
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terminal hydride formation in [FeFe] hydrogenase: understanding the role of the dithiolate bridgehead†
Anjali
Depala
a,
Manon T.
Lachmann
b,
Simone
Morra
 c,
James A.
Birrell
*d and
Patricia
Rodríguez-Maciá
*b
c,
James A.
Birrell
*d and
Patricia
Rodríguez-Maciá
*b
aDepartment of Chemistry, University of Oxford, Inorganic Chemistry laboratory, South Parks Road, Oxford, OX1 3QR, UK
bSchool of Chemistry and Leicester Institute for Structural and Chemical Biology, University of Leicester, University Road, Leicester, LE1 7RH, UK. E-mail: prm28@leicester.ac.uk
cUniversity of Nottingham, Faculty of Engineering, Coates Building, University Park, Nottingham, NG7 2RD, UK
dSchool of Life Sciences, University of Essex, Wivenhoe Park, Colchester, CO4 3SQ, UK. E-mail: james.birrell@essex.ac.uk
First published on 24th March 2025
Abstract
[FeFe]-hydrogenases are highly-active hydrogen-conversion biocatalysts using Earth-abundant metals in their active-site. Understanding their mechanism may enable design of catalysts for renewable energy storage. Here, observation of the crucial Fe-hydride-containing (Hhyd) intermediate in a PDT-variant of [FeFe]-hydrogenase reveals deeper insight into the role of the dithiolate bridgehead in the catalytic mechanism.
Fossil fuel utilisation has led to a surge in atmospheric carbon dioxide (CO2), contributing to climate change.1 We urgently need to transition to renewable energy and find new methods to produce fossil fuel derived chemical precursors. A major challenge is that the currently available catalysts for hydrogen (H2) production are based on precious metals such as platinum or display poor activity and efficiency.2–4 Therefore, we must develop new catalysts using Earth-abundant metals like iron. Nature offers inspiration in the form of hydrogenases: highly active and reversible enzymes for H2 production and oxidation.5 Among the three phylogenetic classes of hydrogenases, [FeFe] hydrogenases are the most active and reversible and, therefore, of greatest technological interest. Their active site is based on Fe, and they achieve catalytic performances comparable to platinum.6 A deeper understanding of [FeFe] hydrogenases’ mechanisms would provide additional design criteria for building new, efficient and affordable catalysts enabling renewable energy storage in the form of H2.
The active site, known as the H-cluster (Fig. 1A), consists of a canonical [4Fe–4S] cluster ([4Fe–4S]H) covalently attached, through the thiolate group of a cysteine amino-acid residue, to a unique [2Fe] site ([2Fe]H), where catalysis occurs.7–9 [2Fe]H comprises two Fe ions, the proximal Fe (Fep), which is closest to [4Fe–4S]H, and the distal Fe (Fed), bridged by a CO and a unique 2-azapropane-1,3-dithiolate (ADT) ligand. Each iron is also coordinated to a terminal CO and a terminal CN− ligand. This leaves an open coordination site on Fed for binding H2. This arrangement of ligands results in the formation of a frustrated Lewis pair (FLP),10 with the open coordination site on Fed acting as a Lewis acid to bind H2 and the NH group of the dithiolate ligand serving as a base. This FLP arrangement polarises the H–H bond to facilitate heterolytic splitting by abstraction of a proton by the NH base as well as through the stabilisation of a terminal hydride by the Fed Lewis acid.
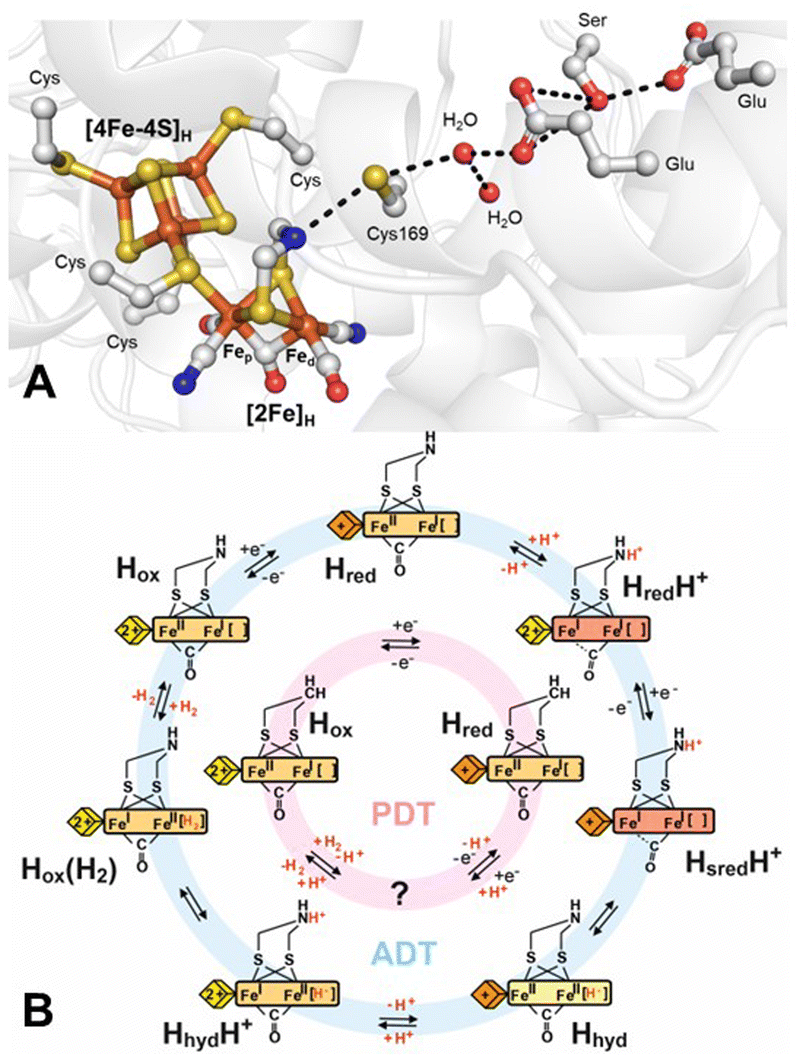 | ||
| Fig. 1 (A) Structure around the H-cluster based on PDB ID 4XDC.11 Cys169 (CrHydA1 nomenclature) of the proton-transfer pathway is labelled along with cysteines ligating the H-cluster and the key amino acids of the proton-transfer pathway. (B) Proposed catalytic cycle for the native ADT ligand-containing H-cluster (blue) and the semi-synthetic PDT ligand-containing H-cluster (pink). The yellow/orange diamond represents [4Fe–4S]H in the 2+ and 1+ states, respectively, while the yellow/orange/red rectangle represents [2Fe]H in the Fep(II)Fed(II), Fep(II)Fed(I) and Fep(I)Fed(I) states, respectively. Terminal CO/CN− ligands are omitted for clarity. | ||
The coordination of the Fe ions by strong-field CO and CN− ligands stabilises low oxidation and low spin states, tuning the electronic structure of the H-cluster for catalytic activity. Both Fe ions can adopt either Fe(II) or Fe(I) oxidation states. [4Fe–4S]H is also redox-active and can alternate between [4Fe–4S]2+ and [4Fe–4S]1+ oxidation states. The “resting” state of the H-cluster, prior to reduction or binding of H2, termed Hox, has an oxidised [4Fe–4S]H and a mixed valent Fep(II)Fed(I) [2Fe]H. During proton reduction (Fig. 1B), the Hox state is thought to become reduced by one electron on [4Fe–4S]H, forming the Hred state. Protonation of the ADT ligand at the amine bridgehead results in the HredH+ state in which an electron has been transferred from [4Fe–4S]H to [2Fe]H generating an Fep(I)Fed(I) [2Fe]H.12 This transfer makes [4Fe–4S]H available for a second reduction, leading to the formation of the HsredH+ state.13 HsredH+ can tautomerise to yield a state in which a terminal hydride (H−) forms on Fed, referred to as Hhyd (also Hhyd:red).14–17 To form the terminal hydride, a proton from the bridging ammonium (NH2+) of the protonated ADT cofactor is transferred to Fed, causing one-electron oxidation of both Fep and Fed, yielding a Fe(II)Fe(II)–H− state. Further protonation of the ADT ligand is then thought to trigger electron transfer from [4Fe–4S]H to [2Fe]H in the HhydH+ state. This state would have the ideal configuration for protonation of the terminal hydride to generate a Hox state with H2 bound. The final stages of catalysis have not yet been experimentally elucidated, and detailed structural, functional, and spectroscopic information is required for a more complete understanding of the catalytic cycle.18
Hhyd has a rich chemistry and hydride intermediates are crucial for activation of small molecules in metalloenzymes such as hydrogenases,5 nitrogenases19,20 and possibly CO dehydrogenases.21 An important open question concerns the conditions under which the Hhyd state can be formed. In [FeFe] hydrogenases, this state was first observed in alanine (A, Ala)/serine (S, Ser) mutants of a cysteine adjacent to the ADT ligand of the proposed proton transfer pathway.15,17,22 Later studies showed that a similar state could be formed in chemical variants of [2Fe]H in which the ADT ligand had been substituted with 2-oxapropane-1,3-dithiolate (ODT).15,23,24 Another method for producing the Hhyd state in the native enzyme involved using high concentrations of the chemical reductant sodium dithionite (NaDT) at low pH.15 The original explanation for the stabilisation of Hhyd in the C → A and C → S mutants and ODT variant was that they were all kinetically compromised for proton transfer. During H2 oxidation, the deprotonation of Hhyd was slow, while during H2 production, the protonation of Hhyd was slow. Following the same argument, low pH was proposed to inhibit the deprotonation of Hhyd during H2 oxidation according to Le Chatelier's principle. However, Hhyd in the cysteine mutants and the ODT variant is thermodynamically stable (it is observed when the enzyme is incubated under H2 in the absence of oxidants), and at low pH, dithionite stimulates H2 production, not H2 oxidation. Therefore, there is no reason to believe that the protonation of Hhyd during H2 production would be slower at low pH – if anything, it should be faster. Instead, our hypothesis is that the variants and conditions that stabilise Hhyd achieve this by altering the electronic structure of the H-cluster, making the Hhyd state, with Fep(II)Fed(II)–H−, more stable than the tautomeric HsredH+ state, with Fep(I)Fed(I)–H+. The relative energies of HsredH+/Hhyd were addressed previously using computational methods.25–27 If the kinetic argument were correct, we would have expected the H-cluster variant containing a propane-1,3-dithiolate (PDT) ligand to behave identically to the ODT variant. However, reduction of the PDT variant with NaDT yields the Hred state, and Hhyd has not yet been observed in this variant.28 Therefore, the open question is: why can’t the PDT variant form the Hhyd state? We propose that it is not for kinetic but thermodynamic reasons and is just a matter of driving force.
While NaDT is already a strong reductant by biological standards (E° = −0.66 V)29 and has, for many reasons, become the reductant of choice for numerous enzymes, it is also known to form various products, some of which may interact directly with the enzyme of interest.30 Therefore, in recent years, it has been proposed to use alternative low-potential reductants, including europium (II) complexes such as Eu(II)-diethylenetriamine pentaacetate (DTPA) (E° = −1.1 V).31–33 Vincent and colleagues were the first to utilise this complex in an enzyme, using it to reduce the Fe-protein of the nitrogenase.33 Since then, Eu(II)-DTPA has been used to reduce iron–sulfur clusters in mitochondrial complex I.31 Nevertheless, so far, Eu(II)-DTPA has not been utilised as a reductant for [FeFe] hydrogenases, but has been used with [FeFe] hydrogenase models.34 Therefore, we investigated whether the PDT variant of [FeFe] hydrogenase could be reduced by Eu(II)-DTPA and whether this would trigger formation of the Hhyd state.
To this end, we used the model [FeFe] hydrogenase from Chlamydomonas reinhardtii (CrHydA1), which contains only the active site H-cluster and lacks any additional redox cofactors (e.g. iron–sulfur clusters). This enzyme was prepared as previously described using heterologous expression in Escherichia coli and in vitro maturation with the synthetic PDT [2Fe] precursor.23,35Fig. 2 compares Fourier transform infrared (FTIR) spectra of CrHydA1PDT under various conditions (a) comparison of FTIR bands for CrHydA1ADT, CrHydA1PDT and CrHydA1ODT is provided in Fig. S1 (ESI†). Under as-isolated conditions (100% N2), CrHydA1PDT is in the Hox state (Fig. 2A) with IR bands at 2090, 2073, 1966, 1942 and 1811 cm−1, almost identical to those reported previously.28 In the presence of 10 mM NaDT, the sample converts to the Hred state (Fig. 2B) with IR bands (2085, 2067, 1964, 1935 and 1798 cm−1) shifted to lower energy by an average of 7 cm−1 compared to Hox. If, instead of NaDT, 10 mM Eu(II)-DTPA is added, the sample is converted to an entirely new state (Fig. 2C) where the IR bands (2089, 2072, 1977, 1961 and 1865 cm−1) are shifted to higher energy, averaging 16 cm−1 higher relative to Hox, consistent with oxidation of [2Fe]H to a [Fe(II)Fe(II)] valence. The addition of a reductant leading to oxidation of [2Fe]H has so far only been observed when oxidative addition of a proton leads to terminal hydride formation on Fed.17 To further investigate whether hydride formation had occurred in CrHydA1PDT on addition of Eu(II)-DTPA, we performed isotope-labelling experiments, in which the same experiment with CrHydA1PDT samples was performed in D2O instead of H2O. It has been previously observed that the bridging CO ligand IR stretch and the terminal Fe–H stretch are vibrationally coupled (the so-called “trans-effect”),17 due to the Fe–H and bridging CO stretches having similar vibrational frequencies and optimal orbital overlap. Addition of Eu(II)-DTPA to CrHydA1PDT samples exchanged into D2O buffer yielded a very similar state to that observed in H2O buffer, except that the bridging CO IR band was shifted to lower energy by 9 cm−1 (Fig. 2D). This is expected since deuterium is heavier than hydrogen. All other bands had the same vibrational frequency in H2O and D2O. This is consistent with the presence of a terminal Fe–H/D on Fed.17,24 The as-isolated enzyme in the presence of D2O had an identical spectrum to that in H2O (Fig. S3, ESI†).
 | ||
| Fig. 2 IR spectra of CrHydA1PDT as isolated (A), reduced with sodium dithionite (B), and reduced with Eu(II)-DTPA in H2O (C) and D2O (D). The IR bands assigned to the Hox, Hred and Hhyd states are coloured blue, orange and purple, respectively. | ||
Two variants of Hhyd have been proposed: Hhyd:ox, which contains an oxidised [4Fe–4S]H, and Hhyd:red, which contains a reduced [4Fe–4S]H.14 We expect the Hhyd state formed under these conditions to be Hhyd:red. To classify the Hhyd state identified in CrHydA1PDT, we measured EPR spectra of Eu(II)-DTPA reduced CrHydA1PDT (Fig. S4, ESI†). Based on the electronic structure Hhyd:ox is expected to be EPR silent while Hhyd:red is expected to exhibit an EPR spectrum consistent with the reduction of a [4Fe–4S] cluster (rhombic S = 1/2 spectrum with gav ≈ 1.9). Eu(II)-DTPA reduced CrHydA1PDT shows a rhombic EPR spectrum with g-values of 2.073, 1.937 and 1.880 (Fig. S5, ESI†). This spectrum supports the hypothesis that the Hhyd state is Hhyd:red-like, with the EPR spectrum being very similar to that of the Hhyd:red state of CrHydA1ADT (g = 2.069, 1.938, 1.880).14
Based on the results presented above, we can draw the following conclusions. The Hhyd state can be formed in the PDT variant of an [FeFe] hydrogenase but requires significantly more negative potentials (E ≪ −600 mV) than those needed for the native ADT enzyme and the ODT variant (Fig. 3). These results do not align with a model that attributes the stabilisation of Hhyd in the ODT variant to kinetic reasons; rather, they are more consistent with a thermodynamic explanation. The ADT, PDT, and ODT ligands differ not only in their ability to undergo protonation at the central atom but also in their electronic structures. Oxygen is more electronegative than nitrogen, which is, in turn, more electronegative than carbon. Consequently, the thiolates of the PDT ligand are more electron-rich than those of ADT or ODT. This electron richness also influences the electron density of the Fe ions, which affects the redox potential of the [2Fe]H site. In all three variants, the reduction potential of [2Fe]H is too negative to be reduced without protonation12 – protonation occurs either at the dithiolate bridgehead (for ADT only) or at Fed. This reduction potential is the least negative for the ODT variant but still requires protonation, which, due to the very low pKa of the ether oxygen, can only occur at Fed, forming a terminal hydride. For the PDT variant, due to the inability to protonate a methylene group, reduction can likewise only occur at Fed, forming a terminal hydride. However, the redox potential is more negative than that of the ODT variant owing to the difference in electronegativity of the bridgehead atom. For the native ADT variant, the redox potential is between those of PDT and ODT, but the pKa of the bridging amine is in the neutral range,12 making protonation more favourable on the amine than on Fed. Nevertheless, a tautomeric equilibrium exists in which the proton can be readily exchanged between the amine and metal. This equilibrium could explain the impressive activity and reversibility of [FeFe] hydrogenases and would explain why these properties are not observed in the active-site variants.
 | ||
| Fig. 3 Simplified representations of the H-cluster showing proposed pathways of Hhyd generation (see Fig. S6 for a more detail, ESI†). [4Fe–4S]H is shown as a diamond, [2Fe]H as a rectangle, the dithiolate ligand as sticks and the CO/CN are omitted. The native, ADT-containing enzyme in Hox is doubly reduced (on [4Fe–4S]H and [2Fe]H) and protonated at the ADT bridgehead yielding Hhydvia Hred or HredH+ intermediates. Both reduction steps happen with redox potentials close to −400 mV. For PDT and ODT, reduction of [4Fe–4S]H happens close to −400 mV (the dithiolate ligand has only a minor influence on the [4Fe–4S]H redox potential). Reduction of [2Fe]H in both the PDT and ODT cases proceeds without protonation of the dithiolate bridgehead, and the redox potential will depend on the stability of Hhyd. Electron withdrawal from the bridgehead group (O in ODT and CH2 in PDT) is predicted to influence the electron density on the Fe ions with O more electron withdrawing than C. Hence, reduction of [2Fe]H and Hhyd formation occurs at less negative potentials in the ODT enzyme than in the PDT enzyme. | ||
Overall, the results presented here highlight the crucial role that the dithiolate ligand plays in the [FeFe] hydrogenase, serving not only as a proton transfer relay that provides rapid proton transfer to and from Fed but also as a key player in tuning the electronic features of the diiron cofactor. This tuning is necessary to balance the requirement for stabilising terminal hydrides during H2 oxidation without overly stabilising them and thereby limiting H2 formation. This implies that ideal synthetic catalysts for reversible H2 conversion will achieve a similar equilibrium. Indeed, this is the essence of the volcano plot:36 metals that stabilise hydrides too much or too little are very poor catalysts for H2 conversion. Nature has positioned [FeFe] hydrogenase at the apex of the volcano plot by selecting the appropriate combination of primary- and secondary-coordination sphere interactions, rivalling precious metals (e.g. platinum) in terms of H2-conversion activity.
PRM thanks Royal Society (RGS\R1\231433), Royal Society of Chemistry (R23-6753486967) and University of Leicester for funding. MTL is supported by a Future 50 Scholarship from University of Leicester. JAB thanks Royal Society (RG\R2\232336), RSC (R22-2594924113), and University of Essex for funding. SM was supported by a Nottingham Research Fellowship from University of Nottingham.
Data availability
Data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- S. Singh, Energy, 2021, 1–17 Search PubMed.
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. Chen, G. T. K. K. Gunasooriya, I. McCrum, R. Mom, N. López and M. T. M. Koper, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS.
- S. Shiva Kumar and V. Himabindu, Mater. Sci. Energy Technol., 2019, 2, 442–454 Search PubMed.
- S. Wang, A. Lu and C.-J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- M. Hambourger, M. Gervaldo, D. Svedruzic, P. W. King, D. Gust, M. Ghirardi, A. L. Moore and T. A. Moore, J. Am. Chem. Soc., 2008, 130, 2015–2022 CrossRef CAS PubMed.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 20002–20014 CrossRef CAS PubMed.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2017, 139, 1440–1443 CrossRef CAS PubMed.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Biochemistry, 2009, 48, 7780–7786 CrossRef CAS PubMed.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8, 16115 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CAS.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CAS.
- M. T. Lachmann, Z. Duan, P. Rodríguez-Maciá and J. A. Birrell, Chem. Sci., 2024, 15, 14062–14080 CAS.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CAS.
- N. Khadka, R. D. Milton, S. Shaw, D. Lukoyanov, D. R. Dean, S. D. Minteer, S. Raugei, B. M. Hoffman and L. C. Seefeldt, J. Am. Chem. Soc., 2017, 139, 13518–13524 CAS.
- P. Amara, J.-M. Mouesca, A. Volbeda and J. C. Fontecilla-Camps, Inorg. Chem., 2011, 50, 1868–1878 CAS.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 Search PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CAS.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CAS.
- A. R. Finkelmann, M. T. Stiebritz and M. Reiher, Chem. Sci., 2014, 5, 215–221 CAS.
- J. H. Artz, O. A. Zadvornyy, D. W. Mulder, S. M. Keable, A. E. Cohen, M. W. Ratzloff, S. G. Williams, B. Ginovska, N. Kumar, J. Song, S. E. McPhillips, C. M. Davidson, A. Y. Lyubimov, N. Pence, G. J. Schut, A. K. Jones, S. M. Soltis, M. W. W. Adams, S. Raugei, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2020, 142, 1227–1235 CAS.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CAS.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CAS.
- S. G. Mayhew, Eur. J. Biochem., 1978, 85, 535–547 CAS.
- M. A. Martini, O. Rüdiger, N. Breuer, B. Nöring, S. DeBeer, P. Rodríguez-Maciá and J. A. Birrell, J. Am. Chem. Soc., 2021, 143, 18159–18171 CAS.
- T. Reda, C. D. Barker and J. Hirst, Biochemistry, 2008, 47, 8885–8893 CAS.
- P. Paengnakorn, P. A. Ash, S. Shaw, K. Danyal, T. Chen, D. R. Dean, L. C. Seefeldt and K. A. Vincent, Chem. Sci., 2017, 8, 1500–1505 CAS.
- K. A. Vincent, G. J. Tilley, N. C. Quammie, I. Streeter, B. K. Burgess, M. R. Cheesman and F. A. Armstrong, Chem. Commun., 2003, 2590–2591 CAS.
- C. Esmieu and G. Berggren, Dalton Trans., 2016, 45, 19242–19248 RSC.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, Chem. Soc. Rev., 2008, 37, 2163–2171 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00860c |
| This journal is © The Royal Society of Chemistry 2025 |