
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo(II)-catalyzed isomerization of enals using hydrogen atom transfer†
Qiqige
Qiqige
a,
Edward
Richmond
*b,
Rocco
Paciello
b,
Mathias
Schelwies
b and
Rylan J.
Lundgren
 *a
*a
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: rylan.lundgren@ualberta.ca
bBASF SE, Synthesis & Homogeneous Catalysis, Carl-Bosch-Strasse 38, 67056 Ludwigshafen am Rhein, Germany. E-mail: edward.richmond@basf.com
First published on 20th March 2025
Abstract
To develop a process for the synthesis of an enal acetate, the so called C5-acetate that is important in the industrial preparation of vitamin A, we show here that Co(II)-based hydrogen atom transfer catalysts under H2 promote such isomerizations in high yield with low catalyst loading (0.1 mol%). D-labelling studies suggest the enal isomerization process and catalyst activation by H2 to be reversible.
The catalytic isomerization of alkenes is a widely studied process.1 Metal-catalyzed isomerization reactions are typically driven by the formation of more thermodynamically stable alkene products. In cases where alkene isomers may have similar stabilities, or the target product is less stable, high yielding processes are not readily achieved. However, recent methods have been developed for the catalytic isomerization of alkenes to less thermodynamically stable products, including terminal-selective,2 strain-inducing,3 enantioselective,4 and Z-selective or E-selective isomerizations.5 Despite advances in catalytic alkene isomerization, some seemingly simple alkene isomerization reactions remain out of reach, including on substrates containing sensitive, reducible functional groups.
We sought a method for the isomerization of conjugated methylenic enals into the corresponding trisubstituted enal. Specifically, we aimed to prepare enal acetate 2 (C5-acetate), a valuable intermediate in the manufacturing of vitamin A6 from the readily available hydroformylation product 1.7 While an array of protocols for the methylenic isomerization of ketones,8 esters,9 amides,10 and styrenes11 have been reported, this type of isomerization with aldehyde substrates was unknown (Fig. 1).12 This is presumably due to challenges associated with conducting metal hydride mediated isomerizations in the presence of a reducible aldehyde unit and the lack of strong thermodynamic driving force to favour the product. Our computational estimates suggest the energy difference between enal 1 and 2 is only ∼1 kJ mol−1 in MeCN (see the ESI,† for details). Given the utility of enals as synthetic intermediates, the discovery of a general, efficient process to isomerize easily synthesized methylenic enals into more highly substituted products would be valuable. Here we show that Co(dmg)2-type catalysts promote such enal isomerizations under H2 to give trisubstituted products in yields >80%, at low catalyst loadings (0.1 mol%), and with minimal alkene or aldehyde reduction.
 | ||
| Fig. 1 Overview of conjugated methylenic isomerizations and challenges of enal isomerization. | ||
Comprehensive reaction screening and mechanistic studies showed the classical isomerization catalyst RhCl3 was a reasonable candidate to promote the desired enal isomerization.13 In buffered ethanol/dioxane the isomerization of enal 1 to target 2 in ∼50% yield was observed (Fig. 2a).14 Further improvements could not be achieved because the enals were found to be in equilibrium and were unstable under the optimized conditions. Reactions under more forcing conditions led to enal hydrogenation, while reactions conducted with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and 2 quickly showed ratios of 1:2 = 33:67 suggesting the reaction under RhCl3 catalysis is not truly at equilibrium. These studies show the challenges associated with developing high yielding isomerizations of sensitive enals like 1 (see the ESI,† for additional screening data). It was not until a hydrogen atom transfer (HAT) approach using Co(dmg)2-type catalysts15,16 was taken that trisubstituted enal 2 could be obtained in good yield (Fig. 2b). Reactions with Co(dmgBF2)2(H2O)2 proceed quickly to ∼90% conversion and 80% yield of 2 under 75 psi H2. Alkene hydrogenation is slow and saturated aldehyde 3 is only observed in >10% well after the isomerization is complete. Reactions could be conducted at 0.1 mol% of Co(II)-catalyst and still gave good yields (83%). The use of other H-atom donors resulted in lower yields (2-propanol: 8% yield; Ph2SiH2:25% yield).
:1 mixture of 1 and 2 quickly showed ratios of 1:2 = 33:67 suggesting the reaction under RhCl3 catalysis is not truly at equilibrium. These studies show the challenges associated with developing high yielding isomerizations of sensitive enals like 1 (see the ESI,† for additional screening data). It was not until a hydrogen atom transfer (HAT) approach using Co(dmg)2-type catalysts15,16 was taken that trisubstituted enal 2 could be obtained in good yield (Fig. 2b). Reactions with Co(dmgBF2)2(H2O)2 proceed quickly to ∼90% conversion and 80% yield of 2 under 75 psi H2. Alkene hydrogenation is slow and saturated aldehyde 3 is only observed in >10% well after the isomerization is complete. Reactions could be conducted at 0.1 mol% of Co(II)-catalyst and still gave good yields (83%). The use of other H-atom donors resulted in lower yields (2-propanol: 8% yield; Ph2SiH2:25% yield).
 | ||
| Fig. 2 Reaction development and mechanistic studies for the isomerization of enal 1. (A) RhCl3 catalyzed reactions and kinetics. (B) Co(dmgBF2)2 catalyzed reactions and kinetics. (C) Labelling studies for the Co-catalyzed enal isomerization using D2. (D) Comparison to related Co-HAT catalysts. aTHF as solvent, 50 °C, 5 h. | ||
Isotope-labeling studies conducted with D2 are consistent with a reaction that is reversible to some extent (Fig. 2c). Under the standard conditions at ∼75 psi, the use of D2 results in the formation of D-2 in 72% yield with one equivalent of D at the newly formed methyl position and 7% D at the alkenyl position. Recovered substrate (7%) is D-labeled to approximately the same extent on the same carbon atoms. The amount of labeling in both 1 and 2 increases even after isomerization to 2 reaches the maximum yield. When the enal product 2 is subjected to the standard isomerization conditions under D2, 9% of 1 is formed with partial D-incorporation. Recovered 2 is labelled at both the methyl and alkenyl positions. The extent of labeling suggests a rate of isomerization that is bracketed by the rate of Co-DAT catalyst formation by reaction with D2 and “Co–D” (the exact structure of the reactive hydride species generated from Co(dmg) catalysts is not resolved17) is generated reversibly. If the active Co-DAT catalyst was generated only in an initial reaction with D2, product and starting material would be labelled in ∼2.5% (i.e. the catalyst loading), while if the “Co–D” was in rapid equilibrium with Co ½ D2, the extent of labelling in product and substrate would converge and both would become completely labelled at the alkenyl and allylic positions. Thus, the lower extent of deuteration of the internal double bond reflects its less preferential reaction with “Co–D”.
Use of the appropriate HAT catalyst was essential to observe productive reactivity. Co(II)-based (dmgBF2) and (dmgH2) complexes could be used as catalysts for enal isomerization, although Co(dmgBF2)2(H2O)2 performed better at reduced catalyst loadings (see the ESI,† for details). Other Co-based complexes established for HAT reactivity failed completely, including related Co(III)dmg type catalysts,2a Cp*Co(II)(bipy)Cl, and Co(III)(salen)Cl complexes,9b further highlighting the unique challenge of enal isomerization (Fig. 2d).
Fig. 3 provides a mechanistic proposal where Co–H mediated HAT generates a tertiary radical from enal 1. This intermediate can undergo H-atom abstraction by Co to generate the isomerized product. Given an estimate of between 0–2 kJ mol−1 (i.e. 1 ± 1 kJ mol−1) difference in energy between the enals, the isomer distribution may be fully under thermodynamic control, however given the higher degree of D-labelling observed at the methyl/exo-methylenic carbon, we suspect the catalyst exhibits some kinetic selectivity for addition to the more sterically accessible alkene in 1vs. that in 2. The poor reactivity of non-methylenic aldehydes supports this assumption.18
 | ||
| Fig. 3 Mechanistic proposal. | ||
We were interested in studying the scope of enal isomerization reactions catalyzed by Co(dmgBF2)2(H2O)2/H2 with other methylenic substrates (Fig. 4). Norton and co-workers have previously reported Co(dmgBF2)2/H2 catalyzed isomerization reactions with arylalkenes and α,β-unsaturated esters.15 Enal isomerization of ester and ether containing substrates were successful (OAc, OBn, and OMe; 1, 4, 5), as was a substrate with a free alcohol group (6). Substrates containing potentially sensitive functional groups like enolizable ketones (7), alkyl chlorides (8), terminal (9) or trisubstituted alkenes (10), or aryl groups (11) were isomerized to the target products in ∼90% yield. Di- and trisubstituted ketones (12, 13) and lactone 14 underwent isomerization in good to moderate yield. Reactions with functionalized arylalkenes proceeded with moderate yields (16–18), as did itaconic acid derivative 19. Notably, the majority of enal or arylalkene substrates gave poor conversions and yields when using RhCl3-based conditions, suggesting the general benefit of an HAT approach in comparison to a metal-hydride based reactivity.
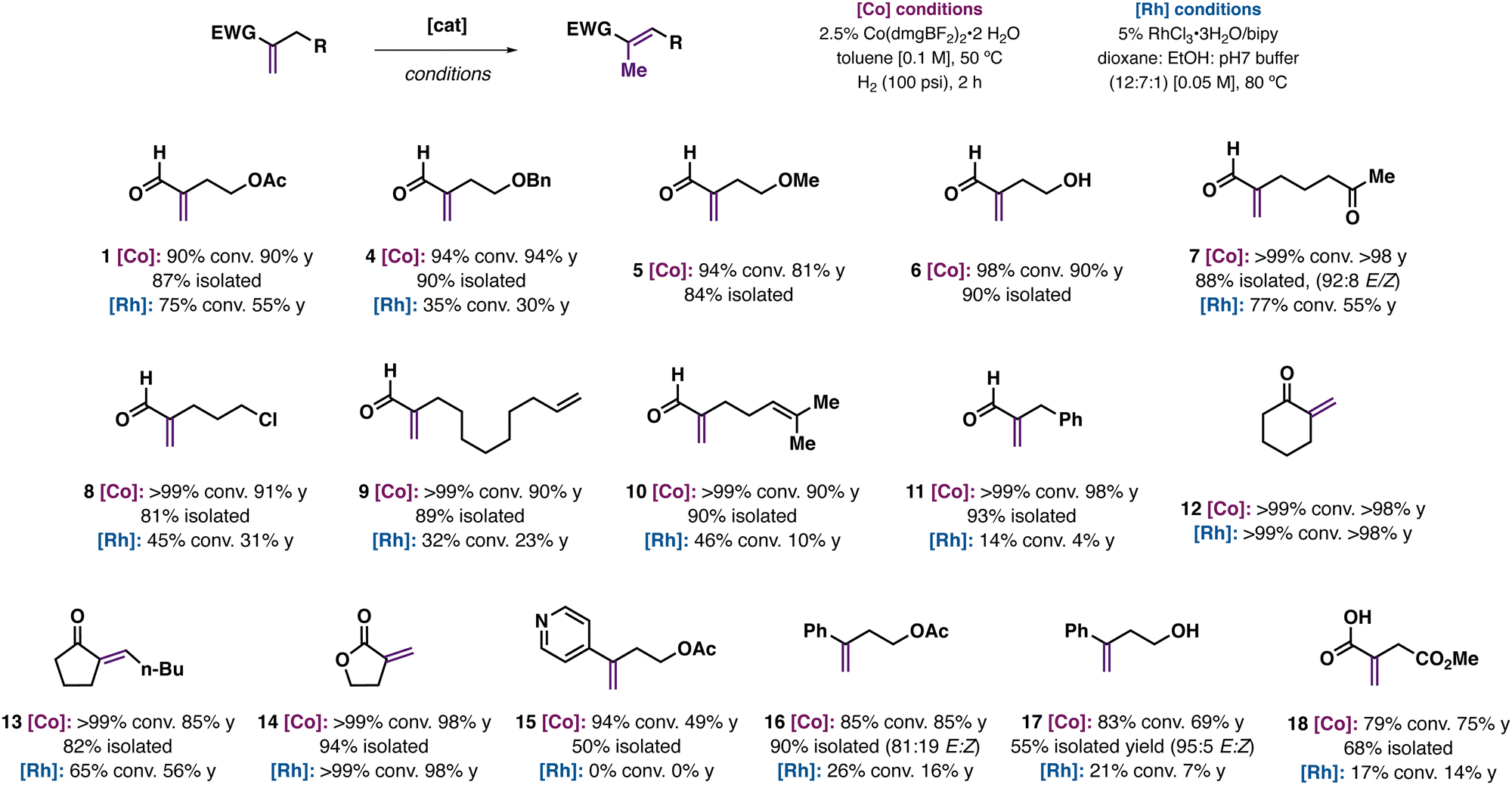 | ||
| Fig. 4 Reaction scope and limitations of the Co(dmgBF2)2-catalyzed isomerization of enals and other alkenes along with select comparisons to conditions using RhCl3. | ||
In summary, we have found using Co(dmgBF2)2-based HAT catalyst systems to be a solution for the high yielding isomerization of methylenic enals and other recalcitrant alkene substrates. This approach allows for an attractive new route to the industrially important C5-acetate in vitamin A synthesis. Our work shows that HAT mechanisms can circumvent problems of substrate reduction and unfavourable equilibrium between isomers while operating under mild reaction conditions.
Support was provided by NSERC Canada (ALLRP 571720-2021) and Canada Foundation for Innovation (IOF 32691). We thank Maximilian Menche (BASF SE) for computational calculations and Lola Zhao (Alberta) for late stage experimental support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
BASF and the University of Alberta have filed a patent application on the reported technology.Notes and references
- (a) J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654 CrossRef CAS PubMed; (b) D. Fiorito, S. Scaringi and C. Mazet, Chem. Soc. Rev., 2021, 50, 1391 RSC; (c) E. Larionov, H. Li and C. Mazet, Chem. Commun., 2014, 50, 9816 RSC; (d) X. Liu, B. Li and Q. Liu, Synthesis, 2019, 1293 Search PubMed; (e) S. Zhang and M. Findlater, Synthesis, 2021, 2787 CAS.
- (a) G. Occhialini, V. Palani and A. E. Wendlandt, J. Am. Chem. Soc., 2022, 144, 145 CAS; (b) K. Zhao and R. R. Knowles, J. Am. Chem. Soc., 2022, 144, 137 CrossRef CAS PubMed.
- V. Palani and A. E. Wendlandt, J. Am. Chem. Soc., 2023, 145, 20053 CAS.
- (a) X. Liu, X. Rong, S. Liu, Y. Lan and Q. Liu, J. Am. Chem. Soc., 2021, 143, 20633 CrossRef CAS PubMed; (b) Y. Liu, L. Zhang, Y. Zhang, S. Cao, X. Ban, Y. Yin, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2023, 145, 18307 CAS; (c) X. Rong, J. Yang, S. Liu, Y. Lan and Q. Liu, CCS Chem., 2023, 5, 1293 CAS.
- (a) T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650 Search PubMed; (b) B. Kweon, L. Blank, J. Soika, A. Messara, C. G. Daniliuc and R. Gilmour, Angew. Chem., Int. Ed., 2024, 63, e20240423 Search PubMed; (c) M. Wienhold, B. Kweon, C. McLaughlin, M. Schmitz, T. J. B. Zähringer, C. G. Daniliuc, C. Kerzig and R. Gilmour, Angew. Chem., Int. Ed., 2023, 62, e202304150 CrossRef CAS PubMed; (d) C. Z. Rubel, A. K. Ravn, H. C. Ho, S. Yang, Z.-Q. Li, K. M. Engle and J. C. Vantourout, Angew. Chem., Int. Ed., 2024, 63, e202320081 CrossRef CAS PubMed; (e) A. Schmidt, A. R. Nödling and G. Hilt, Angew. Chem., Int. Ed., 2015, 54, 801 CrossRef CAS PubMed.
- (a) M. Eggersdorfer, D. Laudert, U. Létinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960 CrossRef CAS PubMed; (b) W. Bonrath, B. Gao, P. Houston, T. McClymont, M.-A. Müller, C. Schäfer, C. Schweiggert, J. Schütz and J. A. Medlock, Org. Proc. Res. Dev., 2023, 27, 1557 CrossRef CAS.
- For example see: S. M. A. De Wildeman, J. G. De Vries and J. A. F. Boogers, WO2012116977a1, DSM Ip Assets B.V., 2012.
- B. W. Disanayaka and A. C. Weedon, Synthesis, 1983, 952 CrossRef CAS.
- (a) G.-Q. Lin and W.-C. Xu, Bioorg. Med. Chem., 1996, 4, 375 CrossRef CAS PubMed; (b) S. W. M. Crossley, F. Barabé and R. A. Shenvi, J. Am. Chem. Soc., 2014, 136, 16788 CrossRef CAS PubMed.
- S. Tekkam, M. A. Alam, S. C. Jonnalagadda and V. R. Mereddy, Chem. Commun., 2011, 47, 3219 RSC.
- (a) S. Zhang, D. Bedi, L. Cheng, D. K. Unruh, G. Li and M. Findlater, J. Am. Chem. Soc., 2020, 142, 8910 CrossRef CAS PubMed; (b) R.-Z. Huang, K. K. Lau, Z. Li, T.-L. Liu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 14647 CAS; (c) L. Chang, C. Cai, R. Chen, J. Chen, Y. Luo and Y. Xia, Org. Chem. Front., 2023, 10, 4643 CAS.
- Reports exist in the patent literature; however no generally applicable homogenous catalyst system exists for the reliable isomerization of methylene enals for example see: (a) T. Li, et al., CN113233979A, Shandong Nhu, 2022; (b) Q. Lyu, Y. Qi, L. Zhou, X. Jiang and M. Zhang, CN110734376A, Xinfa Pharmaceuticals, 2020.
- R. Cramer, Acc. Chem. Res., 1968, 1, 186 CAS.
- X. Bi, W. Xu, Y. Yao, L. Zhou and G. Liang, J. Org. Chem., 2018, 83, 5825 CAS.
- G. Li, J. L. Kuo, A. Han, J. M. Abuyuan, L. C. Young, J. R. Norton and J. H. Palmer, J. Am. Chem. Soc., 2016, 138, 7698 CAS.
- For mechanistic studies of the reaction between Co(dmg)-type catalysts and H2 see: (a) D. P. Estes, D. C. Grills and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 17362 CAS; (b) G. Li, A. Han, M. E. Pulling, D. P. Estes and J. R. Norton, J. Am. Chem. Soc., 2012, 134, 14662 CAS For solvent effects in metal catalyzed HAT see: ; (c) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401 CAS.
- To the best of knowledge and despite efforts by others the (dmg)Co–H species has not been isolated, past reports of (dmg)Co–H species have been proved incorrect: D. C. Lacy, G. M. Roberts and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 4860 CAS.
- Other mechanistic possibilities not ruled out include SH2-type additions: (a) R. L. Sweany and J. Halpern, J. Am. Chem. Soc., 1977, 99, 8335 CAS ; SH2′-type additions: ref. 2a; or C–H oxidative addition processes: ; (b) L.-C. Wang, H.-Y. Jang, Y. Roh, V. Lynch, A. J. Schultz, X. Wang and M. J. Krische, J. Am. Chem. Soc., 2002, 124, 9448 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and experimental data. See DOI: https://doi.org/10.1039/d4cc06075j |
| This journal is © The Royal Society of Chemistry 2025 |