
Crystalline potassium boryl dithiolate and diselenolate†
Yuyang
Dai
b,
Yu
Wang
b,
Liliang
Wang
 *a,
Fan
Qi
*a,
Qianli
Li
c and
Lingbing
Kong
*b
*a,
Fan
Qi
*a,
Qianli
Li
c and
Lingbing
Kong
*b
aCollege of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, Zhejiang, China. E-mail: lwang@hznu.edu.cn; eleven@hznu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China. E-mail: konglb@sdu.edu.cn
cSchool of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252059, China
First published on 25th November 2024
Abstract
Both the B–S and S–S bonds of 1,2,4,3,5-trithiadiborolane 1 could be cleaved by potassium graphite (KC8) to give the first isolable boryl dithiolate DmpB(SK)23, whereas the reduction of 1,2,4-triselena-3,5-diborolane 2 with KC8 afforded the boryl diselenolate DmpB(SeK)(SeSeK) 4, showcasing the unprecedented structural authentication of ligand substituted diselenolate.
There has been broad interest in the study of boracycles due to their versatile applications in synthetic, coordination and materials chemistry.1 The chemistry of boracycles bearing heavier group 16 elements has been much less explored compared with that of BN and BO-heterocycles,2 predominantly because of their higher sensitivity toward air and moisture. Meanwhile, the weak B–Ch bonds provide possible reaction pathways to various organic and inorganic compounds. 1,2,4-Trichalcogena-3,5-diborolanes featuring B2Ch3 five-membered rings are one of the most studied BCh-heterocyclic species (Fig. 1a). The landmark discovery of trithiadiborolane I was made by Schmidt and Siebert in 1964.3 Since then, several synthetic routes to trithiadiborolanes have been developed, mainly involving the reactions of halogenboranes with H2Sx, Na2S2, tBu2S2 and S8etc.4 Remarkably, Tokitoh and Okazaki unveiled the synthesis of 2,4,6-tris[bis(trimethylsilyl)methyl]phenyl (Tbt)-supported trithiadiborolane IIvia the reaction of dimercaptoborane with S8,5 while Braunschweig demonstrated the oxidation of borylene or diborane with S8, providing access to the bis-carbene coordinated trithiadiborolane III.6 In the context of triselenadiborolanes IV, many preparative methods, including selenation of haloboranes or trialkylboranes,7 photolysis of bis(methylseleno)boranes,8 and thermal decomposition of diboryldiselenanes,9 have been successively documented. Trichalcogenadiborolanes exhibit reactivity toward a variety of reagents, such as amines, alkynes and sulfur diimides, enabling the editing of heterocyclic skeletons to forge unusual main group compounds.10
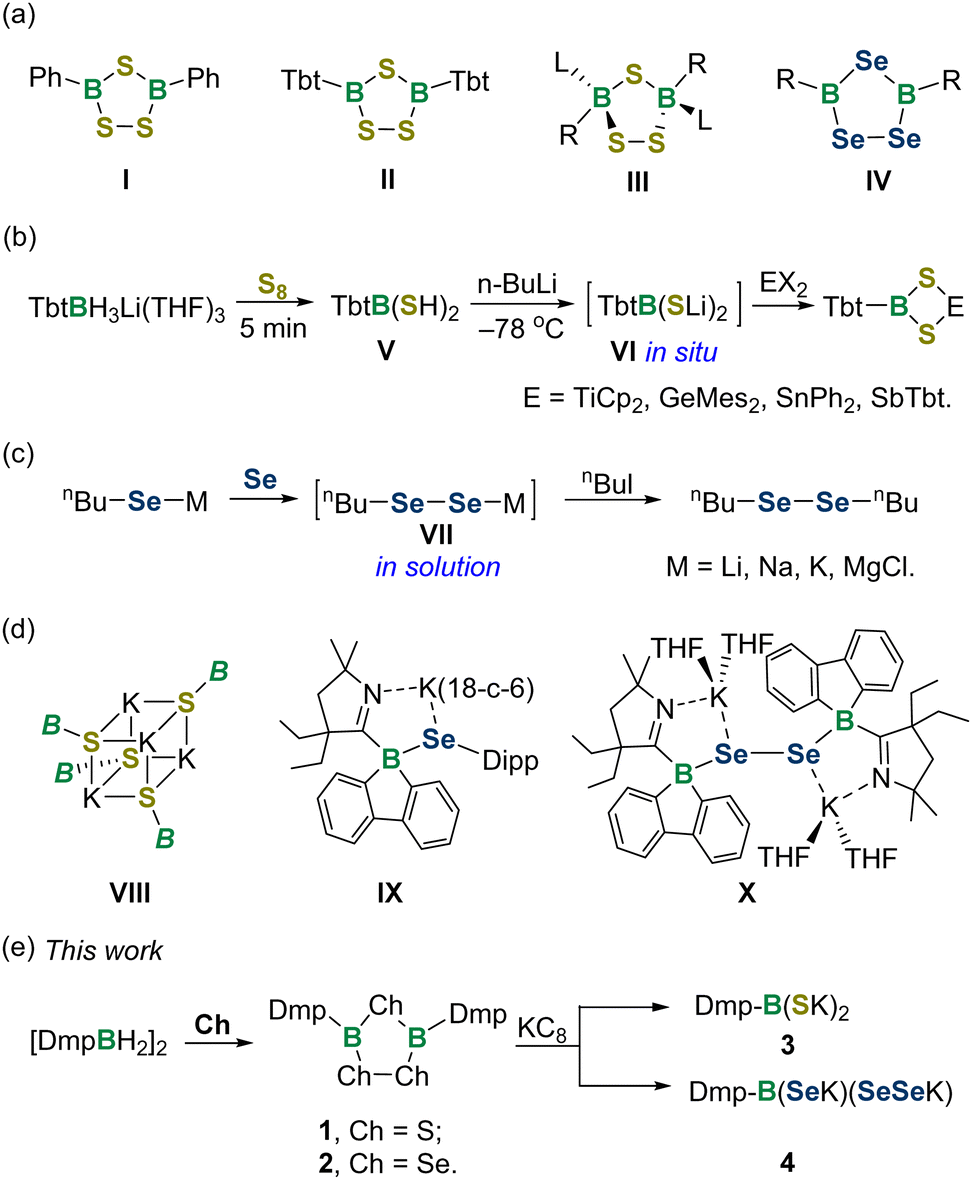 | ||
| Fig. 1 (a) 1,2,4-trichalcogena-3,5-diborolanes I–IV. (b) Synthesis and transformation of boryl dithiolate VI. (c) Synthesis and transformation of diselenolate VII. (d) Boryl-substituted thiolate VIII, selenide IX and diselenide X. (e) This work. B = (CHDippN)2B, Dipp = 2,6-iPr2C6H3; Dmp = 2,6-bis(2,4,6-trimethylphenyl)phenyl. | ||
Boronic acids are versatile reagents and catalysts in synthetic chemistry.11 In sharp contrast, their heavy chalcogen analogues and derivatives are rarely explored. A seminal work was achieved by Tokitoh and Okazaki in 1995, who synthesized dimercaptoborane TbtB(SH)2V by the sulfurization of the corresponding lithium aryltrihydroborate, representing the sole instance in this area (Fig. 1b).12 By way of lithiation of V, the boryl dithiolate VI could be generated in situ and utilized as a versatile reagent for building a series of 1,3,2,4-dithiametallaboretanes bearing four-membered rings.5,13 On the other hand, ligand substituted diselenols (RSeSeH) and their salts (RSeSeM) are essentially unstable, which makes their isolation a formidable challenge.14 Indeed, organic diselenols have eluded isolation thus far, while the sole organic diselenolates VII can be obtained only in solution (Fig. 1c).15
In 2019, Aldridge et al. isolated the tetrameric N-heterocyclic boryl-substituted potassium sulfide VIII,16 while two years later, Wilson and Gilliard described the first examples of boryl ligated selenide IX and diselenide X (Fig. 1d).17 The latter two species feature four-coordinated boron atoms and are produced from reactions of 9-carbene-9-borafluorene monoanion and selenium. Despite these significant strides, the isolable boryl dithiolate and diselenolate have remained elusive so far. Herein, we unveil the reduction of 1,2,4-trichalcogena-3,5-diborolanes 1 and 2 by KC8, providing access to boryl dithiolate 3 and boryl diselenolate 4 (Fig. 1e). Both species are isolated as persistent species and their structural and electronic properties have been investigated by an experimental and computational joint analysis.
A modified synthetic methodology to 1,2,4-trichalcogena-3,5-diborolanes has been developed. Specifically, treatment of the dimeric [DmpBH(μ-H)]2 (Dmp = 2,6-bis(2,4,6-trimethylphenyl)phenyl) with 0.8 equivalent of S8 at 130 °C yielded product 1 in 59% yield (Fig. 2a). The 11B NMR spectrum of 1 revealed a broad singlet at 65.6 ppm, which is close to those for species I (66.4 ppm) and II (69.3 ppm), indicating the three-coordinate boron center. An X-ray crystallographic study revealed 1 to be a 1,2,4,3,5-trithiadiborolane featuring a characteristic planar B2S3 five-membered ring with the sum of internal pentagon angles of 540.0° (Fig. 2b). The B–S bond lengths are slightly shorter than the sum of the covalent radii of boron and sulfur (∼1.87 Å),18 ranging from 1.779(4)–1.811(4) Å.
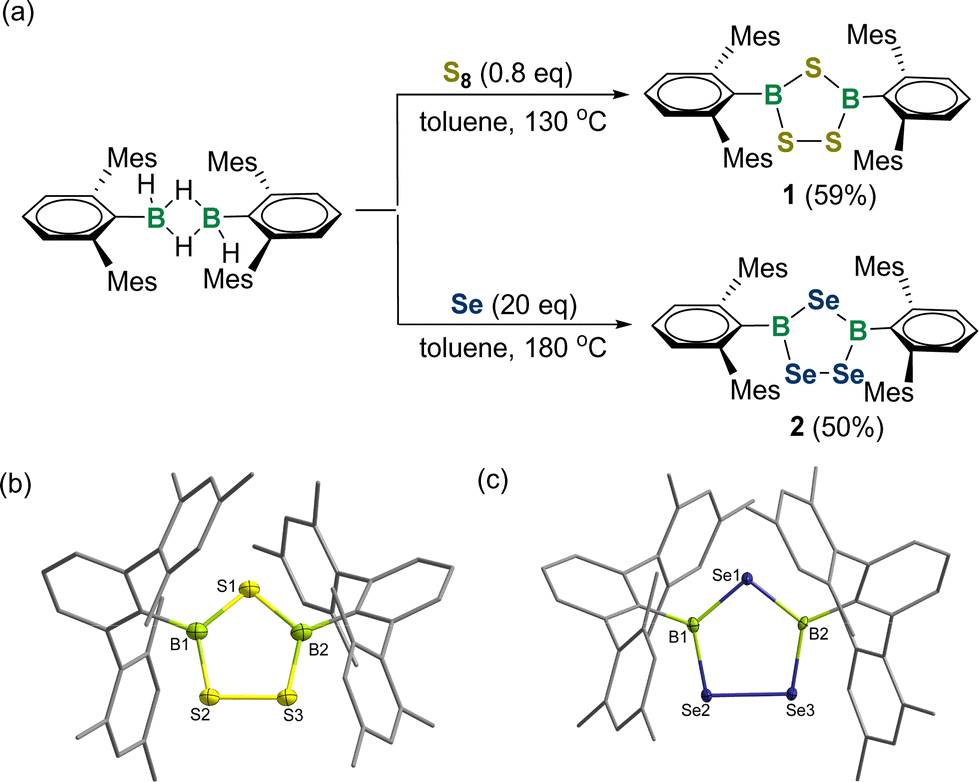 | ||
| Fig. 2 (a) Synthesis of 1,2,4-trichalcogena-3,5-diborolanes 1 and 2. Solid-state structures of 1 (b) and 2 (c). The hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Mes = 2,4,6-Me3C6H2. | ||
Similarly, we carried out the reaction of [DmpBH(μ-H)]2 with 20 equivalents of grey selenium in toluene at 180 °C for 4 days. After workup, a yellow solid of 2 was obtained in 50% yield. The 11B NMR spectrum of 2 exhibited a broad peak at 71.6 ppm, whereas two sharp resonances at 617.8 and 527.6 ppm were detected in the 77Se NMR spectrum. The solid-state structure of 2 confirms its 1,2,4-triselena-3,5-diborolane structure with a planar B2Se3 ring (Fig. 2c). The four B–Se bond lengths (B1–Se1: 1.931(4) Å; B1–Se2: 1.910(4) Å; B2–Se1: 1.935(4) Å; B2–Se3: 1.924(3) Å) compare well with those for Tbt-substituted triselenadiborolane IV (1.913(8)–1.950(9) Å). The Se2–Se3 bond length amounts to 2.335(5) Å, supporting its typical single bond character.
Subsequently, the reduction of 1,2,4-trichalcogena-3,5-diborolanes 1 and 2 was examined (Fig. 3a). Stirring a mixture of 1 with 8 equivalents of potassium graphite (KC8) in THF at ambient temperature resulted in the formation of a white solid of 3 in 56% yield after workup. The 11B NMR spectrum of 3 showed a broad singlet at 72.7 ppm, which is markedly low-frequency-shifted relative to that of precursor 1. Single crystals of 3 were obtained by slow evaporation of a concentrated THF solution at −25 °C and the solid-state structure unambiguously confirmed it to be a dipotassium salt of dimercaptoborane featuring a trimeric structure with a B3S6K6 cage bridged by potassium atoms (Fig. 3c and d). All the three boron atoms in 3 are three-coordinated and adopt a trigonal-planar geometry. The B–S bond lengths amount to 1.795(5)–1.818(6) Å and are comparable to that for species VIII (1.789(4) Å), yet slightly longer than B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond lengths in previously reported cationic, neutral and anionic thioxoboranes (1.710(5)–1.774(1) Å) described by Cui, Frank, Aldridge, Braunschweig and Erker et al.19 The S1 and S4 atoms bind with one boron and four potassium atoms and thus have a distorted square pyramidal geometry, whereas the four S2, S3, S5 and S6 atoms feature pyramidal coordination with one boron and three potassium atoms. The S–K bond lengths in 3 range from 3.024(2) to 3.421(2) Å and some parameters deviate slightly from the data observed for VIII (3.084(1)–3.185(1) Å). Note that compound 3 is stable in both solid and solution phases and represents the first isolable boryl dithiolate.
S double bond lengths in previously reported cationic, neutral and anionic thioxoboranes (1.710(5)–1.774(1) Å) described by Cui, Frank, Aldridge, Braunschweig and Erker et al.19 The S1 and S4 atoms bind with one boron and four potassium atoms and thus have a distorted square pyramidal geometry, whereas the four S2, S3, S5 and S6 atoms feature pyramidal coordination with one boron and three potassium atoms. The S–K bond lengths in 3 range from 3.024(2) to 3.421(2) Å and some parameters deviate slightly from the data observed for VIII (3.084(1)–3.185(1) Å). Note that compound 3 is stable in both solid and solution phases and represents the first isolable boryl dithiolate.
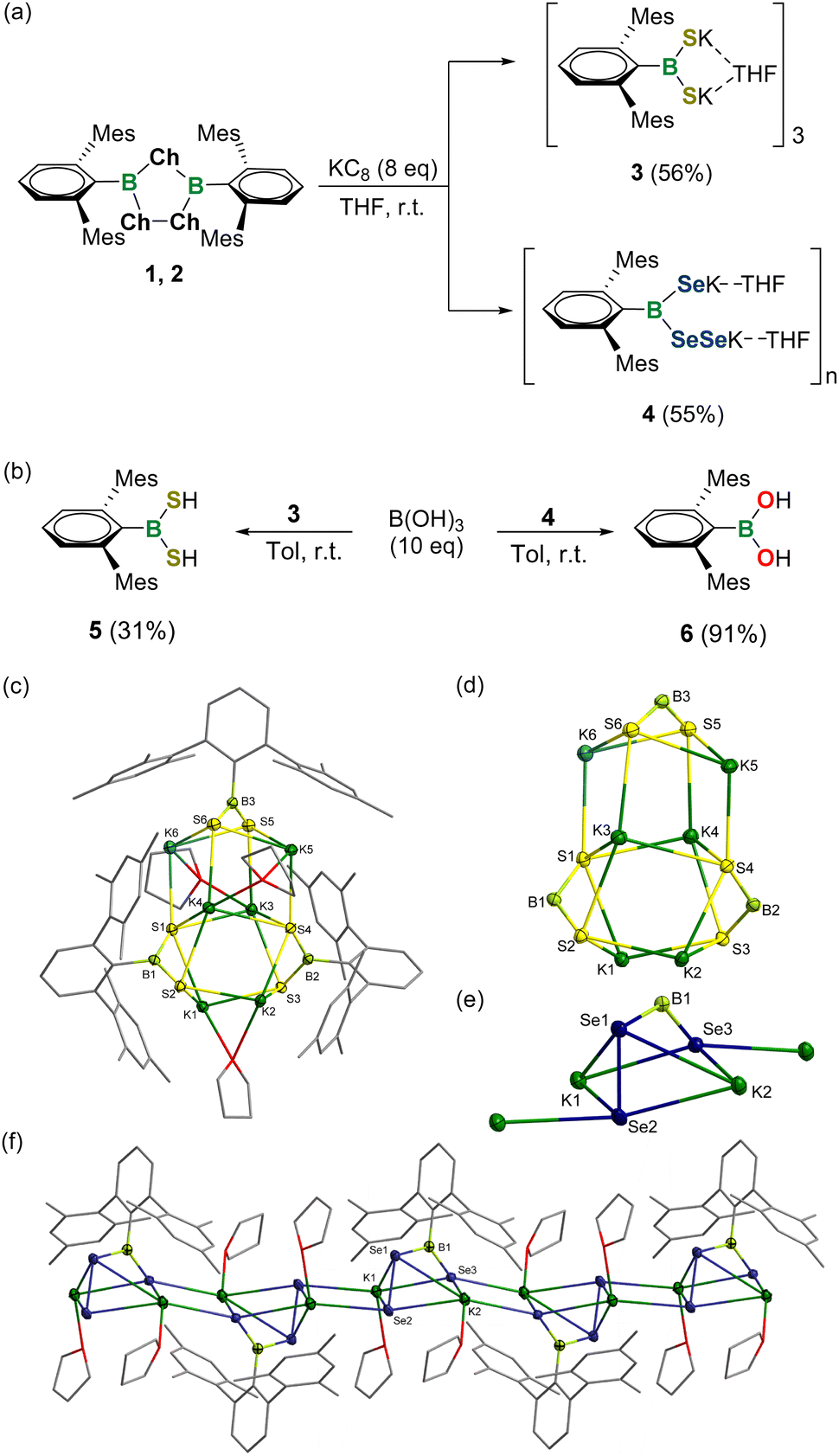 | ||
| Fig. 3 (a) Reduction of 1 and 2 with KC8. (b) Reactions of 3 and 4 with B(OH)3. Solid-state structures of 3 ((c) and (d)) and 4 ((e) and (f)). The hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at the 30% probability level. Mes = 2,4,6-Me3C6H2. | ||
In a similar fashion, the reaction of 2 with 8 equivalents of KC8 afforded yellow crystals of 4 in 55% yield (Fig. 3a). The 11B NMR spectrum of 4 revealed one broad signal at 74.7 ppm, which is shifted downfield compared to that of precursor 2 (72.9 ppm), indicative of the three-coordinate boron center. Single-crystal X-ray diffraction analysis elucidated that 4 is a dipotassium salt of diselaneyl(hydroseleno)borane with a one-dimensional polymeric skeleton linked by Se–K bonds (Fig. 3e and f). Strikingly, the Se–Se bond was maintained after reduction, thereby making 4 a unique species with both seleno and diseleno groups. All the Se atoms in 4 are four-coordinated yet feature different geometries. The Se1 and Se3 atoms are pyramidal and tetrahedral, respectively, while the Se2 has a distorted tetrahedral geometry. The B1–Se3 bond length amounts to 1.879(5) Å and locates in the range typical for the documented selenoboranes (1.876(4)–1.909(2) Å).19 Comparatively, a lengthened B1–Se1 bond length of 1.991(4) Å was observed. But, even so, both B–Se bond lengths are significantly shorter with respect to those for selenide IX (2.129(3) Å) and diselenide X (2.131(7) Å), which might be attributed to the higher coordination number of boron atoms in the latter two species. The Se1–Se2 bond length of 2.347(8) Å in 4 is almost the same as that for species X (2.3807(11) Å) and the reported sum of the Se–Se covalent radii (2.40 Å).18 The Se–K bond lengths range from 3.204 (1) to 3.542(1) Å and also fit well with the data for IX and X (3.2174(14)–3.5103(6) Å).
To probe the electronic structures of 3 and 4, density functional theory (DFT) calculation involving geometry optimization and natural bond orbital (NBO) analyses were performed at the M06-2X/def2-SVP level of theory. The optimized geometry shows excellent agreement with the crystallographically determined parameters of 3. The HOMO–HOMO−4, HOMO−6 and HOMO−7 of 3 are mainly the lone pair of electrons on the six S atoms, whereas the B–S π orbitals can be detected in the HOMO−5 and HOMO−8 (Fig. 4a and Fig. S1, ESI†). The π-bonding scenario could also be implied by natural bonding orbital analysis (Fig. S3, ESI†). The B–S σ-bonds are formed by the sp2.0 hybrid orbital of B atoms interacting with the sp1.7 hybrid orbital of S atoms, whereas the corresponding B–S π-bonds are attributed to the hyperconjugative delocalization from the p-type lone pair on S to the empty p-orbital of B with second-order interactions ranging from 85.2 to 101.4 kcal mol−1. Both B–S σ- and π-bonds are highly polarized. Consequently, the three B atoms carry positive charges of +0.29 to +0.32 a.u., whereas the S atoms are negatively charged (−0.77 to −0.85 a.u.). Given that the NPA charges on the K atoms (∼+0.88 a.u.) are more than those for S atoms, the S−K bonds in 3 are supported to be ionic interactions. Consistently, the quantum theory of atoms in molecules (QTAIM) calculations on 3 gave the topological parameters of (3, −1) bond critical points (BCP) along the S–K bonds with low electron density ρBCP values of 0.0085–0.017 (Fig. S5 and Table S2, ESI†).
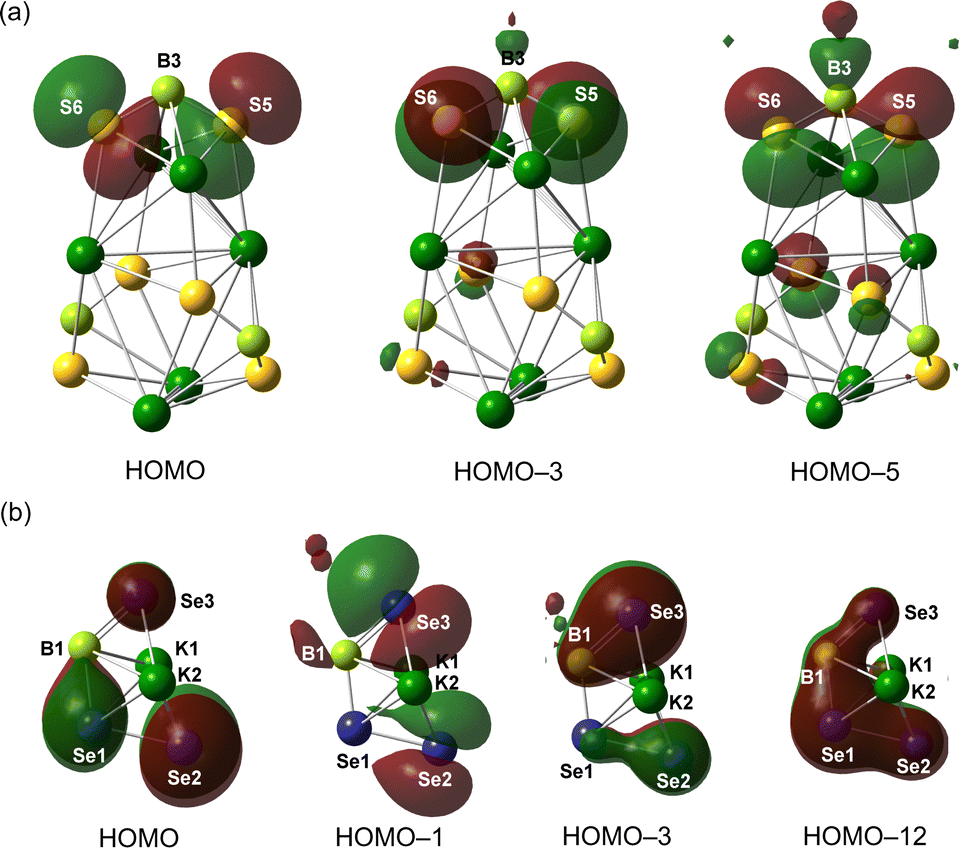 | ||
| Fig. 4 Selected frontier molecular orbitals of 3 (a) and 4 (b) (isovalue = 0.03). The Dmp groups and coordinated solvents are omitted for clarity. | ||
In the analysis of 4, the subunit DmpB(SeK·THF)(SeSeK·THF) has been chosen as the simplified model for the calculation. The HOMO and HOMO−1 reveal a distribution of lone pair electrons on the Se atoms, whereas HOMO−3 and HOMO−12 refer to the B1–Se3 π-bonding orbital and accumulated π electrons over the Se3–B1–Se1–Se2 unit, respectively (Fig. 4b). Natural bonding orbital analysis decisively confirms the B–Se π-bonding (Fig. S4, ESI†). The WBI value for the B1–Se1 bond is recorded as 1.17, lower than that for B1–Se3 (1.58), consistent with their bond length parameters. Comparatively, the Se1–Se2 bond is classified as the typical single bond given its WBI value of 0.98. Furthermore, natural population analysis (NPA) indicates that the charge on Se1 (−0.14 a.u.) is less negative than those for Se2 (−0.75 a.u.) and Se3 (−0.59 a.u.). Considering the positive charges on B (0.090 a.u.) and K atoms (∼+0.91 a.u.), the related B–Se and Se–K bonds in 4 could be classified as polarized covalent and ionic bonds, respectively.
The preliminary reactivity of 3 and 4 toward protic reagents was investigated (Fig. 3b). In the presence of boric acid B(OH)3, 3 could be smoothly protonated to give dimercaptoborane DmpB(SH)25 (11B NMR: 65.0 ppm). In contrast, stirring of 4 with B(OH)3 in toluene at ambient temperature afforded boronic acid DmpB(OH)26 (11B NMR: 29.9 ppm) exclusively, demonstrating its fragile B–Se bonds.
To conclude, we have obtained 1,2,4-trithia-3,5-diborolane 1 and 1,2,4-triselena-3,5-diborolane 2 from the formal oxidation of arylborane with halogens. Subsequent reduction of 1 and 2 with KC8 produced the corresponding dipotassium salts of dimercaptoborane and diselaneyl(hydroseleno)borane. The former boryl dithiolate 3 features a trimeric skeleton, while the latter boryl diselenolate 4 is a one-dimension polymer. Both experimental and computational results reveal the partial double bond characters of B–Ch bonds and predict the nucleophilic properties of the terminal chalcogen atoms in these two ionic compounds. Exploration of the utilization of 3 and 4 in constructing unusual B-heterocycles and unsaturated organoboron compounds is currently underway in our laboratory.
We gratefully acknowledge financial support from the National Natural Science Foundation of China (22101068 and 22271174), the Natural Science Foundation of Zhejiang Province (LTGC24B050005), the Scientific Research Fund of Zhejiang Provincial Education Department (Y202147386) and the Taishan Scholar Project of Shandong Province of China (tsqn202211007).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- S. Kar, S. Bairagi, G. Joshi, E. D. Jemmis, H.-J. Himmel and S. Ghosh, Acc. Chem. Res., 2024, 57, 2901–2914 CrossRef CAS PubMed.
- For selected reviews, see: (a) C. R. McConnell and S.-Y. Liu, Chem. Soc. Rev., 2019, 48, 3436–3453 RSC; (b) B. Su and R. Kinjo, Synthesis, 2017, 2985–3034 CAS; (c) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 4353–4368 CrossRef.
- M. Schmidt and W. Siebert, Angew. Chem., Int. Ed. Engl., 1964, 3, 637–638 Search PubMed.
- For selected examples, see: (a) M. A. Beckett and P. R. Minton, J. Organomet. Chem., 1995, 487, 209–214 CrossRef CAS; (b) M. Schmidt and F. R. Rittig, Angew. Chem., Int. Ed. Engl., 1970, 9, 738–739 CrossRef CAS; (c) M. Schmidt and W. Siebert, Chem. Ber., 1969, 102, 2752–2763 CrossRef CAS.
- M. Ito, N. Tokitoh and R. Okazaki, Organometallics, 1997, 16, 4314–4319 CrossRef CAS.
- (a) S. Liu, M.-A. Légaré, A. Hofmann, A. Rempel, S. Hagspiel and H. Braunschweig, Chem. Sci., 2019, 10, 4662–4666 RSC; (b) D. Auerhammer, M. Arrowsmith, R. D. Dewhurst, T. Kupfer, J. Böhnke and H. Braunschweig, Chem. Sci., 2018, 9, 2252–2260 RSC; (c) H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, C. Schneider and S. Ullrich, Angew. Chem., Int. Ed., 2015, 54, 10271–10275 CrossRef CAS PubMed.
- M. Schmidt, W. Sibert and E. Gast, Z. Naturforschg., 1967, 22b, 557–558 CrossRef.
- M. Ito, N. Tokitoh, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1999, 40, 5557–5560 CrossRef CAS.
- F. Riegel and W. Siebert, Z. Naturforsch., 1974, 29b, 719–722 CrossRef.
- For selected examples, see: (a) H. Nöth and T. Taeger, Z. Naturforsch., 2010, 65b, 173–177 CrossRef; (b) C. Habben and A. Meller, Z. Naturforsch., 1984, 39b, 1022–1026 CrossRef CAS.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC.
- N. Tokitoh, M. Ito and R. Okazaki, Organometallics, 1995, 14, 4460–4462 CrossRef CAS.
- (a) N. Tokitoh, M. Ito and R. Okazaki, Tetrahedron Lett., 1996, 37, 5145–5148 CrossRef CAS; (b) M. Ito, N. Tokitoh and R. Okazaki, Tetrahedron Lett., 1997, 38, 4451–4454 CrossRef CAS.
- F. S. Huziec, Jr and L. J. Guziec, in Patai's Chemistry of Functional Groups, ed. Z. Rappoport, John Wiley & Sons, Ltd., 2013 DOI:10.1002/9780470682531.pat0712.
- A. Krief, T. V. Wemmel, M. Redon, W. Dumont and C. Delmotte, Angew. Chem., Int. Ed., 1999, 38, 2245–2247 CrossRef CAS PubMed.
- Y. K. Loh, K. Porteous, M. Á. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CrossRef CAS PubMed.
- (a) K. E. Wentz, A. Molino, L. A. Freeman, D. A. Dickie, D. J. D. Wilson and R. J. Gilliard, Jr., Inorg. Chem., 2021, 60, 13941–13949 CrossRef CAS PubMed ; See also: ; (b) N. C. Frey, K. K. Hollister, P. Müller, D. A. Dickie, C. E. Webster and R. J. Gilliard, Jr., Inorg. Chem., 2024, 63, 17639–17650 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- For selected examples, see: (a) H. Wang, J. Zhang, H. Hu and C. Cui, J. Am. Chem. Soc., 2010, 132, 10998–10999 CrossRef CAS; (b) S. Liu, M. Legare, A. Hofmann and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 11223–11226 CrossRef CAS; (c) D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264–14268 CrossRef CAS PubMed; (d) Y. K. Loh, K. Porteous, M. Á. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CrossRef CAS PubMed; (e) C. Chen, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, J. Am. Chem. Soc., 2020, 142, 19763–19771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2366072, 2366073, 2388453 and 2388454. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05240d |
| This journal is © The Royal Society of Chemistry 2025 |