
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The need for robust model systems in the study of hybrid interfaces for photocatalysis and photoelectrocatalysis
Mekha P.
Mohandas
 and
Jared P.
Bruce
*
and
Jared P.
Bruce
*
University of Nevada, Las Vegas, Las Vegas, Nevada, USA 89154. E-mail: jared.bruce@unlv.edu
First published on 24th January 2025
Abstract
Small molecule conversion to value-added products using renewable energy sources has emerged as a promising strategy to mitigate our reliance on fossil fuels. Hybrid materials that integrate the strengths of photoabsorbers and co-catalysts (electrocatalysts) are essential for maximizing the efficiency of photochemical (PC) and photoelectrochemical (PEC) systems. In this perspective, we will focus on the need for fundamental studies with a strong emphasis on the importance of beginning with well-defined hybrid interfaces. A particular focus is given to small molecule adsorption studies that correlate surface structure and chemistry to reactivity, highlighting its potential in characterizing complex interfaces. We also make the case for understanding how light and electrochemical environments influence surface structure, adsorption, and reactivity and should be considered in model hybrid system design. Finally, we provide a framework to connect the theory and experiment of model hybrid surfaces to provide a molecular understanding of PC and PEC at these interfaces and accelerate our integration of these materials into real systems capable of meeting our renewable energy needs.
 Mekha P. Mohandas | Mekha received her BSc in Chemistry (2018) from Kuroakose Gregorious College, Pampady, Kerala, India, and her MSc in Applied Chemistry (2020) from CMS College, Kottayam, Kerala, India. She is currently a PhD student at the Department of Chemistry and Biochemistry, University of Nevada, Las Vegas, under the guidance of Dr Jared P. Bruce. Her research interests center on fundamental surface studies of coupled co-catalyst/photoabsorber interfaces for light-mediated reactions. |
 Jared P. Bruce | Jared Bruce is currently an Assistant Professor at the University of Nevada, Las Vegas. He received a PhD at the University of California, Irvine and completed his postdoctoral work at the Fritz Haber Institute of the Max Planck Society in Berlin, Germany. His current work focuses on the surface structure and chemistry of complex interfaces related to photochemistry and photoelectrochemistry. |
1. Introduction
Light-driven chemical conversion of small molecules to fuel is a method that has great promise to alter our reliance on fossil fuels and has been of intense interest over the last few decades. Photocatalysis (PC) and photoelectrocatalysis (PEC), in particular, are two promising methods for addressing energy and environmental challenges associated with consuming fossil fuels. There has been particular focus in the literature to develop materials that can be integrated into these technologies for long-term sustainable fuel production.Heterogenous PC involves using light to initiate chemical reactions on the surface of a material. This process begins with light absorption, leading to the generation and separation of electron–hole pairs. These charge carriers travel to the surface, where they can participate in redox reactions, such as the oxidation of organic pollutants1 or the reduction of carbon dioxide.2 The efficiency of this process depends on many factors, such as the band gap, light absorption properties, charge carrier separation, and surface reactivity of the material.3 Electrocatalysis (EC), on the other hand, accelerates charge transfer reactions at the electrode–electrolyte interface via an external power supply. This process involves reactant adsorption, charge carrier diffusion, surface reactions, and product desorption when converting reactants to products.4 While photocatalysis and electrocatalysis differ in their energy conversion mechanisms, both rely on mass transfer and surface-dependent catalytic processes. A PEC system combines the favorable principles of both PC and EC to enhance its overall light-to-fuel efficiency.5 However, traditional phoroabsorbing materials used in PC and PEC systems, such as TiO2, Si, and InP, often suffer from poor efficiency and lack of durability.6
More recently, there has been a growing interest in coupling various co-catalyst materials to the surface of photoabsorbers in an effort to lower the reaction barrier and increase the rate of reaction while maintaining the favorable light absorption characteristics of the underlying substrate. These coupled co-catalysts serve several vital functions, including enhancing quantum efficiency by suppressing electron–hole recombination at the interface/junction,7 providing new active sites for the desired surface reactions,8 and ensuring long-term stability by protecting the semiconductor from photocorrosion.7,9–11 These new surfaces formed with photoabsorbers and coupled co-catalysts have been called “hybrid” or “co-catalyst” interfaces and represent an emerging set of materials capable of photochemical conversion of small molecules to high value-added products. A wide range of co-catalysts are being investigated to improve the efficiency of photocatalytic activity of semiconductors, like noble metals (e.g., Pt, Ag, Pd, Au),12–15 transition metals (Cu, Ni, Fe),16–18 organometallic compounds (e.g., Co–porphyrin complex),19 carbon-based materials like graphene,20 and biological co-catalysts such as enzymes21 and bacteria.22
PEC, PC, and EC reactions all take advantage of heterogeneous solid–liquid or solid–gas interfaces to perform their catalytic functions. Traditionally, gas–solid interfaces have benefitted from years of careful surface science studies that have significantly transformed our understanding of catalytic reactions that often lead to breakthroughs in industrial processes.23 These fundamental studies are essential for understanding and improving catalytic systems due to the strong correlation between surface structure, chemistry, and stability of these interfaces. For example, the exposure of facets with varying atomic compositions and surface energies directly impacts performance, stability, intermediate formation, and the desired product yield.24 Additionally, the unique interactions between photogenerated charge carriers and specific facets emphasize the significance of surface science understanding.24 Notably, spatial charge differences exist between surfaces with different atomic arrangements, significantly influencing photo/electro-catalytic reaction pathways and the formation of reaction intermediates.24
Extensive research has examined the addition of metal co-catalyst onto semiconductors to enhance its photocatalytic activity. For instance, the addition of Cu to the surface of TiO2 has led to significant improvements, such as a twofold increase in CO2 reduction efficiency.25 The introduction of metals like nickel or ruthenium can also positively impact performance.26,27 Nickel doping can create lattice vacancies, promoting charge carrier separation and reduction reactions.26 Ruthenium doping, meanwhile, can facilitate charge transfer and CO2 adsorption, leading to increased selectivity for specific products.27
A considerable portion of these hybrid material systems are comprised of hybrid nanomaterials, where the newly formed interfaces can induce unique properties, ranging from altered physical characteristics to entirely new functionalities. Many of the unique electronic and optical properties observed in these systems are from their reduced dimensionality and size and the unique chemical and electronic environment at their surfaces. Significant progress been made in using these materials to convert light into fuel.28,29 However, the complex nature of nanoparticle surfaces, characterized by kinks, defects, and multiple facets, makes it challenging to precisely characterize their structure, correlate these features with catalytic activity, and isolate the specific active sites responsible for catalysis.30–33 As a result, a significantly large gap exists in our knowledge of the fundamental surface science of these hybrid materials and their impact on chemical conversion at the interface. Here, well-defined, robust model systems provide a promising approach to address these complexities, elucidating clear structure–function and chemistry–function relationships. While the perspective will not focus on hybrid nanomaterials, there are many excellent reviews that the reader can find.28,29,34–36
Buried interfaces between electrocatalysts and photoabsorbers are another region of the hybrid material that significantly influences device performance.37,38 Thus, the precise engineering of this interface also offers a unique opportunity to optimize the charge transfer kinetics and surface chemistry of hybrid materials. Robust model systems would ideally involve well-defined photoabsorber surfaces and precisely deposited co-catalysts under pristine ultra-high vacuum (UHV) conditions. By controlling the atomic-scale structure and composition of the catalysts, insights into the interfacial reactions, light absorption, charge carrier dynamics, and overall system performance could be obtained. Moreover, the current understanding of hybrid systems, including identifying active sites, largely relies on ex situ characterization and theoretical modeling.39 Experimental evidence directly probing in situ processes (e.g., evolution of reaction intermediates) using the model systems is crucial in advancing this field.39 Importantly, studies have demonstrated that in situ (UHV) and quasi-in situ (inert gas) experiments can offer valuable knowledge regarding photoabsorber/electrolyte interfaces.40 This perspective will explore the current state-of-the-art hybrid surfaces (fabrication and characterization), how the community could benefit from model surface studies to fundamentally understand the reactivity of hybrid materials in light-driven reactions, and where our focus should be, moving forward into engineering and device fabrication that takes advantage of hybrid materials.
2. Semiconductor/co-catalyst (hybrid) interfaces
Integrating electrocatalysts as co-catalysts is a widely adopted strategy in photocatalysis to lower activation energy barriers.7 Some of the common types of electrocatalysts coupled with photoabsorbers are noble metals (Pt, Pd),41,42 transition metals (Cu, Fe),43–45 metal oxides (RuO2, IrO2),7,46 and metal dichalcogenides (MoS2, WS2).47,48 Building upon the advancements in electrocatalyst-modified photoelectrodes, metal–semiconductor interfaces emerge as another promising strategy to enhance charge separation and overall photocatalytic efficiency.29 The Schottky barrier, formed at the metal–semiconductor interface hinders the recombination of photogenerated electron–hole pairs, further promoting charge separation. Due to the difference in their work functions, electrons flow from the material with a lower workfunction to the material with a higher workfunction until the Fermi levels between the two materials align.49 This charge transfer results in the formation of a depletion region near the interface. In n-type semiconductors, the depletion region is characterized by upward band bending, creating an internal electric field that drives photogenerated electrons toward the metal and holes toward the semiconductor bulk. This field effectively separates charge carriers enhancing the efficiency of charge transfer processes. At the same time, in p-type semiconductors the depletion region exhibits downward band bending. The resulting electric field directs holes toward the metal and electrons toward the semiconductor bulk. Fig. 1 schematically depicts the band-bending process at a metal–semiconductor interface formed with an n-type semiconductor.50 This effective separation and selective transfer of charge carriers to the co-catalyst due to the Schottky barrier formation at the co-catalyst/semiconductor interface leads to better utilization of the charge carriers (holes and electrons) for surface reactions (oxidation and reduction), thus improving the overall efficiency of the system.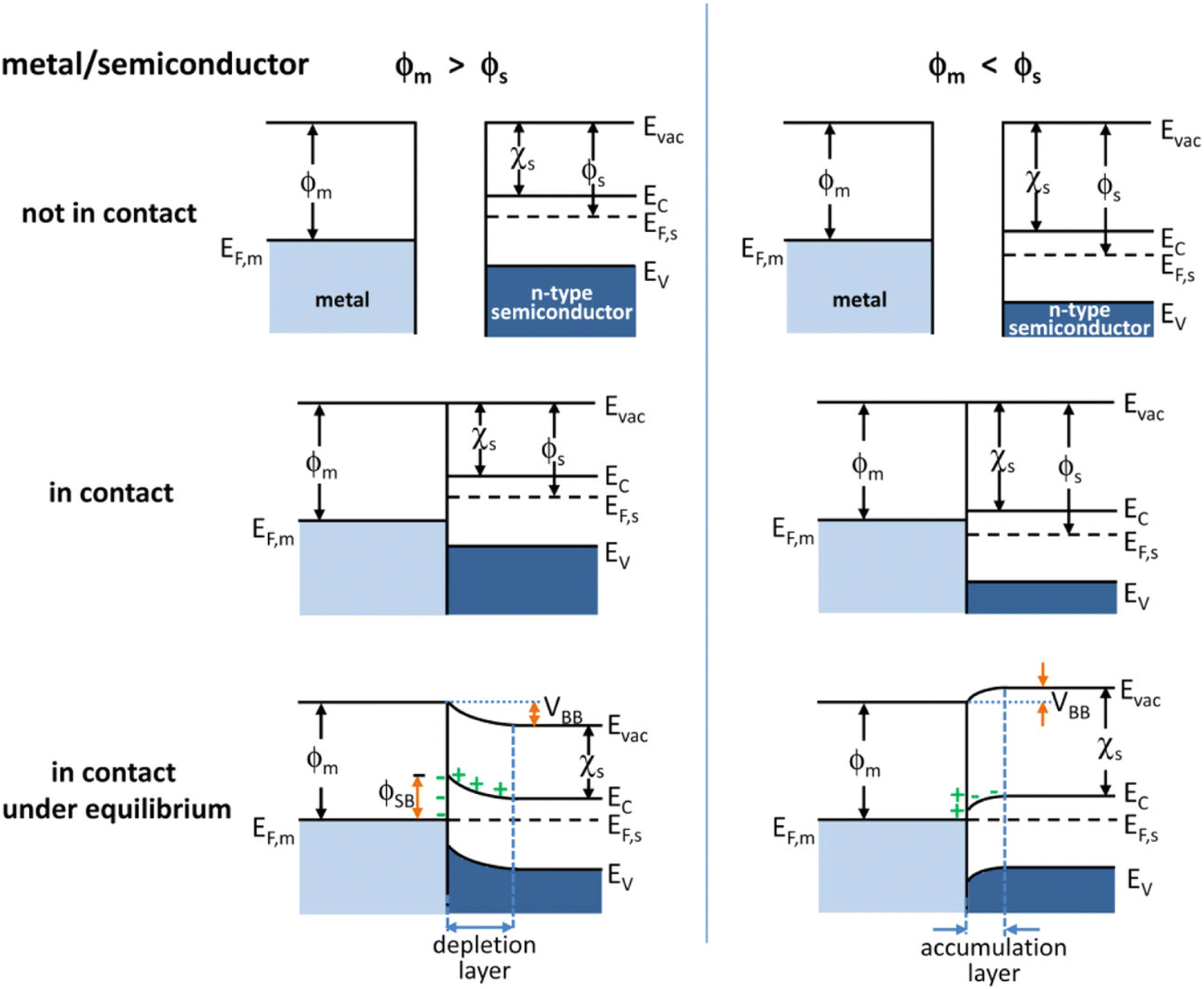 | ||
| Fig. 1 Energy band diagrams of metal and n-type semiconductor contacts. Evac, vacuum energy; Ec, energy of conduction band minimum; Ev, energy of valence band maximum; ϕm, metal work function; ϕs, semiconductor work function; χs, electron affinity of the semiconductor.50 Reprinted with permission from ref. 50. Copyright 2012 American Chemical Society. | ||
2.1 Fabrication and characterization of hybrid Interfaces
A critical aspect of hybrid interface design is understanding how the specific material combination – including the semiconductor type (e.g., TiO2, CdSe, etc.) and the chosen metal (e.g., Au, Pt, etc.) – dictates the resulting structure (electronic and atomic) and properties. The combination of these materials can influence factors like facet reactivity, lattice constant, and surface defects ultimately guiding the growth and morphology.28 The desired properties, whether structural, physical, or chemical, often decide the most suitable synthetic strategy. Due to its importance in heterogeneous catalysis, the growth of metal on oxide surfaces is an intensively explored area. Most importantly, this has received much attention since the metal deposited on an oxide surface can serve as a simple model system for understanding complex commercial catalysts.51 Various surface-sensitive techniques were used to extensively investigate the growth patterns, factors influencing the growth, and electronic properties of these hybrids.52 Surface science techniques conducted within UHV environments allow the precise construction of the hybrid interface and subsequent investigation of composition, structure, and, electronic properties.53 The atomically clean surfaces, a prerequisite for UHV analysis, could be achieved by various techniques, such as electropolishing, chemical etching, sputtering and subsequent annealing.54 These well-defined, single-crystal semiconductor surfaces provide an ideal platform for the deposition of metal atoms. The controlled growth and organization in the deposition are significant in achieving defined hybrid interfaces. Comprehensive characterizations of formed interfaces can establish an accurate correlation between their atomic and electronic structure and chemical reactivity.Well-defined hybrid interfaces can be designed through various deposition methods. This comprises a range of techniques – from thermal deposition to light-induced photo deposition for site-selective growth. Physical vapor deposition (PVD) techniques such as thermal evaporation55–60 and sputtering25,61–63 can be employed for situations requiring accurate film thickness and composition control. Thermal evaporation involves heating the target (e.g., resistively via electron beams or lasers)39 to induce sublimation and subsequent condensation onto a cooler substrate.64 Sputtering, in contrast, bombards the target with high-energy ions (often argon) from a plasma source, ejecting atoms that deposit as a thin film. Sputtering offers distinct advantages over evaporation enabling deposition of high melting point materials, superior control over film stoichiometry, and the ability to deposit insulators. Both of these methods allow the deposition of a wide range of materials while offering excellent control over film uniformity.65,66 For instance, PVD allows for the deposition of a heterojunction layer, enhancing the device's charge separation efficiency.
Chemical vapor deposition (CVD) is another technique that offers similar advantages in precise control and allows for deposition on complex geometries.67 This technique controls a series of gas-phase reactions, transforming precursors into a solid film on a heated substrate.68 CVD is an effective method for constructing metal-decorated structures with clean interfaces, unlike other methods, such as photo-deposition, which often requires surfactants.69 The clean interface contact can maximize the photocatalytic activity. Various advanced CVD methods include atmospheric-pressure CVD (APCVD), low-pressure CVD (LPCVD), and plasma-enhanced CVD (PECVD), which have advantages over the traditional one.70 For example, PECVD uses plasma to activate precursor molecules, lowering the deposition temperature compared to thermal CVD. This helps integrate temperature-sensitive materials into hybrid structures. However, this method has some limitations, including safety concerns with some precursors and limited precursor availability for specific materials.68
Unlike CVD, atomic layer deposition (ALD) relies on the alternating introduction of gaseous precursors, enabling a self-limiting surface reaction mechanism for controlling film thickness.70 This high level of control translates to highly conformal films ensuring constant coverage even on complex, high-aspect-ratio structures. Precursors in ALD must exhibit high reactivity, possess surface termination groups generating volatile byproducts upon reaction with the substrate, and display a self-limiting surface reaction mechanism. Common precursors include metal halides, alkoxides, hydrides, and metal organics, while oxygen and nitrogen sources are employed for depositing oxides and nitrides, respectively.70 The uniformity and conformality of deposited films with ALD ensure consistent performance across the overall surface.71 The technique's low-temperature nature minimizes thermal stress on sensitive substrates and promotes film adhesion. However, the slow growth rate compared to other methods and the wastage of a significant portion (up to 60%) of the precursor material are drawbacks of ALD.71
Photodeposition, also known as photoreduction or photochemical chemical deposition, is another method employed in fabricating hybrid surfaces. It uses light-mediated control to initiate and progress deposition.72 Photodeposition offers a simple, environmentally friendly alternative to complex material synthesis methods requiring only basic reaction setups and mild conditions.
New interfaces created through these deposition procedures require various surface-sensitive techniques that can precisely define the interface between a substrate and deposited materials. These techniques, which rely on different fundamental principles, are extensively discussed in numerous books57,73–78 and reviews.79–84 Techniques like low-energy electron diffraction (LEED),58 Auger electron spectroscopy (AES), and scanning tunneling microscopy (STM)85 play a central role in characterizing substrate surfaces prior to deposition. LEED analysis provides visualization of diffraction patterns and the crystalline structure of the surface, typically employing electrons of energy 10–500 eV.86 Visual inspection of the LEED pattern alone provides insights into the surface unit cell and long-range order,58 revealing details such as surface defects86 and surface reconstructions that can significantly influence the growth mode of metal islands.87 Using AES as a complementary technique to LEED can characterize the surface elemental composition, revealing vital information like oxygen vacancies and surface contaminants.88 LEED imaging, as illustrated in Fig. 2 for ultra-thin Cu films on TiO2(110), can study the geometrical structure and growth of metal films on metal oxide surfaces.89 As Cu coverage increases beyond 7 Å, substrate spots diminish while the hexagonal Cu pattern intensifies.89 Notably, the rectangular TiO2(110) diffraction pattern completely disappeared for Cu coverages exceeding 50 Å. The LEED study revealed initial layer-by-layer growth followed by Cu atom agglomeration upon reaching a critical thickness and the preferential alignment of Cu atoms along the [110] direction.
 | ||
| Fig. 2 The evolution of LEED patterns from a TiO2(110) surface as a function of deposition thickness dCu of Cu deposited at room temperature. (a) dCu = 7 Å, Ep = 123.4 eV; (b) dCu, = 15 Å, Ep = 124.2 eV; (c) dCu, = 50 Å, Ep = 122.8 eV; (d) the (c) surface flash heated at 160 °C for 5 min. Ep = 119.6 eV.89 Adapted with permission from ref. 89. Copyright 1989 Elsevier. | ||
Analogous to LEED, in situ reflection high-energy electron diffraction (RHEED) measurements provide real-time data on the progressive changes in the film's microscopic structure as it grows.90 It also gives the relative positions of atoms on the surface, like LEED, with diffraction of electrons in the 10–100 keV range (high energy electrons).91 It provides detailed information about the early stages of metal growth, including epitaxial relationships and crystallographic parameters up to the formation of complete overlayers.92 RHEED's high surface sensitivity allows for precise investigation of strain relaxation processes. It can accurately measure lateral lattice parameter changes and monitor growth mode through intensity oscillations of RHEED spots.93
At the same time, STM provides unparalleled atomic-scale resolution, visualizing surface defects and adatom positions. It directly reveals local processes during early growth stages and enables the study of kinetics and morphology of both single adatoms and larger islands.94 As a direct imaging technique, STM is best suited for studying the nucleation behavior of metals on model catalysts. An STM study of the initial growth stages of Fe/GaAs(110) revealed the preferential adsorption sites of Fe adatoms on the As-rich surface, which provides crucial information on their diffusion and nucleation behavior.95 This technique's ability to directly visualize film morphology under various deposition parameters, including temperature and subsequent annealing treatments, makes it invaluable for investigating the evolution and structure of interfaces.89 Variable-temperature STM studies can probe growth from submonolayer to multilayer regimes. This technique has revealed surface alloy formation between Cu and Ag, even at temperatures as low as 300 K.96
Due to its sensitivity to the outermost atomic layer, low-energy ion scattering (LEIS) is employed to elucidate model surfaces’ structural and compositional evolution.97 Using LEIS and STM, the growth mode of Ag metal on TiO2 was investigated. A linear correlation between the LEIS peak area and metal exposure indicates layer-by-layer growth, while a nonlinear relationship suggests a 3D growth.98
Insights into the electronic excitations at the interface, revealing the interaction between deposited metal atoms and surface defect sites, can be gained from electron energy-loss spectroscopy (EELS) analysis. EELS measures the energy distribution of electrons that have lost energy upon interacting with a sample.99 EELS analysis revealed a strong interaction between deposited copper and the MgO surface during the initial stages of film growth.100 Combining LEED and EELS results, the growth mode was elucidated, and Mg2+ vacancy sites were identified as nucleation centers, further confirming the strong interaction observed between copper and the MgO surface.100
X-ray photoelectron spectroscopy (XPS) is a spectroscopic technique for studying interface electronic properties that utilizes X-ray photons to eject core electrons from a surface. XPS is widely used to monitor growth processes by tracking changes in signal intensities, including the diminishing of substrate signals and the emergence of adsorbate signals.93 Unlike EELS, which probes electronic excitations using a beam of electrons, XPS directly analyses the core-level electron binding energies.88 It's an excellent technique for studying the chemical interactions between the deposited metal and substrate since the binding energy shifts directly indicate changes in the electronic properties of both metal and substrate.98 The electron transfer and band-bending phenomena that occur at the interface can be investigated using this technique.101,102 The initial growth mode and subsequent morphological evolution of metal films on oxide substrates were studied as a function of substrate temperature, deposition rate, and surface defects, and a kinetic model for this process was developed with the combined application of XPS and LEIS.59 The balance of interfacial and surface energies plays a significant role in determining the growth mode of metal islands on a substrate material.103 These surface-sensitive techniques have enabled a deep understanding of the surface structure and its evolution, including the synergy between metal adatoms, their diffusion on the semiconductor surface, and the subsequent nucleation and growth of metal islands in model systems.104 An in-depth understanding of the growth processes at the metal–semiconductor interface can help with its precise fabrication. The deposition and annealing temperatures influence the metal film's thermal stability and morphology.105 As metal atoms get deposited on the semiconductor surface, their interaction strength with the underlying atoms dictates their mobility.106 Factors like surface contaminants and the kinetic energy of the incoming metal atoms can significantly affect growth behavior. The substrate temperature can also significantly influence the orientation relationship of metal with the support.107 Metal island sizes, densities, and size distributions can also be found to be controlled by deposition temperature and deposition rate for a variety of different metals deposited on oxide surfaces.51 Numerous studies explored the growth of metal on well-defined metal oxide surfaces, highlighting its importance.90,108,109
A clear picture of the structure–function relationship can be explained by correlating the atomic structure with the comprehensively characterized surface by employing surface-sensitive techniques with reactivity work. The interaction of the reactant with the catalyst surface is of primary importance in catalytic reaction systems. Recent studies have revealed that the stability of reaction intermediates on the catalyst surface dictates the reaction pathway and product selectivity.110 Small-molecule probes for surface adsorption studies give detailed insights into surface–adsorbate interaction, including surface structure, active sites, and reaction mechanisms that are often challenging to study with other techniques. The next section will discuss how small molecules are used for current model systems and their applicability to hybrid systems.
3. Small molecule probes for investigating surface chemistry
Small molecule adsorption is a well-established technique for investigating the surface chemistry and reactivity of catalyst materials. In hybrid interfaces, adsorption is advantageous because it allows for precisely characterizing the local atomic arrangement, chemistry and electronic properties of the complex surfaces.111 Various adsorption techniques, such as temperature programmed desorption (TPD), temperature programmed reaction (TPR), and reflection absorption infrared spectroscopy (RAIRS), provide complementary information. Various probe molecules are used, each providing distinct information about the surfaces. Carbon monoxide (CO) is a popular probe due to its unique interactions with surfaces arising from its electronic and vibrational characteristics.112 The lone pair electrons in CO readily engage with metal surfaces, serving as a potent tool for studying surface electronic properties.113 A large body of work has been done on CO adsorption on well-defined single crystal surfaces using techniques such as RAIRS and TPD.114–119RAIRS offers a detailed analysis of a molecule's vibrational spectrum upon adsorption, providing insights into its bonding geometry and surface electronic properties.111,120 A characteristic peak in the RAIRS spectrum corresponds to a specific adsorption configuration on a flat, uniform surface. In contrast, adsorption on defects or steps can shift the peak position or cause additional peaks to appear.121 This difference arises from the change in the bonding between the molecule and the substrate.122 These spectral patterns distinctively identify different adsorption sites on the surface, each characterized by specific binding energies and geometric configurations. TPD provides complementary information to RAIRS and involves monitoring the desorption process as the sample temperature increases. The concentration of desorbing molecules is observed as a function of temperature, revealing the strength of the interaction between the adsorbed molecule and the surface.123–125 A single desorption peak in the TPD profile signifies a homogeneous surface with a uniform binding energy for the adsorbate.93 Conversely, a broader profile with multiple peaks indicates a heterogeneous surface with diverse binding sites.126 TPD enables the kinetic study of various rate processes on solid surfaces, making it invaluable for investigating surface phenomena such as adsorbate-induced surface reconstruction.127
A comprehensive understanding of the surface structure and its interaction with CO molecules, which is then correlated with known catalytic functionalities of the material, can be achieved by combining TPD and RAIRS. For example, studies comparing CO adsorption on Pt(111) and Pt(100) surfaces have revealed a higher tendency for CO dissociation on Pt(100), a crucial step in many catalytic reactions, and is attributed to the presence of undercoordinated surface atoms on Pt(100) that weaken the CO bond.128 The investigations on the influence of surface composition on CO adsorption behavior offer another valuable dimension to this characterization technique. Studies employing RAIRS and TPD have explored how the presence of additional metals can modify the CO adsorption behavior on bimetallic surfaces. For instance, compared to pure Cu(100), CO adsorption on Pd–Cu(100) bimetallic surfaces exhibits a lower wavenumber peak in the RAIRS spectrum, indicating a stronger CO–metal interaction. The corresponding TPD profile has a higher desorption temperature, further confirming the enhanced binding strength (Fig. 3).112 The IR observations provide insights into how the electronic structure of bimetallic catalysts can influence CO back-donation and, consequently, the CO bond strength.129 Correlating specific CO adsorption behaviors with known catalytic functionalities can help analyse how surface structure influences reaction mechanisms. For example, the previously mentioned study comparing Pt(111) and Pt(100) surfaces showed a higher CO dissociation on Pt(100) due to weaker CO binding.128 This aligns with undercoordinated surface atoms, facilitating CO dissociation at lower temperatures. O desorption behavior from TPD can be connected with the catalyst's effectiveness for reactions requiring CO dissociation as a step in the reaction pathway.
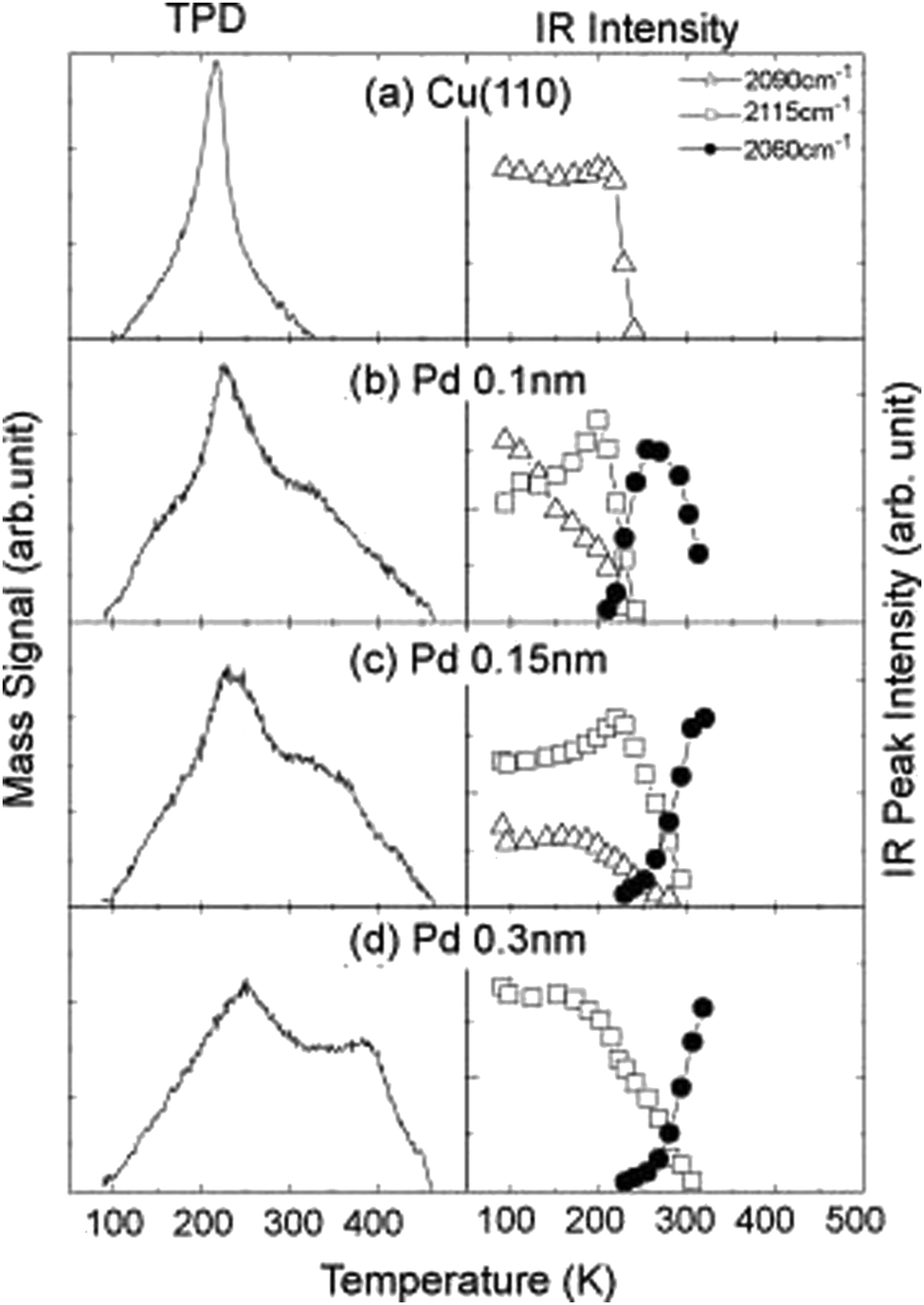 | ||
| Fig. 3 The TPD spectra (left) and RAIRS peak intensity changes (right) with elevating the substrate temperature for the 1.0-Langmuir (L)-CO exposed surfaces: (a) clean Cu(110), (b) 0.1-nm-thick-, (c) 0.15-nm-thick-, and (d) 0.3-nm-thick-Pd/Cu(110). Heating rate for TPD measurements: 3 K s−1.112 Reprinted with permission from ref. 112. Copyright 2008 Elsevier. | ||
By incorporating molecules with diverse chemical properties, the probe molecule beyond CO can offer complementary information on surface chemistry and catalysis. Nitric oxide (NO) is an alternative probe molecule with a weaker dipole moment.122 TPD examined NO dissociation on Pt(100), Pt(411), and Pt(211) surfaces to study the influence of surface structure (beyond orbital symmetry) on NO dissociation. These surfaces possess nearly identical orbital symmetries but differ in active site densities. The TPD profiles revealed compelling differences despite comparable NO dissociation (66–70%). The study showed that while orbital symmetry plays a role, factors like site density and local atomic arrangements (e.g., steps vs. terraces) significantly influence binding energies and reactivity.130 Thermal desorption can be used to characterize surface defects by analysing the desorption profiles.131 TPD studies on Cu(100) revealed the dependence on surface defects in water adsorption. Clean Cu(100) surfaces exhibit a single desorption peak at 162 K, indicative of three-dimensional, ice-like water clusters forming even at low coverages. This suggests an interaction between water and the pristine Cu(100) surface.132 However, introducing defects through sputtering creates additional binding sites, leading to a new desorption peak at 177 K attributed to water clusters bound at these defect sites. These water clusters exhibited stronger hydrogen bonding than those on defect-free surfaces, as revealed by high-resolution electron energy loss spectroscopy (HREELS). This observation aligns with the notion that defect sites can enhance molecule–surface interactions by providing localized regions with altered electronic properties.133 A comparative analysis of TPD studies on Pt(553) and TiO2(110) surfaces can further emphasize the influence of surface composition on water adsorption.134,135 Pre-adsorbed oxygen atoms (Oad) on Pt(553) significantly influenced the water desorption profile. Multiple peaks suggest the formation of various water/OH hydrogen-bonded networks, revealing the synergy between water and pre-adsorbed species. Adsorbed oxygen can act as anchoring sites for water molecules, altering their bonding configurations and desorption behavior.134 In contrast, TPD results on TiO2(110) suggest that water primarily adsorbs molecularly at low exposures, with minimal dissociation to form hydroxyl groups.133 This behavior contrasts with Pt(553), where water dissociation is more prevalent at high coverages.134 These observations emphasize the dependence of water interaction on the surface chemical nature. The co-adsorption study of CO and H2O on Au(310) illustrated how water adsorption could modify the surface reactivity towards other adsorbates with isotopic labeling (13CO). The presence of water hinders CO oxidation at lower temperatures, likely due to forming a hydration shell around reactive oxygen species generated from water dissociation.126 The initial stages of water adsorption on weakly interacting surfaces were studied using STM on Au(111), which showed the formation of planar and amorphous water monolayer films at low temperatures. Interestingly, water clusters were only observed on top of this amorphous film, suggesting its role as a precursor for multilayer formation.136 Another example of TPD's ability to investigate surface reactivity comes from the observation of water dissociation on SrTiO3(100) surfaces. Molecular adsorption on stoichiometric surfaces suggests the role of bridging oxygen atoms in facilitating water dissociation.137 Bridging oxygen atoms aids water dissociation by forming hydrogen bonds with water molecules, weakening the O–H bonds. This effect is more pronounced on surfaces like TiO2(100), where multiple hydrogen bonding interactions can occur. In contrast, surfaces like TiO2(110) and SrTiO3(100) exhibit weaker water adsorption and limited dissociation due to the absence or reduced number of bridging oxygen atoms.137 TPD has been employed to investigate various molecular adsorption and desorption behaviors on catalyst surfaces, including O2,127,138–140 H2,141–143 N2,144–146 CH3OH,135,147,148 and others.
Insights on how these small molecules interact with catalyst surfaces are crucial in heterogeneous catalysis. Extensive research has demonstrated the potential of adsorption studies in elucidating surface structure and reactions. While this section focuses on non-light-activated heterogeneous systems, the information from these studies is valuable in investigating complex hybrid materials using desorption techniques. The distinct desorption behavior revealed by CO-TPD corresponded to different primary surface sites that are important for reactant activation.53,149 Analyzing the position and intensity of the TPD peaks can distinguish the type of reaction sites. Additionally, H2-TPD provides valuable insights into the interaction between metal and semiconductor components. For example, H2-TPD studies on Cu/ZnO catalysts, where hydrogen spillover from Cu to ZnO, provide evidence for metal–support interactions.150 The emergence of a low-temperature desorption peak and the enhancement of a high-temperature peak in Cu-containing catalysts suggest a spillover mechanism. Hydrogen dissociates on Cu or at the Cu–ZnO interface, spilling over to ZnO, where it can adsorb and recombine.150 Similarly, in Mn–Ce oxides supported on TiO2 for formaldehyde oxidation, H2-TPD indicated a greater abundance of readily reducible surface oxygen species, crucial for the reaction.151 NH3-TPD revealed that the acidic site distribution correlated with the observed catalytic behavior.53,152 Correlating TPD results with data from complementary characterization techniques like XPS, AES, and FT-IR provides a comprehensive understanding of how surface structure and composition influence photocatalytic activity.
XPS identifies surface metal species’ oxidation state and electronic structure on hybrid catalysts. In the case of Cu/ZnO catalysts for CO2 hydrogenation, XPS analysis identified the presence of metallic Cu (Cu0) as the primary active species for CO2 hydrogenation. Cu (LMM) AES confirmed the identification of Cu0 in the CO2 hydrogenation study, a technique that can differentiate Cu0 from Cu+ based on subtle differences in their Auger electron energy.149 Fe/ZnO catalysts decorated with hydroxyl (–OH) and alkyl (–R) groups for CO2 hydrogenation, CO2-TPD revealed the creation of strong basic sites upon modification.153 Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis identified surface-bound carbonate species formed during CO2 adsorption, offering valuable clues about the reaction mechanism.150 Similarly, in the study on Cu(111) and Zn–Cu(111) surfaces for methanol adsorption, XPS revealed similar chemical states for methanol on both surfaces. However, the presence of Zn on Zn–Cu(111) surfaces, as evidenced by XPS, modified the interaction between methanol and the surface, potentially by altering the electronic properties or introducing new binding sites, emphasizing the importance of surface composition in influencing small molecule adsorption (Fig. 4).152 Studies on the influence of reduction environments highlight the importance of understanding the spatial arrangement of active sites within the hybrid material and its identification.154 CO-TPD, N2O-titration, and FTIR revealed a dual-site reaction pathway.155 Similarly, reduction modifies the surface morphology, potentially influencing adsorption behavior as identified through TPD analysis.154
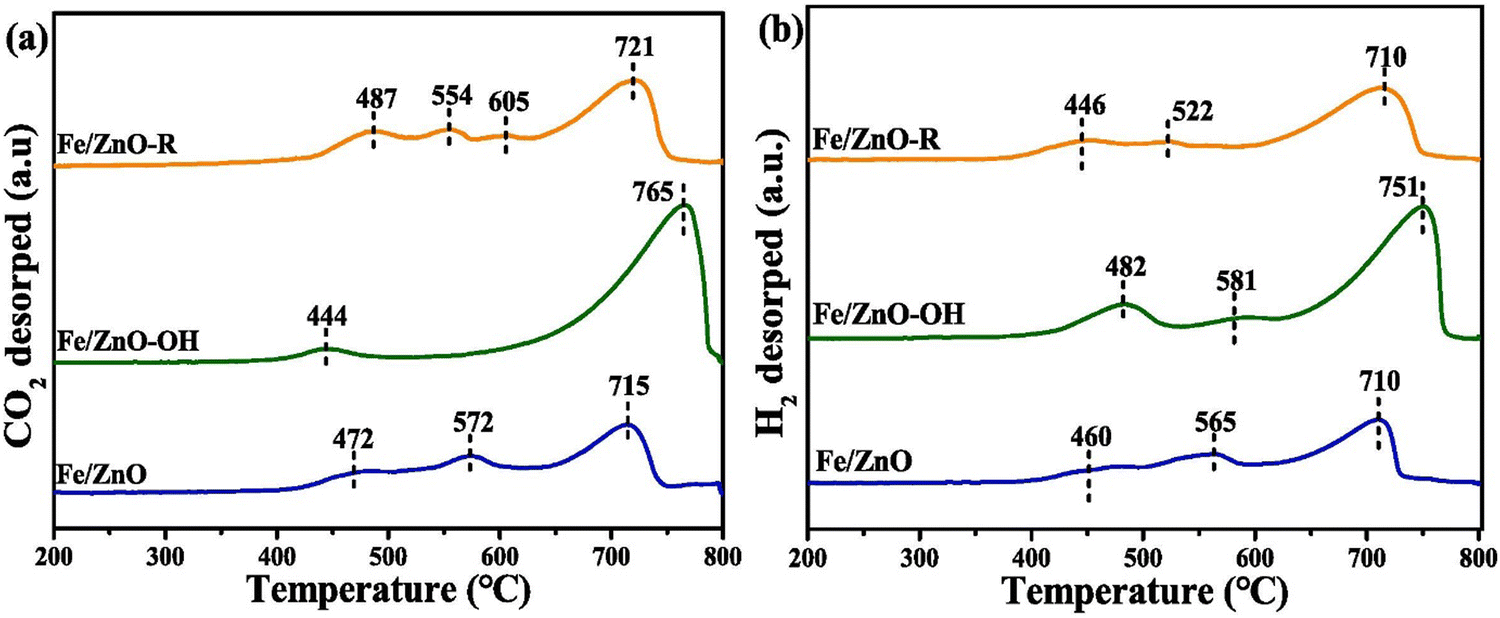 | ||
| Fig. 4 CO2-TPD (a) and H2-TPD (b) profiles of CO-activated catalysts.152 Reprinted with permission from ref. 152. Copyright 2024 Elsevier. | ||
The investigation of Pd/Ag(111) single-atom alloys (SAAs) using CO-RAIRS and TPD also exemplifies the power of small molecule adsorption in characterizing complex hybrid surfaces. The study establishes CO as a probe molecule to identify isolated Pd atoms and quantify their concentration on the Ag surface. The observed CO desorption peaks and their dependence on Pd coverage and temperature reveal the dynamics of Pd aggregation and subsurface diffusion. Additionally, H2-TPD experiments provide insights into H2 dissociation on Pd/Ag(111) SAAs.156 By correlating TPD data with surface composition information from synchrotron X-ray photoelectron spectroscopy (SXPS), the researchers elucidated the dependence of water and CO interactions on ceria oxidation state and the influence of Rh deposition by investigating the adsorption behavior of water and CO on ceria thin films with varying oxidation states.157 Rhodium (Rh) significantly impacted how water interacts with ceria. The observation from TPD is corroborated by SXPS analysis, suggesting minimal interaction between water and oxidized ceria, while reduced ceria can store oxygen derived from water dissociation.157 Rh on oxidized ceria did not alter water adsorption. Still, it facilitated H2 production from hydroxyls on reduced ceria at lower temperatures, implying that Rh promotes oxygen removal from hydroxyls, potentially through hydrogen spillover. CO adsorption studies revealed that the oxidation state of ceria also influences the interaction between CO and co-adsorbed water. Co-adsorbed water enhances CO dissociation and its subsequent reaction with surface oxygen. CO desorption temperature and the extent of dissociation varied with the oxidation state. Notably, co-adsorbed water did not affect the desorption of molecular CO from Rh sites, indicating a lack of interference with CO adsorption on Rh.157
4. Photoabsorbers – surface adsorption studies
The initial step in photocatalysis is the adsorption of reactant molecules onto the catalyst surface. For example, in photocatalytic CO2 reduction, chemisorption can occur via several mechanisms: Lewis acid sites accepting electrons from the CO2 oxygen atom, Lewis basic sites donating electrons to a CO2 carbon atom, or a combination of both, leading to a mixed-coordination structure.158 Following adsorption, multi-electron transfer processes can reduce the molecule to various products such as CO, formic acid, formaldehyde, methanol, methane, ethylene, or ethanol.159 UHV-based surface science studies that use well-defined single crystals can provide direct evidence for the elementary processes of photocatalytic chemistry at the model interfaces.160Small molecule adsorption studies on photocatalytic reactions offer valuable insights into the interaction between reactants, intermediates, and the photocatalyst surface. An important example of this is shown by Friend and co-workers, wherein they employed TPR to assess the photocatalytic oxidation of formaldehyde on rutile TiO2(110) single crystals with a focus on the influence of surface reduction and oxygen adatoms and their results demonstrate a strong correlation between the presence of oxygen adatoms and efficient formate production, the primary product of formaldehyde photo-oxidation on rutile TiO2(110).161 This work highlights the critical role of surface and near-surface defects in photocatalytic formaldehyde oxidation (FCO). TPR revealed that oxygen adatoms (Oad) on highly reduced TiO2 surfaces significantly enhance FCO efficiency. Conversely, bridging oxygen vacancies and subsurface Ti3+ defects hinder the reaction by trapping photogenerated holes.161 The oxygen adatoms on rutile TiO2 have also been shown to play an important role in facilitating the photochemistry of ketones via complex formation and photoactivation on this surface.131 The observed reaction pathway indicated an excited-state mechanism, suggesting the importance of considering excited-state dynamics when studying reactions on TiO2.131 Another study from the same group reported the formation of methyl formate, a product not previously detected in the photo-oxidation of methanol.162 This study demonstrated that temperature plays a significant role in determining the product distribution. At lower temperatures (240 K), the formation of methyl formate is favoured, while at higher temperatures (300 K), formaldehyde is the primary product suggesting that the reaction mechanism involves a competition between different pathways. Oxygen adatoms were found to be take part in the activation of methanol, leading to the formation of methoxy species; however, subsequent photochemical steps involving the oxidation of methoxy and formaldehyde do not require the presence of oxygen adatoms. Instead, the photogenerated holes on the TiO2 surface drive these reactions. Notably, methyl formate formed at low temperatures, was found to be trapped on the surface, which in turn can significantly influence the overall product distribution and the efficiency of the photocatalytic process.162 The significant influence of temperature on reaction selectivity in the photocatalytic oxidation of isobutanol was elucidated with combined TPD and reactivity studies.163 At lower temperatures, the primary photoproduct, isobutanal, can undergo further photooxidation to form propane and CO. However, at higher temperatures isobutanal desorbs before undergoing secondary reactions leading to a different product distribution. This temperature-dependent product selectivity is because the thermal desorption of the initial photoproduct competes with a subsequent photochemical reaction. In the case of isobutanol, the products formed at 240 K significantly differ from those formed at 300 K, emphasizing the necessity of elucidating the elementary steps involved in photochemical reactions.163
Studies employing STM have revealed light-induced surface reconstructions at the atomic scale in transition metal oxides (TMOs), particularly in TiO2. These reconstructions can significantly impact the adsorption behavior of small molecules, thereby influencing photocatalytic reaction rates and selectivities.164 One such study investigated the effect of UV radiation on the (1 × 1) and (1 × 2) terminations of the TiO2(110) single-crystal surface.165 Their findings highlight the dependence of surface structure on UV-induced defect formation. The (1 × 1) surface exhibited remarkable resilience to UV irradiation, with no statistically significant changes observed in surface morphology or pre-existing oxygen vacancy densities. This suggests that under these conditions, UV photons might lack the energy threshold required to dislodge bridging oxygen atoms on the (1 × 1) surface. In contrast, the (1 × 2) surface substantially responded to UV exposure. The characteristic STM revealed the emergence of well-defined line defects propagating along specific crystallographic directions. Due to their distinct topographic profile, these defects are readily distinguishable. The study quantified the efficiency of UV light in creating these defects on the (1 × 2) surface by estimating a cross-section of 10−23.5 ± 0.2 cm2 photon−1. The authors propose a mechanism involving UV-generated excitons, where holes become trapped by oxygen anions in the added rows specific to the (1 × 2) reconstruction while the photogenerated electrons become delocalized. This partial neutralization triggers molecular oxygen's movement and subsequent release. The increased electrical conductivity of the (1 × 2) phase compared to the (1 × 1) phase is hypothesized to enhance exciton-electron delocalization and reduce electron–hole recombination (Fig. 5).165
 | ||
| Fig. 5 Effect of UV irradiation on the TiO2(110)-(1 × 1) surface. (a) 30 × 30 nm2 empty-state STM image of the clean TiO2(110)-(1 × 1) surface at a sample bias voltage of +2.5 V and a tunneling current of 0.035 nA; (b) 10 × 10 nm2 close-up empty-state STM image of the clean TiO2(110)-(1 × 1) surface (V = +2.5 V, IT = 0.035 nA) displaying oxygen vacancies; (c) 30 × 30 nm2 empty-state STM image of the irradiated (60 min, 2.7 W cm−2) TiO2(110)-(1 × 1) surface at a sample bias voltage +2.5 V and a tunneling current of 0.035 nA; (d) 10 × 10 nm2 close up empty-state STM image of the irradiated (60 min, 2.7 W cm−2) TiO2(110)-(1 × 1) surface (V = +2.5 V, IT = 0.035 nA) showing no morphological changes. Effect of UV irradiation on the TiO2(110)-(1 × 2) surface. (e) 100 × 100 nm2 empty-state STM image of the clean TiO2(110)-(1 × 2) surface at a sample bias voltage +3.5 V and a tunneling current of 0.035 nA; (f) 30 × 30 nm2 close-up empty-state STM image of the clean TiO2(110)-(1 × 2) surface (V = +3.5 V, IT = 0.035 nA) displaying a typical cross-linking structure; (g) 100 × 100 nm2 empty-state STM image of the irradiated (60 min, 2.7 W cm−2) TiO2(110)-(1 × 2) surface at a sample bias voltage +3.5 V and a tunneling current of 0.035 nA; (h) 30 × 30 nm2 close up empty-state STM image of the irradiated (60 min, 2.7 W cm−2) TiO2(110)-(1 × 2) surface (V = +3.5 V, IT = 0.035 nA) showing morphological changes.165 Reprinted with permission from ref. 165. Copyright 2003 Elsevier. | ||
Several reviews comprehensively summarize and discuss the surface photocatalytic chemistry of various molecules, including hydrogen, water, oxygen, carbon monoxide, alcohols, aldehydes, ketones, and carboxylic acids.166–169 These findings offer valuable insights into understanding and controlling the adsorption and desorption of small molecules on photocatalyst surfaces. The light-induced creation of surface defects can alter the surface electronic structure and introduce new adsorption sites for small molecules. Depending on the specific defect type and its interaction with the adsorbate, the adsorption strength and geometry can be significantly affected. This, in turn, can influence photocatalytic processes' reaction kinetics and product selectivity. For instance, oxygen vacancies might create stronger adsorption sites for specific reactant molecules, promoting their activation and facilitating the photocatalytic reaction.140 A recent study investigated the influence of temperature and methanol surface coverage on the kinetics of its photocatalytic reaction on the rutile TiO2(110)-(1 × 1) surface.148 This reaction is proposed to be a step-wise reaction with the initiation of the photocatalytic oxidation from methanol, and the rate of the response is affected by temperature, light intensity, and the coverage of molecules. The combined application of STM and TPD resolved conflicting observations regarding product formation, underscoring the significance of the reverse reaction. The study also revealed that light intensity can be optimized and that the kinetics of response transition from 1D to 2D fractal behavior as coverage increases.148
Due to the complex physical, chemical, and electrical processes involved in solar energy conversion reactions, an in-depth understanding of the mechanism is essential for designing efficient materials for PC and PEC. Small molecule probes are shown to be valuable tools for this purpose. Au clusters on rutile–TiO2(110) surfaces catalyze photocatalytic hydrogen production from methanol.170 Using deuterated methanol, the study revealed that Au clusters efficiently dissociate methanol at low temperatures (below 150 K) (Fig. 6). The adsorption and dissociation of deuterated methanol on Au/TiO2 surfaces. By analyzing the desorption spectra, the researchers observed that gold clusters significantly lowered the temperature required for methanol dissociation, allowing efficient dissociation at temperatures as low as 150 K, revealing that gold clusters promote the activation of methanol molecules and reduce the energy barrier for bond breaking.170
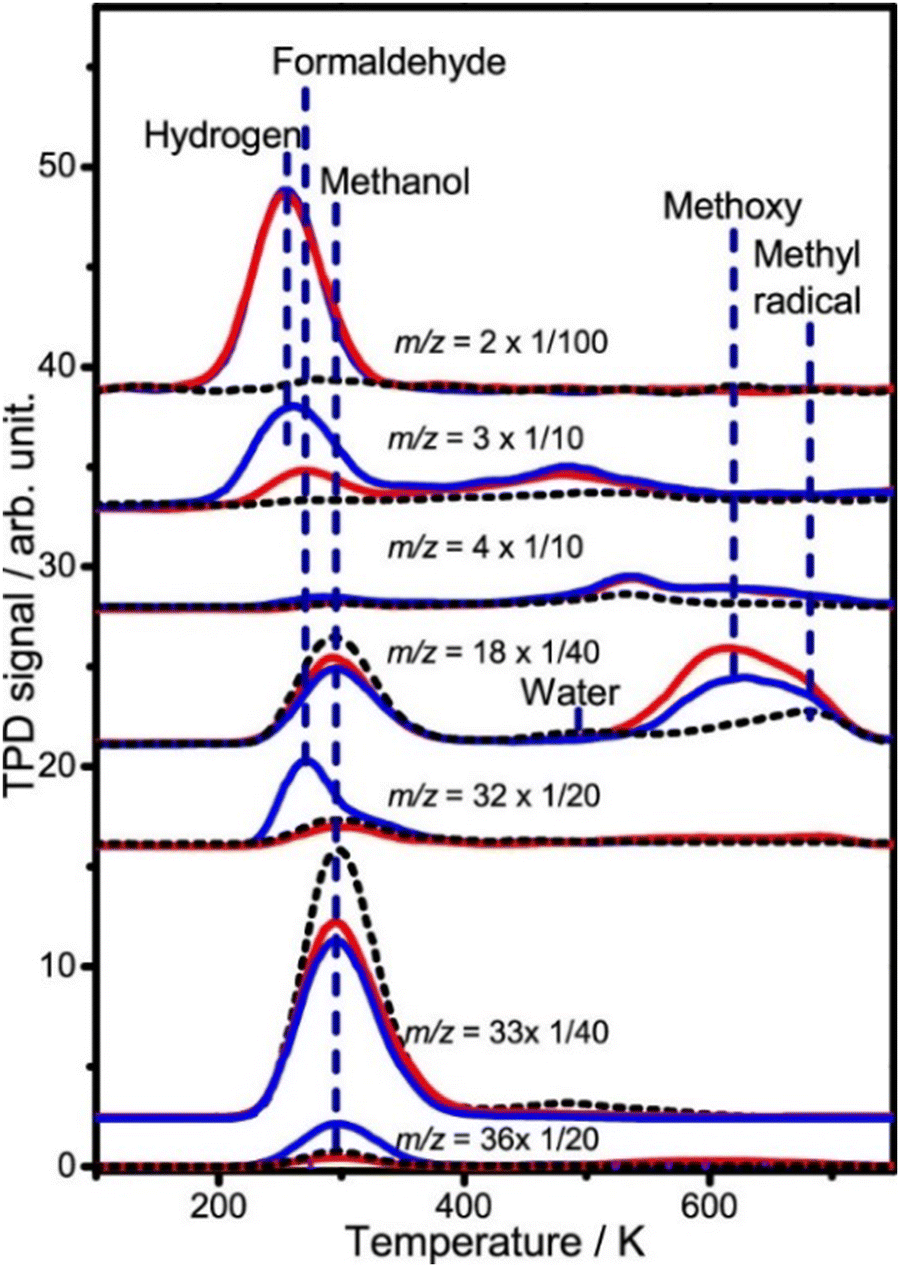 | ||
| Fig. 6 TPD spectra acquired at m/z = 2 (H2+), 3 (HD+), 4 (D2+), 18 (CD3+, H2O+), 32 (CD2O+), 33 (CD2OH+), and 36 (CD3OD+) after adsorbing 0.65 ML CD3OH on Au/R–TiO2(110) surfaces followed by irradiating the surface for 0 (red lines) and 10 min (blue lines) at 100 K. The black dash lines show the TPD traces of these masses collected after adsorbing 0.65 ML CD3OH on the clean R–TiO2(110) at 100 K. The Au/R–TiO2(110) surface was prepared via depositing 0.1 ML Au atoms on R–TiO2(110), followed by annealing to 800 K for 1 min. The photon flux of 355 nm light is 1.3 × 1017 photons cm−2 s−1.171 Reprinted with permission from ref. 170. Copyright 2020 American Chemical Society. | ||
Another important study on Pt/TiO2(110) also shows the significance of detailed surface structure investigations using small molecule adsorption.160 TPD (Fig. 7) and UPS analyses demonstrated that Pt deposition alters the surface structure and modifies reactant/product interactions. Pt preferentially adsorbs at oxygen vacancy sites, facilitating dissociative methanol adsorption and promoting water desorption at lower temperatures. This enhances hydrogen production and potentially hinders H recombination with formaldehyde, leading to increased reaction.160
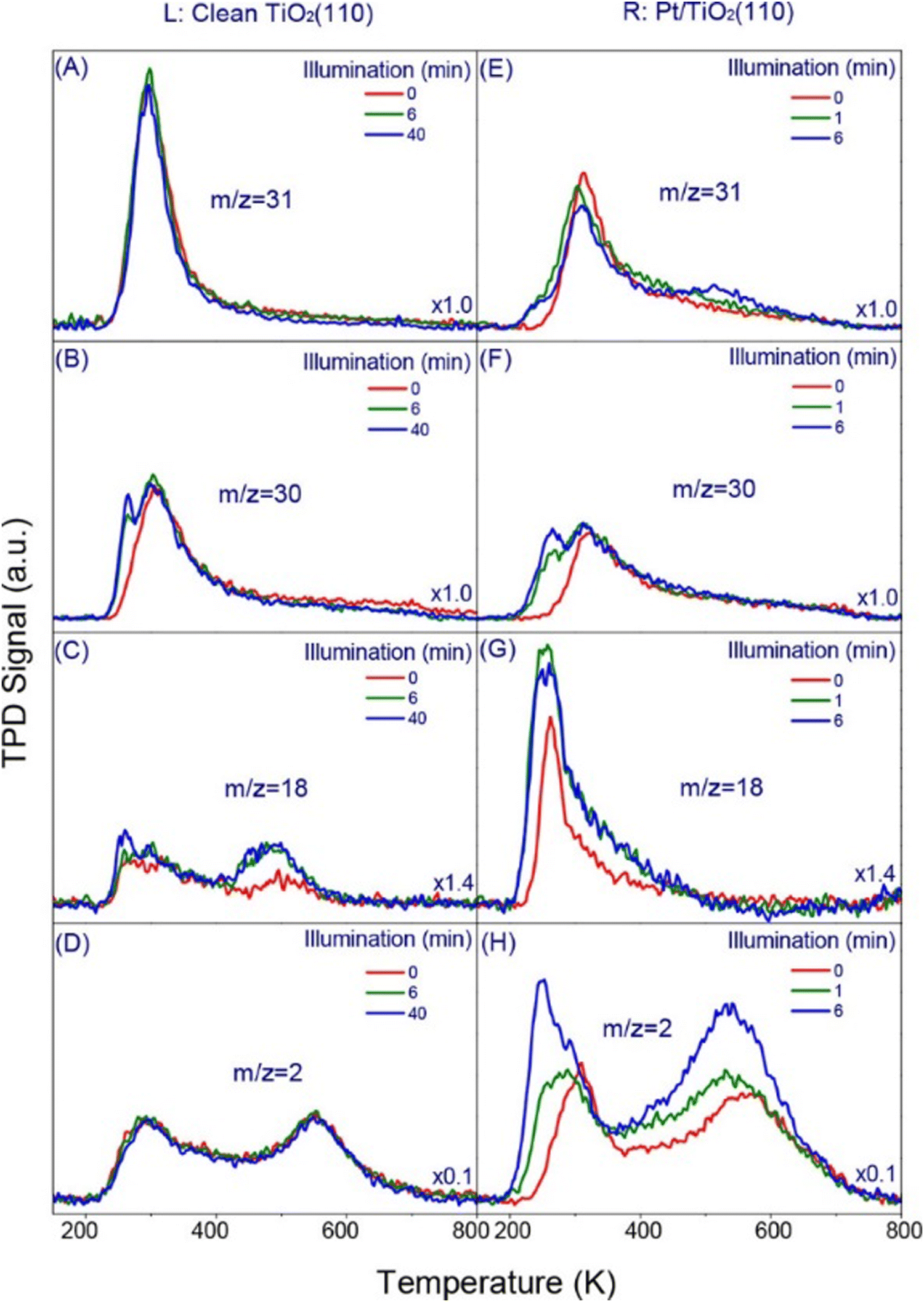 | ||
| Fig. 7 TPD spectra from 0.5 ML methanol covered (L) clean and (R) 0.058 ML Pt-loaded TiO2(110), respectively, as a function of 380 nm light illumination. The average flux of UV irradiation is 1.6 × 1018 photons cm−2 s−1. Four masses (m/z = 31 (A and E), 30 (B and F), 18 (C and G), and 2 (D and H)) representing the main products and/or their fragmentation from the photocatalytic chemistry of methanol on clean and Pt-loaded TiO2(110) are shown. The scales in all the figures are the same while the signal in each figure is multiplied by a value specified at the right bottom corner. Please note the representative illumination time in the left and right panel are different.160 Reprinted with permission from ref. 160. Copyright 2018 American Chemical Society. | ||
To distinguish between thermally driven and photo-driven reaction pathways, a combination of isothermal photoreactions, TPD, and post-irradiation TPDs were used in the photocatalytic conversion of methanol on Pt/TiO2(110).147 Interestingly, the photo-oxidation of methoxy on TiO2 was independent of Pt co-catalyst loading within the investigated range (1%), suggesting that photo-oxidation is the rate-determining step in hydrogen evolution from methanol, contradictory to previous observations.160 TPD experiments in this study offer kinetic evidence that the photocatalyst's surface reaction kinetics are significant in governing the overall conversion and selectivity observed during photocatalytic reactions.147
Furthermore, modulating Pt co-catalyst facet geometry and employing F− pre-admission improves hydrogen generation from water splitting.171 F− pre-adsorption on Pt(111) facets effectively inhibits H2/O2 recombination, boosting hydrogen generation. This is due to F− preferentially occupying top sites on Pt(111), blocking nearby bridge sites crucial for H2 and O2 adsorption and activation. DFT calculations and experimental results confirm this mechanism. In contrast, pristine Pt/TiO2 suffers from rapid recombination, leading to negligible H2 production. However, F− incorporation, especially on Pt(111), significantly enhances photocatalytic activity by suppressing recombination. Isotope labeling experiments verify that the generated H2 and O2 originate solely from water splitting. XPS and TPD of H2 and O2 confirmed weaker H2/O2 adsorption on F/Pt(111)/TiO2 compared to F/Pt(100)/TiO2.171 All this research highlights the growing interest in using small molecule adsorption techniques to understand light-induced reactions using well-defined hybrid systems.
5. Electrocatalysts – surface adsorption studies
Throughout the earlier portions of this perspective, there has been a significant focus on studies under UHV conditions; however, adsorption at the metal or semiconductor/liquid interface is deeply consequential to the desired catalytic or photocatalytic outcome. Reactant adsorption is the initial step in surface redox reactions, generally occurring upon contact between semiconductor photoelectrodes and electrolytes.172 Several factors, such as the surface interaction with solvent molecules, must be considered when discussing adsorption behavior. Importantly, adsorption at the electrode surface is a replacement reaction and a high electric field localized close to the electrode surface is possible even with a low external voltage.173 Various factors affect this adsorption process, including solvent (solvent displacement, desolvation),174 applied potential,174 pH,175 and temperature176 (Fig. 8). The atomic arrangement of the surface significantly impacts the adsorption of reactant molecules.177 This initial adsorption influences subsequent adsorption events and can affect the pathways for charge carrier transport.178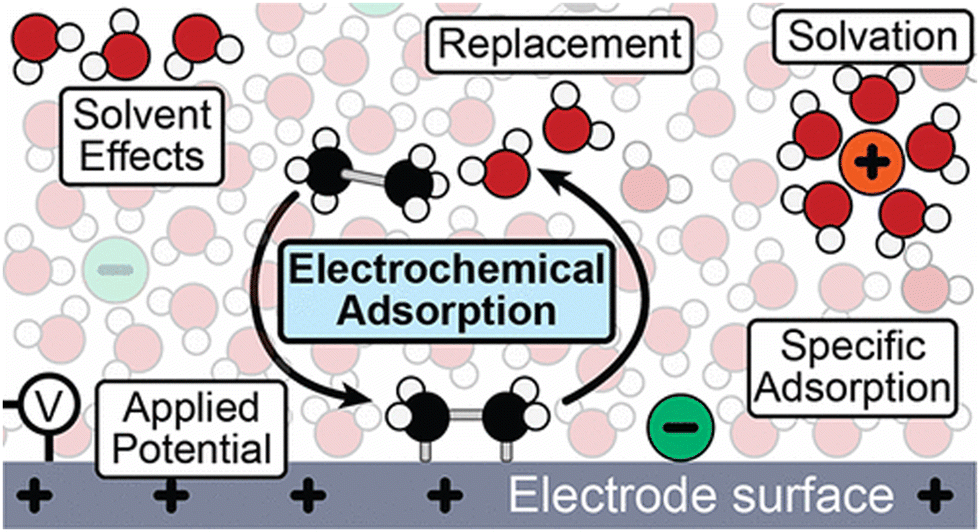 | ||
| Fig. 8 Schematic of the electrochemical interface showing the interactions that must be considered in electrochemical adsorption processes. Not to scale.174 Reprinted with permission from ref. 174. Copyright 2024 American Chemical Society. | ||
Well-defined single crystal electrodes are essential in systematically studying the structure–reactivity relationship.179 Numerous studies have demonstrated the effect of surface adsorption in electrochemical reactions employing model surfaces.180–182 Studies conducted by Feliu and co-workers have revealed the electrochemical behavior of surface adsorption on well-defined platinum surfaces.183 The voltammetric profile of adsorbed CO on Pt(111) in the acidic solution has revealed complex adsorption behaviors of CO and sulfate anions influenced by surface order and charge density.184 It showed two distinct distributions: type I, where small, easily oxidizable CO islands allow for small CO-free domains at low coverage, and type II, appearing at high coverage that coexists with type I at intermediate coverage. Type II forms large, compact CO domains with significant CO-free (111) areas, even at similar overall CO coverage to type I. Both types exhibit a mixture of linearly and bridge-bonded CO, with linear bonding dominating when hydrogen adsorption sites are entirely blocked. The maximum CO coverage appeared to be one CO molecule per Pt atom.184 The desorption of sulfate anions on Pt(111) electrodes is unusual due to its sensitivity to surface order and unique charge density.185 Two distinct processes occur that may be influenced by capacitive and faradaic processes. The charge densities involved in sulfate anion desorption were investigated by studying CO adsorption and comparing it to voltammetric measurements. While the exact nature of these processes remains unclear, the impact of surface order on the bonding of sulfate anions to the Pt(111) surface was elucidated.185 In investigating the oxidation of CO on a Pt single crystal surface, it was also found that CO preferentially adsorbs on step sites, blocking hydrogen adsorption.186 Surfaces with higher step densities, such as Pt(553) and Pt(554), exhibited lower overpotentials for CO oxidation compared to Pt(111). The increased catalytic activity of Pt(553) and Pt(554) is due to the increased formation of oxygen-containing species on step sites.186 Step sites on Pt(111) surfaces promotes the formation of oxygen-containing species, thereby accelerating the electrochemical oxidation of CO adlayers.187 The kinetics of this CO oxidation followed a Langmuir–Hinshelwood mechanism involving the reaction between adsorbed CO and OH species. At low CO coverages, the reaction proceeds through a different mechanism, possibly involving direct oxidation of CO by water molecules. Additionally, surface defects can act as active sites, influencing the kinetics of CO oxidation.187 Another recent study provides a detailed analysis of the electrochemical behavior of a Pt(100) single-crystal electrode.188 By employing techniques like graphene modification and studying stepped surfaces, distinct adsorption/desorption processes for hydrogen, hydroxyl, and anions at specific crystallographic sites are identified. Larger cations, like K+ and Cs+, can interact with adsorbed hydroxyl species, affecting their adsorption energy and peak potential. Anions, such as sulfate, can specifically adsorb on the Pt(100) surface, competing with hydroxyl adsorption and shifting the corresponding peaks to higher potentials.188
EC single-crystal studies are vital in interpreting the reaction mechanisms in electrochemical environments. For example, the methanol oxidation reaction (MOR) on Pt single-crystal electrodes using cyclic voltammetry (CV) and isotopic labeling with CD3OH revealed that the reaction mechanism involves the adsorption of methanol aided by OH species, followed by the rate-determining step of C–H bond cleavage.189 Step sites, particularly (110) steps, improved the reaction rate by promoting OH adsorption. The kinetic isotope effect (KIE) observed using CD3OH confirms the importance of C–H bond breaking in the reaction pathway.189 CV measurements were used to characterize the adsorption/desorption of hydrogen and OH species on the Pt surface and to monitor methanol oxidation and its intermediates.189 The adsorption of OH species on low-coordinated Pt step sites at low potentials shows that OH adsorbs on these sites regardless of the step geometry or pH, with higher coverage on (110) steps than (100) steps. The potential of zero total charge is influenced by the adsorption of species on the step sites, particularly for surfaces with longer terraces.190 One latest study investigated the effect of pH and KIE with CV to understand another alcohol oxidation reaction, formic acid oxidation (FAOR).191 This study employed acetate adsorption as a model for formate adsorption (owing to the similarities between the adsorption behavior) to better understand adsorption at the interface and the role of competing adsorbates like the acetate in the FAOR mechanism.191
Bimetallic surfaces – a step towards hybrid surfaces – have also been shown to exhibit improved electrocatalytic activity. Sub-monolayer coverage of Sn on platinum single-crystal electrodes in acidic media enhances ethanol oxidation.192 Platinum surface structure dictates the optimum Sn coverage, the Sn/Pt(110) system with the highest activity. Pt surface structure was also found to decide its further oxidation from acetaldehyde, with the C–C bond breaking on Pt(110), forming CO2, whereas Pt(111) lacked such sites, forming acetic acid.192 A detailed study on the Pt/Ru(111) system under electrochemical conditions investigated surface adsorption by elucidating a correlation between voltammetric behavior and the electrode surface process. It revealed the altered adsorption behavior of Pt(111) with Ru deposition (Fig. 9). Notably, the electrochemical behavior of Pt/Ru(111) is similar to Ru(0001).193 Interestingly, recent CV investigation results on the influence of glassware on Cu single-crystal electrodes have shown that the dissolved glassware can significantly alter the results, highlighting the need to avoid them in EC analysis.194
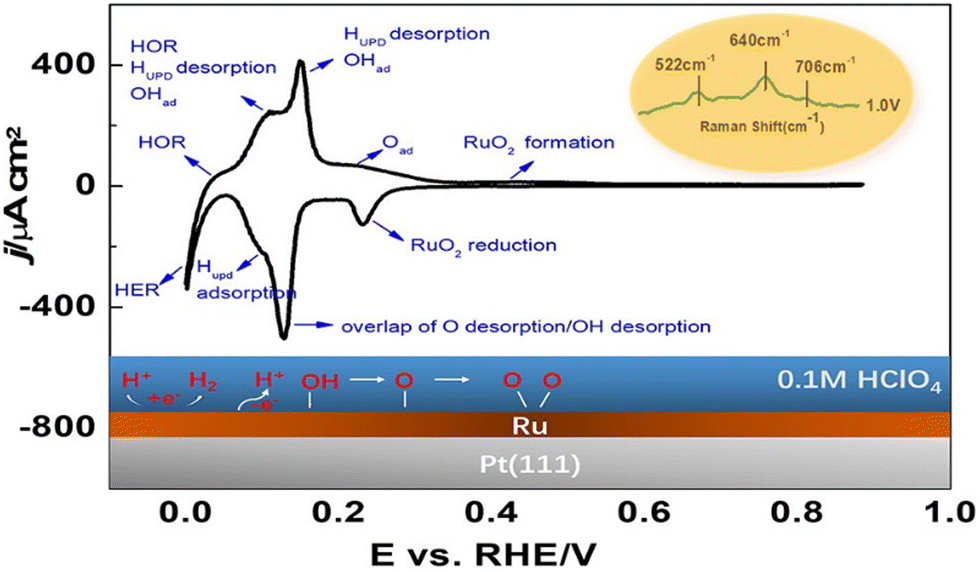 | ||
| Fig. 9 Depicted surface processes on Ru/Pt(111) in 0.1 M HClO4.193 Reprinted with permission from ref. 193. Copyright 2022 American Chemical Society. | ||
A combination of experimental CV analysis and ab initio simulations illustrated the facet-dependent hydroxyl (OH) feature, a clear electrochemical distinction between different Cu crystallographic planes.194 The structure- and electrolyte-reactivity correlations are comprehensively summarised, discussing the effect of metal nanoparticle size, loading, composition, stability, support, defects, and crystal facets.110 Unifying factors have been illustrated in the reactivity of surfaces in UHV and electrochemical environments.195 However, in-depth research is still needed to comprehensively understand electrode surface reactivity, considering the influence of additional factors on surface adsorption.
These works highlight the significant role of electrode surface adsorption and the importance of well-defined materials in understanding the crucial surface reactions in electrocatalysis. Many studies emphasize the critical role of atomic-scale surface structure with in-depth investigations in understanding photo and electrocatalysis on single-crystal metal surfaces.159,196–198
6. Photoelectrocatalysts – surface studies
While significant progress has been made in the fields of PC and EC surface studies, PEC studies involving well-defined surfaces remain relatively limited. As in the case of PC and EC reactions, single crystal studies in PEC allow for precise control over the exposed surface orientation, eliminating the influence of grain boundaries and other structural defects and allowing for a clear understanding of the relationship between facet orientation, electronic properties, and catalytic performance.199 Single crystal studies in PEC are mostly employed to study charge transfer dynamics and stability. For instance, studies on SrTiO3 have shown that the presence of hydroxide ions on the surface enhances PEC hydrogen evolution by promoting charge transfer and the generation of reactive oxygen species.200 Favorable surface states on the semiconductor promote charge transfer and separation, improving the process. Conversely, unfavorable surface states trap charge carriers, leading to recombination.200 The facet dependence on charge transfer has also been shown in PEC reactions.201 The orientation of a crystal facet significantly influences its ability to facilitate charge separation and transfer. Variations in work function have been attributed to the facet-dependent water oxidation activity of SiTiO3. The (111) facet, with its higher work function, exhibits superior hole accumulation, leading to enhanced photocurrent and photovoltage, followed by (110) and (100) facets.199 This efficient charge separation prevented recombination and enhanced the photocatalytic activity.199Defects can introduce new electronic states that broaden the light absorption range, but they can also act as recombination centers, hindering charge carrier separation and transport. For example, oxygen vacancies in hematite can improve light absorption but can also compromise stability.202 Balancing the benefits of defect engineering with the need for long-term stability is crucial for optimizing PEC performance. ZnO single crystals provide another example. Despite the wider light absorption range due to native defects, as-grown ZnO exhibits higher photocurrent density and efficiency than the oxygen-annealed crystals,203 which is due to reduced electron–hole recombination in this system. However, these defects also make the crystals susceptible to photocorrosion. While oxygen annealing improved the crystal's stability, it reduced its light absorption and overall efficiency.203 Likewise, the polar ZnO(0001) surface, stabilized by hydrogen adsorption, exhibits higher water-splitting activity but is susceptible to photo corrosion.204 In contrast, the nonpolar ZnO(10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) surface is more stable but less active.204
0) surface is more stable but less active.204
Similar to electrocatalytic systems, PEC systems often use adsorption studies to understand how surface interactions influence reactivity. Materials with high surface areas and specific functional groups, such as Lewis acid sites, use NH3 desorption analysis to probe the surfaces and, thus, the strength of adsorption of reactance molecules for PEC reactions. The increased adsorption concentrates reactants near the photocatalyst surface, enhancing the overall efficiency of these systems.205 For instance, improved chemisorption of reactant N2 molecule was observed on hybrid Au-coupled TiO2, attributed to increased oxygen vacancies and the unique morphology on this surface.206 This strong N2 adsorption facilitates the transfer of photogenerated electrons to adsorbed N2 molecules and promotes subsequent reduction reactions, thus enhancing the efficiency of this PEC system.206 The PEC analysis, linear sweep voltammetry (LSV) results showed increased photocurrent densities under N2 atmosphere, revealing efficient charge separation and injection. Lower charge transfer resistance was observed in EIS measurements, suggesting improved interfacial charge transfer kinetics.206
Most electrocatalyst-modified (hybrid) photoelectrodes have shown increased photocurrent density at a given potential, reduced onset over potential, and improved charge carrier separation, transport, and surface reaction kinetics.45,207Fig. 10(b) and (c) show how adding an electrocatalyst (NiOx) improves the performance of a photoanode (hematite) with photoelectrochemical impedance spectroscopy (PEIS).208 The NiOx-coated hematite photoanodes exhibited significantly lower interface resistance than bare hematite, showing the NiOx layer acts as an efficient electrocatalyst, facilitating charge transfer at the interface and reducing the energy barrier for the water oxidation reaction. The increased charge storage capacity due to the NiOx layer is reflected in the higher interface capacitance values observed in the EIS data.208 Similarly, the deposition of cobalt borate (CoBi) onto BiVO4 photoanodes has been shown to improve their performance significantly.209 The onset potential for the PEC water splitting reaction was reduced by 320 mV, leading to a higher current density and fill factor, suggesting that the CoBi co-catalyst effectively suppresses charge recombination (Fig. 10(d)). EIS confirmed that CoBi accelerates the water oxidation reaction and enhances charge transfer at the semiconductor–electrolyte interface. Similarly, the deposition of cobalt borate (CoBi) onto BiVO4 photoanodes has been shown to improve their performance significantly.209 The figure shows the amperometric I–V curve of BiVO4 and CoBi/BiVO4 photoanodes under continuous illumination. Loading of CoBi significantly improved the photocurrent. Additionally, the onset potential for the PEC water splitting reaction was reduced by 320 mV, leading to a higher current density and fill factor, suggesting that the CoBi co-catalyst effectively suppresses charge recombination. EIS confirmed that CoBi not only accelerates the water oxidation reaction but also enhances charge transfer at the semiconductor–electrolyte interface.209 However, optimizing the amount of catalyst is crucial, as excessive loading can hinder light absorption or increase the parasitic absorption and resistance.207 A thinner layer may compromise the overall photoelectrochemical efficiency. Ideally, co-catalysts should exhibit high activity for surface reactions to maximize catalytic performance.207
 | ||
| Fig. 10 Photoelectrochemical charge transport and charge transfer plots. (a) Schematic representations of the different photoanodes under study and associated equivalent circuit (EC) used to fit the electrochemical impedance spectroscopy (EIS) data. The EC of α-Fe2O3/NiOx-H is implemented with an additional Warburg element (ZW) if compared with α-Fe2O3 and α-Fe2O3/NiOx-L electrodes. Electrical parameters are defined as the resistance and capacitance of the electrode/electrolyte interface (Rinterface and Cinterface, respectively) and of the space-charge layer (Rbulk and Cbulk, respectively); Rs is the resistance associated with Ohmic losses in the electrolyte. (b) Trend of resistance at the electrode/electrolyte interface Rinterfacevs. the potential of bare hematite, α-Fe2O3/NiOx-L, and α-Fe2O3/NiOx-H electrodes. (c) Trend of capacitance at the electrode/electrolyte interface Cinterfacevs. the potential of bare hematite, α-Fe2O3/NiOx-L, and α-Fe2O3/NiOx-H electrodes.208 Reprinted with permission from ref. 207. Copyright 2020 American Chemical Society. (d) Amperometric I–t curves of the BiVO4 and CoBi/BiVO4 electrodes at 0.4 V vs. SCE in 0.5 M Na2SO4 (adjusted to pH 9) and 0.2 M sodium borate (buffered at pH 9) electrolyte. Light source: 300 W Xe lamp (l > 420 nm).209 Reprinted from ref. 208 with permission from the PCCP Owner Societies. | ||
These hybrid systems show great promise for developing efficient photoelectrodes. However, a comprehensive understanding of these hybrid materials’ surface structure and chemistry remains incomplete. Very few studies have investigated the surface structure of these hybrid systems, especially in the context of PEC processes. Thus, moving forward, extending these investigations to robust model systems to facilitate a deeper understanding of complex hybrid materials is essential. Implementing small molecule adsorption studies, which have demonstrated significant promise in characterizing complex systems, could be a valuable tool for investigating these model systems. By carefully controlling irradiation parameters, surface structure, and electrochemical environment, these systems hold immense potential for studying PEC reactions and advancing their applications.
7. Conclusion and perspectives
Numerous efforts are being made to develop hybrids that use hybrid surfaces for various PC and PEC reactions. While significant progress has been made in understanding the electronic properties of metal–semiconductor hybrid photocatalysts, the crucial role of atomic surface structure and chemistry in dictating reaction activity remains largely unexplored. Single-crystal studies highlight the profound influence of atomic-scale surface modifications on product selectivity, but such investigations still need to be done for hybrid materials. This knowledge gap can be addressed by utilizing small molecule adsorption as a powerful probe. Techniques like TPD, TPR, and RAIRS offer established methods for surface science analysis; however, their application to hybrid photocatalysts has been limited.A critical future perspective lies in establishing a better correlation between surface structure, adsorption behavior, and photocatalytic reactivity at the atomic level. This necessitates integrating small molecule adsorption studies with complementary techniques like RAIRS, STM, and theoretical calculations. Such a comprehensive approach will provide a deeper understanding of how light interacts with the hybrid surface, potentially inducing structural changes that influence reaction pathways and product selectivity. This research avenue holds immense promise by bridging the gap between theory210,211 and experiment. Notably, the limitations of purely theoretical approaches in accurately describing PEC systems have been recently highlighted.212 Well-defined experimental models can offer precise input parameters for computational modeling there by a robust foundation for theoretical studies of PEC systems. This emphasizes the potential of model systems with precisely controlled interfaces and characterized surface and electronic structures to gain insights into the complex synergistic phenomena occurring at the interfaces in photocatalytic and photoelectrocatalytic reactions.
Successful implementation and investigation of model hybrid systems hold immense promise for advancing our fundamental understanding of PC and PEC processes. This knowledge will undoubtedly inform the design and development of next-generation PC and PEC systems with enhanced efficiencies, leading to better solar energy conversion and a clean fuel future.
Author contributions
Preparation, creation and/or presentation of the published work was completed by MPM and JPB. Critical review, revision, and supervision was completed by JPB.Data availability
No primary research results, software, or code have been included and no new data were generated or analysed as a part of this review.Conflicts of interest
We have no conflicts of interest to declare.Acknowledgements
MPM and JPB would like to thank UNLV for startup funds that has supported this work.References
- C. Domínguez, J. García, M. A. Pedraz, A. Torres and M. A. Galán, Catal. Today, 1998, 40, 85–101 CrossRef.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 1–21 CrossRef.
- N. M. Gupta, Renewable Sustainable Energy Rev., 2017, 71, 585–601 CrossRef CAS.
- J. Li, J. Ren, S. Li, G. Li, M. M.-J. Li, R. Li, Y. S. Kang, X. Zou, Y. Luo, B. Liu and Y. Zhao, Green Energy Environ., 2024, 9, 859–876 CrossRef CAS.
- F. O. Ochedi, D. Liu, J. Yu, A. Hussain and Y. Liu, Environ. Chem. Lett., 2021, 19, 941–967 CrossRef CAS.
- T. Wang and J. Gong, Angew. Chem., Int. Ed., 2015, 54, 10718–10732 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- N. Liu, K. Tang, D. Wang, F. Fei, H. Cui, F. Li, J. Lei, D. Crawshaw, X. Zhang and L. Tang, Sep. Purif. Technol., 2024, 332, 125873 CrossRef CAS.
- Q. Dang, L. Wang, J. Liu, D. Wang, J. Chai, M. Wu and L. Tang, J. Water Process Eng., 2023, 53, 103609 CrossRef.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- K. Kočí, K. Matějů, L. Obalová, S. Krejčíková, Z. Lacný, D. Plachá, L. Čapek, A. Hospodková and O. Šolcová, Appl. Catal., B, 2010, 96, 239–244 CrossRef.
- O. Ishitani, C. Inoue, Y. Suzuki and T. Ibusuki, J. Photochem. Photobiol., A, 1993, 72, 269–271 CrossRef CAS.
- I. Rossetti, A. Villa, M. Compagnoni, L. Prati, G. Ramis, C. Pirola, C. L. Bianchi, W. Wang and D. Wang, Catal. Sci. Technol., 2015, 5, 4481–4487 RSC.
- A. N. Kadam, T. G. Kim, D. S. Shin, K. M. Garadkar and J. Park, J. Alloys Compd., 2017, 710, 102–113 CrossRef CAS.
- P.-Y. Liou, S.-C. Chen, J. C. S. Wu, D. Liu, S. Mackintosh, M. Maroto-Valer and R. Linforth, Energy Environ. Sci., 2011, 4, 1487–1494 RSC.
- M. Ismael, J. Environ. Chem. Eng., 2020, 8, 103676 CrossRef CAS.
- G. Zhao, H. Pang, G. Liu, P. Li, H. Liu, H. Zhang, L. Shi and J. Ye, Appl. Catal., B, 2017, 200, 141–149 CrossRef CAS.
- W. Tu, Y. Zhou, Q. Liu, S. Yan, S. Bao, X. Wang, M. Xiao and Z. Zou, Adv. Funct. Mater., 2013, 23, 1743–1749 CrossRef CAS.
- A. Bachmeier and F. Armstrong, Curr. Opin. Chem. Biol., 2015, 25, 141–151 CrossRef CAS PubMed.
- K. K. Sakimoto, S. J. Zhang and P. Yang, Nano Lett., 2016, 16, 5883–5887 CrossRef CAS PubMed.
- C. M. Friend, Sci. Am., 1993, 268, 74–79 CrossRef CAS.
- Q. Li, J. Li, H. Bai and F. Li, Chin. J. Catal., 2024, 58, 86–104 CrossRef CAS.
- O. Mekasuwandumrong, N. Jantarasorn, J. Panpranot, M. Ratova, P. Kelly and P. Praserthdam, Ceram. Int., 2019, 45, 22961–22971 CrossRef CAS.
- T. Billo, F.-Y. Fu, P. Raghunath, I. Shown, W.-F. Chen, H.-T. Lien, T.-H. Shen, J.-F. Lee, T.-S. Chan, K.-Y. Huang, C.-I. Wu, M. C. Lin, J.-S. Hwang, C.-H. Lee, L.-C. Chen and K.-H. Chen, Small, 2018, 14, 1702928 CrossRef PubMed.
- Y. Zhou, Q. Zhang, X. Shi, Q. Song, C. Zhou and D. Jiang, J. Colloid Interface Sci., 2022, 608, 2809–2819 CrossRef CAS PubMed.
- U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS.
- M. Volokh and T. Mokari, Nanoscale Adv., 2020, 2, 930–961 RSC.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308–318 CrossRef CAS PubMed.
- C. L. Bentley, M. Kang and P. R. Unwin, J. Am. Chem. Soc., 2019, 141, 2179–2193 CrossRef CAS PubMed.
- H. Li, L. Li and Y. Li, Nanotechnol. Rev., 2013, 2, 515–528 CAS.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- Y. Fu, J. Li and J. Li, Nanomaterials, 2019, 9, 359 CrossRef CAS PubMed.
- J. Hu, C. Zhai and M. Zhu, Chin. Chem. Lett., 2021, 32, 1348–1358 CrossRef CAS.
- J. Chen, D. Hao, W. Chen, Y. Liu, Z. Yin, H. Hsu, B. Ni, A. Wang, S. W. Lewis and G. Jia, Chin. J. Chem., 2023, 41, 3050–3062 CrossRef CAS.
- C. Heske, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 829–835 CrossRef CAS.
- L. Weinhardt, M. Blum, O. Fuchs, S. Pookpanratana, K. George, B. Cole, B. Marsen, N. Gaillard, E. Miller, K.-S. Ahn, S. Shet, Y. Yan, M. M. Al-Jassim, J. D. Denlinger, W. Yang, M. Bär and C. Heske, J. Electron Spectrosc. Relat. Phenom., 2013, 190, 106–112 CrossRef CAS.
- Y. Ding, J. Chen, X. Lian, Z. Tian, X. Geng, Y. Wang, Y. Liu, W. Wang, M. Wang, Y. Xiao, T. Jin, M. Sun, Z. Yang, K. H. L. Zhang, J.-Q. Zhong and W. Chen, Appl. Catal., B, 2024, 343, 123508 CrossRef CAS.
- T. Mayer, K. Schwanitz, B. Kaiser, A. Hajduk, M. V. Lebedev and W. Jaegermann, J. Electron Spectrosc. Relat. Phenom., 2017, 221, 116–133 CrossRef CAS.
- E. Kemppainen, A. Bodin, B. Sebok, T. Pedersen, B. Seger, B. Mei, D. Bae, P. C. K. Vesborg, J. Halme, O. Hansen, P. D. Lund and I. Chorkendorff, Energy Environ. Sci., 2015, 8, 2991–2999 RSC.
- W. Yang, Y. Xiong, L. Zou, Z. Zou, D. Li, Q. Mi, Y. Wang and H. Yang, Nanoscale Res. Lett., 2016, 11, 283 CrossRef PubMed.
- J. Zhao, S. Xue, J. Barber, Y. Zhou, J. Meng and X. Ke, J. Mater. Chem. A, 2020, 8, 4700–4734 RSC.
- H. Yu, L. Xu, P. Wang, X. Wang and J. Yu, Appl. Catal., B, 2014, 144, 75–82 CrossRef CAS.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef PubMed.
- J. M. Spurgeon, J. M. Velazquez and M. T. McDowell, Phys. Chem. Chem. Phys., 2014, 16, 3623 RSC.
- P. D. Tran, S. S. Pramana, V. S. Kale, M. Nguyen, S. Y. Chiam, S. K. Batabyal, L. H. Wong, J. Barber and J. Loo, Chem. – Eur. J., 2012, 18, 13994–13999 CrossRef CAS PubMed.
- M. Patel, T. T. Nguyen, J. Kim, J. Kim and Y. K. Kim, FlatChem, 2022, 33, 100361 CrossRef CAS.
- L. Yuan, Z. Geng, J. Xu, F. Guo and C. Han, Adv. Funct. Mater., 2021, 31, 2101103 CrossRef CAS.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- J. Zhou and D. A. Chen, Surf. Sci., 2003, 527, 183–197 CrossRef CAS.
- N. Lopez and J. K. Nørskov, Surf. Sci., 2002, 515, 175–186 CrossRef CAS.
- Z. Chen, J. Li, S. Wang, J. Zhao, J. Liu, J. Shen, C. Qi and P. Yang, Chemosphere, 2023, 316, 137797 CrossRef CAS PubMed.
- R. G. Musket, W. McLean, C. A. Colmenares, D. M. Makowiecki and W. J. Siekhaus, Appl. Surf. Sci., 1982, 10, 143–207 CrossRef CAS.
- B. Ghebouli, S.-M. Chérif, A. Layadi, B. Helifa and M. Boudissa, J. Magn. Magn. Mater., 2007, 312, 194–199 CrossRef CAS.
- C. Chang, J. Appl. Phys., 1990, 67, 566–569 CrossRef CAS.
- R. A. Bennett, P. Stone and M. Bowker, Faraday Discuss, 1999, 114, 267–277 RSC.
- M. Bowker, P. Stone, R. Bennett and N. Perkins, Surf. Sci., 2002, 511, 435–448 CrossRef CAS.
- S. C. Parker, A. W. Grant, V. A. Bondzie and C. T. Campbell, Surf. Sci., 1999, 441, 10–20 CrossRef CAS.
- Z. Wu, Y. Li and W. Huang, J. Phys. Chem. Lett., 2020, 11, 4603–4607 CrossRef CAS PubMed.
- G. Zhu, M. Han, B. Xiao and Z. Gan, Molecules, 2023, 28, 4786 CrossRef CAS PubMed.
- W. Kreuzpaintner, M. Störmer, D. Lott, D. Solina and A. Schreyer, J. Appl. Phys., 2008, 104, 114302 CrossRef.
- J. M. Purswani, T. Spila and D. Gall, Thin Solid Films, 2006, 515, 1166–1170 CrossRef CAS.
- S. S. Mundra, S. S. Pardeshi, S. S. Bhavikatti and A. Nagras, Mater. Today: Proc., 2021, 46, 1229–1234 CAS.
- P. Migowski and A. F. Feil, Recyclable Catal., 2016, 3, 1–12 Search PubMed.
- A. Baptista, F. Silva, J. Porteiro, J. Míguez and G. Pinto, Coatings, 2018, 8, 402 CrossRef.
- J.-O. Carlsson and P. M. Martin, in Handbook of Deposition Technologies for Films and Coatings, ed. P. M. Martin, William Andrew Publishing, Boston, 3rd edn, 2010, pp. 314–363 Search PubMed.
- S. T. Nguyen, C. Q. Nguyen, N. N. Hieu, H. V. Phuc and C. V. Nguyen, Phys. Chem. Chem. Phys., 2024, 26, 9657–9664 RSC.
- H. Shi, X. Wang, M. Zheng, X. Wu, Y. Chen, Z. Yang, G. Zhang and H. Duan, Adv. Mater. Interfaces, 2016, 3, 1600588 CrossRef.
- J. E. Crowell, J. Vac. Sci. Technol., A, 2003, 21, S88–S95 CrossRef CAS.
- M. Z. Ansari, I. Hussain, D. Mohapatra, S. A. Ansari, R. Rahighi, D. K. Nandi, W. Song and S.-H. Kim, Adv. Sci., 2024, 11, 2303055 CrossRef CAS PubMed.
- M. Yamamoto, Y. Minoura, M. Akatsuka, S. Ogawa, S. Yagi, A. Yamamoto, H. Yoshida and T. Yoshida, Phys. Chem. Chem. Phys., 2020, 22, 8730–8738 RSC.
- K. Oura, V. G. Lifshits, A. A. Saranin, A. V. Zotov and M. Katayama, Surface Science: An Introduction, Springer Science & Business Media, 2013 Search PubMed.
- D. P. Woodruff, Modern Techniques of Surface Science, Cambridge University Press, 2016 Search PubMed.
- J. B. Pendry, in Interaction of Atoms and Molecules with Solid Surfaces, ed. V. Bortolani, N. H. March and M. P. Tosi, Springer, US, Boston, MA, 1990, pp. 201–211 Search PubMed.
- W. Braun, Applied RHEED: Reflection High-Energy Electron Diffraction During Crystal Growth, Springer Science & Business Media, 1999 Search PubMed.
- P. Laukkanen, J. Sadowski and M. Guina, in Semiconductor Research: Experimental Techniques, ed. A. Patane and N. Balkan, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–21 Search PubMed.
- P. van der Heide, X-ray Photoelectron Spectroscopy: An introduction to Principles and Practices, John Wiley & Sons, 2011 Search PubMed.
- F. Jonat and J. A. S. Jr.
- N. J. C. Ingle, A. Yuskauskas, R. Wicks, M. Paul and S. Leung, J. Phys. D: Appl. Phys., 2010, 43, 133001 CrossRef.
- T. E. Feuchtwang and P. H. Cutler, Phys. Scr., 1987, 35, 132–140 CrossRef CAS.
- F. Hofer, F. P. Schmidt, W. Grogger and G. Kothleitner, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 109, 012007 Search PubMed.
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS.
- E. Taglauer and W. Heiland, Appl. Phys., 1976, 9, 261–275 CAS.
- E. Madej, J. Korecki and N. Spiridis, J. Chem. Phys., 2020, 152, 054712 CrossRef CAS PubMed.
- M. Henzler, Appl. Surf. Sci., 1982, 11–12, 450–469 CrossRef CAS.
- F. T. Wagner and P. N. Ross, Surf. Sci., 1985, 160, 305–330 CrossRef CAS.
- J.-M. Pan, B. L. Maschhoff, U. Diebold and T. E. Madey, Surf. Sci., 1993, 291, 381–394 CrossRef CAS.
- P. J. Møller and M.-C. Wu, Surf. Sci., 1989, 224, 265–276 CrossRef.
- I. Hashim, B. Park and H. A. Atwater, Appl. Phys. Lett., 1993, 63, 2833–2835 CrossRef CAS.
- P. J. Estrup and E. G. McRae, Surf. Sci., 1971, 25, 1–52 CrossRef CAS.
- K. Mašek, J. Beran and V. Matolín, Appl. Surf. Sci., 2012, 259, 34–38 CrossRef.
- A. de Siervo, R. Paniago, E. A. Soares, H.-D. Pfannes, R. Landers and G. G. Kleiman, Surf. Sci., 2005, 575, 217–222 CrossRef CAS.
- C. Polop, J. L. Sacedón and J. A. Martín-Gago, Surf. Sci., 1998, 402–404, 245–248 CrossRef CAS.
- P. M. Thibado, E. Kneedler, B. T. Jonker, B. R. Bennett, B. V. Shanabrook and L. J. Whitman, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, R10481 CrossRef CAS PubMed.
- P. T. Sprunger, E. Lægsgaard and F. Besenbacher, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 8163–8171 CrossRef CAS PubMed.
- F. Pesty, H.-P. Steinrück and T. E. Madey, Surf. Sci., 1995, 339, 83–95 CrossRef CAS.
- K. Luo, T. P. Clair, X. Lai and D. W. Goodman, J. Phys. Chem. B, 2000, 104, 3050–3057 CrossRef CAS.
- R. F. Egerton, Rep. Prog. Phys., 2008, 72, 016502 CrossRef.
- J.-W. He and P. J. Møller, Surf. Sci., 1986, 178, 934–942 CrossRef CAS.
- L. Huang, F. Peng and F. S. Ohuchi, Surf. Sci., 2009, 603, 2825–2834 CrossRef CAS.
- M. C. Patterson, X. Nie, F. Wang, R. L. Kurtz, S. B. Sinnott, A. Asthagiri and P. T. Sprunger, J. Phys. Chem. C, 2013, 117, 18386–18397 CrossRef CAS.
- J. Zhou, Y. C. Kang and D. A. Chen, Surf. Sci., 2003, 537, L429–L434 CrossRef CAS.
- J. A. Venables, Surf. Sci., 1994, 299–300, 798–817 CrossRef CAS.
- D. L. Carroll, M. Wagner, M. Rühle and D. A. Bonnell, J. Mater. Res., 1997, 12, 975–983 CrossRef CAS.
- D. A. Chen, M. C. Bartelt, R. Q. Hwang and K. F. McCarty, Surf. Sci., 2000, 450, 78–97 CrossRef CAS.
- F. Cosandey, L. Zhang and T. E. Madey, Surf. Sci., 2001, 474, 1–13 CrossRef CAS.
- G. R. Harp, R. F. C. Farrow, R. F. Marks and J. E. Vazquez, J. Cryst. Growth, 1993, 127, 627–633 CrossRef CAS.
- K. H. Ernst, A. Ludviksson, R. Zhang, J. Yoshihara and C. T. Campbell, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 13782–13796 CrossRef CAS PubMed.
- R. M. Arán-Ais, D. Gao and B. Roldan Cuenya, Acc. Chem. Res., 2018, 51, 2906–2917 CrossRef PubMed.
- C. Yang and C. Wöll, Surf. Sci., 2024, 749, 122550 CrossRef CAS.
- T. Wadayama, H. Osano, H. Yoshida, S. Oda and N. Todoroki, Appl. Surf. Sci., 2008, 254, 5380–5384 CrossRef CAS.
- E. Zahidi, M. Castonguay and P. McBreen, J. Am. Chem. Soc., 1994, 116, 5847–5856 CrossRef CAS.
- X. Yu, Z. Zhang, C. Yang, F. Bebensee, S. Heissler, A. Nefedov, M. Tang, Q. Ge, L. Chen, B. D. Kay, Z. Dohnálek, Y. Wang and C. Wöll, J. Phys. Chem. C, 2016, 120, 12626–12636 CrossRef CAS.
- M. A. Chesters, J. Electron Spectrosc. Relat. Phenom., 1986, 38, 123–140 CrossRef CAS.
- R. J. Madix, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1980, vol. 29, pp. 1–53 Search PubMed.
- R. G. Greenler, K. D. Burch, K. Kretzschmar, R. Klauser, A. M. Bradshaw and B. E. Hayden, Surf. Sci., 1985, 152–153, 338–345 CrossRef CAS.
- M. M. M. Jansen, J. Gracia, B. E. Nieuwenhuys (Hans) and J. W. Niemantsverdriet, Phys. Chem. Chem. Phys., 2009, 11, 10009 RSC.
- M. M. M. Jansen, F. J. E. Scheijen, J. Ashley, B. E. Nieuwenhuys and J. W. Niemantsverdriet (Hans), Catal. Today, 2010, 154, 53–60 CrossRef CAS.
- R. Ranjan and M. Trenary, in Springer Handbook of Advanced Catalyst Characterization, ed. I. E. Wachs and M. A. Bañares, Springer International Publishing, Cham, 2023, pp. 53–73 Search PubMed.
- V. K. Agrawal and M. Trenary, Surf. Sci., 1991, 259, 116–128 CrossRef CAS.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51–94 CrossRef CAS.
- S. Bhatia, J. Beltramini and D. D. Do, Catal. Today, 1990, 7, 309–438 CrossRef CAS.
- V. Rakić and L. Damjanović, in Calorimetry and Thermal Methods in Catalysis, ed. A. Auroux, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 131–174 Search PubMed.
- S. Lv, X. Liu and X. Shen, Surf. Sci., 2022, 718, 122015 CrossRef CAS.
- M. E. van Reijzen, M. A. van Spronsen, J. C. Docter and L. B. F. Juurlink, Surf. Sci., 2011, 605, 1726–1731 CrossRef CAS.
- K. Yagi, D. Sekiba and H. Fukutani, Surf. Sci., 1999, 442, 307–317 CrossRef CAS.
- B. Hammer, O. H. Nielsen and J. K. Nrskov, Catal. Lett., 1997, 46, 31–35 CrossRef CAS.
- T. Makino and M. Okada, Surf. Sci., 2014, 628, 36–40 CrossRef CAS.
- J. M. Gohndrone and R. I. Masel, Surf. Sci., 1989, 209, 44–56 CrossRef CAS.
- S. C. Jensen, K. R. Phillips, M. Baron, E. C. Landis and C. M. Friend, Phys. Chem. Chem. Phys., 2013, 15, 5193 RSC.
- R. Brosseau, M. R. Brustein and T. H. Ellis, Surf. Sci., 1993, 294, 243–250 CrossRef CAS.
- M. A. Henderson, Surf. Sci., 1996, 355, 151–166 CrossRef CAS.
- M. J. T. C. van der Niet, A. den Dunnen, L. B. F. Juurlink and M. T. M. Koper, Phys. Chem. Chem. Phys., 2011, 13, 1629–1638 RSC.
- G. S. Herman, Z. Dohnálek, N. Ruzycki and U. Diebold, J. Phys. Chem. B, 2003, 107, 2788–2795 CrossRef CAS.
- N. Ikemiya and A. A. Gewirth, J. Am. Chem. Soc., 1997, 119, 9919–9920 CrossRef CAS.
- L.-Q. Wang, K. F. Ferris and G. S. Herman, J. Vac. Sci. Technol., A, 2002, 20, 239–244 CrossRef CAS.
- V. K. Ocampo-Restrepo, S. Vijay, G. T. K. K. Gunasooriya and J. K. Nørskov, Phys. Chem. Chem. Phys., 2024, 26, 17396–17404 RSC.
- D. H. Parker and B. E. Koel, J. Vac. Sci. Technol., A, 1990, 8, 2585–2590 CrossRef CAS.
- E. Lira, J. Ø. Hansen, P. Huo, R. Bechstein, P. Galliker, E. Lægsgaard, B. Hammer, S. Wendt and F. Besenbacher, Surf. Sci., 2010, 604, 1945–1960 CrossRef CAS.
- P. J. Berlowitz and D. W. Goodman, Surf. Sci., 1987, 187, 463–480 CrossRef CAS.
- T. Genger, O. Hinrichsen and M. Muhler, Catal. Lett., 1999, 59, 137–141 CrossRef CAS.
- J. Zhang, Y. Pan, D. Feng, L. Cui, S. Zhao, J. Hu, S. Wang and Y. Qin, Adv. Mater., 2023, 35, 2300902 CrossRef CAS PubMed.
- J. C. L. Cornish and N. R. Avery, Surf. Sci., 1990, 235, 209–216 CrossRef CAS.
- Z. Dohnálek, J. Kim, O. Bondarchuk, J. M. White and B. D. Kay, J. Phys. Chem. B, 2006, 110, 6229–6235 CrossRef PubMed.
- B. Chen, Y. Ma, L. Ding, L. Xu, Z. Wu, Q. Yuan and W. Huang, Chin. J. Catal., 2013, 34, 964–972 CrossRef CAS.
- C. A. Walenta, C. Courtois, S. L. Kollmannsberger, M. Eder, M. Tschurl and U. Heiz, ACS Catal., 2020, 10, 4080–4091 CrossRef CAS.
- H. Feng, S. Tan, H. Tang, Q. Zheng, Y. Shi, X. Cui, X. Shao, A. Zhao, J. Zhao and B. Wang, J. Phys. Chem. C, 2016, 120, 5503–5514 CrossRef CAS.
- S. Natesakhawat, J. W. Lekse, J. P. Baltrus, P. R. Ohodnicki, B. H. Howard, X. Deng and C. Matranga, ACS Catal., 2012, 2, 1667–1676 CrossRef CAS.
- S. Natesakhawat, P. R. Ohodnicki, B. H. Howard, J. W. Lekse, J. P. Baltrus and C. Matranga, Top. Catal., 2013, 56, 1752–1763 CrossRef CAS.
- Z. Zheng, C. Zhang, J. Li, D. Fang, P. Tan, Q. Fang and G. Chen, J. Hazard. Mater., 2024, 474, 134710 CrossRef CAS PubMed.
- C. Wang, X. Gao, J. Zhang, Q. Ma, S. Fan and T.-S. Zhao, Appl. Surf. Sci., 2024, 658, 159820 CrossRef CAS.
- F. Zhang, R. X. Wang, A. Tian, F. Li, J. Liu and H. Yang, Vacuum, 2024, 220, 112766 CrossRef CAS.
- R. N. d’Alnoncourt, X. Xia, J. Strunk, E. Löffler, O. Hinrichsen and M. Muhler, Phys. Chem. Chem. Phys., 2006, 8, 1525–1538 RSC.
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS.
- M. Muir and M. Trenary, J. Phys. Chem. C, 2020, 124, 14722–14729 CrossRef CAS.
- Lj Kundakovic, D. R. Mullins and S. H. Overbury, Surf. Sci., 2000, 457, 51–62 CrossRef CAS.
- Y. He, L. Yin, N. Yuan and G. Zhang, Chem. Eng. J., 2024, 481, 148754 CrossRef CAS.
- K.-L. C. Nguyen, J. P. Bruce, A. Yoon, J. J. Navarro, F. Scholten, F. Landwehr, C. Rettenmaier, M. Heyde and B. R. Cuenya, ACS Energy Lett., 2024, 9, 644–652 CrossRef CAS PubMed.
- Q. Hao, Z. Wang, T. Wang, Z. Ren, C. Zhou and X. Yang, ACS Catal., 2019, 9, 286–294 CrossRef CAS.
- T. Cremer, S. C. Jensen and C. M. Friend, J. Phys. Chem. C, 2014, 118, 29242–29251 CrossRef CAS.
- K. R. Phillips, S. C. Jensen, M. Baron, S.-C. Li and C. M. Friend, J. Am. Chem. Soc., 2013, 135, 574–577 CrossRef CAS PubMed.
- C. Courtois, C. A. Walenta, M. Tschurl, U. Heiz and C. M. Friend, J. Am. Chem. Soc., 2020, 142, 13072–13080 CrossRef CAS PubMed.
- X. Ma, Y. Shi, Z. Cheng, X. Liu, J. Liu, Z. Guo, X. Cui, X. Sun, J. Zhao, S. Tan and B. Wang, Nat. Commun., 2024, 15, 2326 CrossRef CAS PubMed.
- S. Mezhenny, P. Maksymovych, T. L. Thompson, O. Diwald, D. Stahl, S. D. Walck and J. T. Yates, Chem. Phys. Lett., 2003, 369, 152–158 CrossRef CAS.
- Q. Guo, C. Zhou, Z. Ma, Z. Ren, H. Fan and X. Yang, Chem. Soc. Rev., 2016, 45, 3701–3730 RSC.
- L. Wu, C. Fu and W. Huang, Phys. Chem. Chem. Phys., 2020, 22, 9875–9909 RSC.
- M. A. Henderson and I. Lyubinetsky, Chem. Rev., 2013, 113, 4428–4455 CrossRef CAS PubMed.
- S. C. Jensen and C. M. Friend, Top. Catal., 2013, 56, 1377–1388 CrossRef CAS.
- F. Li, X. Chen, Q. Guo and X. Yang, J. Phys. Chem. C, 2020, 124, 26965–26972 CrossRef CAS.
- M. Wang, H. Liu, J. Ma and G. Lu, Appl. Catal., B, 2020, 266, 118647 CrossRef CAS.
- H. Gerischer, in Solar Energy Conversion, ed. B. O. Seraphin, Springer Berlin Heidelberg, Berlin, Heidelberg, 1979, vol. 31, pp. 115–172 Search PubMed.
- Studies in Surface Science and Catalysis, ed. J. Bénard, Y. Berthier, F. Delarnare, E. Hondros, M. Huber, P. Marcus, A. Masson, J. Oudar and G. E. Rhead, Elsevier, 1983, vol. 13, pp. 211–244 Search PubMed.
- C. Lucky and M. Schreier, ACS Nano, 2024, 18, 6008–6015 CrossRef CAS PubMed.
- S. Liu, Z. Wang, S. Qiu and F. Deng, Carbon Lett., 2024, 34, 1269–1286 CrossRef CAS.
- R. Gómez, J. M. Orts, B. Álvarez-Ruiz and J. M. Feliu, J. Phys. Chem. B, 2004, 108, 228–238 CrossRef.
- N. Lahiri, D. Song, X. Zhang, X. Huang, K. A. Stoerzinger, O. Q. Carvalho, P. P. Adiga, M. Blum and K. M. Rosso, J. Am. Chem. Soc., 2023, 145, 2930–2940 CrossRef CAS PubMed.
- A. Litke, Y. Su, I. Tranca, T. Weber, E. J. M. Hensen and J. P. Hofmann, J. Phys. Chem. C, 2017, 121, 7514–7524 CrossRef CAS PubMed.
- R. Rizo, S. Pérez-Rodríguez and G. García, ChemElectroChem, 2019, 6, 4725–4738 CrossRef CAS.
- A. Ma. Gómez-Marín, R. Rizo and J. M. Feliu, Catal. Sci. Technol., 2014, 4, 1685–1698 RSC.
- V. Grozovski, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, ChemPhysChem, 2011, 12, 1641–1644 CrossRef CAS PubMed.
- V. Briega-Martos and Y. Yang, Acc. Mater. Res., 2024, 5, 518–522 CrossRef CAS.
- J. M. Feliu and E. Herrero, EES Catal., 2024, 2, 399–410 RSC.
- J. M. Feliu, J. M. Orts, A. Femandez-Vega, A. Aldaz and J. Clavilier, J. Electroanal. Chem. Interf. Electrochem., 1990, 296, 191–201 CrossRef CAS.
- J. M. Feliu, J. M. Orts, R. Gómez, A. Aldaz and J. Clavilier, J. Electroanal. Chem., 1994, 372, 265–268 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, E. Herrero, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2000, 487, 37–44 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2002, 524–525, 242–251 CrossRef CAS.
- X. Chen, K. Ojha and M. T. M. Koper, J. Phys. Chem. Lett., 2024, 15, 4958–4964 CrossRef CAS PubMed.
- P. Jordá-Faus, C. A. Pressley, J. M. Feliu, R. M. Arán-Ais and E. Herrero, Electrochim. Acta, 2024, 498, 144634 CrossRef.
- R. Rizo, L. Chico-Mesa, R. M. Arán-Ais, V. Climent, E. Herrero and J. M. Feliu, ACS Electrochem., 2024 DOI:10.1021/acselectrochem.4c00107.
- Z. Wei, M.-K. Zhang, Y.-H. Yu, J. Cai, Y.-X. Chen, J. M. Feliu and E. Herrero, ACS Catal., 2024, 14, 8983–8995 CrossRef CAS.
- R. Rizo, M. J. Lázaro, E. Pastor and M. T. M. Koper, ChemElectroChem, 2016, 3, 2196–2201 CrossRef CAS.
- A. Yu, R. Rizo, F. J. Vidal-Iglesias, R. M. Arán-Ais, Y.-X. Chen, E. Herrero and J. M. Feliu, ACS Sustainable Chem. Eng., 2022, 10, 14826–14834 CrossRef CAS.
- A. Tiwari, T. Maagaard, I. Chorkendorff and S. Horch, ACS Energy Lett., 2019, 4, 1645–1649 CrossRef CAS.
- P. Waszczuk, G.-Q. Lu, A. Wieckowski, C. Lu, C. Rice and R. I. Masel, Electrochim. Acta, 2002, 47, 3637–3652 CrossRef CAS.
- F. Scholten, K.-L. C. Nguyen, J. P. Bruce, M. Heyde and B. Roldan Cuenya, Angew. Chem., Int. Ed., 2021, 60, 19169–19175 CrossRef CAS PubMed.
- J. Kim, Y. Yu, T. W. Go, J.-J. Gallet, F. Bournel, B. S. Mun and J. Y. Park, Nat. Commun., 2023, 14, 3273 CrossRef CAS PubMed.
- Y. Qiao and B. Seger, Curr. Opin. Chem. Eng., 2024, 43, 100999 CrossRef.
- S. Assavachin, C. Xiao, K. Becker and F. E. Osterloh, Energy Environ. Sci., 2024, 17, 3493–3502 RSC.
- F. T. Wagner and G. A. Somorjai, J. Am. Chem. Soc., 1980, 102, 5494–5502 CrossRef CAS.
- Y. Di, C. Ma, Y. Fu, X. Dong, X. Liu and H. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 8405–8416 CrossRef CAS PubMed.
- R. Fernández-Climent, S. Giménez and M. García-Tecedor, Sustainable Energy Fuels, 2020, 4, 5916–5926 RSC.
- Y. Suhak, K. Izdebska, P. Skupiński, A. Wierzbicka, A. Reszka, P. Sybilski, B. J. Kowalski, A. Mycielski, Z. R. Zytkiewicz, M. Soszko and A. Suchocki, Mater. Chem. Phys., 2014, 143, 1253–1257 CrossRef CAS.
- D. Dworschak, C. Brunnhofer and M. Valtiner, ACS Appl. Mater. Interfaces, 2020, 12, 51530–51536 CrossRef CAS PubMed.
- P. Zhang, X. Gu, N. Qin, Y. Hu, X. Wang and Y. Zhang, J. Hazard. Mater., 2023, 441, 129896 CrossRef CAS PubMed.
- S. Chang and X. Xu, Inorg. Chem. Front., 2020, 7, 620–624 RSC.
- S. Manna, A. K. Satpati, C. N. Patra and A. K. Tyagi, ACS Omega, 2024, 9, 6128–6146 CrossRef CAS PubMed.
- F. Malara, M. Fracchia, H. Kmentová, R. Psaro, A. Vertova, D. Oliveira De Souza, G. Aquilanti, L. Olivi, P. Ghigna, A. Minguzzi and A. Naldoni, ACS Catal., 2020, 10, 10476–10487 CrossRef CAS.
- C. Ding, J. Shi, D. Wang, Z. Wang, N. Wang, G. Liu, F. Xiong and C. Li, Phys. Chem. Chem. Phys., 2013, 15, 4589 RSC.
- C.-H. Lin, J. Rohilla, H.-H. Kuo, C.-Y. Chen, T.-F. Mark Chang, M. Sone, P. P. Ingole, Y.-C. Lo and Y.-J. Hsu, Sol. RRL, 2024, 8, 2300948 CrossRef CAS.
- A. A. Mamun, A. Billah and M. A. Talukder, Int. J. Hydrogen Energy, 2024, 59, 982–990 CrossRef CAS.
- A. J. Kaufman, A. C. Nielander, G. J. Meyer, S. Maldonado, S. Ardo and S. W. Boettcher, Nat. Catal., 2024, 7, 615–623 CrossRef CAS.
| This journal is © the Owner Societies 2025 |