
Polythiophene side chain chemistry and its impact on advanced composite anodes for lithium-ion batteries†
Han
Li‡
 a,
Haoze
Ren‡
a,
Zeyuan
Sun
a,
Siyu
Qin
a,
Armando Rodriguez
Campos
bc,
Esther S.
Takeuchi
bcde,
Amy C.
Marschilok
bcde,
Kenneth J.
Takeuchi
bcde and
Elsa
Reichmanis
*a
a,
Haoze
Ren‡
a,
Zeyuan
Sun
a,
Siyu
Qin
a,
Armando Rodriguez
Campos
bc,
Esther S.
Takeuchi
bcde,
Amy C.
Marschilok
bcde,
Kenneth J.
Takeuchi
bcde and
Elsa
Reichmanis
*a
aDepartment of Chemical and Bimolecular Engineering, Lehigh University, Bethlehem, PA 18015, USA. E-mail: elr420@lehigh.edu
bInstitute of Energy: Sustainability, Environment and Equity, Stony Brook University, Stony Brook, NY 11794, USA
cDepartment of Chemistry, Stony Brook University, Stony Brook, NY 11794, USA
dDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, NY 11794, USA
eInterdisciplinary Science Department, Brookhaven National Laboratory, Upton, NY 11973, USA
First published on 28th November 2024
Abstract
In the development of high-performance lithium-ion batteries (LIBs), the design of polymer binders, particularly through manipulation of side-chain chemistry, plays a pivotal role in optimizing electrode stability, ion transport, and adaptability to the volume changes during cycling. In particular, poly[3-(potassium-4-butanoate)thiophene-2,5-diyl] (P3KBT) increases magnetite and silicon capacity and cycling stability. This work explores the impact of polythiophene alkyl sidechain length on anode characteristics, aiming to enhance performance in LIBs. P3KBT and its alkyl chain alternatives, poly[3-(potassium-5-pentanoate)thiophene-2,5-diyl] (P3KPT) and poly[3-(potassium-6-hexanoate)thiophene-2,5-diyl] (P3KHT) were systematically investigated over 300 charge–discharge cycles. The experiments were designed to assess how varying side-chain length affects the stability, ion transport, and capacity retention of the electrodes. The results revealed that P3KHT, with its longer alkyl chain, exhibited superior capacity retention and reduced charge-transfer resistance after 300 cycles compared to its shorter chain analogs. The findings demonstrate that tailored side chains can improve ion transport, structural integrity, and capacity retention, addressing critical challenges in LIBs such as capacity fade and electrode degradation. This research contributes to the development of next-generation LIBs with enhanced performance and reliability.
The rapid advancement of lithium-ion batteries (LIBs) is critical to meet the increasing demand for high-performance energy storage systems.1 Introducing high theoretical capacity anode active materials, such as silicon and magnetite (Fe3O4), is seen as an efficient, eco-friendly way to enhance battery energy density.2,3 However, practical implementation of these materials faces significant challenges due to issues such as low electrical conductivity, dramatic volume changes during cycling, and uncontrolled formation of the solid electrolyte interphase (SEI).4 For instance, Fe3O4, a representative high-capacity metal oxide insertion-conversion active material, undergoes complex reactions with greater volume changes upon (dis)charge, unlike the lower structural disruption in graphite due to its layered structure for ion (de)intercalation.5
Electrode issues often stem from interactions between material interfaces that are in turn, closely tied to the properties of the carbon additive6–8 and the binder component.9 Ideally, the binder should serve as a conduit for both ions and electrons, help maintain electrode integrity despite volume changes during (de)lithiation, and suppress undesirable side reactions.10 However, commercialized binders like carboxymethyl cellulose (CMC) and polyvinylidene fluoride (PVDF) fall short.11 Their insulating characteristics require the incorporation of large amounts of conductive carbon additives, which block ion transfer and increase electrode tortuosity.12 Weak physicochemical interactions between all of the materials used to fabricate the electrode lead to structural damage and capacity degradation, particularly in systems where the active material undergoes significant, repeated volume contraction/expansion.13
Kwon et al. proposed a thiophene-based polymer, namely poly[3-(potassium-4-butanoate)thiophene-2,5-diyl] (P3KBT, previously referred to as PPBT) as a binder for LIB anodes.14 While P3KBT does not impact specific capacity,14 upon electrochemical doping, P3KBT was shown to exhibit significantly enhanced electronic conductivity vs. PVDF.4 Further, the water-soluble polymer forms stable covalent bonds with high-capacity anode materials, enhancing electrode materials interactions and positively impacting capacity retention. Kwon et al. also speculated that the presence of P3KBT carboxylate moieties also facilitates ionic conductivity,14 while Das, et al. demonstrated advantages associated with the incorporation of a poly(3,4-propylenedioxythiophene)-based mixed ion-electron conductor into composite electrodes.15
Recent breakthroughs exploring the mixed conduction behavior of carboxyl-alkyl functionalized polythiophenes in aqueous electrolytes as a function of alkyl spacer length revealed that varying spacer length significantly impacts electrochemical properties and structural stability.16 Here, we build on this discovery to explore the use of a series of carboxyl-alkyl polythiophenes, namely P3KBT, poly[3-(potassium-5-pentanoate)thiophene-2,5-diyl] (P3KPT) and poly[3-(potassium-6-hexanoate)thiophene-2,5-diyl] (P3KHT), as the binder component in Fe3O4 composite anodes (Fig. 1a). Electrodes formulated with PVDF served as a control. Details associated with materials and electrode fabrication and characterization can be found in the experimental section of the ESI.† By tailoring side-chain chemistry, we aim to develop composite anodes with superior ion transport characteristics, structural integrity, and capacity retention, addressing critical challenges in LIB technology and contributing to next-generation high-performance, reliable LIBs.17
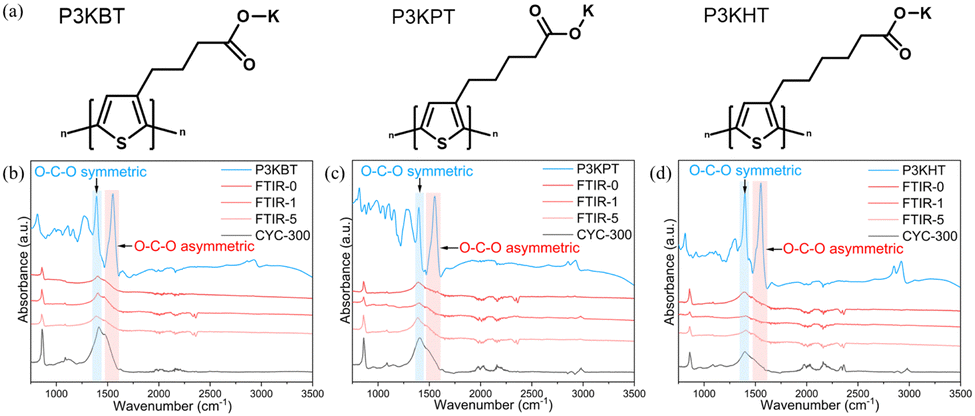 | ||
| Fig. 1 (a) Chemical structures of P3KBT, P3KPT, and P3KHT. FT-IR spectra of Fe3O4 composite electrodes after 0, 1, 5, and 300 cycles of (b) P3KBT, (c) P3KPT, and (d) P3KHT binder. | ||
To investigate the molecular interactions between the active material and polymeric binders, FT-IR spectroscopy provided valuable insight into the chemical interactions that occur between the various electrode components. The respective FT-IR analyses of electrodes comprising P3KBT, P3KPT, and P3KHT as the binder after 0, 1, 5, and 300 cycles are provided in Fig. 1b–d. Cycling was conducted at a rate of 0.3C in a voltage range of 0.01–3 V vs. Li/Li+. The vibrational bands at 1550 cm−1 and 1400 cm−1 correspond to the carboxylate O–C–O asymmetric and symmetric stretching modes, respectively,18 and the peak around 860 cm−1 corresponds to the C–H out-of-plane bending deformation of the thiophene ring.19 These bands appeared consistently across all three derivatives and their Fe3O4 composites from 0 cycles to 300 cycles, supporting the successful and stable interaction of the polymers with Fe3O4. The standard carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching peak typically observed around 1720 cm−1 (Fig. S1, ESI†) was absent. In contrast, within the composite, the polythiophene carboxylate O–C–O asymmetric and symmetric stretching vibrations merged into a single peak around 1400 cm−1 with a shoulder at approximately 1480 cm−1, suggesting the formation of a chemical bond between Fe3O4 and the polymers (structure highlighted in blue in Fig. S1, ESI†). The observed interactions between the binder and high-capacity active materials are consistent with previously reported factors influencing electrode stability.20
O stretching peak typically observed around 1720 cm−1 (Fig. S1, ESI†) was absent. In contrast, within the composite, the polythiophene carboxylate O–C–O asymmetric and symmetric stretching vibrations merged into a single peak around 1400 cm−1 with a shoulder at approximately 1480 cm−1, suggesting the formation of a chemical bond between Fe3O4 and the polymers (structure highlighted in blue in Fig. S1, ESI†). The observed interactions between the binder and high-capacity active materials are consistent with previously reported factors influencing electrode stability.20
To further investigate the impact of molecular interactions between the active material and polymeric binders, field-emission scanning electron microscopy (FE-SEM) before and after cycling was utilized to probe electrode morphological features. Examination of Fig. 2 and Fig. S2 (ESI†) demonstrates that before cycling, the Fe3O4-P3KHT electrode surface is smoother with limited cracks, suggesting that Fe3O4-P3KHT may form a more stable structure. Significantly, after 300 cycles, the P3KHT electrode surface appears crack-free. In contrast, after cycling the Fe3O4-P3KBT system exhibits a thick SEI layer with a significant number of cracks, while the P3KPT analog's appearance is intermediate. The results suggest that P3KHT provides for a more stable structural configuration among the three alternatives, most likely facilitated by the binding of the carboxylated polythiophene to the active material surface (vide supra).
The impact of the carboxylated polythiophene side-chain length on composite electrode electrochemical performance was examined using a half-cell configuration. The anodes consisted of Fe3O4 (10 nm particles) active material,21,22 Super-P carbon additives, and polymer binder in a weight ratio of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:15. Both the capacity and rate capability of the three polythiophene-based electrodes exceeded that of the PVDF control. Although the rate capability performance (Fig. 3a) shows no significant differences between the three polythiophene analogs, the cycling performance (Fig. 3b and Fig. S3 and S4, ESI†) is distinctly different. The Fe3O4-P3KPT anode exhibited a higher capacity in the first 30 cycles, which mirrored the rate capability results. Notably, when P3KHT was used as the binder, the composite showed high capacity retention over 300 cycles, vs. Fe3O4-P3KBT, Fe3O4-P3KPT, and Fe3O4-PVDF. Further, the large cracks evident in the P3KBT-based electrode after cycling (Fig. 2d), suggested the significant potential for loss of electrical contact with the current collector and helped rationalize its rapid degradation when cycling continued past 30 cycles. Added insight into the impact of binder structure on performance was gained through electrochemical impedance spectroscopy (EIS) measurements conducted on cells before and after cycling, within a frequency range of 0.1 MHz to 0.1 Hz. The diameter of the high-frequency semicircle represents the charge-transfer resistance (Rct) of the electrode, while the intercept on the real axis can be assigned to the ohmic resistance (Rs). Prior to cycling, Fe3O4-P3KBT exhibited the lowest Rct, while Fe3O4-P3KHT presented the highest Rct value (Fig. 3c and Table S1, ESI†). After cycling however, the trend was reversed (Fig. 3d and Table S2, ESI†). Notably, Rct for the Fe3O4-P3KHT electrodes decreased significantly with respect to the other polythiophene analogs, especially Fe3O4-P3KBT, and even more so when compared to the PVDF control. From the electrochemical studies, Fe3O4-P3KHT far outperforms the butanoate and pentanoate alternatives. Conceivably, the longer alkyl side chain facilitates more stable bonding to the active material with resultant structural integrity due to the longer side chain. This in turn limits the negative impacts of stress and volume changes induced by cycling, thereby significantly enhancing the electrode durability and performance.
:15:15. Both the capacity and rate capability of the three polythiophene-based electrodes exceeded that of the PVDF control. Although the rate capability performance (Fig. 3a) shows no significant differences between the three polythiophene analogs, the cycling performance (Fig. 3b and Fig. S3 and S4, ESI†) is distinctly different. The Fe3O4-P3KPT anode exhibited a higher capacity in the first 30 cycles, which mirrored the rate capability results. Notably, when P3KHT was used as the binder, the composite showed high capacity retention over 300 cycles, vs. Fe3O4-P3KBT, Fe3O4-P3KPT, and Fe3O4-PVDF. Further, the large cracks evident in the P3KBT-based electrode after cycling (Fig. 2d), suggested the significant potential for loss of electrical contact with the current collector and helped rationalize its rapid degradation when cycling continued past 30 cycles. Added insight into the impact of binder structure on performance was gained through electrochemical impedance spectroscopy (EIS) measurements conducted on cells before and after cycling, within a frequency range of 0.1 MHz to 0.1 Hz. The diameter of the high-frequency semicircle represents the charge-transfer resistance (Rct) of the electrode, while the intercept on the real axis can be assigned to the ohmic resistance (Rs). Prior to cycling, Fe3O4-P3KBT exhibited the lowest Rct, while Fe3O4-P3KHT presented the highest Rct value (Fig. 3c and Table S1, ESI†). After cycling however, the trend was reversed (Fig. 3d and Table S2, ESI†). Notably, Rct for the Fe3O4-P3KHT electrodes decreased significantly with respect to the other polythiophene analogs, especially Fe3O4-P3KBT, and even more so when compared to the PVDF control. From the electrochemical studies, Fe3O4-P3KHT far outperforms the butanoate and pentanoate alternatives. Conceivably, the longer alkyl side chain facilitates more stable bonding to the active material with resultant structural integrity due to the longer side chain. This in turn limits the negative impacts of stress and volume changes induced by cycling, thereby significantly enhancing the electrode durability and performance.
 | ||
| Fig. 2 Top view FE-SEM images of (a) Fe3O4-P3KBT, (b) Fe3O4-P3KPT, (c) Fe3O4-P3KHT electrodes before cycling; and (d) Fe3O4-P3KBT, (e) Fe3O4-P3KBT, and (f) Fe3O4-P3KHT after cycling. | ||
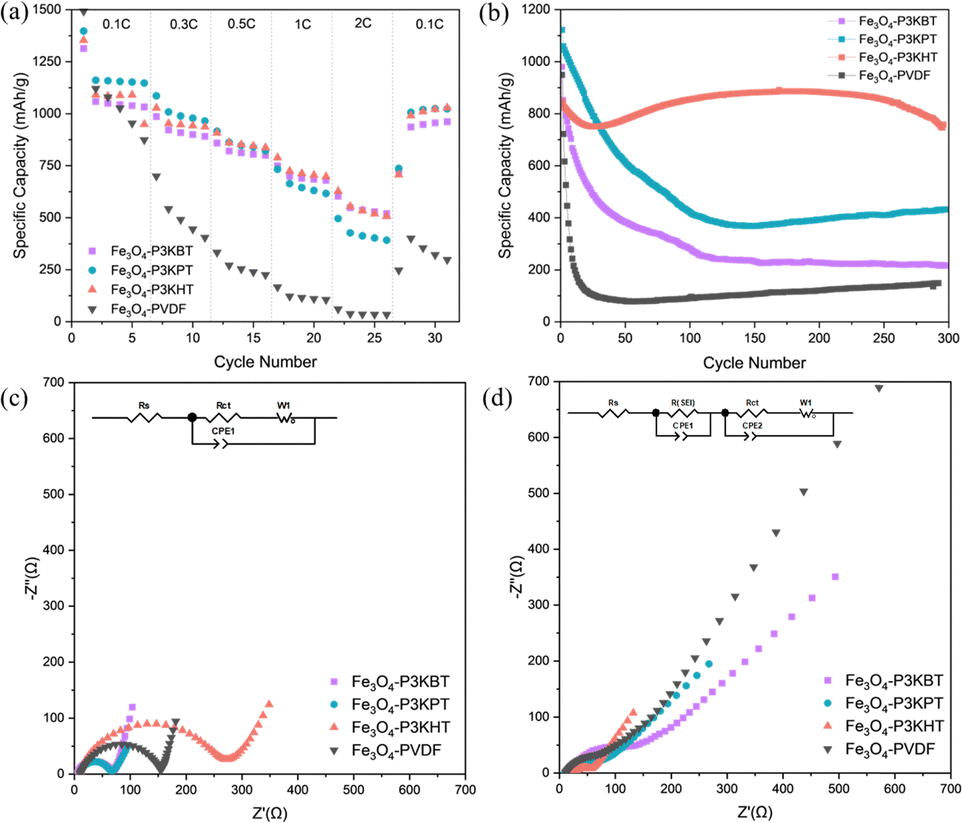 | ||
| Fig. 3 (a) Specific capacity of Fe3O4 electrodes with the different binders. (b) Cycling performance of corresponding electrodes at 0.3C between 0.01 and 3 V. EIS Nyquist curve and corresponding equivalent circuit for (c) before cycling and (d) after 300 cycles in the frequency range from 0.1 MHz to 0.1 Hz. | ||
Cyclic voltammetry (CV) was performed in the potential window of 0.01 to 3 V versus Li/Li+ to characterize the basic electrochemical behavior of electrodes. After 10 cycles (Fig. S5, ESI†), all three electrodes exhibited the same trend: note that the first cycle showed specific Fe3O4 anodic and cathodic peaks. Subsequently, different CV scan rates were applied to both polymer thin films and composite electrodes to provide insight into electrode kinetics and study the relationship between the electrochemical process and resultant electrode performance. The electrodes fabricated with the respective thin polymer films were prepared by spray coating the aqueous polymer solution onto a copper foil substrate. Fig. 4 provides the plot of log (Ipc) vs. log(ν), obtained from the results of anodic peak currents of the CV curves (Fig. S6 and S7, ESI†) acquired with scan rates ranging from 0.1 mV s−1 to 2 mV s−1. The result provided insight into electrode kinetics and helped to further elucidate differences in the performance of the three binders.23 The kinetic data was characterized by analyzing the voltammetric response of the polymeric binders at various scan rates according to Ipc = aνb, where I is the peak current, v is the scan rate, and b is the desired kinetic parameter. When the b value is ∼ 0.5, the electrochemical process is predominantly ion diffusion controlled, while a b value of ∼1 points to an electrochemical process that is primarily charge transfer controlled. When the electrochemical process is controlled by charge transfer, it implies that ion diffusion is less restricted, suggesting improved ion transport behaviour. In the present case, a higher b value indicates faster lithium diffusion kinetics.24 After analyzing the data from Tables S3 and S4 (ESI†), the b values of the pristine polymer films were found to be in the order of P3KBT (0.899) > P3KPT (0.862) > P3KHT (0.833), while the corresponding b value of the respective composite electrodes was calculated as Fe3O4-P3KBT (0.430) < Fe3O4-P3KPT (0.694) < Fe3O4-P3KHT (0.738). The higher b value obtained for P3KBT could be attributed to its shorter side chain, which likely enhances its ability to ionize, thereby increasing its solubility and limiting the formation of semicrystalline aggregates in the pure polymer film. These factors in turn, may reduce the barriers to ion transport. The higher value of b obtained for Fe3O4-P3KHT suggests that incorporation of P3KHT into advanced high-capacity composite anodes may be advantageous for enhancing electrochemical performance vs. its shorter side chain alternatives.
 | ||
| Fig. 4 Plot of log (Ipc) vs. log (ν), from the results of anodic peak currents of CV curves with different scan rates of (a) P3KBT, P3KPT, P3KHT, and (b) corresponding Fe3O4 electrodes. | ||
Grazing incidence wide-angle X-ray scattering (GIWAXS) results provide additional insight. As reported by Patel et al.,25 the P3KHT lamellar stacking distance is significantly increased compared to the shorter side chain analogs, P3KPT and P3KBT. Further, Shih and Chueh et al.26 found that larger lamellar stacking distance increased interchain spacing volume to better accommodate applied strain due to better dissipation of mechanical forces in the amorphous regions. Compared to Shih and Chueh's results, those obtained by Patel et al. present the same trends and match the electrochemistry results (vide supra), where P3KHT, with its longer side chains and larger lamellar stacking distance, exhibited enhanced electrode performance, perhaps due to its ability to dissipate mechanical forces associated with expansion/contraction of the active material during lithiation/delithiation.27 Additionally, the larger lamellar stacking distance exhibited by P3KHT may contribute to the increased volume associated with the hydrophilic amorphous component of the binder, which may create ion transport pathways.
When incorporated into high-capacity composite Fe3O4-based anodes, we demonstrated that the alkyl side chain length of carboxyl-alkyl polythiophene binders significantly impacts electrochemical performance. With its longer alkyl chain that provides larger lamellar stacking and better performance in dissipating mechanical forces, P3KHT provides superior performance in terms of ion transport, electrode structural integrity, and capacity retention over 300 cycles, outperforming its shorter side chain analogs and underscoring its potential to address the challenges of capacity fade and electrode degradation in next-generation LIBs. Our results highlight the crucial role of side-chain engineering in polymer binder design and offer a pathway to enhance high-capacity LIB performance and lifetime. This research contributes to the development of more reliable and longer-lasting LIBs, which are essential for the future of energy storage technologies.
This work was performed as part of the Center for Mesoscale Transport Properties, an Energy Frontier Research Center supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award #DE-SC0012673. E. R. also appreciates support from Lehigh University through funds associated with the Carl Robert Anderson Chair in Chemical Engineering. E. S. T. acknowledges support as the William and Jane Knapp Chair for Energy and the Environment at Stony Brook University.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts to declare.Notes and references
- D. Gueon, M. A. Gonzalez, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Reichmanis, Acc. Mater. Res., 2023, 4, 156–167 CrossRef CAS.
- H. Ren, H. Li, P. Barry, Z. Wang, A. R. Campos, E. S. Takeuchi, A. C. Marschilok, S. Yan, K. J. Takeuchi and E. Reichmanis, Chem. Mater., 2024, 36, 9299–9319 CrossRef CAS.
- H. Li and T. J. Webster, Nanomedicine, Woodhead Publishing, 2023, pp. 1–18 DOI:10.1016/b978-0-12-818627-5.00020-8.
- Y. H. Kwon, M. M. Huie, D. Choi, M. Chang, A. C. Marschilok, K. J. Takeuchi, E. S. Takeuchi and E. Reichmanis, ACS Appl. Mater. Interfaces, 2016, 8, 3452–3463 CrossRef CAS PubMed.
- Z. Xu, X. Shi, X. Zhuang, Z. Wang, S. Sun, K. Li and T. Y. Zhang, Research, 2021, 9842391 CAS.
- R. Tian, N. Alcala, S. J. K. O’Neill, D. V. Horvath, J. Coelho, A. J. Griffin, Y. Zhang, V. Nicolosi, C. O’Dwyer and J. N. Coleman, ACS Appl. Energy Mater., 2020, 3, 2966–2974 CrossRef CAS.
- J. Entwistle, R. Ge, K. Pardikar, R. Smith and D. Cumming, Renewable Sustainable Energy Rev., 2022, 166, 112624 CrossRef CAS.
- D. Park, P. C. Sherrell, F. Xie and A. V. Ellis, J. Mater. Chem. A, 2024, 12, 4884–4892 RSC.
- K. Minnici, Y. H. Kwon, L. M. Housel, G. D. Renderos, J. F. Ponder, C. Buckley, J. R. Reynolds, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Reichmanis, ACS Appl. Energy Mater., 2019, 2, 7584–7593 CrossRef CAS.
- M. A. Gonzalez, W. H. Freer, M. Wang, S. Jeon, T. Fuller, E. S. Takeuchi, K. J. Takeuchi, A. Marschilok and E. Reichmanis, J. Phys. Chem. C, 2022, 126, 19603–19617 CrossRef CAS.
- R. Na, K. Minnici, G. Zhang, N. Lu, M. A. Gonzalez, G. Wang and E. Reichmanis, ACS Appl. Mater. Interfaces, 2019, 11, 40034–40042 CrossRef CAS.
- H. Ren, Y. Wang, D. Cao, W. Gedney, T. Ji, X. Sun and H. Zhu, Energy Environ. Mater., 2023, 6, e12584 CrossRef CAS.
- M. M. Huie, D. C. Bock, A. M. Bruck, K. R. Tallman, L. M. Housel, L. Wang, J. Thieme, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, ACS Appl. Mater. Interfaces, 2019, 11, 7074–7086 CrossRef CAS.
- Y. H. Kwon, K. Minnici, M. M. Huie, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Reichmanis, Chem. Mater., 2016, 28, 6689–6697 CrossRef CAS.
- P. Das, B. Zayat, Q. Wei, C. Z. Salamat, I.-B. Magdău, R. Elizalde-Segovia, D. Rawlings, D. Lee, G. Pace, A. Irshad, L. Ye, A. Schmitt, R. A. Segalman, T. F. Miller, S. H. Tolbert, B. S. Dunn, S. R. Narayan and B. C. Thompson, Chem. Mater., 2020, 32, 9176–9189 CrossRef CAS.
- Z. Sun, B. Khau, H. Dong, C. J. Takacs, S. Yuan, M. Sun, B. Mosevitzky Lis, D. Nguyen and E. Reichmanis, Chem. Mater., 2023, 35, 9299–9312 CrossRef CAS.
- G. Pace, O. Nordness, K. Asham, R. J. Clément and R. A. Segalman, Chem. Mater., 2022, 34, 4672–4681 CrossRef CAS.
- Y. H. Kwon, J. J. Park, L. M. Housel, K. Minnici, G. Zhang, S. R. Lee, S. W. Lee, Z. Chen, S. Noda, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and E. Reichmanis, ACS Nano, 2018, 12, 3126–3139 CrossRef CAS.
- T. Zhang, Y. Yuan, X. Cui, H. Yin, J. Gu, H. Huang and J. Shu, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 751–761 CrossRef CAS.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- S. Zhu, A. C. Marschilok, E. S. Takeuchi and K. J. Takeuchi, Electrochem. Solid-State Lett., 2009, 12, A91 CrossRef CAS.
- S. Zhu, A. C. Marschilok, E. S. Takeuchi, G. T. Yee, G. Wang and K. J. Takeuchi, J. Electrochem. Soc., 2010, 157, A1158 CrossRef CAS.
- K. Minnici, Y. H. Kwon, J. O'Neil, L. Wang, M. R. Dunkin, M. A. Gonzalez, M. M. Huie, M. V. de Simon, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Reichmanis, ACS Appl. Mater. Interfaces, 2019, 11, 44046–44057 CrossRef CAS PubMed.
- M. Gonzalez, K. Minnici, B. Risteen, L. Wang, L. M. Housel, G. D. Renderos, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok, T. F. Fuller and E. Reichmanis, ACS Appl. Energy Mater., 2021, 4, 9836–9847 CrossRef CAS.
- G. L. Grocke, B. X. Dong, A. D. Taggart, A. B. F. Martinson, J. Niklas, O. G. Poluektov, J. W. Strzalka and S. N. Patel, ACS Polym Au, 2022, 2, 275–286 CrossRef CAS.
- Y.-H. Shih, G.-L. Chen, P.-H. Liu, K.-W. Tseng, W.-Y. Lee, W.-C. Chen, L. Wang and C.-C. Chueh, ACS Appl. Electron. Mater., 2024, 6, 1797–1808 CrossRef CAS.
- Y. Zhang, Y. Shi, L. Weng, C. Xu, C. Gao, B. Chen, J. Zhou and R. Cai, Int. J. Solids Struct., 2021, 233, 111179 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06117a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |