
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFemto-microsecond electron transfer and intermediates in Al/Fe CO2 photoreduction systems through optical and X-ray spectroscopy†
Maxime
Sauvan
 a,
Ashok
Ugale
a,
Lucia
Velasco
ab,
Asterios
Charisiadis
a,
Fan
Ma
c,
Xiaoyi
Zhang
d,
Jia-Wei
Wang
*c and
Dooshaye
Moonshiram
*a
a,
Ashok
Ugale
a,
Lucia
Velasco
ab,
Asterios
Charisiadis
a,
Fan
Ma
c,
Xiaoyi
Zhang
d,
Jia-Wei
Wang
*c and
Dooshaye
Moonshiram
*a
aInstituto de Ciencia de Materiales de Madrid (ICMM-CSIC), Sor Juana Inés de la Cruz, 3, Madrid, 28049, Spain. E-mail: dooshaye.moonshiram@csic.es
bDepartamento de Química Física, Universidad Complutense de Madrid, Avenida Complutense s/n, Madrid, E-28040, Spain
cSchool of Chemical Engineering and Technology, Sun Yat-sen University, Zhuhai, 519082, China. E-mail: wangjw89@mail.sysu.edu.cn
dX-ray Science Division, Argonne National Laboratory, 9700 S. Cass Avenue, Lemont IL, 60439, USA
First published on 13th February 2025
Abstract
This study reveals the reaction pathways of 2 Al–Fe earth-abundant photocatalytic systems for CO2 reduction through femto-nanosecond optical transient absorption and microsecond X-ray spectroscopies. Time-resolved experimental findings with time-dependent density functional theory illustrate the formation of an elusive FeI octahedral species with bound aqua ligands and lifetimes of 23–29 μs.
The increasing burning of fossil fuels has over the past decades resulted in an increasing CO2 concentration that is responsible for today's global warming. The photochemical transformation of CO2 into natural gases or liquid fuels, using water as an abundant electron donor, could not only generate renewable fuels1 but also mitigate greenhouse gas emissions. The reduction of CO2 is, however, not a trivial process and requires proton-assisted multiple electron transfer steps2 that can further outcompete H2 formation. Multielectron–proton reactions can effectively be achieved through redox-photosensitizer, catalytic systems3 that can couple transient one-electron excited states in the presence of an electron donor to the catalyst. An efficient and selective CO2 reduction catalyst with multiple electrons can subsequently react with CO2 and release reduction products while avoiding side proton reduction reactions. Rare noble photosensitizers composed of Re,4 Os5 or Ru6 centres have extensively been employed in homogeneous CO2-reduction photocatalysis. The lack of efficient noble-metal-free photosensitizers has further prompted the development of exclusively earth-abundant metal complexes, such as binuclear Cu(I) complexes7 composed of two tetradentate ligands which achieved photocatalytic CO2 reduction in the presence of either Fe or Mn catalysts.8 Such families of Cu photosensitizers have, however, been known to display the formation of a flattened geometry within their excited states that enables nucleophilic attack by solvent molecules or counter anions. As a result, low-energy excited states known as an ‘‘exciplex’’9,10 that decay to the ground state within microsecond time scales are formed, limiting their stabilities during photocatalysis.
In the search for a larger library of economical CO2 photocatalytic systems, multimolecular assemblies composed of a [FeII(qpy)(OH2)2]2+ (qpy = 2,2′:6′,2′′:6′′,2′′′-quaterpyridine) catalyst and Al-based photosensitizers featuring monoanionic 2-pyridyloyrrolide ligands11,12 with varying number of methyl ligands denoted here as Al-1 and Al-2 (Scheme 1) were further explored in the presence of a BIH (1,3-dimethyl-2-phenyl-2,3,dihydro-1H-benzo[d]imidazole) electron donor and TEOA (triethanolamine) (Scheme 1). Al-based complexes represent for instance an interesting class of earth-abundant photosensitizers with an earth abundance of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm13 and have been less explored for photophysical and photochemical applications. The noble-metal-free Al/Fe systems were further shown to display ligand–ligand charge transfer (LLCT) excited states,11 impressive turnover numbers of 10250 for CO formation and higher robustness than benchmarking Cu and Ru photosensitizers, thus providing a promising platform for CO2 reduction. A complete photocatalytic system composed of Ru or Cu based photosensitizers with the Fe catalyst under the same experimental conditions for instance showed much lower turnover numbers of 401 and 72, respectively.11 Importantly, the excited states were found to be a singlet in nature while the triplet excited states displayed non-emissive behaviors.
000 ppm13 and have been less explored for photophysical and photochemical applications. The noble-metal-free Al/Fe systems were further shown to display ligand–ligand charge transfer (LLCT) excited states,11 impressive turnover numbers of 10250 for CO formation and higher robustness than benchmarking Cu and Ru photosensitizers, thus providing a promising platform for CO2 reduction. A complete photocatalytic system composed of Ru or Cu based photosensitizers with the Fe catalyst under the same experimental conditions for instance showed much lower turnover numbers of 401 and 72, respectively.11 Importantly, the excited states were found to be a singlet in nature while the triplet excited states displayed non-emissive behaviors.
 | ||
| Scheme 1 Structure of the Al photosensitizers and Fe catalyst studied in a multimolecular system with a BIH electron donor and a TEOA proton donor and acceptor. | ||
Although the photophysics of the Al photosensitizers (Scheme 1) together with their catalytic activities were probed, the kinetics along with the electronic and structural conformation of the Fe catalyst upon photoexcitation with the Al photosensitizers were elusive, yet of crucial importance for the development of cost-effective photocatalytic systems for durable CO2 reduction. In this regard, femto-nanosecond optical transient absorption (OTA) and pico-microsecond time-resolved X-ray absorption spectroscopy (tr-XAS, Fig. S1, ESI†) are employed herein to investigate the formation of the Al excited states and electron transfer events to generate the active FeI species. The structural conformation of the FeI reduced state responsible for CO2 reduction is further corroborated with time-dependent density functional theory calculations (TD-DFT). This work provides valuable insights into the overall mechanistic scenario of a family of earth-abundant molecular photocatalytic systems crucial for the development of artificial photosynthetic assemblies for CO2 utilization.
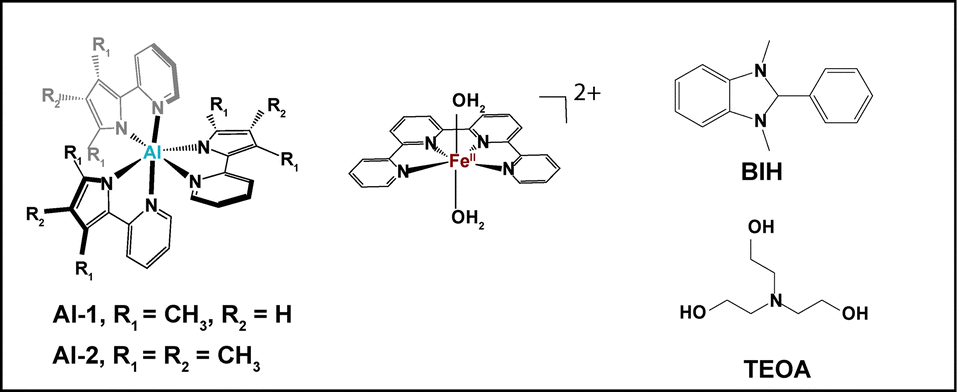
Prior to investigating a full photocatalytic system through time-resolved spectroscopy, steady state X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements were carried out to compare the coordination behaviours and structural conformations of the Fe catalyst in solid vs. solution (Fig. 1). The XAS measurements were conducted at 15 K with a defocused beam to minimize radiation damage (Fig. 1). The XANES spectra of the Fe catalyst in the solid and solution states display K-edge energies of 7123.1 eV and 7123.6 eV, respectively at half height and normalized fluorescence of 0.6 consistent with an FeII oxidation state14 (Fig. 1A). Although the Fe complex maintains the same oxidation state in both the solid and solution states, a small shift in the XANES rising edge of 0.5 eV (Fig. 1A) together with a decrease in the pre-edge intensities (Fig. 1A inset) is observed upon dissolution in a mixture of 0.93:0.07 CH3CN:DMF suggesting a change in the local Fe symmetries. A small amount of DMF was employed to match the same solvent experimental conditions as OTA and tr-XAS experiments elaborated below.
 | ||
| Fig. 1 (A) Normalized Fe K-edge XANES spectra of 1 mM of the FeQPY catalyst in solid (black) and a solution mixture of 0.93:0.07 CH3CN:DMF. Inset: Zoomed-in view of the pre-edge features. (B) Experimental Fourier transforms of k2-weighted Fe EXAFS of the FeQPY solid and solution complex (1 mM). Inset: Back Fourier transforms experimental (solid lines) and fitted (dashed lines) Re[χ(k)]. Experimental spectra were calculated for k values of 2.118 to 13.443 Å−1. | ||
The FeII catalyst in a 3d6 high spin distorted octahedral geometry has 4 unpaired electrons, 2 in each one of its t2g and eg levels thus leading to broad multiplet pre-edge features with peak energies at 7112.6 and 7114.2 eV (Fig. 1A inset). The presence of pre-edge features corresponds to the weak 1s to 3d quadrupole transitions which gain intensity through the dipole excitations of the core electrons into the valence 3d states hybridized with the N/C ligand p orbitals.15 Interestingly, the Fe catalyst in solution displays a small decrease in its pre-edge intensity confirming a more centrosymmetric coordination environment than that in the solid state (Fig. 1A inset). Indeed, complexes in a centrosymmetric coordination have been shown to exhibit weaker pre-edge features than non-centrosymmetric complexes.14 The trends in the pre-edge features further rule out the formation of a distorted FeII complex bound to only one water or solvent molecule.
To further assess the structural conformation of the Fe catalyst upon dissolution, DFT optimization calculations were carried out using the BP86 functional in the gas phase and through the conductor-like polarizable models for the solid and solution complexes, respectively, as elaborated in the ESI.† Several possible geometries were theoretically explored and the best agreement between both the pre-edge and rising edge features was found for an FeII complex bound strongly to two aqua ligands in solution (Fig. S2, ESI†).
The EXAFS spectrum of the catalyst in the solid and solution states further displays a prominent peak I corresponding to the average contribution of the Fe–N/O bond distances (Fig. 1B). The EXAFS fits for the extraction of the actual distances are shown in Fig. 1B inset, Fig. S2 and Table S1 (ESI†). Analysis of the first peak for the Fe solid complex clearly resolves 6 Fe–N distances at 2.15 Å (Table S1 and Fig. S3, ESI†), in perfect agreement with its X-ray diffraction (XRD) analysis and DFT calculations conducted in the gas phase (Table S2, Appendix, ESI†). By contrast, the Fe complex in CH3CN:DMF reveals six slightly shortened Fe–N bond distances at 2.13 Å in agreement with the DFT structure of the optimized complex in solution phase (Table S1 and Fig. S3, ESI†). The shortened Fe–N distances and six-coordinated geometry explain its shifted XANES spectra vs. its solid spectrum, and thus slightly larger energy is needed to eject a core 1s electron. The extracted bond distances for the catalyst in solution additionally rules out the formation of a FeII complex ligated with a single CH3CN, single aqua or a square planar FeII geometry whereby averaged Fe–N bond distances of 2.09, 2.10 and 2.08 Å (Table S2, Appendix, ESI†) would be respectively expected. Importantly theoretical methods including geometrical optimizations and XANES simulations reproduce the experimental EXAFS analysis well showing that they can be employed to deduce the structural conformation of the photoinduced catalytic species.
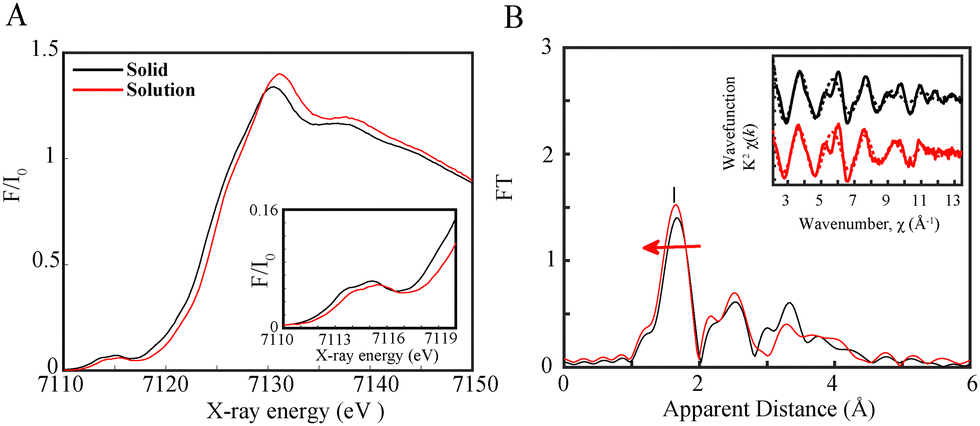
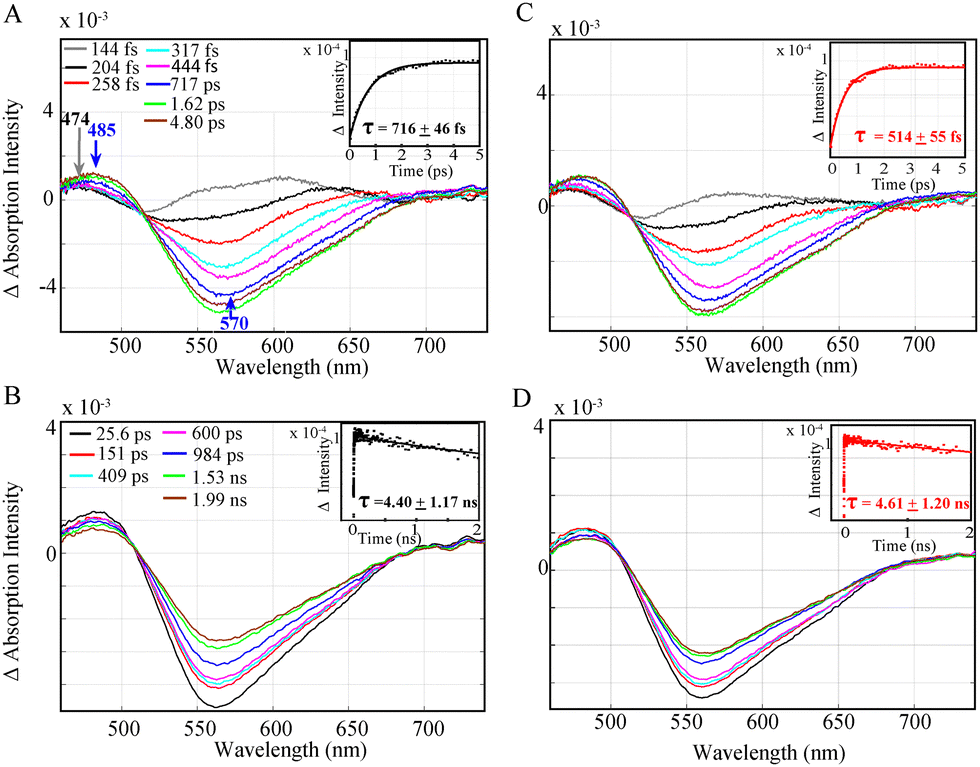
Following steady state XAS spectra of the catalyst, optical transient absorption was carried out to probe the electron transfer events in both Al photosensitizers separately (Fig. 2) as well as in a complete photocatalytic system with the Fe catalyst, BIH and TEOA (Fig. 3). The TEOA can in this case function both as a proton acceptor for the deprotonation reaction of BIH and as a proton donor for the facilitation of the CO2 reduction reaction as previously shown.11 The Al-1 and Al-2 photosensitizers with (2-(3,5-dimethyl-1H-pyrrol-2-yl)pyridine) and (2-(3,4,5,trimethyl,1H,pyrrol,2-yl)pyridine) ligands containing 2 and 3 methyl groups respectively were designed to serve as π-acceptors for the stabilization of the metal reduced states in a photocatalytic system.
 | ||
| Fig. 2 OTA spectra of a solution of (A) 0.3 mM Al-1, (C). 0.3 mM Al-2 in 0.93:0.07 CH3CN:DMF upon light excitation at 355 nm between pump-probe delays from 144 fs up to 4.80 ps. Insets (A) and (C): kinetic decay profiles at λ = 485 nm after excitation at λexc = 355 nm illustrating the formation of the singlet excited state. OTA spectra of a solution of (B) 0.3 mM Al-1, (D) 0.3 mM Al-2 in 0.93:0.07 CH3CN:DMF upon light excitation at 355 nm between 25.6 ps and 1.99 ns. Insets (B) and (D): kinetic decay profiles at λ = 485 nm after excitation at λexc = 355 nm illustrating the intersystem crossing rate from the singlet to the triplet. | ||
 | ||
| Fig. 3 OTA spectra of a complete photocatalytic system of (A) 0.3 mM Al-1 and (B) 0.3 mM Al-2 with 0.3 mM Fe catalyst, 30 mM BIH and 30 mM TEOA in 0.93:0.07 CH3CN:DMF. Insets (A) and (B): quenching of the Al− singlet excited state by BIH to generate the reduced optically silent Al− reduced species. | ||
Both Al-1 and Al-2 were known to exhibit a broad absorption band above 350 nm peaking around 393 and 405 nm, respectively.11 Upon laser excitation at 355 nm, a broad negative absorption between 520 nm and 700 nm with a maximum peak at 570 nm is observed due to stimulated emission (Fig. 2A and C). A broad positive peak at around 485 nm can further be seen corresponding to the formation of the singlet excited state. The singlet excited state Al* was found to occur faster and within 514 ± 55 fs in Al-2 (Fig. 2C and Fig. S3, ESI†) vs. 716 ± 46 fs in Al-1 (Fig. 2A and Fig. S4, ESI†) due to the presence of more electron donating methyl groups which play an important role in populating the excited state. By contrast, the intersystem crossing rate from the singlet to the triplet excited state occurs at a slower nanosecond rate within 4.40 ± 1.17 ns (Fig. 2B) and 4.61 ± 1.20 ns (Fig. 2D) in Al-1 and Al-2, respectively, in agreement with previous findings.11 Our femto-sub nanosecond laser measurement did not allow observation of the microsecond-lived triplet absorption found11 in the range of 430 to 500 nm.
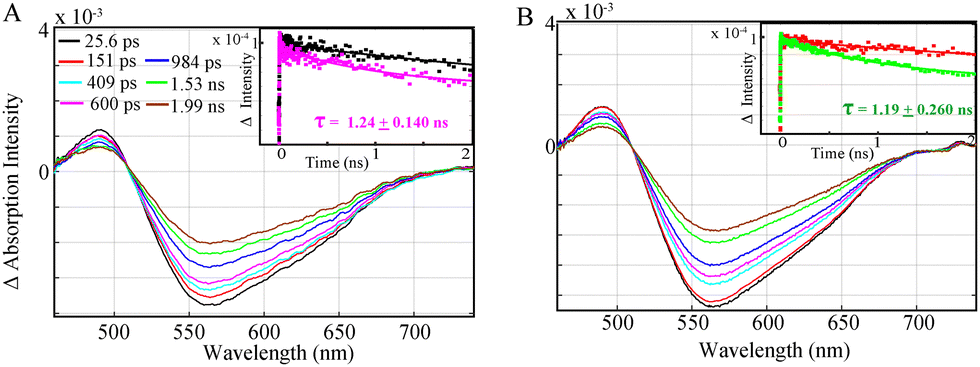
Nevertheless, the study of a complete photocatalytic system with the Fe catalyst, excess BIH and TEOA shows that an emissive singlet excited state is effectively quenched for Al-1 and Al-2 within 1.24 ± 0.140 ns (Fig. 3A) and 1.19 + 0.260 ns (Fig. 3B) respectively, thus illustrating the reductive quenching of the photosensitizers singlet excited states to be the dominant pathway for photoinduced catalysis. Previous work11 has shown that the lifetimes of the Al photosensitizers could only be dynamically quenched by BIH rather than the Fe catalyst. However, while the OTA measurements effectively illustrate the quenching of the singlet excited states, the formation of the reduced Al− state or photocatalytic FeI species could not be observed due to their overlapping absorption bands with the stimulated emission together with the lower extinction coefficient of the photocatalyst.
Time-resolved XANES was thus applied to monitor the kinetics and electronic configurations of the photo-induced FeI in a complete photocatalytic system. The multimolecular Al/Fe photocatalytic systems were optically pumped at 400 nm and probed with X-ray pulses from 100 ps to ∼25 μs (Fig. 4 and Fig. S5A, ESI†). Upon light excitation, low lying LLCT initially occurs to form a singlet excited state followed by the BIH electron donor quenching to generate a reduced Al− state. This process is followed by an electron transfer from the Al− reduced state to the FeII catalyst, generating the elusive FeI species (Fig. 4A).
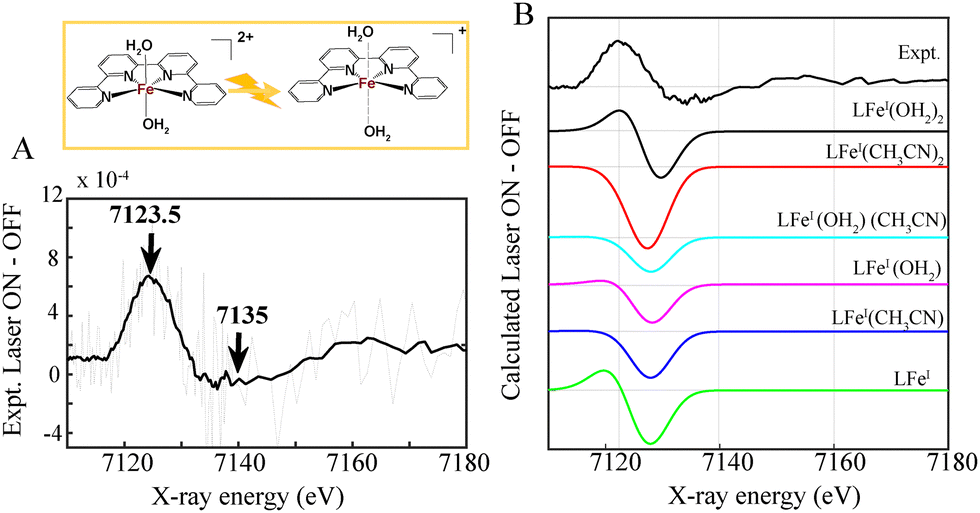 | ||
| Fig. 4 Top: formation of a photoreduced FeI species with 2 loosely coordinated molecules for a multimolecular assembly composed of the 2.5 mM Al-1 or Al-2 photosensitizer, 1 mM FeQPY catalyst, 100 mM BIH and 100 mM TEOA in a solution mixture of 0.93:0.07 CH3CN:DMF. A small amount of DMF is added to improve the solubility of the Al-based photosensitizers used in 2.5 mM concentrations. (A) Experimental laser ON–OFF spectra corresponding to formation of FeI at 14.2 μs (B). Experimental laser ON–OFF spectrum (black) in comparison with theoretical XANES simulations corresponding to the formation of a [LFeI–(OH2)2] (black), [LFeI–(CH3CN)2], [LFeI–(OH2) (CH3CN)] (cyan) octahedral complex, a square bipyramidal [LFeI–OH2] (magenta), [LFeI–CH3CN] (blue) and a [LFeI] square planar photoreduced species (blue) from [LFeII–(OH2)2]. | ||
The time-resolved XANES spectra obtained by subtracting the laser-ON and laser-OFF (dark) spectra provided valuable information about the photo-induced dynamics, electronic and structural nature of the photoinduced FeI. The time resolved XANES spectrum at an averaged delay of 14.2 μs between the laser pump excitation and X-ray probing for both photocatalytic mixtures composed of Al-1 and Al-2 is shown in Fig. 4A. The transient signals display a time-dependent broad peak at 7123.5 eV together with a dip at 7148 eV which is related to the characteristic formation of the reduced FeI species and ground state bleaching of the FeII ground state, respectively (Fig. 4A). These two transitions further show that the FeII K-edge energy shifts to lower energy upon photoexcitation thus confirming the effective electron transfer process from the reduced Al− state to the FeI catalyst. It is also important to note that no transient signals were observed in the absence of the BIH electron donor thus showing that the electron donor is indeed essential to quench the singlet excited state Al* to generate Al− which thereby transfers an electron to the Fe catalyst.
It should be mentioned that the excited state fractions of the photoreduced FeI was too low (less than 5%), to reconstruct the EXAFS and structural conformation. Tr-XANES spectra were thus next combined with TD-DFT to extract the structural conformation of the photo-generated FeI species (Fig. 4B). Several FeI geometries were considered namely an FeI complex coordinated with 2 aqua (black), another with 2 CH3CN (red) or with 1 aqua and 1 CH3CN (cyan) ligand (Fig. 4B). A square bipyramidal FeI complex with a single aqua (magenta) or CH3CN (blue) ligand, as well as a square planar FeI geometry (green), were also considered. The best agreement between the experimental trends and best energy matching was obtained for an FeI species with 2 elongated aqua molecules at 2.29 Å (Fig. 4 and Table S2, Appendix, ESI†) which would allow coordination and activation of a CO2 molecule. The progressions and decays of the transient signals for both photocatalytic systems composed of Al-1 and Al-2 photosensitizers were further analysed at successive time points ranging from 2.75 to 25.7 μs (Fig. S5, ESI†). The FeI reduced species was shown to be formed within 1.02–1.72 μs and decay within 23.2–28.9 μs in both photocatalytic systems (Fig. S5, ESI†). In summary, we report the application of femto-picosecond optical as well as pico-microsecond X-ray spectroscopy together with TD-DFT calculations to monitor the electronic and structural dynamics of the elusive FeI species in two sets of earth-abundant robust and selective Al–Fe multimolecular systems for CO2 reduction (Scheme 2). Upon light excitation in a complete photocatalytic system, LLCT initially triggers the formation of a singlet excited state within 716 fs and 514 fs, respectively, in Al-1 and Al-2 followed by an electron transfer to BIH within 1.2 ns to form the reduced Al− state (Scheme 2 and Fig. 2A, B and 3). The Al* excited state is notably formed faster in Al-2 due to the presence of more electron donating methyl groups. The reduced Al− species subsequently transfers an electron to the FeII catalyst within 1.1–1.7 μs to form a long-lived FeI species with lifetimes of 23 to 29 μs (Scheme 2 and Fig. 4 and Fig. S5, ESI†). The singlet excited states were further shown to display intersystem crossing rates of 4.4–4.6 ns in the absence of BIH and TEOA (Fig. 2C and D and Scheme 2) showing that the reductive quenching of the singlet is the predominant pathway. These findings report for the first time the mechanistic scenario and structure of the elusive FeI species in a fuly noble metal-free molecular system for CO2 reduction and thus constitute an important step for the future design of robust and durable earth-abundant photosensitizer/catalytic assemblies.
D. M. acknowledges funding from Spanish Ministerio de Ciencia, Innovación y Universidades grants (TED2021-1327 57B-I00, PID2022-143013OB-I00).
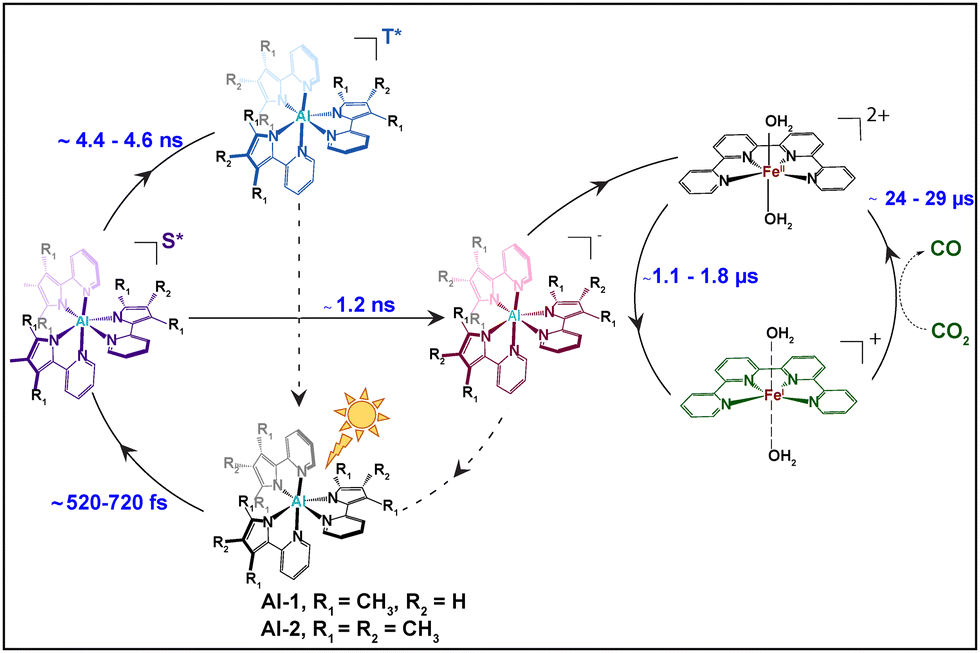 | ||
| Scheme 2 Mechanistic pathways for the photoexcitation reaction of Al photosensitizers Al-1 and Al-2 followed by electron transfer reaction to the Fe-based CO2 reduction catalyst. | ||
Data availability
The data supporting this article have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- F. Droghetti, A. Amati, F. Pascale, A. Crochet, M. Pastore, A. Ruggi and M. Natali, ChemSusChem, 2024, 17, e202300737 CrossRef CAS PubMed.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- H. Ishida, K. Fujiki, T. Ohba, K. Ohkubo, K. Tanaka, T. Terada and T. Tanaka, J. Chem. Soc. Dalton Trans., 1990, 2155–2160 RSC.
- H. Takeda, Y. Monma and O. Ishitani, ACS Catal., 2021, 11, 11973–11984 CrossRef CAS.
- H. Takeda, H. Kamiyama, K. Okamoto, M. Irimajiri, T. Mizutani, K. Koike, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2018, 140, 17241–17254 CrossRef CAS PubMed.
- S.-P. Luo, E. Mejía, A. Friedrich, A. Pazidis, H. Junge, A.-E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419–423 CrossRef CAS PubMed.
- L. Velasco, L. Llanos, P. Levín, A. Vega, J. Yu, X. Zhang, L. Lemus, D. Aravena and D. Moonshiram, Phys. Chem. Chem. Phys., 2021, 23, 3656–3667 RSC.
- J. W. Wang, F. Ma, T. Jin, P. He, Z. M. Luo, S. Kupfer, M. Karnahl, F. Zhao, Z. Xu, T. Jin, T. Lian, Y. L. Huang, L. Jiang, L. Z. Fu, G. Ouyang and X. Y. Yi, J. Am. Chem. Soc., 2023, 145, 676–688 CrossRef CAS PubMed.
- V. Caliskanyürek, A. Riabchunova, S. Kupfer, F. Ma, J.-W. Wang and M. Karnahl, Inorg. Chem., 2024, 63, 15829–15840 CrossRef PubMed.
- J. W. Morgan and E. Anders, Proc. Natl. Acad. Sci., 1980, 77, 6973–6977 CrossRef CAS PubMed.
- T. E. Westre, P. Kennepohl, J. G. DeWitt, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1997, 119, 6297–6314 CrossRef CAS.
- R. Sarangi, Coord. Chem. Rev., 2013, 257, 459–472 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, EXAFS analysis, time-resolved XAS kinetics, and DFT optimization. See DOI: https://doi.org/10.1039/d4cc06404f |
| This journal is © The Royal Society of Chemistry 2025 |