-like aggregates are fragmented into fibrils and rods (Fig. 2c–e). The SEM images indicate that when the Y4L4 cage is polymerized with an alkene:thiol ratio of 1:1, cage–cage linkages are favored, preventing aggregation into large chunks as seen for the Y4L4 cages and instead forming nanoscale fibrils. In contrast, with a 10-fold excess of thiol, cages terminating in excess thiol are favored, resulting in a lower degree of cross-linking and yielding 1D micron-scale rods due to cage aggregation. These thiol–ene polymerization reactions demonstrate that varying the ratio of alkene to thiol can produce different morphological structures. While the Y4L4 cage is soluble in polar organic solvents, including acetonitrile and methanol, the cross-linked cages are insoluble in common solvents, making them potential candidates for solid-state adsorbents in future rare-earth separations.
Driven by our curiosity about the selectivity of rare-earth binding in the cages, and given that the networks of cages are insoluble in most common solvents, we performed a series of small-molecule binding studies to investigate metal ion selectivity. We combined L with binary mixtures of ions to test whether the cages self-assemble to yield homo-metallic or mixed-metal cages. When ligand L (1.0 equivalent) was combined with an equimolar mixture of La(OTf)3 (1.0 equivalent) and Ho(OTf)3 (1.0 equivalent), L selectively assembled with Ho(OTf)3 to form the Ho4L4 cage, while no La4L4 or mixed-metal cages were observed in the ESI-TOF-MS spectrum (Fig. 3a). Similar behavior was observed in the reaction of L with La(OTf)3 and Eu(OTf)3: Eu4L4 cages were exclusively formed, with no La4L4 or mixed-metal cages observed (Fig. 3b). Thus, reaction with L leads to metal self-sorting behavior, in which only the smaller metal ion is incorporated into the cage. This result is consistent with previous reports of cooperative binding within tetrahedral cages8,9 and is a promising finding for lanthanide separations.
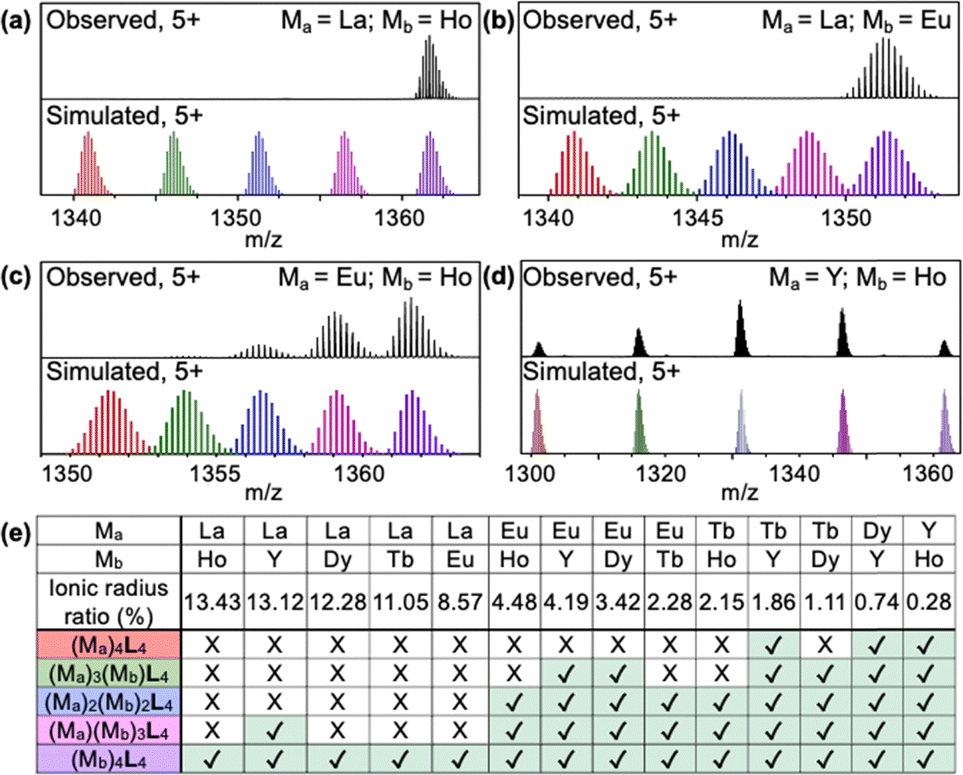 | ||
| Fig. 3 Selective self-assembly of L with binary mixtures of rare-earth metals Ma and Mb, where Ma is larger in size than Mb. ESI-TOF-MS spectra for reaction of L with (a) La/Ho, (b) La/Eu, (c) Eu/Ho, and (d) Y/Ho (black), above simulated isotopic patterns corresponding to [(Ma)4L4(OTf)7]5+ (red), [(Ma)3(Mb)L4(OTf)7]5+ (green), [(Ma)2(Mb)2L4(OTf)7]5+ (blue), [(Ma)(Mb)3L4(OTf)7]5+ (pink), and [(Mb)4L4(OTf)7]5+ (purple). (e) Summary of the results of mixed-metal self-assembly experiments. | ||
Europium and lanthanum ions differ significantly in ionic radius, but many critical rare-earth ions are much more similar to each other, such as Eu3+, Tb3+, and Dy3+.16 For example, La3+ is 8.57% larger than Eu3+, while Eu3+ is only 3.42% larger than Dy3+.17 (More detail for the calculation of radius ratios between metal ions can be found in the ESI† and Table S1) This challenge prompted us to investigate the limits of the polymerizable ligand L for separations of lanthanides with similar sizes. We conducted mixed-metal self-assembly experiments for rare-earth ions with extremely similar ionic radii: Eu3+, Tb3+, Dy3+, Y3+, and Ho3+. For the reaction of L with a mixture of Eu3+ and Ho3+, the pure Ho4L4 cage was observed, along with mixed-metal cages containing one or two Eu3+ ions (Eu1Ho3L4 and Eu2Ho2L4; Fig. 3c). For a mixture of Y3+ and Ho3+, all five possible combinations (Ho4L4, YHo3L4, Y2Ho2L4, Y3HoL4, and Y4L4) were observed (Fig. 3d). The MS spectra and simulated isotopic patterns for all mixed-metal self-assembly experiments are in Fig. S33–S56 (ESI†), and the results are summarized in Fig. 3e.
Taken together, the results of the mixed-metal self-assembly experiments provide several general conclusions about the self-sorting (or cooperative binding)8 of metal ions in cages formed with L. For the combinations of metals studied here, complete self-sorting to yield homo-metallic cages occurs when the larger metal ion has an ionic radius at least 8.57% greater than that of the smaller ion. Although minor La1Y3L4 forms when the ionic radius difference is 13.12%, this may be attributed the similar electron configuration between La3+ and Y3+. When the difference in radii is only 4.48–2.15%, metal self-sorting occurs to a more limited extent, yielding cages with formulas (Mb)4L4, (Ma)(Mb)3L4, and (Ma)2(Mb)2L4, where Ma is larger than Mb. When the difference in radii is less than 1.86%, assembly with L does not lead to any differentiation between the two metals, and cages of all possible metal combinations are formed. These findings show that small changes to the size ratio of two rare-earth ions can dictate their self-sorting behavior. To test the potential of the cage networks for rare-earth separations, we removed the Y3+ ions from the Y4L4-based networks and subjected the demetallated networks to ion adsorption experiments. While the networks show a modest preference for La3+ ions over Y3+ ions in single-ion experiments, Y3+ adsorption significantly exceeds La3+ adsorption in mixed-ion experiments, suggesting that the networks preserve cage-templated binding sites even after demetallation (Fig. S57, S58 and Table S2, ESI†).
In summary, we designed a polymerizable ligand L and combined it with rare-earth ions to yield a series of metal–organic cages. We found that these cages can be polymerized through thiol–ene click reactions to yield cross-linked networks of cages. SEM images show that the network made with a 1:1 ratio of alkenes to thiols forms nanoscale fibrils, while the network made with an excess of thiols exhibits 1D micron-scale rods, suggesting the morphologies of cage networks can be adjusted by the ratio of alkene to thiol. The ESI-TOF-MS results from mixed-metal experiments show that ligand L selectively binds smaller rare-earth ions and rejects larger ones when the difference in their ionic radii exceeds 8.57%, but the selectivity of L becomes limited when the difference in radii is less than 4.48%. In future work, we will test the performance of these polymer networks for batch separations of lanthanide ions.
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under award number DE-SC0024088. We thank Dr. Fu Chen and the Analytical NMR Service & Research Center at UMD for the use of NMR spectrometers (supported by award NSF-1726058). We thank Dr. Yue Li and the Mass Spectrometry Facility at UMD for the use of the Bruker Maxis-II QTOF mass spectrometer (supported by award NSF-2018860). We acknowledge the use of the Xenocs Xeuss Small Angle X-ray instrument (supported by award NSF-1228957). We acknowledge the Maryland NanoCenter and its Advanced Imaging & Microscopy (AIM) Lab.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Buchholz and T. Brandenburg, Chem. Ing. Tech., 2018, 90, 141–153 CrossRef
.
- S. Massari and M. Ruberti, Resour. Policy, 2013, 38, 36–43 CrossRef
- T. Dutta, K.-H. Kim, M. Uchimiya, E. E. Kwon, B.-H. Jeon, A. Deep and S.-T. Yun, Environ. Res., 2016, 150, 182–190 CrossRef
- S. R. Golroudbary, I. Makarava, A. Kraslawski and E. Repo, Sci. Total Environ., 2022, 832, 155022 CrossRef
- K. Binnemans, P. T. Jones, B. Blanpain, T. V. Gerven and Y. Pontikes, J. Cleaner Prod., 2015, 99, 17–38 CrossRef
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2012, 42, 1728–1754 RSC
- X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374–6458 CrossRef
- X.-Z. Li, L.-P. Zhou, L.-L. Yan, Y.-M. Dong, Z.-L. Bai, X.-Q. Sun, J. Diwu, S. Wang, J.-C. Bünzli and Q.-F. Sun, Nat. Commun., 2018, 9, 547 CrossRef PubMed
- Q.-Q. Yan, L.-P. Zhou, H.-Y. Zhou, Z. Wang, L.-X. Cai, X.-Q. Guo, X.-Q. Sun and Q.-F. Sun, Dalton Trans., 2019, 48, 7080–7084 Search PubMed
- M. Sanguinetti, S. Sanfilippo, D. Castagnolo, D. Sanglard, B. Posteraro, G. Donzellini and M. Botta, ACS Med. Chem. Lett., 2013, 4, 852–857 CrossRef
- J.-T. Ou, M. K. Brown, J. D. Kaff, R. W. Hughes, D. L. Huber, B. S. Sumerlin, K. I. Winey and M. K. Taylor, ACS Appl. Polym. Mater., 2024, 6, 11420–11426 CrossRef
- Y. Zhang, Y. Zhao, C. Zhang, X. Luo and X. Liu, CrystEngComm, 2022, 24, 4496–4499 RSC
- C.-L. Liu, R.-L. Zhang, C.-S. Lin, L.-P. Zhou, L.-X. Cai, J.-T. Kong, S.-Q. Yang, K.-L. Han and Q.-F. Sun, J. Am. Chem. Soc., 2017, 139, 12474 CrossRef PubMed
- L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550–8555 CrossRef PubMed
- W. R. Barrionuevo and F. M. Lanças, Bull. Environ. Contam. Toxicol., 2002, 69, 123–128 CrossRef PubMed
- U.S. Department of Energy, 2011, Critical Materials Strategy. https://www.energy.gov/sites/default/files/DOE_CMS2011_FINAL_Full.pdf Search PubMed.
- R. D. Shannon, Acta Cryst., 1976, 32, 751–767 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, synthetic procedures, and supplementary figures, including 1H NMR, 13C NMR, ESI-Mass spectra, and X-ray scattering data. See DOI: https://doi.org/10.1039/d4cc05827e |
| This journal is © The Royal Society of Chemistry 2025 |