
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT investigation of the impact of inner-sphere water molecules on RE nitrate binding to internal pore and external surface of MCM-22†
Tamalika
Ash
 *ab,
Yong
Han
ac,
James W.
Evans
ac and
Theresa L.
Windus
*ad
*ab,
Yong
Han
ac,
James W.
Evans
ac and
Theresa L.
Windus
*ad
aAmes National Laboratory, U.S. Department of Energy, Ames, Iowa 50011, USA. E-mail: tamalika@iastate.edu
bDepartment of Aerospace Engineering, Iowa State University, Ames, Iowa 50011, USA
cDepartment of Physics & Astronomy, Iowa State University, Ames, Iowa 50011, USA
dDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: twindus@iastate.edu
First published on 2nd December 2024
Abstract
The impact of inner-sphere water molecules on the binding of rare earth (RE) nitrates to MCM-22 aluminosilicates is analyzed. We used cluster models of MCM-22 to investigate the binding phenomena through localized-basis density functional theory (DFT) calculations. We also conducted plane-wave DFT calculations for a few selected binding configurations using the entire periodic MCM-22 unit cell to check for consistency. Two different MCM-22 cluster models are developed to represent an internal pore and an external surface. Starting with pure silica MCM-22, we substituted one Si with Al and added a H atom on the O bridging the Si and Al to create a Brønsted acid site (BAS), ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–{OH}–Al. Specifically, we investigated the binding of two RE nitrate aqua complexes, [X(NO3)3(H2O)n] where n = 4 (3) for X = Nd (Yb) via the reaction X(NO3)3(H2O)n + Si–{OH}–Al → Si–{OX(NO3)2(H2O)n}–Al + HNO3 at BASs, and via an analogous reaction at silanol sites, Si–{OH}. The above analysis just includes the inner coordination sphere H2O. Actually, for the Nd (Yb) complex, after binding at the T1 and T2 sites (T1 site) within the internal pore, one of the H2O molecules leaves this inner sphere. The binding strength at BASs and silanol sites is calculated from the energy change during the above reactions. One finds that Nd complexes prefer binding at the internal pore, while Yb complexes have a comparable binding preference both at the internal pore and external surface. The cluster calculations show good agreement with periodic calculations, implying that the cluster models are suitable for binding studies. Compared to the binding of non-hydrated RE nitrates, the explicit H2O molecules have a minimal impact on overall binding energy trends, but they do increase individual binding energy values. This study also demonstrated the stronger binding affinity of BASs over silanol sites.
Si–{OH}–Al. Specifically, we investigated the binding of two RE nitrate aqua complexes, [X(NO3)3(H2O)n] where n = 4 (3) for X = Nd (Yb) via the reaction X(NO3)3(H2O)n + Si–{OH}–Al → Si–{OX(NO3)2(H2O)n}–Al + HNO3 at BASs, and via an analogous reaction at silanol sites, Si–{OH}. The above analysis just includes the inner coordination sphere H2O. Actually, for the Nd (Yb) complex, after binding at the T1 and T2 sites (T1 site) within the internal pore, one of the H2O molecules leaves this inner sphere. The binding strength at BASs and silanol sites is calculated from the energy change during the above reactions. One finds that Nd complexes prefer binding at the internal pore, while Yb complexes have a comparable binding preference both at the internal pore and external surface. The cluster calculations show good agreement with periodic calculations, implying that the cluster models are suitable for binding studies. Compared to the binding of non-hydrated RE nitrates, the explicit H2O molecules have a minimal impact on overall binding energy trends, but they do increase individual binding energy values. This study also demonstrated the stronger binding affinity of BASs over silanol sites.
1. Introduction
Rare-earth elements (REEs) are the set of fifteen lanthanides (La to Lu) along with chemically similar scandium (Sc) and yttrium (Y). They are recognized as critical materials because of their widespread applications in modern technologies and renewable energy solutions.1,2 Also of concern is the potential hindrance to global economic and technological progress due to supply limitations.3 They have numerous applications in the fields of medicine;4–8 biology;4–6 imaging techniques4–6 and hi-tech industries.9–12 Regolith-hosted ion adsorption deposits are a great source of REEs, where the REE adsorption is believed to occur via ionic interactions associated with dehydroxylation of surface hydroxyls.13,14 However, the detailed binding mechanism and energetics of REEs binding on clay surfaces have been studied for only a few cases. Borst et al.15 showed that the REEs are adsorbed on clays, especially on kaolinite as easily leachable 8- to 9-coordinated outer-sphere hydrated complexes. Chatterjee et al.16 showed that the synthetic aluminosilicate MCM-22 can adsorb substantial amounts of Yb(III), and that the Yb(III) adsorption increases with the increase of Al-content in MCM-22. MCM-22 zeolite, an ionic clay mimic, is selected for the REE adsorption study due to the challenges in analysis for natural clay adsorbents due to defects, impurities, a mixture of phases, and local fluctuations in solution-phase conditions. Moreover, the clay mimics are well-characterized,17–19 easy to handle under experimental conditions and tunable with respect to substitution20 and pillaring.21,22 Substitutions in clay materials can play a significant role in enhancing the REE adsorption. To further elucidate the RE nitrate binding phenomena, in our recent published article,23 we thoroughly explored the binding of X(NO3)3 (where, X = Nd, Yb) on MCM-22 zeolites employing two computational strategies: localized-basis density functional theory (DFT) and plane-wave DFT analyses. In harmony with the observation of Chatterjee et al.,16 our analysis showed that the Brønsted acid sites (BASs), generated by the substitution of Si with Al, have a much stronger binding affinity towards X(NO3)3 compared to the silanol sites, establishing the important role of BASs for REE adsorption. However, in this regard, we considered X(NO3)3 complexes, where the coordination environment of the REE, i.e., X, is unsaturated.In the present study, we explore the effect of the coordination environment of the REEs on the binding phenomena. In doing so, we consider neodymium nitrate aqua, [Nd(NO3)3(H2O)4] and ytterbium nitrate aqua, [Yb(NO3)3(H2O)3] complexes, where four and three H2O molecules are directly coordinated to Nd and Yb-centers forming inner-sphere complexes with a total coordination number (CN) of 10 and 9, respectively (Fig. 1). In this context, it is important to note that we focus solely on the inner-sphere REE complexes and exclude the H-bonded waters that form outer-sphere complexes.
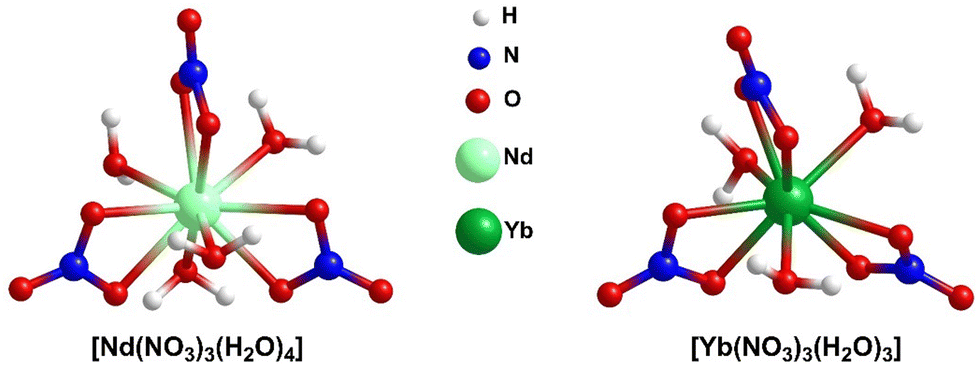 | ||
| Fig. 1 Lowest energy optimized geometries for [Nd(NO3)3(H2O)4] and [Yb(NO3)3(H2O)3]. | ||
A thorough investigation of the binding of [X(NO3)3(H2O)n] complexes to MCM-22 is performed employing extensive DFT analyses. To do so, we consider two different cluster models, internal pore and external surface, to describe binding in these distinct environments. One difference between the internal pore and external surface clusters is that only BASs are present in the internal pores, whereas both BASs and silanol sites are present on the external surfaces. In Section 2.2, we explain the construction of these MCM-22 cluster models in detail. The binding of [X(NO3)3(H2O)n] at BAS schematically corresponds to –
| Si–{OH}–Al + [X(NO3)3(H2O)n] → Si–{OX(NO3)2(H2O)n}–Al + HNO3 | (1) |
For the binding at the silanol site, a similar mechanism applies –
| Si–{OH} + [X(NO3)3(H2O)n] → Si–{OX(NO3)2(H2O)n} + HNO3 | (2) |
Si–{OH}–Al represents the BAS motif, and Si–{OH} represents the silanol site.
A crystalline MCM-22 unit cell has 72 tetrahedrally coordinated crystallographic Si sites, usually termed as T-sites, of which 8 are symmetrically inequivalent and traditionally labeled as T1–T8 sites.24–28 Following our previous work,23 here also, we consider Al substitution at T1, T2, and T3 sites in both the internal pore and external surface using the labeling scheme from ref. 24. These three T-sites are selected as they create BASs accessible to REE species in the interlayer regions. The other T-sites, from T4–T8 are embedded within the zeolite structure, and are less accessible to guest molecules.
In Section 2, we describe the development of the MCM-22 models used in our analysis, adopting the above notation for T-sites. We also describe the DFT methodology which is employed. In Section 3, we focus on presenting the results obtained from localized-basis DFT calculations using a cluster model for RE nitrate binding at BASs at an internal pore. To validate these results, we also present results from plane-wave DFT calculations in a periodic model incorporating the entire MCM-22 unit cell for RE nitrate binding at selected BASs. In this work, we prepare a larger internal pore cluster model (compared to the previous study23 without H2O) to explicitly account for the interactions of H2O molecules with MCM-22 (Fig. 2). In Section 4, we summarize the results obtained for REEs binding at an external surface of MCM-22. In both cases, we compare the results obtained for binding of Nd and Yb nitrates. In Section 5, we provide a discussion in support of our findings and our conclusions.
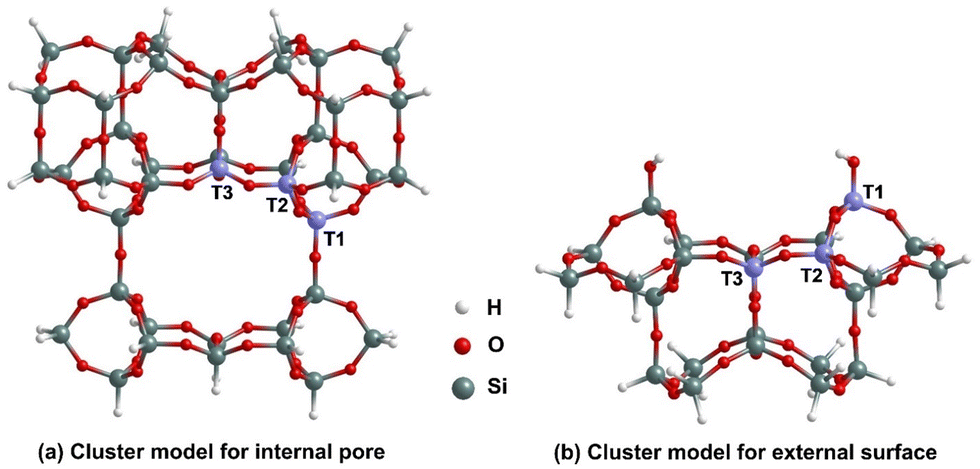 | ||
| Fig. 2 Cluster models for (a) the internal pore, and (b) the external surface binding sites for RE nitrates in MCM-22 highlighting the T1, T2 and T3 sites. | ||
2. MCM-22 models and DFT methodology
2.1. Consideration of RE nitrate aqua complexes
To account for the impact of explicit water molecules on the RE nitrate binding to MCM-22, the aqua complexes of RE nitrates have been considered. Neodymium nitrate is found to exist as a hexa-aqua complex in the crystal structure, with four water molecules directly attached to the metal center and two additional water molecules hydrogen bonded to the inner-sphere complexes, represented as [Nd(NO3)3(H2O)4]·2H2O.29 Similarly, ytterbium nitrate exists as penta-aqua complex with the molecular formula [Yb(NO3)3(H2O)3]·2H2O.30 However, for the present study, we have focused on the inner-sphere REE complexes, as depicted in Fig. 1. Experimentally, during adsorption studies, solvent water molecules would also engage in hydrogen bonding with the inner-sphere coordination complex, extending the number of outer-sphere H2O molecules just beyond two. In our current study, we aim to specifically examine the effect of water molecules directly coordinated to the Nd/Yb center on binding, rather than the influence of outer-sphere H2O molecules, which are anticipated to have a small influence. For this reason, we have excluded the two outer-sphere H2O molecules.2.2. Construction of MCM-22 cluster models
To accurately capture the effect of the local environment of MCM-22 on the RE nitrate binding, two cluster models, named internal pore and external surface are considered. In Fig. 2, we illustrated the structures of these cluster models before any Al substitution. The internal pore cluster model includes forty-one Si atoms, while the external surface cluster model comprises twenty-four Si atoms, with molecular formulas Si41O77H42 and Si24O38H24, respectively. Compared to the cluster model of the internal pore used in our previous article,23 a larger cluster is required here to more accurately account for the interaction between H2O molecules and MCM-22 associated with [X(NO3)3(H2O)n] binding at the 10-membered ring (MR) crossing windows or channels. The peripheral Si atoms are held fixed in their crystallographic positions corresponding to the crystalline MCM-22 system and are terminated with H atoms. This constraint avoids major restructuring of the cluster to an artificial configuration not representing the desired portion of MCM-22. Following earlier studies,27,28,31,32 the Si–H bond distances are fixed to 1.47 Å, aligned along the bond direction corresponding to the next lattice oxygen in the periodic model for crystalline MCM-22. The rest of the atoms of the cluster models are fully relaxed. As with previous studies, Si at T1, T2, and T3 sites are substituted with Al for the internal pore cluster model. For the external surface cluster model, the Si at T2 and T3 sites are substituted with Al. In both cases H is added to the O atom bridging the Al with an adjacent Si to produce the complete BASs. The details of atomic coordinates are provided in the SI. Here, it is important to note that although the T1, T2, and T3 sites are positioned on the edge of the cluster models, in the periodic model for crystalline MCM-22, these sites are on the circumference of a large pore and thus have minimal interactions with the excluded parts. The key difference between the two cluster models, as shown in Fig. 2, is that the internal pore model has only BASs, while the external surface model includes both BASs and silanol sites. MCM-22 materials have been synthesized experimentally with pure silica (ITQ-1) and with Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratios from 42:1 to 25:1.16 Thus, in all the latter cases, there is more than one Al per unit cell, but at least for higher Si:Al ratios, the BASs should not interact.
:Al ratios from 42:1 to 25:1.16 Thus, in all the latter cases, there is more than one Al per unit cell, but at least for higher Si:Al ratios, the BASs should not interact.
2.3. Computational details for localized-basis DFT analysis
All the electronic structure calculations for the cluster models are performed using the NWChem 7.0.233 quantum chemistry program. For analysis of energetics including geometry optimization, the PBE0 functional34,35 in conjunction with the def2-svp basis set, developed by Ahlrichs and coworkers36,37 for main group elements, and Stuttgart RSC Segmented basis set and ECP with 28 core electrons for REE,38,39 have been used. The PBE0 functional incorporates a mix of Perdew–Burke–Ernzerhof (PBE) and Hartree–Fock exchange energy in a 3:1 ratio, along with the full PBE correlation energy. For REE modeling, the use of the ECP basis set reduces the calculation expense and accounts for the scalar relativistic effects. The van der Waals interactions are included by employing Grimme's dispersion (D3).40 To ensure selected peripheral Si and H atoms remained in the correct positions, we used the ‘fix atom’ keyword under the ‘constraints’ directive during geometry optimization in NWChem. The default self-consistent field (SCF) convergence and geometry optimization criteria in NWChem have been used.
2.4. Construction of periodic MCM-22 models
The experimental synthesis of MCM-22 involves a precursor, MCM-22P, which consists of a periodic array of layers or slabs of aluminosilicate material (with a period of around 27.9 Å along the direction perpendicular to the layers), where the surfaces of each layer or slab are fully hydroxylated. MCM-22 forms during calcination by dehydroxylating these internal surfaces, resulting in the periodicity perpendicular to the layers decreasing by about 2.7 Å. Our model of MCM-22 used for analysis of RE nitrate binding at internal pores includes as the computational supercell an entire unit cell of the bulk MCM-22 material (with periodic boundary conditions). Thus, the dimensions of the computational cell are a, b = 14.5 Å in the plane of the layers, and c = 25.2 Å in the orthogonal direction. For the pure silica version of MCM-22, referred to as ITQ-1, this includes 72 Si and 144 O. We replaced one Si by Al for studies involving RE nitrate binding at BASs. For analysis of RE nitrate binding at an external surface of MCM-22, the model for the external surface is simply taken as the fully hydroxylated surface of the MCM-22P precursor, but expanding the vacuum region adjacent to this surface relative to MCM-22P. Specifically, the computational unit cell includes a single layer or slab of the precursor and a vacuum region with thickness no smaller than 20 Å along the direction orthogonal to the slab surfaces (again with periodic boundary conditions). Thus, now the dimension of the computational unit cell is increased to about 45.0 Å orthogonal to the layers.2.5. Computational details for plane-wave DFT analysis
For the plane-wave DFT analysis, the VASP code41 is used with the projector augmented wave potentials42 developed by the VASP group. To include the van der Waals (vdW) interactions, we choose the optB88-vdW functional43 in the electron–electron exchange-correlation component. Previous DFT calculations for the bulk properties of silica polymorphs, various vdW materials or biomaterials,43–48 as well as various small molecules consisting of C, H, O and N,49 indicate the reliability of this DFT method with the optB88-vdW functional. In this work, we use this plane-wave DFT method to compare with the localized atomic basis DFT analysis.In our plane-wave DFT analyses for RE nitrate binding both within the bulk and at the surface of MCM-22, the computational supercells are selected as indicated in Section 2.4 with a k mesh of 2 × 2 × 1.50 In the structural optimization for single molecules,49 HNO3, [Nd(NO3)3(H2O)4], or [Yb(NO3)3(H2O)3], we use a periodic cubic box with any side length of 30.0 Å and with a k mesh of 1 × 1 × 1. In all plane-wave DFT calculations, the energy cutoff is always taken to be 600 eV, and the energy convergence tolerance is taken to be less than 0.01 eV Å−1 for the force on each atom. All atoms are fully relaxed during the structure optimization.
2.6. RE nitrate aqua complexes binding energies
Schematic representations of the binding reactions of [X(NO3)3(H2O)n] binding at the BASs and silanol sites are given in eqn (1) and (2). The equations for the corresponding RE nitrate binding energies for both cluster and periodic MCM-22 models are as follows:For binding at BASs, one has
| BE(BAS) = E[Si–{OX(NO3)2(H2O)n}–Al] + E[HNO3] − E[Si–{OH}–Al] − E[X(NO3)3(H2O)n] | (3) |
For binding at the silanol site (for external surface), one has
| BE(silanol) = E[Si–{OX(NO3)2(H2O)n}] + E[HNO3] − E[Si–{OH}] − E[X(NO3)3(H2O)n] | (4) |
3. RE nitrate binding at an internal pore of MCM-22
3.1. Localized-basis DFT analysis
The binding of [X(NO3)3(H2O)n] to Tj BASs within the cavity formed by –O–Si–O– linkages (see Fig. 3), which represent the 10 MR crossing window interlayer space in the periodic model, has been considered. Our previous work23 considered the analogous RE nitrate binding process without the explicit H2O.The BASs at the internal pore are termed bulk-Tj and after binding with [X(NO3)3(H2O)n] complexes, they are referred to as bulk-Tj-X.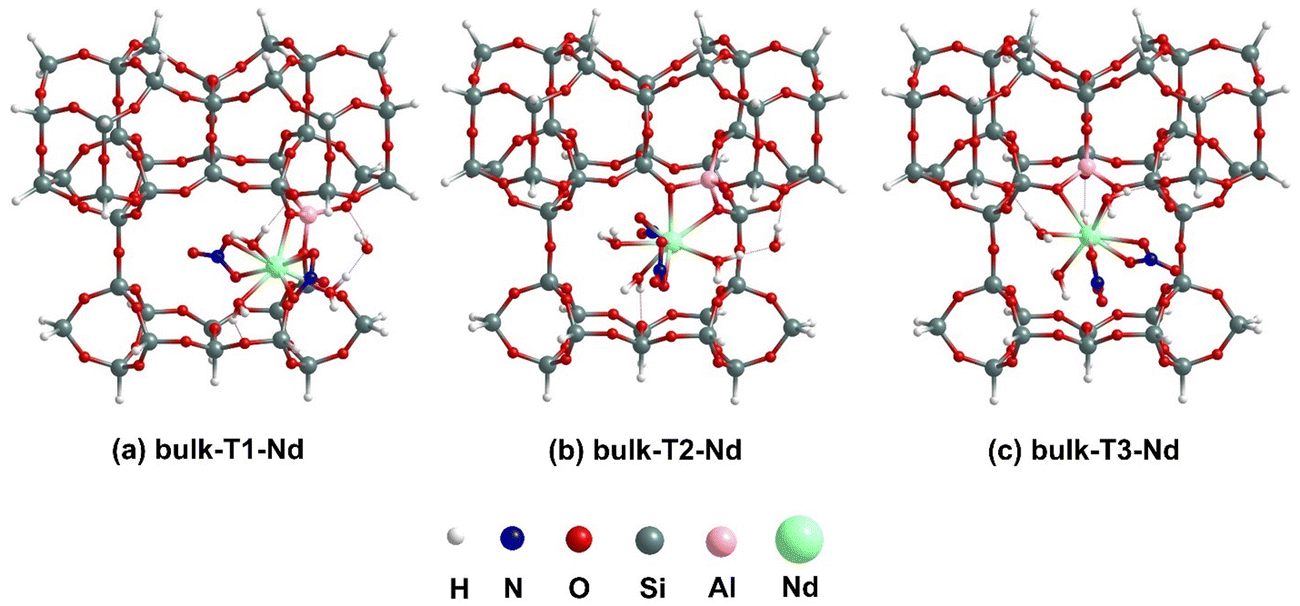 | ||
| Fig. 3 Lowest energy optimized geometries for [Nd(NO3)3(H2O)4] binding to T1, T2 and T3 BASs at internal pore using cluster models. | ||
In Fig. 3, we have illustrated the lowest-energy optimized structures for the binding of [Nd(NO3)3(H2O)4] complex at bulk-Tj BASs for the internal pore cluster model. In the case of bulk-T1-Nd, the binding of [Nd(NO3)3(H2O)4] to the cluster causes one H2O molecule to leave the inner-coordination sphere and bridge between the oxygen of an –Al–O–Si– linkage of the cluster (1.82 Å) and one of the directly bound H2O molecules (1.55 Å) through a hydrogen bonding interaction, resulting in a total coordination number (CN) of 9 for Nd. Similarly, for bulk-T2-Nd also, the CN around Nd becomes 9 after binding, with the leaving H2O molecule dangling between the directly bound H2O molecule and cluster with hydrogen bonding distances of 1.61 and 1.86 Å, respectively. Unlike the previous two scenarios, for bulk-T3-Nd, the geometry around the Nd center remains unchanged before and after binding to the cluster, maintaining a 10 coordinated complex. As given in eqn (1), the binding of [Nd(NO3)3(H2O)4] complex to the cluster results in the release of one molecule of nitric acid in all three cases. The structures of bulk-Tj-Nd also show that the H2O molecules attached to the Nd center form hydrogen bonds with the oxygen of –O–Si–O– linkage of the cluster, with bond distances varying between 1.78–2.00 Å. Analyzing the Nd–Al bonds formed after binding, it is evident that the bond distances are approximately 3.40 Å, while the Nd–O bond distances vary significantly from 2.45 to 2.64 Å. The binding energy values in Table 1 indicate that binding at the T1 site is slightly stronger (−2.38 eV) than at the T3 site (−2.33 eV), which is, in turn, stronger than at the T2 site (−1.92 eV).
| Model/analysis-internal pore BAS | Bulk-T1-X | Bulk-T2-X | Bulk-T3-X |
|---|---|---|---|
| Cluster/localized basis DFT X = Nd | −2.38 | −1.92 | −2.33 |
| Cluster/localized basis DFT X = Yb | −2.32 | −1.79 | −2.07 |
The lowest-energy optimized structures for the binding of [Yb(NO3)3(H2O)3] complexes at bulk-Tj BASs in the internal pore are shown in Fig. S1 (ESI†). In the bulk-T1-Yb structure, similar to bulk-T1-Nd, one of the H2O molecules leaves the inner-coordination sphere but forms hydrogen bonds with the oxygen of –Al–O–Si– of the cluster and the bonded NO3, with distances of 2.01 and 1.98 Å, respectively, resulting in an 8 coordinated complex. The optimization of the bulk-T2-Yb structure shows that it is a 9-coordinated complex, where all three attached H2O molecules remain intact after binding, similar to the precursor [Yb(NO3)3(H2O)3] complex. In the bulk-T3-Yb structure, the coordination environment around the Yb also remains 9 after binding, comprising two NO3, three H2O and two new bonds with the oxygens of –Al–O–Si– linkage of the cluster. For all three optimized bulk-Tj-Yb structures, two out of the three H2O molecules attached to the Yb center interact with the oxygen of a –Si–O–Si– linkage of the cluster through hydrogen bonding interactions, with distances ranging from 1.74 to 1.99 Å. Yb being smaller in size, the Yb–Al bond lengths are approximately 3.20 Å, smaller than the Nd–Al bond lengths. Similar to Nd–O distances, the Yb–O distances also vary widely, from 2.31 to 2.45 Å. The binding energy strengths, as shown in Table 1, follow the trend: bulk-T1-Yb (−2.32 eV) > bulk-T3-Yb (−2.07 eV) > bulk-T2-Yb (−1.79 eV). Notably, the binding energy values for the bulk-Tj-Yb complexes are lower than those for the bulk-Tj-Nd complexes, indicating that Nd complexes bind more strongly at the internal pore of MCM-22 than Yb complexes.
3.2. Plane-wave DFT analysis
We further studied the binding of [X(NO3)3(H2O)n] (X = Nd, Yb) at the bulk-T3 BAS in the internal pore employing plane-wave DFT calculations to provide further support for the validity of the results obtained from localized-basis calculations. Fig. 4(a) shows the configuration of [Nd(NO3)3(H2O)4] binding at the bulk-T3 BAS site, while the [Yb(NO3)3(H2O)3] binding configuration is shown in Fig. S2(a) in the ESI.† Similar to cluster calculations, the [X(NO3)3(H2O)n] binding occurs through interactions between the X-center and Al, as well as various nearby O atoms in MCM-22. The Nd–Al and Yb–Al distances are calculated to be 3.44 and 3.22 Å, respectively, consistent with the cluster calculations. The variation in Nd–O (2.60–2.70 Å) and Yb–O (2.41–2.45 Å) distances is also evident from the plane-wave DFT calculations. Hydrogen bonding interactions between the attached H2O molecules and oxygen of –Si–O–Si– linkage are 1.86–2.00 Å for Nd-bound cluster and 1.80–1.90 Å for Yb-bound cluster. Therefore, in terms of geometrical parameters, the localized-basis calculation aligns well with the plane-wave calculations. Table 2 compares the binding energies for [X(NO3)3(H2O)n] binding at the bulk-T3 BAS using both cluster and periodic models. For the periodic model, the binding energy for [Nd(NO3)3(H2O)4] binding at T3 site is −2.12 eV and for [Yb(NO3)3(H2O)3] binding, it is −1.76 eV. The slight discrepancy between the binding energy results for the two methods is expected due to the different types of models employed for the MCM-22 material (periodic vs. cluster), and also the different functionals used in the DFT analysis. However, the stronger binding of [Nd(NO3)3(H2O)4] to the BAS compared to [Yb(NO3)3(H2O)3] is consistent in both calculations for the bulk-T3 BAS. It should be noted that, similar to our previous article,23 the results from our cluster calculations align well with those from periodic calculations, where now we have incorporated explicit H2O molecules in treating RE nitrate binding. This indicates that the results from our cluster calculations can be accurately translated to periodic calculations. | ||
| Fig. 4 Binding of [Nd(NO3)3(H2O)4] complexes at BASs at (a) internal pore and (b) external surfaces of MCM-22. The overall reaction mechanism indicating all reactants and products is shown by using plus signs and arrows. All structures are optimized from our plane-wave DFT calculations. Nd nitrate binding reactions with an Al atom at the T3 site of the MCM-22 reactant. | ||
| Model/analysis-internal pore BAS | Bulk-T3-Nd | Bulk-T3-Yb |
|---|---|---|
| Cluster/localized basis DFT | −2.33 | −2.07 |
| Periodic/plane-wave DFT | −2.12 | −1.76 |
4. RE nitrate binding at the external surface of MCM-22
4.1. Localized-basis DFT analysis
The lowest-energy optimized structures for the binding of [Nd(NO3)3(H2O)4] complexes at a silanol site and at the surf-Tj BASs for the external surface cluster models are shown in Fig. 5. The coordination environment around the Nd center remains unchanged before and after binding for all three surface structures, maintaining a total CN of 10. For the surf-silanol-Nd, the Nd–Si distance is calculated to be 3.47 Å, whereas for surf-Tj–Nd, the Nd–Al distances are approximately 3.31 Å. The Nd–O distance in surf-silanol-Nd is 2.31 Å; however, the Nd–O distances for binding at BASs vary over a large range (2.53–2.64 Å) like the internal pore cluster models. The H2O molecules coordinated to the Nd center are interacting with the cluster via the oxygen of a –Si–O–Si– linkage and an OH of a silanol group through hydrogen bonding interaction (1.75–1.99 Å). In addition, the bound nitrates also interact with the Si–OH moiety of the cluster through their oxygen ends via hydrogen bonding of distances 1.76–1.90 Å. In terms of binding energy, the surf-silanol-Nd structure experiences the lowest binding energy (−0.26 eV) compared to the surf-Tj-Nd even after considering explicit water molecules to satisfy the coordination environment of Nd. Analogous to our previous analysis without explicit H2O,23 the surf-T3-Nd has a stronger binding (−2.23 eV) than surf-T2-Nd (−2.04 eV). Therefore, it is apparent that regardless of the coordination environment around Nd center, Nd complexes bind more strongly to BASs compared to silanol sites.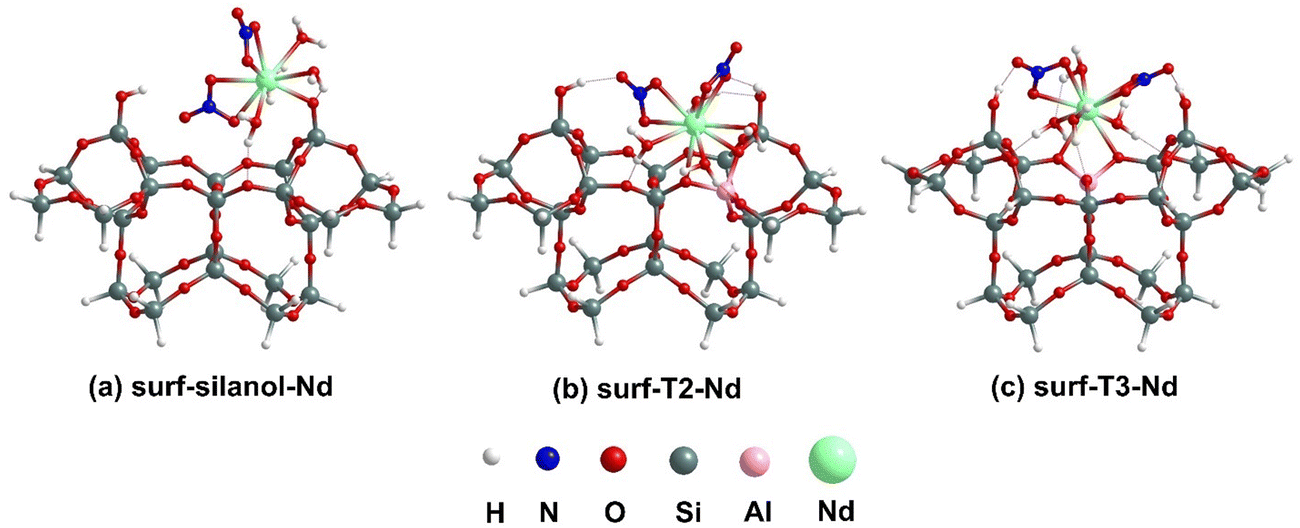 | ||
| Fig. 5 Lowest energy optimized geometries for [Nd(NO3)3(H2O)4] binding to silanol site and T2, and T3 BASs at external surface using cluster models. | ||
The lowest-energy optimized geometries for the binding of [Yb(NO3)3(H2O)3] complexes at the silanol site and surf-Tj BASs on the external surface are shown in Fig. S3 (ESI†). Similar to the Nd-bound surface cluster, the coordination number around the Yb center remains unchanged before and after binding, maintaining it at 9. The Yb–Si distance for surf-silanol-Yb is 3.24 Å, which is 0.06 Å longer than the Yb–Al distances (∼3.18 Å) for surf-Tj-Yb. The Yb–O distance for surf-silanol-Yb is 2.17 Å, while it varies between 2.37 and 2.42 Å for surf-Tj-Yb. The interaction between the coordinated H2O molecules and the cluster is evident from the hydrogen bonding between the oxygen of the –Si–O–Si– linkage and the OH of the silanol group (1.69–1.83 Å). Additionally, the nitrates attached to the Yb center interact with the cluster via the silanol group, forming O⋯HO hydrogen bonds with distances ranging from 1.86 to 1.93 Å. Here also, the surf-silanol-Yb has the lowest binding energy (−0.54 eV) among all Yb-bound surface geometries. Interestingly, unlike Nd-bound surface complexes, here the binding at surf-T2 site (−2.29 eV) is stronger than surf-T3 site (−2.19 eV) by ∼0.10 eV. Moreover, the binding at surf-T2 is energetically comparable with bulk-T1, suggesting that for Yb complexes, both bulk-T1 and surf-T2 sites are equally competitive. Notably, when comparing the binding of Nd and Yb complexes at the external surface, it is evident that Yb experiences stronger binding at the external surface than Nd (Table 3).
| Model/analysis-external silanol/BAS | Surf-silanol-X | Surf-T2-X | Surf-T3-X |
|---|---|---|---|
| Cluster/localized basis DFT X = Nd | −0.26 | −2.04 | −2.23 |
| Cluster/localized basis DFT X = Yb | −0.54 | −2.29 | −2.19 |
4.2. Plane-wave DFT analysis
For the external surface, we also studied the binding of [X(NO3)3(H2O)n] (X = Nd, Yb) at the surf-T3 BAS using plane-wave DFT calculations to compare the results from the two DFT methods. Fig. 4(b) shows the configuration of [Nd(NO3)3(H2O)4] binding at the surf-T3 BAS site, while [Yb(NO3)3(H2O)3] binding is given in Fig. S2(b) in the ESI.† Similar to the cluster models, the binding at the external BAS occurs through the interaction of the X-center with Al and nearby O atoms. The calculated Nd–Al and Yb–Al distances are 3.37 Å and 3.24 Å, respectively, which are consistent with the cluster calculations. The variation in Nd–O (2.58–2.60 Å) and Yb–O (2.41 Å) distances is also in a similar range to that observed in the localized-basis calculations. Additionally, the hydrogen bonding interactions between the attached H2O molecules and oxygen of –Si–O–Si– linkage and the silanol group are also found in a similar range. Therefore, in terms of geometrical parameters, the structures optimized using localized-basis calculation are comparable to that of plane-wave DFT calculations. Table 4 compares the binding energies for [X(NO3)3(H2O)n] binding at the surf-T3 BAS using both cluster and periodic models. The binding energy for [Nd(NO3)3(H2O)4] binding at surf-T3 is −2.01 eV, and for [Yb(NO3)3(H2O)3] binding, it is −1.61 eV using plane-wave DFT calculations. Again, a discrepancy in binding energy between the two methods occurs because of the adoption of two different models for the MCM-22 material (periodic vs. cluster) and the use of two different DFT functionals for the calculations. The plane-wave DFT shows a larger difference between the Nd and Yb binding energies than the localized basis cluster models. However, the trends are similar. Therefore, for surface calculations, the results for Nd complex binding show reasonable agreement between our cluster and periodic calculations, but for Yb complex binding, there is some discrepancy in the binding energy value between the two methods.| Model/analysis-external surface BAS | Surf-T3-Nd | Surf-T3-Yb |
|---|---|---|
| Cluster/localized basis DFT | −2.23 | −2.19 |
| Periodic/plane-wave DFT | −2.01 | −1.61 |
5. Discussion and conclusion
We have performed a comprehensive DFT analysis of the binding of RE nitrate aqua complexes, [X(NO3)3(H2O)n] (X = Nd, Yb) to MCM-22. This study primarily utilized localized-basis DFT calculations with cluster models, but we further validated our results using plane-wave DFT calculations with periodic models for a few selected binding configurations. The energetics obtained from the plane-wave DFT calculations align well with those from localized-basis calculations in most cases. This consistency suggests that our cluster models are adequate representations of the periodic MCM-22 models for binding studies. For both RE nitrate aqua complexes, as observed in our previous article,23 the binding at BASs is significantly stronger than at silanol sites, aligning with the observed trend of increased RE adsorption with higher Al content.16Considering cluster models, for Nd-complexes, the maximum binding strength at BASs in the internal pore (at bulk-T1) is around −2.38 eV, which is slightly stronger than the maximum binding strength at BASs on the external surface (at surf-T3, −2.23 eV). Thus Nd-complexes exhibit a slight preference to adsorb within the internal pores (although this could be countered by transport limitations). In the case of Yb-complexes, the localized-basis calculations show that the binding strength is comparable at both bulk-T3 BAS in the internal pore (−2.32 eV) and surf-T2 BAS on the external surface (−2.29 eV), i.e., there is a comparable preference for binding at internal pores and external surfaces of MCM-22. To investigate the reason behind the difference in behavior for Nd versus Yb, we have performed a geometrical parameter analysis, provided in Table S1 (ESI†). It shows that for bulk-T1-Nd and surf-T3-Nd structures, the Nd–Al distances are relatively consistent in both the structures. However, the Nd–O distances are quite different; 2.45 and 2.61 Å in bulk-T1-Nd structure and 2.53 and 2.55 Å in surf-T3-Nd structure. So, in the former case, one of the Nd-O distances is longer, whereas for the later both the Nd–O distances are quite long. These longer Nd–O distances in the surface structure correspond to weaker interactions, which might be the reason behind the lower binding strength of Nd complexes on the external surface compared to the internal pore. In contrast, for Yb complexes, both the Yb–Al and Yb–O distances remain comparable in the internal pore and on the external surface. This similarity in bond distances might account for the similar binding strengths observed for Yb complexes in both the internal pore and external surface.
Another noticeable observation is that the binding of Nd complexes at the internal pore is stronger than Yb complexes, whereas comparable binding strengths or the reverse trend applies on the external surface. This different trend cannot be explained by any single factor as several factors, such as structural difference between Nd and Yb complexes, including the number of coordinated H2O molecules, the accommodation of these complexes within internal pores or on external surfaces, hydrogen bonding between coordinated H2O molecules and the cluster, and the orientation of nitrate and H2O molecules post-optimization influence the binding and so the binding energy.
A comparison between X(NO3)323 binding and [X(NO3)3(H2O)n] binding shows that the binding energy values increase in the presence of explicit H2O. This enhancement can be attributed to the formation of hydrogen bonding interactions between the bound H2O molecules and MCM-22. See Table 5 which compares behavior for Nd(NO3)3 and [Nd(NO3)3(H2O)4] from the cluster model, and see the ESI† for additional results. On the other hand, trends of binding energies, e.g., comparing different binding sites, remain substantially the same, i.e., saturating the coordination environment around the RE cations does not affect such trends. However, the binding scenario can be more complicated for the RE nitrate aqua complex, e.g., as reflected in a change in CN due to the loss of a water molecule from the first coordination sphere for binding at the internal pore, but not at the external surface.
| Internal pore Nd(NO3)3 | −1.97 (T1) | −1.69 (T2) | −1.75 (T3) |
| Internal pore [Nd(NO3)3(H2O)4] | −2.38 (T1) | −1.92 (T2) | −2.33 (T3) |
| External surface Nd(NO3)3 | −0.19 (silanol) | −1.33 (T2) | −1.59 (T3) |
| External surface [Nd(NO3)3(H2O)4] | −0.26 (silanol) | −2.04 (T2) | −2.23 (T3) |
Overall, our modeling approach provides a realistic representation of the binding of RE nitrates on MCM-22 clay mimics. This comprehensive analysis provides valuable insights into the binding mechanisms of RE complexes on MCM-22, a topic that has been relatively underexplored until now.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division. TA & YH were supported by the Geo-inspired Separation of Rare-earth Elements project, and JWE & TLW by the Computational & Theoretical Chemistry project. The research was performed at Ames National Laboratory, which is operated by Iowa State University under contract No. DE-AC0207CH11358. DFT calculations were performed with a grant of computer time at the National Energy Research Scientific Computing Center (NERSC). NERSC is a DOE Office of Science User Facility supported by the Office of Science of the U.S. DOE under Contract No. DE-AC02-05CH11231.References
- E. Alonso, A. M. Sherman, T. J. Wallington, M. P. Everson, F. R. Field, R. Roth and R. E. Kirchain, Environ. Sci. Technol., 2012, 46, 3406–3414 CrossRef CAS PubMed.
- S. Massari and M. Ruberti, Resour. Policy, 2013, 38, 36–43 CrossRef.
- M. A. de Boer and K. Lammertsma, ChemSusChem, 2013, 6, 2045–2055 CrossRef CAS.
- P. Ascenzi, M. Bettinelli, A. Boffi, M. Botta, G. De Simone, C. Luchinat, E. Marengo, H. Mei and S. Aime, Rend. Sedute Accad. Naz. Lincei, Cl. Sci. Fis., Mat. Nat., 2020, 31, 821–833 CrossRef.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- C. Bouzigues, T. Gacoin and A. Alexandrou, ACS Nano, 2011, 5, 8488–8505 CrossRef CAS PubMed.
- J. Wang and S. Li, Front. Med., 2022, 9, 946100 CrossRef.
- Z. Wei, Y. Liu, B. Li, J. Li, S. Lu, X. Xing, K. Liu, F. Wang and H. Zhang, Light: Sci. Appl., 2022, 11, 175 CrossRef CAS PubMed.
- P. C. Dent, J. Appl. Phys., 2012, 111, 07A721 CrossRef.
- J. M. D. Coey, Engineering, 2020, 6, 119–131 CrossRef CAS.
- J. Guo, S. Chen, W. Chen, X. Huang and T. Cui, Front. Electron. Mater., 2022, 2, 906213 CrossRef.
- X. Zhong, Y. Sun, T. Iitaka, M. Xu, H. Liu, R. J. Hemley, C. Chen and Y. Ma, J. Am. Chem. Soc., 2022, 144, 13394–13400 CrossRef CAS PubMed.
- X. Feng, O. Onel, M. Council-Troche, A. Noble, R.-H. Yoon and J. R. Morris, Appl. Clay Sci., 2021, 201, 105920 CrossRef CAS.
- S. Qiu, T. Qiu, H. Yan, Q. Long, H. Wu, X. Li and D. Zhu, Colloids Surf., A, 2022, 642, 128596 CrossRef CAS.
- A. M. Borst, M. P. Smith and A. A. Finch, et al. , Nat. Commun., 2020, 11, 4386 CrossRef CAS PubMed.
- P. Chatterjee, Y. Han, T. Kobayashi, K. K. Verma, M. Mais, R. K. Behera, T. H. Johnson, T. Prozorov, J. W. Evans and I. I. Slowing, et al. , ACS Appl. Mater. Interfaces, 2023, 15, 54192–54201 CrossRef CAS.
- G. G. Juttu and R. F. Lobo, Microporous Mesoporous Mater., 2000, 40, 9–23 CrossRef CAS.
- W. J. Roth and D. L. Dorset, Microporous Mesoporous Mater., 2011, 142, 32–36 CrossRef CAS.
- W. J. Roth, J. Čejka, R. Millini, E. Montanari, B. Gil and M. Kubu, Chem. Mater., 2015, 27, 4620–4629 CrossRef CAS.
- K. Okumura, M. Hashimoto, T. Mimura and M. Niwa, J. Catal., 2002, 206, 23–28 CrossRef CAS.
- A. Corma, U. Díaz, T. García, G. Sastre and A. Velty, J. Am. Chem. Soc., 2010, 132, 15011–15021 CrossRef CAS PubMed.
- Y. Han, P. Chatterjee, S. B. Alam, T. Prozorov, I. I. Slowing and J. W. Evans, Phys. Chem. Chem. Phys., 2023, 25, 4680–4689 RSC.
- Y. Han, T. Ash, P. Chatterjee, W. Huang, T. L. Windus and J. W. Evans, J. Phys. Chem. C, 2024, 128, 6309–6318 CrossRef CAS.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910–1913 CrossRef CAS PubMed.
- G. Sastre, V. Fornes and A. Corma, J. Phys. Chem. B, 2000, 104, 4349–4354 CrossRef CAS.
- D. Zhou, Y. Bao, M. Yang, N. He and G. Yang, J. Mol. Catal. A: Chem., 2006, 244, 11–19 CrossRef CAS.
- Y. Li, W. Guo, W. Fan, S. Yuan, J. Li, J. Wang, H. Jiao and T. Tatsumi, J. Mol. Catal. A: Chem., 2011, 338, 24–32 CrossRef CAS.
- Y. Wang, D. Zhou, G. Yang, S. Miao, X. Liu and X. Bao, J. Phys. Chem. A, 2004, 108, 6730–6734 CrossRef CAS.
- D. J. Rogers, N. J. Taylor and G. E. Toogood, Acta Crystallogr., 1983, 39, 939–941 Search PubMed.
- National Center for Biotechnology Information, PubChem Compound Summary for CID 16211525, Ytterbium(III) nitrate pentahydrate, PubChem, 2024, https://pubchem.ncbi.nlm.nih.gov/compound/Ytterbium_III_-nitrate-pentahydrate Search PubMed.
- A. Zheng, L. Chen, J. Yang, M. Zhang, Y. Su, Y. Yue, C. Ye and F. Deng, J. Phys. Chem. B, 2005, 109, 24273–24279 CrossRef CAS.
- S. Wang, Z. Wei, Y. Chen, Z. Qin, H. Ma, M. Dong, W. Fan and J. Wang, ACS Catal., 2015, 5, 1131–1144 CrossRef CAS.
- E. Aprà, E. J. Bylaska, W. A. de Jong, N. Govind, K. Kowalski, T. P. Straatsma, M. Valiev, H. J. Van Dam, Y. Alexeev and J. Anchell, et al. , J. Chem. Phys., 2020, 152, 184102 CrossRef.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- Y. Han, K. C. Lai, A. Lii-Rosales, M. C. Tringides, J. W. Evans and P. A. Thiel, Surf. Sci., 2019, 685, 48–58 CrossRef CAS.
- Y. Han, M. C. Tringides, J. W. Evans and P. A. Thiel, Phys. Rev. Res., 2020, 2, 013182 CrossRef CAS.
- Y. Han, J. W. Evans and M. C. Tringides, Appl. Phys. Lett., 2021, 119, 033101 CrossRef CAS.
- X. Wang and Y. Han, Comput. Mater. Sci., 2023, 224, 112153 CrossRef CAS.
- Y. Han, M. Kolmer, M. C. Tringides and J. W. Evans, Carbon, 2023, 205, 336–344 CrossRef CAS.
- Y. Han, AIP Adv., 2022, 12, 125009 CrossRef CAS.
- Y. Han, P. Chatterjee, S. B. Alam, T. Prozorov, I. I. Slowing and J. W. Evans, Phys. Chem. Chem. Phys., 2023, 25, 4680–4689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03424d |
| This journal is © the Owner Societies 2025 |