
Unraveling the effect of reagent vibrational excitation on the scattering mechanism of the benchmark H + H2 → H2 + H hydrogen exchange reaction in the coupled 12E′ ground electronic manifold†
Jayakrushna
Sahoo‡
,
Sugata
Goswami§
 and
S.
Mahapatra
*
and
S.
Mahapatra
*
School of Chemistry, University of Hyderabad, Hyderabad 500 046, India. E-mail: susanta.mahapatra@uohyd.ac.in
First published on 14th November 2024
Abstract
The hydrogen exchange reaction, H + H2 → H2 + H, along with its isotopic variants, has been the cornerstone for the development of new and novel dynamical mechanisms of gas-phase bimolecular reactions since the 1930s. The dynamics of this reaction are theoretically investigated in this work to elucidate the effect of reagent vibrational excitation on differential cross sections (DCSs) in a nonadiabatic situation. The dynamical calculations are carried out using a time-dependent quantum mechanical method, both on the lower adiabatic potential energy surface and employing a two-state coupled diabatic theoretical model to explicitly include all the nonadiabatic couplings present in the 12E′ ground electronic manifold of the H3 system. Towards this effort, the Boothroyd–Keogh–Martin–Peterson (BKMP2) surface of the lower adiabatic component is coupled with the double many-body expansion (DMBE) surface of the upper one. The smooth variation of energy along the D3h seam of the conical intersections is explicitly confirmed. The coupled two-state calculations are performed only for H2 (v = 3–4, j = 0), as the minimum of the conical intersections becomes energetically accessible for these vibrational levels in the considered energy range. Initial state-selected total and state-to-state DCSs are calculated to elucidate various mechanistic aspects of reagent vibrational excitation. The latter enhances the forward scattering and makes the backward scattering less prominent. Important roles of collision energy in the vibrational energy disposal of both forward- and backward-scattered products are examined. Analysis of the state-to-state DCSs of the vibrationally excited reagents reveals an important correlation among scattering angle, and the product rotational angular momentum and its helicity state. Such an analysis establishes a novel mechanism for the forward scattering of the reaction.
I. Introduction
The hydrogen exchange reaction, H + H2 (v, j) → H2 (v′, j′) + H, along with its isotopic (D) counterparts, has been the most important of all chemical reactions for understanding bond breaking and making at a microscopic level in both theoretical and experimental reaction dynamics. Numerous studies on this reaction have laid the foundation for developing various concepts like potential energy surfaces (PESs),1–3 transition states,4,5 reactive resonances,6–9 quantized bottleneck states (QBSs)10–16 and, of late, nonadiabatic quantum dynamics.17–22 Even after almost a century of investigation, this so-called simplest chemical reaction still continues to bring surprises to the community with new observations and discoveries. To name a few, the signature of a geometric phase (GP) in differential reaction cross sections,20,23–31 quantum interference between the distinct topological paths around the conical intersections (CIs) in the energy-resolved differential cross sections (DCSs),32 the appearance of Feshbach resonance below the reaction barrier,33,34 and glory scattering in the forward-scattered DCSs35,36 are noteworthy. This reaction has also served as a benchmark system for the investigations of electronic nonadiabatic effects in reaction dynamics17–21,37–43 and GP effects in the ultracold regime.44–48 Apart from its immense breadth and rigor, this reaction has also been found to have presence and importance in an astrophysical context for the cooling process of the early universe and in ortho–para H2 conversion.49–53The involvement of three light nuclei under the influence of a potential provided only by three electrons makes theoretical investigation relatively simple and accurate. A number of PESs54–62 have been constructed by employing various methods and used in different dynamical investigations (see ref. 16 and 63 and the references therein). Moreover, the availability of advanced experimental methods16,64,65 and isotopic substitutions of the reagent H2 have made the measurement of the energy-resolved state-to-state DCSs feasible.32 In fact, the agreement between theory and experiment has become closer than ever,30–32,65–67 encouraging both theorists and experimentalists towards further investigations. Though the reactive system seems to be very simple, experimental outcomes made theorists look into the dynamics more critically and indeed the findings are apparently not simple!68
It is generally believed that the H + H2 reaction is direct in nature, following a collinearly dominated rebound mechanism, mainly producing backward-scattered products. Nevertheless, forward scattering was found for low rotational product states at relatively high collision energies and was thought to be due to quantum mechanical Feshbach resonances.63 However, later studies showed that the forward scattering also appears in the quasi-classical trajectory (QCT) dynamics and therefore its origin is obviously not quantum mechanical in nature.69,70 Rather, it is because of a time-delayed mechanism,13,71,72 where the delay was found to be due to slowing down of the colliding partners near the top of the effective barrier of the QBSs corresponding to higher total angular momentum.13 Such slowing down provides enough time for the triatomic complex to rotate and to consequently form forward-scattered products. Yuan et al.65 were the first to measure the fast oscillatory state-to-state DCSs in the forward direction of the H + HD (v = 0, j = 0) → H2 (v′, j′) + D reaction at 1.35 eV collision energy (Ecol) by using a D-atom near-threshold ionization velocity map imaging technique with an estimated angular resolution of ≈1.5°. Later, in a theoretical investigation, Xiahou and Connor35,36 showed that the forward-scattering oscillations measured by Yuan et al.65 mainly involve forward glory scattering, and the fast angular oscillations actually originate from quantum interference between the nearside and farside scattering, which was predicted earlier.73–75
Backward scattering takes place due to head-on collisions with little rotational excitation, whereas sideways scattering results from glancing collisions producing rotationally excited products.67,76,77 This phenomenon is known as negative j′–θ correlation78 and has been explained by a simple hard-sphere scattering model.67,76,77 However, a combined theoretical and experimental investigation of the H + D2 → HD + D reaction by Jankunas et al. at Ecol = 1.97 eV showed an anomalous trend, opposite to the usual negative j′–θ, for vibrationally hot product HD (v′ = 4).79 This surprising behaviour could not be explained by the purely repulsive minimum energy path (MEP) of the H + H2 reaction involving a barrier of ∼0.42 eV (see Fig. 1 of ref. 68). Rather, it was due to the lack of enough recoil energy between the products while passing over the centrifugal barrier of the vibrationally adiabatic potential corresponding to v = 4.68 A subsequent study by Sneha et al.78 at Ecol = 3.26 eV confirmed this reasoning, where the usual negative j′–θ trend was recovered as the products had enough recoil energy to overcome the centrifugal barrier. It is important to note here the significant role of vibrationally adiabatic potentials in the scattering mechanism of this reaction, which has also been useful in explaining the behaviour of the barrier resonances and QBSs.12,15,68,80
It was pointed out by Jankunas et al.68 that either enhancement of the relative translational energy or internal excitation of the reagent diatom can facilitate the nonadiabatic dynamics of this reaction. A great deal of interest has also been invested to prepare the reagent H2 and its isotopologues at highly vibrationally excited levels, where crossed-molecular-beam experiments can be carried out under appropriate experimental conditions to explore the effect of reagent vibration on the dynamics of this reaction.81–85
Although a few theoretical investigations25–27,37,44,45,86,87 have already been carried out with internally excited reagent H2 by employing different PESs, there is still a requirement for comprehensive quantum dynamics investigations at the state-to-state level with internally excited reagent diatoms. Such a thorough investigation can further reveal the dynamics of this seemingly simple reaction in great detail. We have taken up such an investigation recently by calculating total and state-to-state reaction probabilities, ICSs and product level distributions up to Ecol = 1.25 eV for the H + H2 (v = 0–4, j = 0–3) → H2 (v′, j′) + H reaction in its electronic ground state.88 The BKMP2 PES59 and a numerically exact Hamiltonian have been employed in the investigation, where the emphasis was on finding the effect of reagent vibration on reaction probabilities, ICSs and product level distributions.88 The onset of the reaction was found to shift towards low collision energies with reagent vibrational excitation and finally the reaction with a classical barrier of ≈0.42 eV became barrierless for reagent H2 (v = 4, j = 0). Vibrational adiabaticity was found to be maintained where the total angular momentum quantum number (J) was zero, whereas it was lost when contributions from J > 0 were included in the ICSs. The collision and reagent vibrational energies affected the product vibrational distribution (in terms of the ICS) in opposite ways. An overall enhancement in reactivity with reagent vibrational excitation was observed.88 All these findings together revealed that the dynamics of the H-exchange reaction with vibrationally excited reagent diatoms is different and somehow complicated compared to that occurring with reagent H2 (v = 0, j = 0). To the best of our knowledge, our previous investigation88 is the first and to date the most comprehensive description of the effect of reagent vibration on the state-to-state reaction probabilities and ICSs of the H-exchange reaction in its electronic ground state.
The present investigation is a further effort and continuation in this endeavor and can be considered as an extension of our earlier work.88 Total and product state-resolved DCSs of the H + H2 (v = 0–4, j = 0) → H2 (v′, j′) + H reaction are calculated here for collision energies up to 1.25 eV. The dynamics are carried out by employing a time-dependent quantum mechanical (TDQM) methodology89,90 based on the real wave packet (RWP) formalism of Gray and Balint-Kurti.91 It is well known that H3 is a (E ⊗ e)-Jahn–Teller system having a seam of CIs between the ground (12A′) and first-excited (22A′) electronic states along the D3h symmetry configurations. The energetic minimum of the CI seam becomes accessible at a total energy of ∼2.73 eV59 with respect to the H + H2 ground-state asymptote. In the present case, the CI becomes accessible at Ecol ≈ 1.0 eV for H2 (v = 3, j = 0) and ≈ 0.57 eV for H2 (v = 4, j = 0). In this situation, the adiabatic formalism breaks down, especially for the state-to-state DCSs,20,30,92 and the dynamics need be treated with inclusion of electronic nonadiabatic effects. This is done in the present work by carrying out the state-to-state TDQM calculations by using a two-state coupled diabatic theoretical model.17 The nonadiabatic coupled surface dynamics calculations are carried out for reagent H2 (v = 3–4, j = 0) and the DCSs obtained are used to elucidate the scattering mechanism. The DCSs for H2 (v = 0–2, j = 0) are obtained from single-surface adiabatic dynamics calculations on the electronic ground state. The dependence of reagent vibrational excitation on the DCSs is examined. As the DCS is one of the most subtle dynamical observables among them all, the present investigation provides a better understanding of the dynamics of the H-exchange reaction in conjunction with our earlier findings.88
We note that the calculations are carried out by considering the three hydrogen nuclei as distinguishable, since the main aim here is to understand the underlying mechanism of the reactive encounter of H with H2.
The rest of the article is organized in the following way. The theoretical methodology followed in the present investigation is described briefly in Section II, followed by a detailed discussion of the results in Section III. The results of the investigation are summarized in Section IV with an outlook.
II. Theoretical and computational details
A. Adiabatic dynamics on the electronic ground PES
The adiabatic single-surface dynamical calculations are carried out on the electronic ground BKMP2 PES.59 A TDQM method, based on the real wave packet (RWP) approach,91 as implemented in the DIFFREALWAVE algorithm,89,90 is employed here. This algorithm has been successful in explaining the dynamics of various atom–diatom reactions.88,89,93–101 Comprehensive details of this method are available in the literature;89–91,93,96 only a brief description of the essentials is provided below.An initial wave packet (WP) is prepared at the reagent asymptote in the body-fixed (BF) reagent Jacobi coordinates. The WP is then transformed to the product Jacobi coordinates where only its real part is propagated in time. The time propagation of the WP is carried out using a Chebyshev polynomial-based three-term iterative equation. At each step of the iteration, the action of the Hamiltonian operator on the WP is evaluated using the procedures described in ref. 89, 90 and 102. The Coriolis couplings among all the angular momentum substates of each total angular momentum, J, are considered explicitly by parallelizing each set of calculations over different computer processors using an MPI library.103,104 The propagated WP is analyzed at the product asymptote by projecting it onto the specific product ro-vibrational states at every iteration step. The scattering matrix (S-matrix) elements, 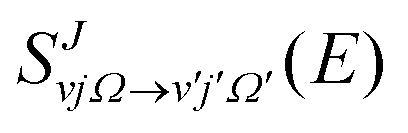 , are then calculated using the techniques explained in ref. 89, 91 and 102 in the helicity representation, wherein the reaction observables are obtained. The quantities Ω and Ω′ represent the projection of J onto the BF z-axis of the reagent and the product Jacobi coordinate system, respectively.
, are then calculated using the techniques explained in ref. 89, 91 and 102 in the helicity representation, wherein the reaction observables are obtained. The quantities Ω and Ω′ represent the projection of J onto the BF z-axis of the reagent and the product Jacobi coordinate system, respectively.
The state-to-state DCSs are given by:
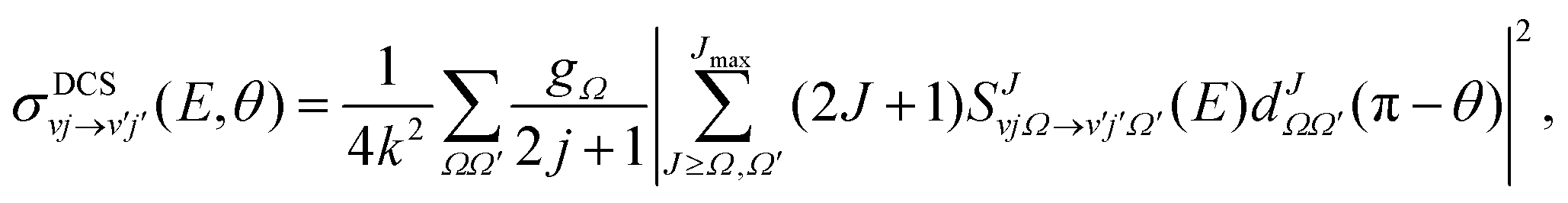 | (1) |
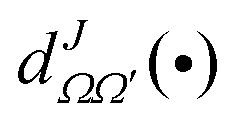 are the elements of the reduced Wigner rotation matrix,105,106 and
are the elements of the reduced Wigner rotation matrix,105,106 and 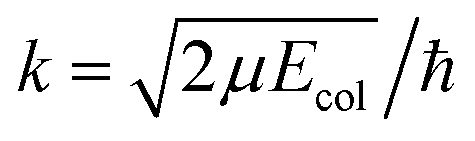 , with μ being the atom–diatom reduced mass of the reagent channel. The term gΩ equals 1 and 2 for Ω = 0 and Ω > 0, respectively. The initial state-selected total DCSs are obtained by summing up the state-to-state DCSs over the v′ and j′ quantum numbers. In eqn (1), the definition of the scattering angle, θ, as given by Zhang and Miller107 (the “π − θ” convention) is used. This assumes θ = 0° and 180° as forward and backward scattering, respectively.
, with μ being the atom–diatom reduced mass of the reagent channel. The term gΩ equals 1 and 2 for Ω = 0 and Ω > 0, respectively. The initial state-selected total DCSs are obtained by summing up the state-to-state DCSs over the v′ and j′ quantum numbers. In eqn (1), the definition of the scattering angle, θ, as given by Zhang and Miller107 (the “π − θ” convention) is used. This assumes θ = 0° and 180° as forward and backward scattering, respectively.
It is important to analyze the contribution of each partial wave to the DCS in order to understand the details of the scattering of products at a certain angle arising from different ranges of partial waves or equivalently impact parameters. These J-dependent partial DCSs must ensure full coherence between all Js and are calculated at a particular collision energy from the S-matrix elements as:
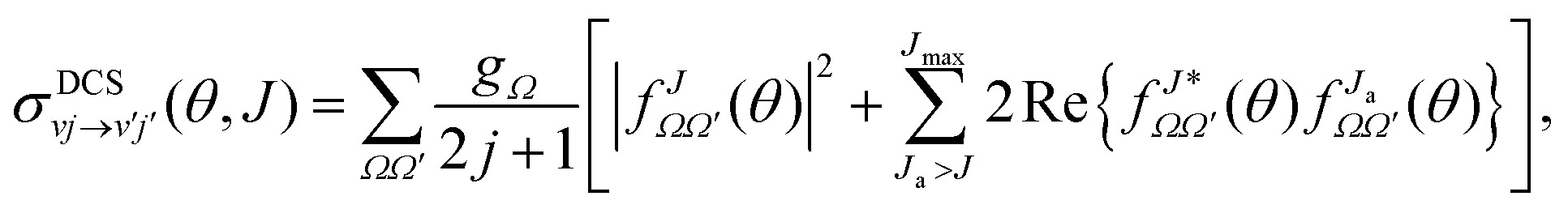 | (2) |
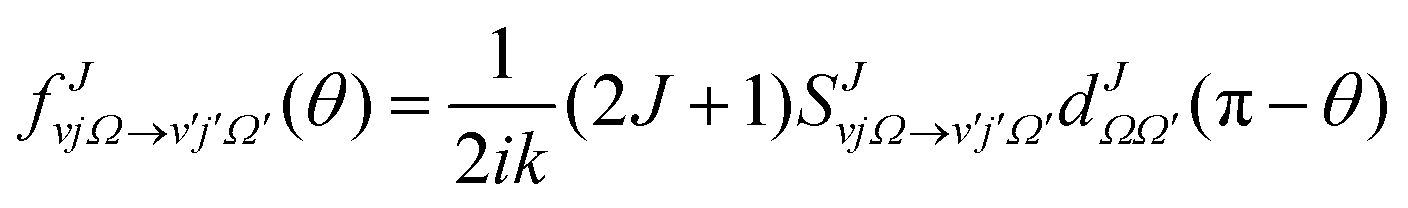 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, which is not multiplied in the above equation. This is because in the present work we mostly analyzed products scattered at θ = 0° and 180°, for which the QM GDF exactly becomes zero. The QM GDF is an important tool to describe the quantum mechanical correlation between the scattering angle, θ, and the total angular momentum, J.108–110
θ, which is not multiplied in the above equation. This is because in the present work we mostly analyzed products scattered at θ = 0° and 180°, for which the QM GDF exactly becomes zero. The QM GDF is an important tool to describe the quantum mechanical correlation between the scattering angle, θ, and the total angular momentum, J.108–110
B. Nonadiabatic dynamics in the coupled diabatic representation
The nonadiabatic state-to-state quantum scattering calculations are carried out here by extending the DIFFREALWAVE algorithm to the nonadiabatic framework, where both the ground and excited electronic PESs of H3 and all the nonadiabatic couplings between them are included in a modified version of the coupled-state diabatic theoretical model of Mahapatra et al.17In the electronically diabatic representation, the Hamiltonian for the ground (12E′) electronic manifold of the H + H2 reactive system is given by a 2 × 2 matrix as:
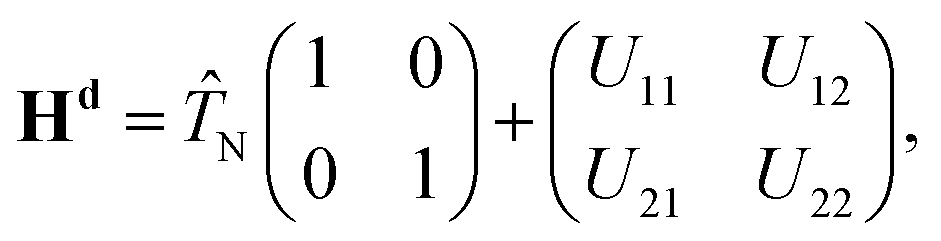 | (4) |
![[T with combining circumflex]](https://www.rsc.org/images/entities/i_char_0054_0302.gif) N is the nuclear kinetic energy operator and U11 and U22 are the two diabatic electronic PESs coupled by the diabatic coupling elements U12 = U21. The N operator in the present work is expressed in terms of the Jacobi coordinates of the product arrangement channel. The readers are referred to ref. 88 and 89 for a detailed expression of N. The diabatic potential energy matrix elements are obtained by diabatizing the diagonal adiabatic potential energy matrix through the following similarity transformation:111–113
N is the nuclear kinetic energy operator and U11 and U22 are the two diabatic electronic PESs coupled by the diabatic coupling elements U12 = U21. The N operator in the present work is expressed in terms of the Jacobi coordinates of the product arrangement channel. The readers are referred to ref. 88 and 89 for a detailed expression of N. The diabatic potential energy matrix elements are obtained by diabatizing the diagonal adiabatic potential energy matrix through the following similarity transformation:111–113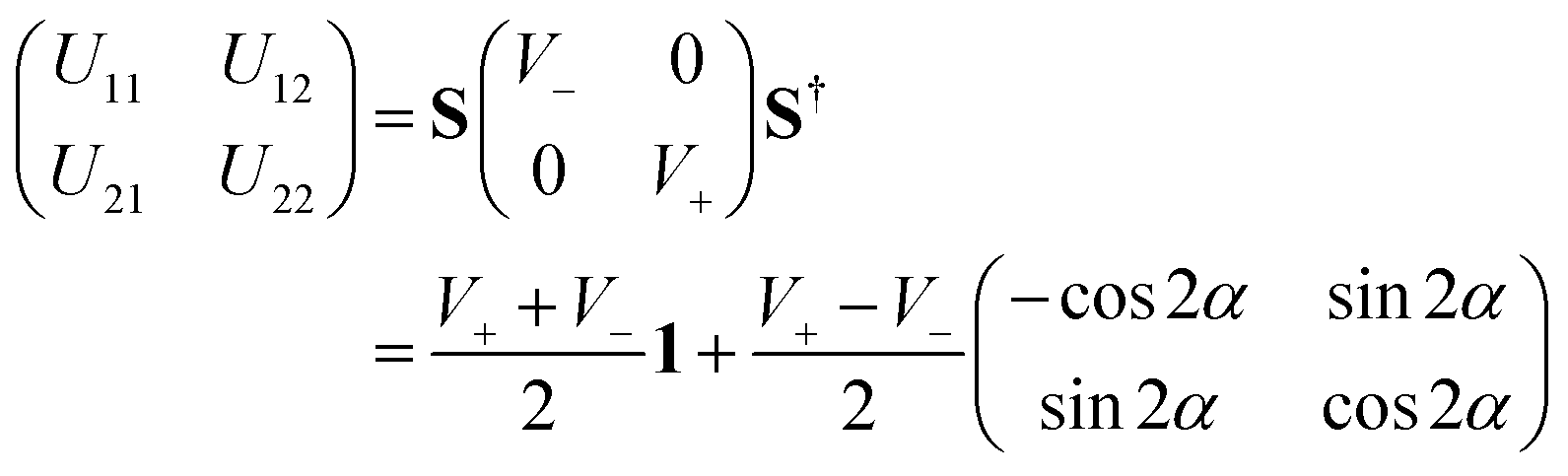 | (5) |
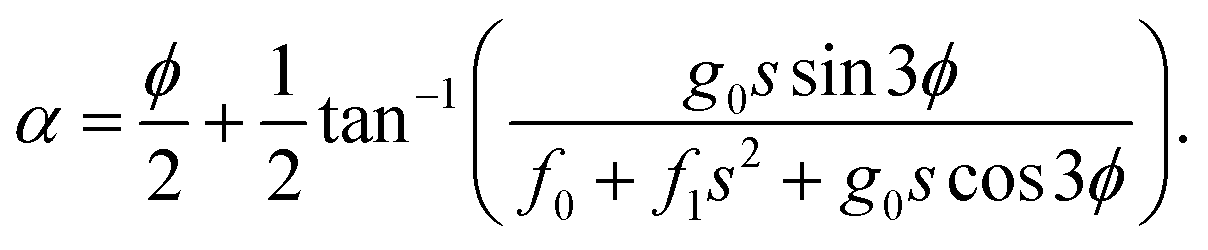 | (6) |
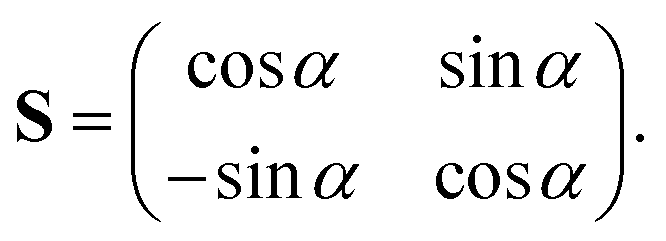 | (7) |
The ADT angle is considered here up to the quadratic coupling term and avoids the unwanted CIs occurring at nonzero values of s.57,114 Here, s stands for the normalized radial distance from the D3h CI in its two-dimensional vibrational subspace and s = 0 gives the seam of the CI. It is necessary to mention here that up to the linear coupling approximation, the ADT angle is equal to half the pseudorotation angle, ϕ (defined as the direction of the ε-type vibration in its two-dimensional vibrational subspace of the D3h symmetry point group), which eliminates the leading singular part of the derivative coupling.112 The additional quadratic term on the right-hand side of eqn (6) eliminates the remaining non-singular part21 and depends on the two adiabatic PESs. This term becomes important near the collinear geometry.62
Inclusion of the quadratic term in the ADT angle makes the diabatic representation more accurate for dynamical studies, as the primary aim of present work is to understand the underlying mechanism of this reaction with a vibrationally excited reagent. However, a sole assessment of the effect of this quadratic term is not carried out in the present contribution, since it was already examined in ref. 22. The readers are referred to eqn (5) and (38)–(41) of ref. 57 for the relevant expressions for s and ϕ. In eqn (6), the quantities f0, f1 and g0 are functions of the nuclear coordinate corresponding to the symmetric stretching (breathing) vibrational mode of the D3h configuration of H3 and are calculated by taking the derivatives of the difference between the two adiabatic PESs (see eqn (50)–(53) of ref. 57).
In the present work, the Boothroyd–Keogh–Martin–Peterson (BKMP2)59 surface is used for the lower adiabatic component (V−) and the upper part of the double many-body expansion (DMBE)57 PES is used for the upper adiabatic component (V+). In order to ensure the degeneracy along the CI seam, a small correction term is introduced to the upper DMBE surface. We would like to point out here that since the coupled two-state dynamical calculations are performed in a diabatic representation, where the smooth diabatic potentials are obtained from the adiabatic PESs through similarity transformation (see eqn (5)), the use of two different functional forms for the upper (DMBE) and lower (BKMP2) adiabatic PESs does not cause any numerical difficulty.
Following the TDQM methodology, an initial WP is prepared in the asymptotic reagent channel of the repulsive lower adiabatic PES V−. This is because the upper adiabatic PES V+ does not have any dissociative asymptotes in the considered energy range. The WP is first prepared in the BF reagent Jacobi coordinate system (R, r and γ) and then transformed to the product Jacobi coordinate (R′, r′ and γ′) system. The real part of the adiabatic WP can be expressed as a column matrix:
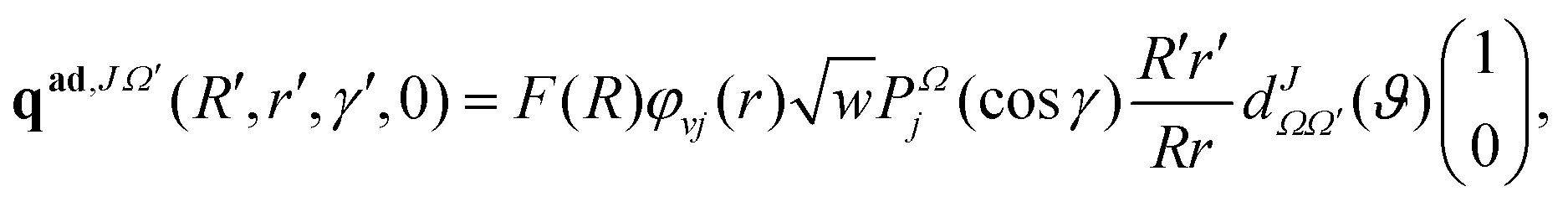 | (8) |
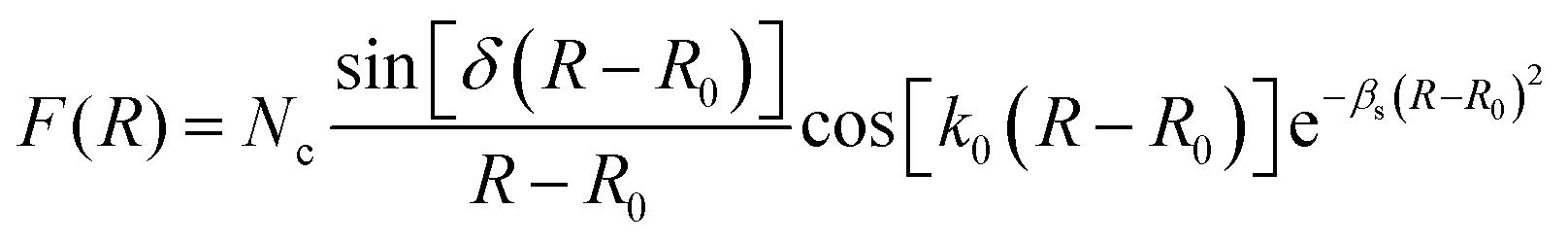 | (9) |
γ) represent the ro-vibrational wave function of the reagent diatom and the associated Legendre polynomials, respectively. The imaginary part of the adiabatic WP (pad,JΩ′) is similar to the real counter part, qad,JΩ′, except the function cos[k0(R − R0)] in F(R) is replaced by −sin[k0(R − R0)]. It is noteworthy to mention here that in the RWP formalism only the real part of the WP is propagated in space and time to obtain the desired reaction observables, while the imaginary part of the WP is required only once at the beginning of the time propagation.91 As a consequence, the RWP methodology offers a significant reduction in computational overheads as compared to the complex WP propagation schemes.
The initial real and imaginary parts of the WP defined in the above equations (see eqn (8) and (9)) are transformed to the diabatic representation by using the ADT matrix (see eqn (7)) prior to the propagation. In the diabatic representation, these functions read as:
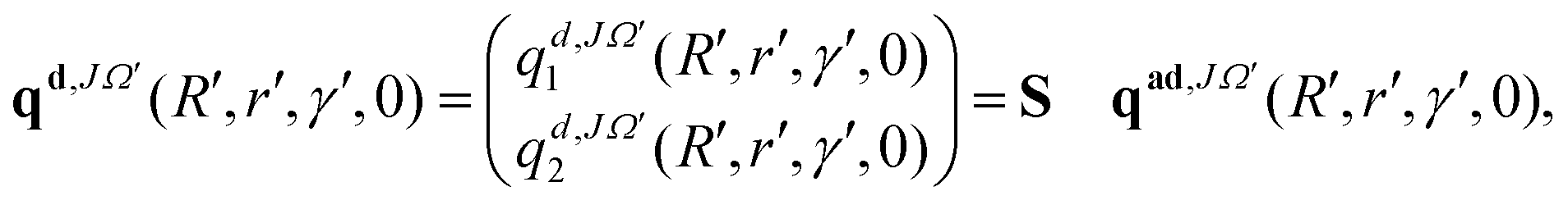 | (10) |
 | (11) |
The dynamics are carried out afterwards in the coupled diabatic representation in the RWP theoretical framework. The time propagation of the diabatic WPs is carried out using Chebyshev polynomial-based iterative equations with the inclusion of an absorption function, as explained in ref. 22. The action of the 2 × 2 diabatic Hamiltonian matrix Hd on the real (qd,JΩ′) and imaginary (pd,JΩ′) parts of the diabatic WP column vectors is carried out via a straightforward matrix multiplication. The action of the kinetic energy operator and the diabatic potentials on the WP is carried out in the same way as in case of the single-surface adiabatic calculations (see Section II A). The Coriolis coupling terms are treated here accurately for each J. It is important to note here that the computational time for the WP propagation in the diabatic representation (involving both the electronic states) doubles as compared to that for the adiabatic single-surface propagation, making the nonadiabatic calculations more demanding computationally as compared to the single-surface adiabatic calculations.
At each step of the iterations, the propagated diabatic WP is transformed to the adiabatic representation by using the ADT matrix before it is analyzed. This is given by:
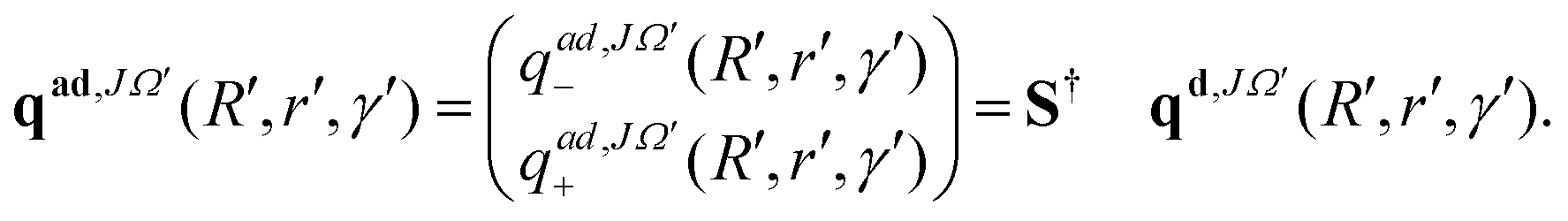 | (12) |
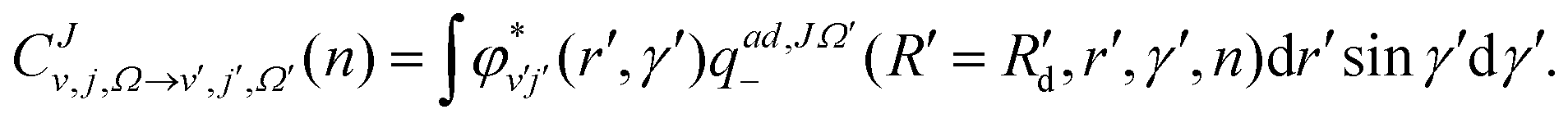 | (13) |
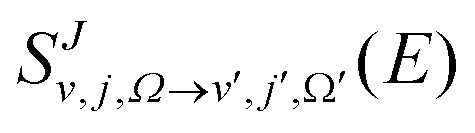 in the helicity representation. The state-to-state DCSs and the J-dependent partial DCSs can be obtained by using eqn (1) and (2).
in the helicity representation. The state-to-state DCSs and the J-dependent partial DCSs can be obtained by using eqn (1) and (2).
III. Results and discussion
Total and product state-resolved DCS results of the H + H2 (v = 0–4, j = 0) → H2 (v′, j′) + H reaction are shown and discussed in this section. The product vibrational level distributions in terms of the DCS at fixed θ are also presented at a few collision energies to elucidate the energy disposal mechanism of scattering angle-resolved products. The effect of vibrational excitation of reagent H2 on the scattering mechanism is discussed in detail.The numerical parameters used in the present TDQM calculations are the same as those optimized in our earlier study.88 It is to be noted that numerous test calculations were carried out to ensure numerical convergence of the TDQM parameters and some of the results were compared with the available literature results in our earlier study88 for benchmarking the present results. Moreover, in order to validate the correctness of the present DCSs, a few of them for reagent H2 (v = 0, j = 0) are compared with those available in the literature and are shown in Fig. S1–S4 of the ESI.† We note that the parameters used in the coupled surface calculations for reagent H2 (v = 3–4, j = 0) are the same as those used in the uncoupled surface calculations.88 All required partial waves are included in the calculations to obtain a converged DCS up to Ecol = 1.25 eV for each individual vibrational level of reagent H2.88 Although the present contribution does not aim towards rigorous discussion of the electronic nonadiabatic effects on the dynamics, the coupled and uncoupled surface results are shown together whenever found necessary. In this article, uncoupled refers to the calculations on the single adiabatic ground state PES (V−) and coupled refers to the calculations involving both the electronic state PESs (V− and V+) in a diabatic electronic representation.
A. Effect of reagent vibrational excitation on total DCSs
In order to show the effect of reagent vibrational excitation on the overall dynamics, the initial state-selected total DCSs for the H + H2 (v = 0–4, j = 0) reaction are shown in Fig. 1 as a function of the center-of-mass scattering angle, at a few selected collision energies. The DCSs for each reagent vibrational level are plotted in each panel to show the effect of reagent vibrational excitation. The DCSs for reagent H2 (v = 0–2, j = 0) (see panels a–c) and H2 (v = 3–4, j = 0) (see panels d and e) are obtained from uncoupled and coupled surface calculations, respectively. It can be seen from Fig. 1(a) that for H2 (v = 0, j = 0), the total DCS is dominated by backward scattering and forward scattering appears only at higher collision energies. With an increase in the vibrational excitation of the reagent diatom, the forward scattering increases and becomes more dominant than the backward scattering for H2 (v = 3–4, j = 0) at higher collision energies. This can be seen more clearly in Fig. S5 of the ESI,† where the total DCSs for H2 (v = 0–4, j = 0) are shown in terms of three-dimensional perspective plots as a function of both scattering angle and collision energy. Furthermore, Fig. S5 (ESI†) reveals that the threshold for the forward scattering appears at higher energy as compared to that of the backward scattering for reagent H2 (v = 0–3, j = 0). The difference between the thresholds of the forward and backward DCSs is found to decrease with vibrational excitation of the reagent diatom. The threshold energies for both the forward and backward DCSs for reagent H2 (v = 4, j = 0) tend to zero, suggesting a barrierless nature of the reaction for a highly vibrationally excited reagent. The forward scattering for reagent H2 (v = 0–4, j = 0) increases with an increase in the collision energy; however, the backward scattering increases initially and then decreases slowly at higher collision energies after attaining a maximum (see Fig. S5(a)–(e), ESI†). Hence, it can be said that the increase in reactivity in the ICSs with reagent vibrational excitation, as found in our earlier study,88 mainly comes from the forward-scattered products. It can be seen that for H2 (v = 2–4, j = 0), the variation of the forward- and backward-scattered DCSs as a function of collision energy is similar for different vibrational levels of reagent H2 irrespective of the magnitude of the DCS.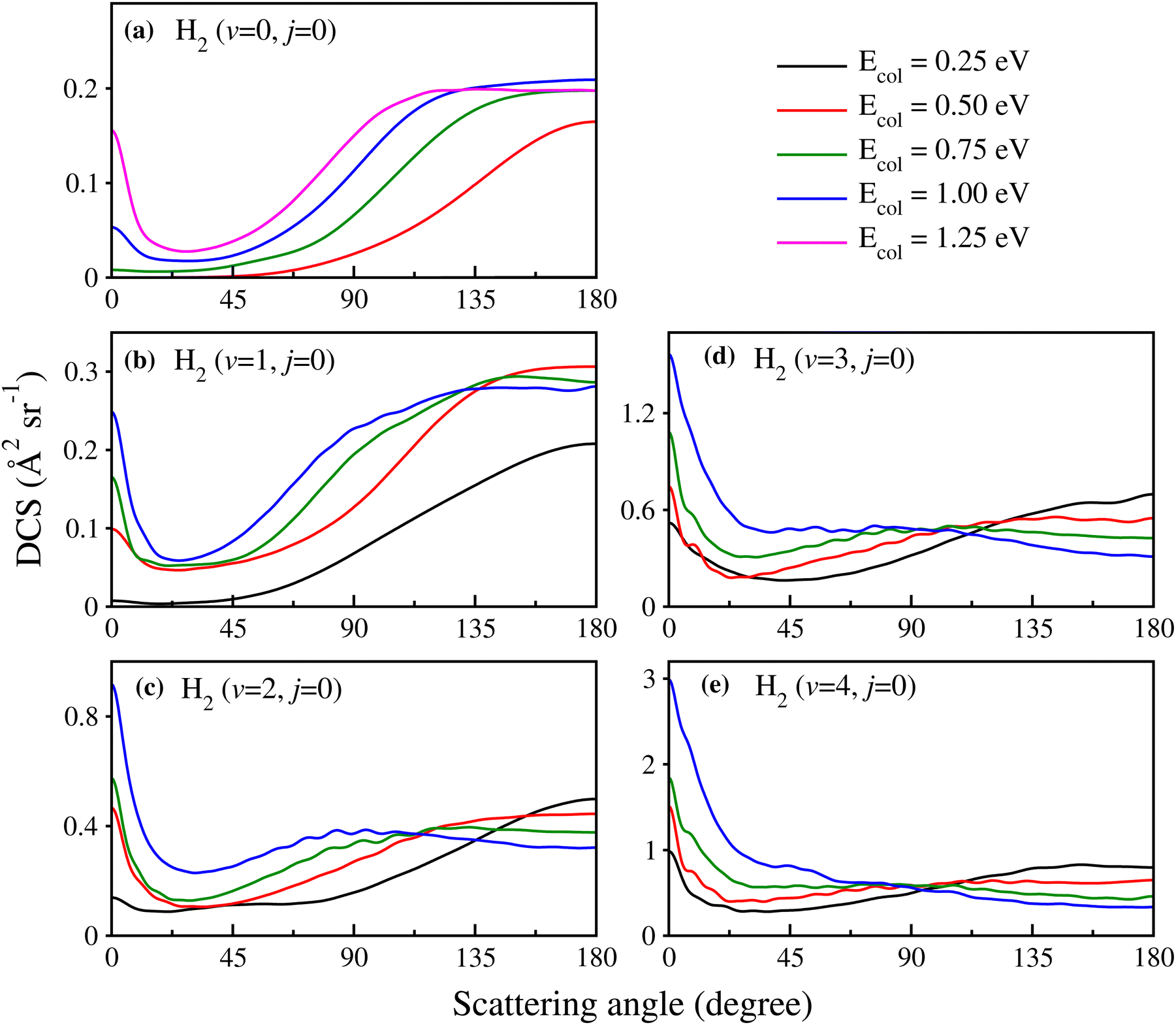 | ||
Fig. 1 Initial state-selected total DCSs of the 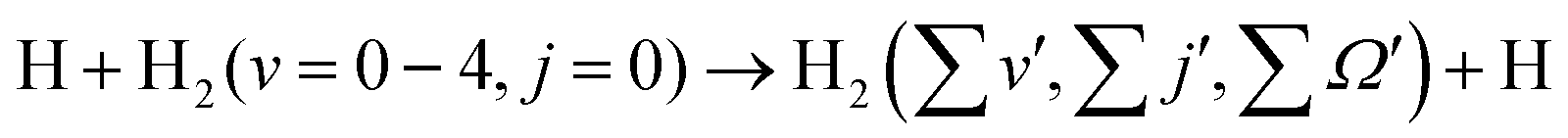 reaction (indicated in each panel) as a function of center-of-mass scattering angle (θ) at Ecol = 0.25, 0.50, 0.75, 1.0 and 1.25 eV, shown by lines of different colours as indicated in the top right corner of the figure. The DCSs for reagent H2 (v = 0–2, j = 0) are obtained from uncoupled surface calculations (panels a–c) and those for H2 (v = 3–4, j = 0) are obtained from coupled surface calculations (panels d and e). reaction (indicated in each panel) as a function of center-of-mass scattering angle (θ) at Ecol = 0.25, 0.50, 0.75, 1.0 and 1.25 eV, shown by lines of different colours as indicated in the top right corner of the figure. The DCSs for reagent H2 (v = 0–2, j = 0) are obtained from uncoupled surface calculations (panels a–c) and those for H2 (v = 3–4, j = 0) are obtained from coupled surface calculations (panels d and e). | ||
Although it is well understood that for reagent H2 (v = 0, j = 0) the forward scattering in the hydrogen exchange reaction stems from a time-delayed mechanism involving contributions from higher total angular momentum,13,71,72 the question still remains whether the overall scattering mechanism of the reaction with a vibrationally excited reagent will be any different from that mentioned above! Therefore, in order to understand the origin of the forward and backward scattering, the J-dependent partial DCSs are calculated for each vibrational level of the reagent H2. The total (summed over final v′, j′ and Ω′ states) J-dependent partial DCSs at θ = 0° and 180° (corresponding to forward and backward scattering, respectively) are shown in Fig. 2 for reagent H2 (v = 0, 2, 4, j = 0). These partial DCSs are shown in terms of colour map plots as a function of Ecol and J in order to understand the contribution of each partial wave at different collision energies. The J-dependent partial DCSs for reagent H2 (v = 1, 3, j = 0) are shown in Fig. S6 of the ESI.†
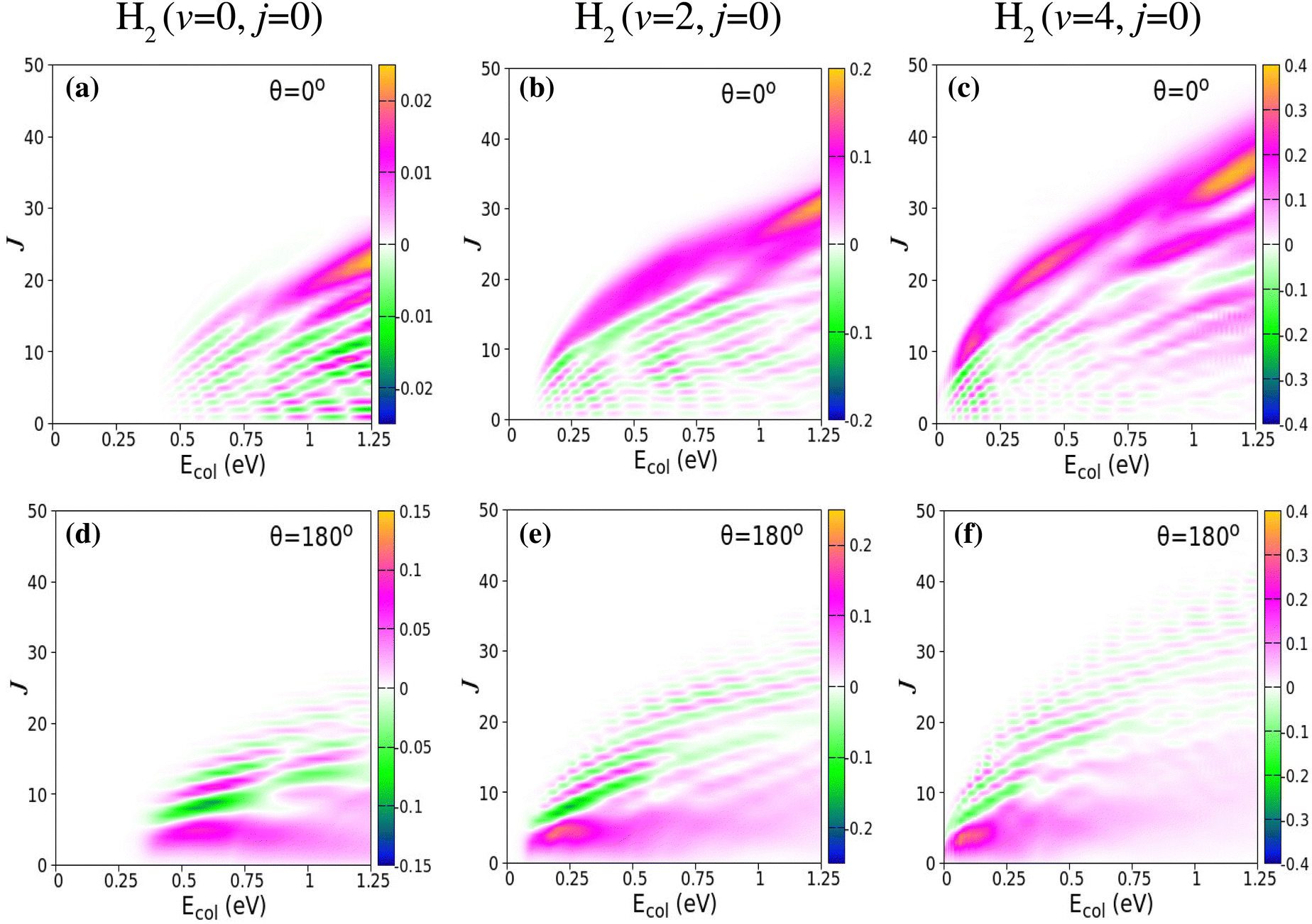 | ||
Fig. 2 Total (summed over final states) J-dependent partial DCSs (Å2 sr−1) of the 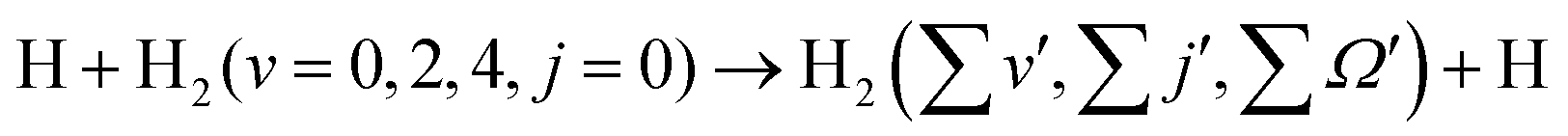 reaction shown as a function of Ecol (abscissa) and J (ordinate) for θ = 0° (panels a–c) and 180° (panels d–f) corresponding to forward and backward scattering, respectively. The partial DCSs for reagent H2 (v = 0,2, j = 0) are obtained from uncoupled surface calculations and those for H2 (v = 4, j = 0) are obtained from coupled surface calculations. reaction shown as a function of Ecol (abscissa) and J (ordinate) for θ = 0° (panels a–c) and 180° (panels d–f) corresponding to forward and backward scattering, respectively. The partial DCSs for reagent H2 (v = 0,2, j = 0) are obtained from uncoupled surface calculations and those for H2 (v = 4, j = 0) are obtained from coupled surface calculations. | ||
It can be seen from Fig. 2 and Fig. S6 (ESI†) that the forward scattering mainly originates from the higher partial waves and backward scattering from the lower partial waves. Most interestingly, this is same for each vibrational level of the reagent. In the case of forward scattering, the partial waves closer to Jmax (the maximum value of J to obtain a converged cross section at a particular Ecol) contribute significantly to the DCSs. Note that the value of the Jmax increases with increasing Ecol. In addition, the contribution of higher Js towards forward scattering increases with increasing collision energy, whereas the contribution of lower Js decreases with increasing collision energy for backward scattering. It is known from the characteristics of the opacity function of the H + H2 reaction that as collision energy increases, the contribution of any particular higher J towards the overall reactivity increases.88,115 However, for any lower value of J, it remains almost the same with the change in collision energy.88,115 For example, the (2J + 1) weighted opacity functions for H2 (v = 4, j = 0) are plotted in Fig. 3. For J = 28, the weighted partial wave contributions (2J + 1)P(J;E) to the cross section are 16.66, 31.83 and 39.21 at Ecol = 0.75, 1.0 and 1.25 eV (marked by a vertical dotted line in Fig. 3). However, the (2J + 1)P(J;E) values are 14.96, 15.96 and 16.31 for J = 9 at the same collision energies (see Fig. 3). Moreover, as the collision energy increases, more and more higher partial waves contribute to the overall reactivity. Since the higher Js mainly contribute to forward scattering, the DCS at θ = 0° increases with increasing collision energy. In contrast, the backward-scattered DCS decreases with increasing collision energy, as it emerges from lower Js. The same analogy can be applied towards the increase in forward scattering with increasing reagent vibrational excitation. In this case, at a fixed value of Ecol, the weighted partial wave contribution for any particular higher J increases significantly with increasing reagent vibrational excitation, whereas such enhancement is less significant for any low values of J (see Fig. 3). In addition, the maximum number of J to obtain the converged cross section at a particular Ecol increases with increasing reagent vibrational excitation.88 This observation also supports the higher threshold of forward scattering than backward for reagent H2 (v = 0–3, j = 0).
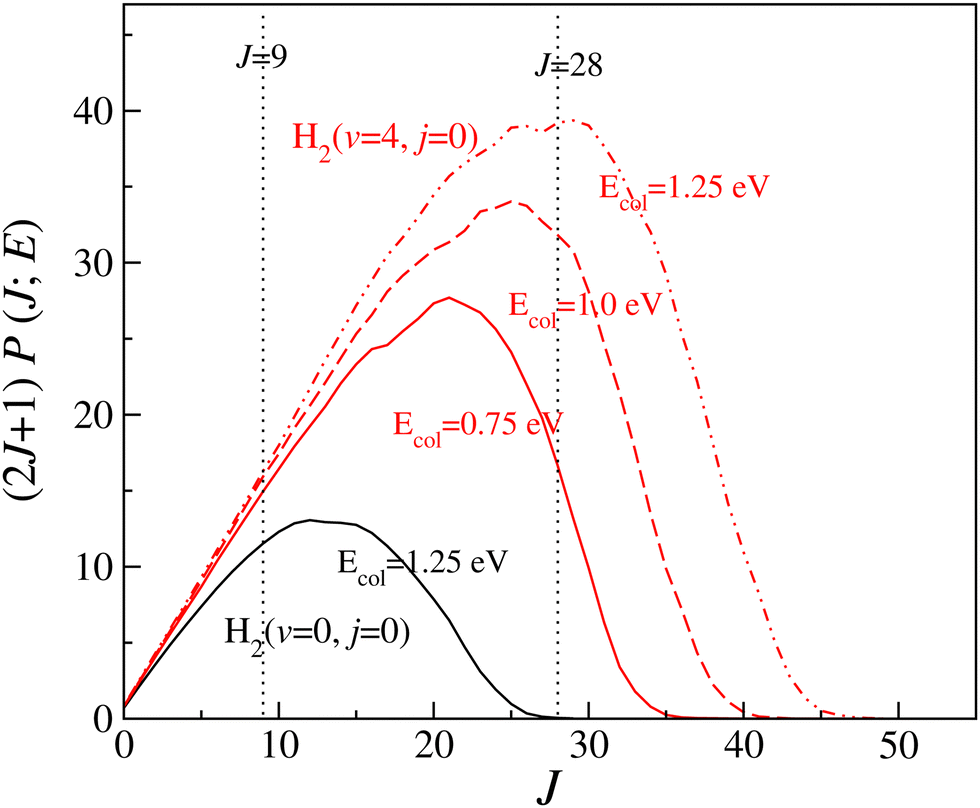 | ||
| Fig. 3 (2J + 1) weighted opacity functions for H2 (v = 0, j = 0) at Ecol = 1.25 eV (black) and for H2 (v = 4, j = 0) at Ecol = 0.75 (solid red), 1.0 (dashed red) and 1.25 eV (dot-dashed red). | ||
We reiterate here that in the calculation of J-dependent partial DCSs, the cross terms are included in eqn (2) in order to ensure full coherence among all the partial waves. This may lead to constructive or destructive interferences among the partial waves, corresponding to the positive and negative values of the J-dependent partial DCSs. This can be seen from Fig. 2 and Fig. S6 (ESI†) as magenta and green coloured stripes. It can also be seen that, in contrast to a small set of J, a broad range of J actually contributes to the forward- and backward-scattered DCS through interference. However, their collective effect becomes minimal once they are summed up. It can be observed that the interference at both forward and backward scattering is strong in the case of H2 (v = 0, j = 0) and becomes weaker with increasing reagent vibrational excitation.
B. Product vibrational-level-resolved DCSs
The effect of reagent vibrational excitation on the product vibrational-level-resolved DCSs is presented and discussed in this section. The v′-resolved DCSs are shown in Fig. 4 in terms of the product vibrational level distribution for fixed values of the scattering angle, θ at 0° and 180°, corresponding to forward and backward scattering, respectively, at four different collision energies as indicated in the legend of the figure. For reagent H2 (v = 3,4, j = 0), the solid lines in panels (c), (d), (g) and (h) represent the DCSs obtained from coupled surface calculations. The uncoupled surface results are also shown by dashed lines of the same colour for the sake of comparison. It can be seen from the figure that for reagent H2 (v = 1, j = 0), both the forward- and backward-scattered products are formed predominantly at the v′ = 1 level maintaining partial vibrational adiabaticity, except at Ecol = 0.5 eV and 1.0 eV for backward and forward scattering, respectively. In these two instances, the scattered products are predominantly found at the v′ = 0 level. However, with increasing reagent vibrational excitation, a few important observations can be made, as follows.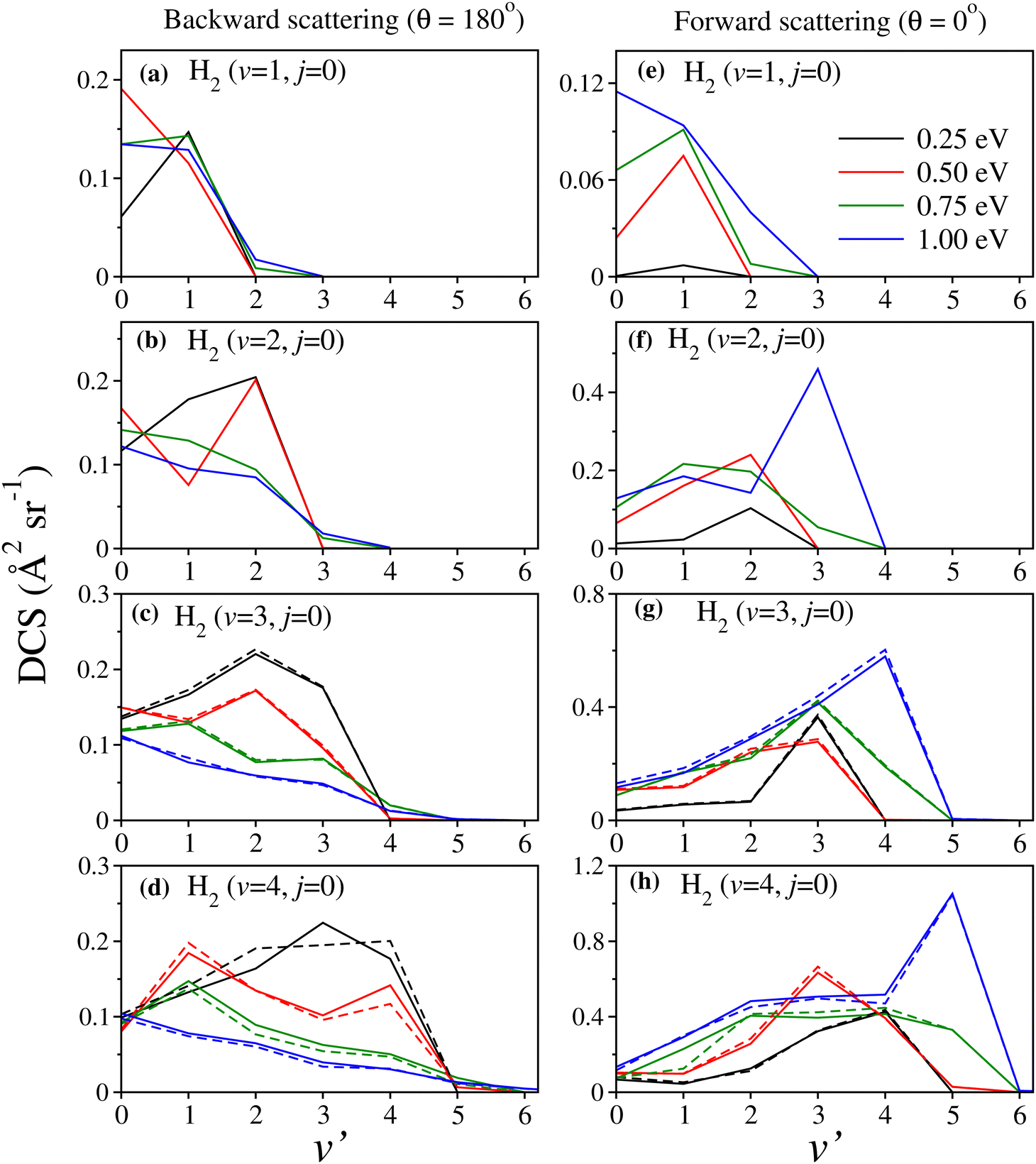 | ||
Fig. 4 Product vibrational level distributions in terms of DCSs at θ = 180° (panels a–d) and 0° (panels e–h) for the  reaction at Ecol = 0.25, 0.50, 0.75 and 1.0 eV, shown by lines of different colours as mentioned in the upper right corner of panel (e). In addition to the coupled surface results (solid lines) for H2 (v = 3–4, j = 0), the uncoupled surface results are also shown as dashed lines in panels (c), (d), (g) and (h) for comparison. The uncoupled surface results for reagent H2 (v = 1–2, j = 0) are shown in panels (a), (b), (e) and (f) by solid lines. reaction at Ecol = 0.25, 0.50, 0.75 and 1.0 eV, shown by lines of different colours as mentioned in the upper right corner of panel (e). In addition to the coupled surface results (solid lines) for H2 (v = 3–4, j = 0), the uncoupled surface results are also shown as dashed lines in panels (c), (d), (g) and (h) for comparison. The uncoupled surface results for reagent H2 (v = 1–2, j = 0) are shown in panels (a), (b), (e) and (f) by solid lines. | ||
At fairly low values of collision energy, both forward- and backward-scattered products are mostly formed at the higher v′ levels (v′ being the same as or close to v) in the case of highly vibrationally excited reagents. On the other hand, at higher collision energies, the backward-scattered products predominantly formed at the lower v′ levels exhibiting a statistical vibrational distribution, whereas the forward-scattered products are predominantly formed at the higher v′ levels. In the case of the latter, the most probable v′ in some instances is even greater than the corresponding v. As can be seen from Fig. 4(f)–(h), at Ecol = 1.0 eV the most probable v′ for reagent H2 (v = 2–4, j = 0) is v + 1. This interestingly showcases a certain type of opposite behavior between the forward- and backward-scattered products in vibrational energy disposal at higher collision energies.
To complement this strong dependence of collision energy on the forward- and backward-scattered product vibrational distributions of vibrationally excited reagents, the distributions at θ = 30° and 150°, corresponding to forward and backward scattering, respectively, are also calculated. The results are shown in Fig. S7 (ESI†) at the same values of collision energy as in Fig. 4. Similar findings can be found from the vibrational distributions at θ = 30° and 150°. The most important results are: the effect of the reduction of vibrational excitation of backward-scattered products in the case of highly vibrationally excited reagents at higher collision energies, and formation of vibrationally excited products both in backward and forward directions at low collision energies.
Another interesting observation from the angle-resolved product vibrational level distributions (see Fig. 4 and Fig. S7, ESI†) is the negligibly small nonadiabatic effects in the DCSs for reagent H2 (v = 3–4, j = 0) at different collision energies. In our earlier work,22 it was shown that electronic nonadiabatic effects manifest as “out-of-phase” oscillations in the DCSs due to the GP. The diminishing nonadiabatic and, hence, GP effects (since the upper adiabatic state (V+) plays a very minor role in the reaction dynamics17,22,30) in the extreme forward (θ = 0°) and backward (θ = 180°) scattering directions are due to the summation over the final j′ and Ω′ quantum numbers, as discussed in our earlier work.22 However, we must note that some non-negligible nonadiabatic effects were observed in the v′-resolved DCSs, but only in the sideways scattering direction (see Fig. 8 of ref. 22). It is also interesting to note from Fig. 4 and Fig. S7 (ESI†) that such diminutions of nonadiabatic effects are persistent across the collision energies considered in this work.
In order to understand the partial wave contribution to the v′-resolved DCSs, the v′-resolved J-dependent partial DCSs are calculated at θ = 0° and 180°. The results are shown in Fig. S8 and S9 (ESI†) as a function of Ecol and J for some of the most probable v′ levels of the product (chosen from Fig. 4). It is found that, similar to the total DCSs, the forward scattering in v′-resolved DCSs mainly originates from the higher partial waves and the backward scattering from the lower partial waves.
While Fig. 4 represents the vibrational level distribution of the angle-resolved products, the v′-resolved DCSs are shown in Fig. S10 (ESI†) for reagent H2 (v = 0–4, j = 0) at two different collision energies, Ecol = 0.5 and 1.0 eV. These DCSs are plotted as a function of θ to provide a comparison between the magnitude of the forward- and backward-scattering reaction products. As can be seen from the figure, backward scattering dominates the reaction at the lower vibrational level of the reagent, forming the product at lower v′ levels. However, with successive reagent vibrational excitation, the products formed at the excited vibrational levels mainly give dominant contributions to the DCS. These vibrationally excited products are predominantly forward scattered, at both the indicated energies. The forward scattering mainly originates from the higher partial waves or high-impact-parameter collisions, as shown in Fig. S8 and S9 (ESI†). In such a scenario, when the attacking atom comes closer to the reagent diatom with a high impact parameter, it approaches the collinear transition-state structure from the sideways direction.72 The high orbital angular momentum slows down the triatomic complex and makes it rotate in the same direction of the approach. Within this short period of time, the triatomic complex undergoes symmetric stretching motions, forming vibrationally excited products in the forward direction.72
C. Energy disposal into angle-resolved products
It is found from the vibrational distribution of the product that an increase in the collision energy in the case of a vibrationally excited reagent reduces the vibrational excitation of the backward-scattered products to a large extent, but not in the case of the forward-scattered products. In consequence, this raises the question of where does the lost vibrational energy go for the total energy to be conserved. In order to understand this, the energy disposal in angle-resolved products, that is, the average fraction of the total available energy entering into product vibration , rotation
, rotation 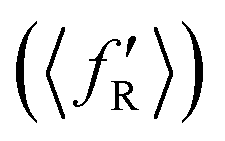 and relative translation
and relative translation 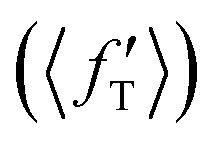 , is calculated individually for products scattered at θ = 0° and 180°. The 〈f′〉 values are calculated here using eqn (5)–(9) of ref. 100 with the ICSs replaced by DCSs. The results are shown in Fig. 5 for reagent H2 (v = 4, j = 0), where the 〈f′〉 values are plotted as a function of collision energy. It can be seen that in the case of forward-scattering (see Fig. 5(a)) a larger portion of the available energy goes into product vibration, being almost 70% at Ecol = 0.25 eV and varying to 52% at Ecol = 1.25 eV. This is consistent with the fact that the forward-scattered products are formed at the vibrationally excited levels, both at lower and higher collision energies, in the case of vibrationally excited reagents, as seen in Fig. 4. The average fraction of available energy entering into products’ relative translation is less as compared that entering into vibration and varies around an average of 30% throughout the collision energies considered. The fraction of energy entering into product rotation is the least among them all and varies from 4% to ∼18% (see Fig. 5(a)).
, is calculated individually for products scattered at θ = 0° and 180°. The 〈f′〉 values are calculated here using eqn (5)–(9) of ref. 100 with the ICSs replaced by DCSs. The results are shown in Fig. 5 for reagent H2 (v = 4, j = 0), where the 〈f′〉 values are plotted as a function of collision energy. It can be seen that in the case of forward-scattering (see Fig. 5(a)) a larger portion of the available energy goes into product vibration, being almost 70% at Ecol = 0.25 eV and varying to 52% at Ecol = 1.25 eV. This is consistent with the fact that the forward-scattered products are formed at the vibrationally excited levels, both at lower and higher collision energies, in the case of vibrationally excited reagents, as seen in Fig. 4. The average fraction of available energy entering into products’ relative translation is less as compared that entering into vibration and varies around an average of 30% throughout the collision energies considered. The fraction of energy entering into product rotation is the least among them all and varies from 4% to ∼18% (see Fig. 5(a)).
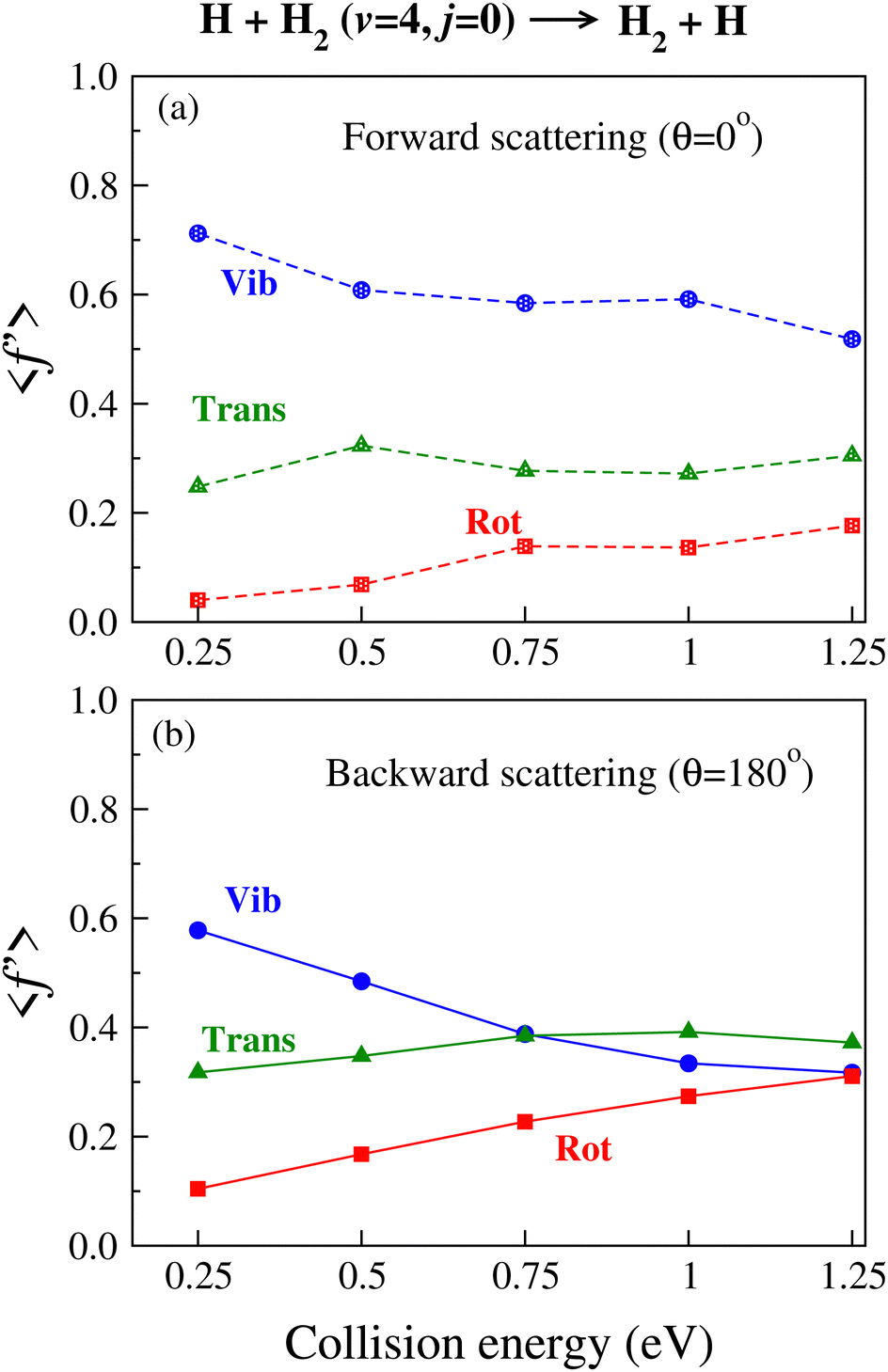 | ||
| Fig. 5 Average fractions of the total available energy entering into product vibration, rotation and relative translation for the products scattered at (a) θ = 0° (dashed lines) and (b) θ = 180° (solid lines) for the H + H2 (v = 4, j = 0) → H2 + H reaction as a function of collision energy. The 〈f′〉 values are shown by lines of different colours and symbols. The abbreviations Vib, Rot and Trans represent product vibration, rotation and relative translation, respectively. | ||
The situation is different for backward scattering (see Fig. 5(b)). The fraction of available energy going into product vibration has significantly lowered as compared to that in forward scattering and the reduction is severe at higher collision energies. It can also be seen from Fig. 5(b) that the 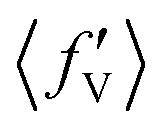 ,
, 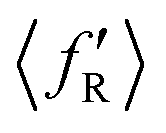 and
and 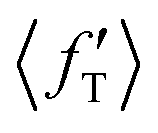 values are close to each other at Ecol = 1.25 eV. This showcases an almost equal partitioning of the total available energy into product vibration, rotation and relative translation in the case of the backward-scattered products at higher collision energies. Such equipartitioning can be related to the statistical vibrational distribution of the backward-scattered products at higher collision energies, as can be seen from Fig. 4(b)–(d). In addition, the fraction of energy going to product rotation and relative translation has increased for backward-scattered products compared to forward scattering. There is more gain in product rotation than in the relative translation. This comes at the expense of a reduction in vibrational energy disposal in the backward-scattered products. Hence, it can be assumed that the loss in vibrational energy disposal for backward-scattered products is mostly compensated by product rotation and a little by the relative translation. Similar findings are also noticed for reagent H2 (v = 3, j = 0) (see Fig. S11, ESI†). We note that the 〈f′〉 values are also calculated for other values of scattering angle both in the forward and backward directions and their corresponding energy disposal is also similar to what is found for θ = 0° and 180°.
values are close to each other at Ecol = 1.25 eV. This showcases an almost equal partitioning of the total available energy into product vibration, rotation and relative translation in the case of the backward-scattered products at higher collision energies. Such equipartitioning can be related to the statistical vibrational distribution of the backward-scattered products at higher collision energies, as can be seen from Fig. 4(b)–(d). In addition, the fraction of energy going to product rotation and relative translation has increased for backward-scattered products compared to forward scattering. There is more gain in product rotation than in the relative translation. This comes at the expense of a reduction in vibrational energy disposal in the backward-scattered products. Hence, it can be assumed that the loss in vibrational energy disposal for backward-scattered products is mostly compensated by product rotation and a little by the relative translation. Similar findings are also noticed for reagent H2 (v = 3, j = 0) (see Fig. S11, ESI†). We note that the 〈f′〉 values are also calculated for other values of scattering angle both in the forward and backward directions and their corresponding energy disposal is also similar to what is found for θ = 0° and 180°.
It can be understood from the above discussion that there are two scenarios depending on whether the highly vibrationally excited reagent diatom encounters a slow collision or a relatively fast collision with the attacking atom. It was found in earlier studies of this reaction and also shown in Section III A that with increasing reagent vibrational excitation, the threshold for the reaction decreases,88 and for H2 (v = 4, j = 0) it behaves as barrierless with zero threshold45,68 (see Fig. S5, ESI†). On account of these, when the reaction accesses collisions with higher partial waves (or equivalently with a high impact parameter), dynamical barriers can be formed corresponding to the higher orbital angular momentum, even for the highly vibrationally excited reagent. This can cause slowing down of the incoming atom to some extent as a consequence of converting some of its relative translational energy to centrifugal energy. The products formed in such cases are forward-scattered (see Fig. S8 and S9, ESI†), as reported in ref. 13, 71 and 72. It can be conjectured that a slow collision is very much unlikely to disrupt the vibrational motion of the reagent diatom as compared to a fast collision. Hence, the products formed from the slow collisions (low collision energy) are more likely to retain the reagent diatom's vibrational energy in the case of both forward and backward scattering. The disruption in fact is more plausible when the fast collisions are mediated by low impact parameters (or lower partial waves), where they barely face any dynamical barriers. This explains why at higher collision energies the reagent vibrational energy could not efficiently be disposed of into product vibration in the case of backward-scattered products (see Fig. 4(b)–(d), 5(b) and Fig. S7(b)–(d), ESI†), which are dominated by lower partial waves. However, in the case of forward scattering, dominated by higher partial waves, the dynamical centrifugal barriers may slow down the fast moving attacking atom where the disruption of reagent vibrational motion would not be effective and the reagent vibrational energy can be efficiently disposed of into the product vibration (see Fig. 4(f)–(h), 5(a) and Fig. S7(f)–(h), ESI†).
D. Effect of reagent vibration on product rotational-level-resolved DCSs
The effect of reagent vibration on the product rotational-level-resolved DCSs is presented and discussed in this section. The j′-resolved state-to-state DCSs are presented here in terms of colour contour plots as a function of θ along the abscissa and j′ along the ordinate. The j′-resolved DCSs of the H + H2 (v = 0–4, j = 0) → H2 (v′ = 0, j′) + H reaction are shown for Ecol = 0.5 eV and 0.75 eV in Fig. 6 and 7, respectively. These figures show the DCSs for a particular product vibrational manifold and for different reagent vibrational levels to indicate the effect of reagent vibrational excitation on j′-resolved DCSs.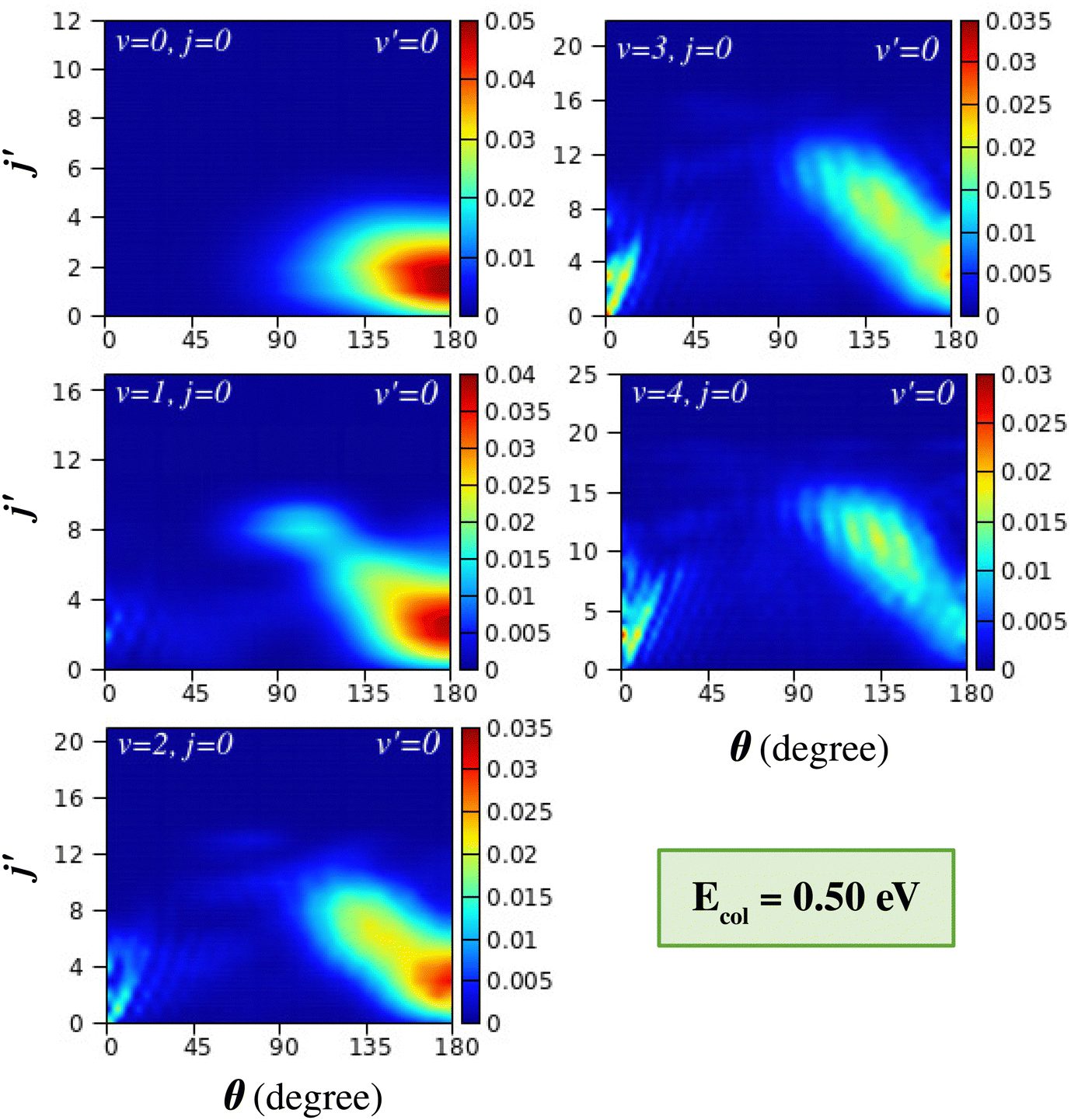 | ||
| Fig. 6 Product rotational-level-resolved state-to-state DCSs of the H + H2 (v = 0–4, j = 0) → H2 (v′ = 0, j′) + H reaction as a function of θ (abscissa) and j′ (ordinate) at Ecol = 0.5 eV. The results for reagent H2 (v = 0–2, j = 0) are obtained from uncoupled surface calculations and those of H2 (v = 3–4, j = 0) are from coupled surface calculations. | ||
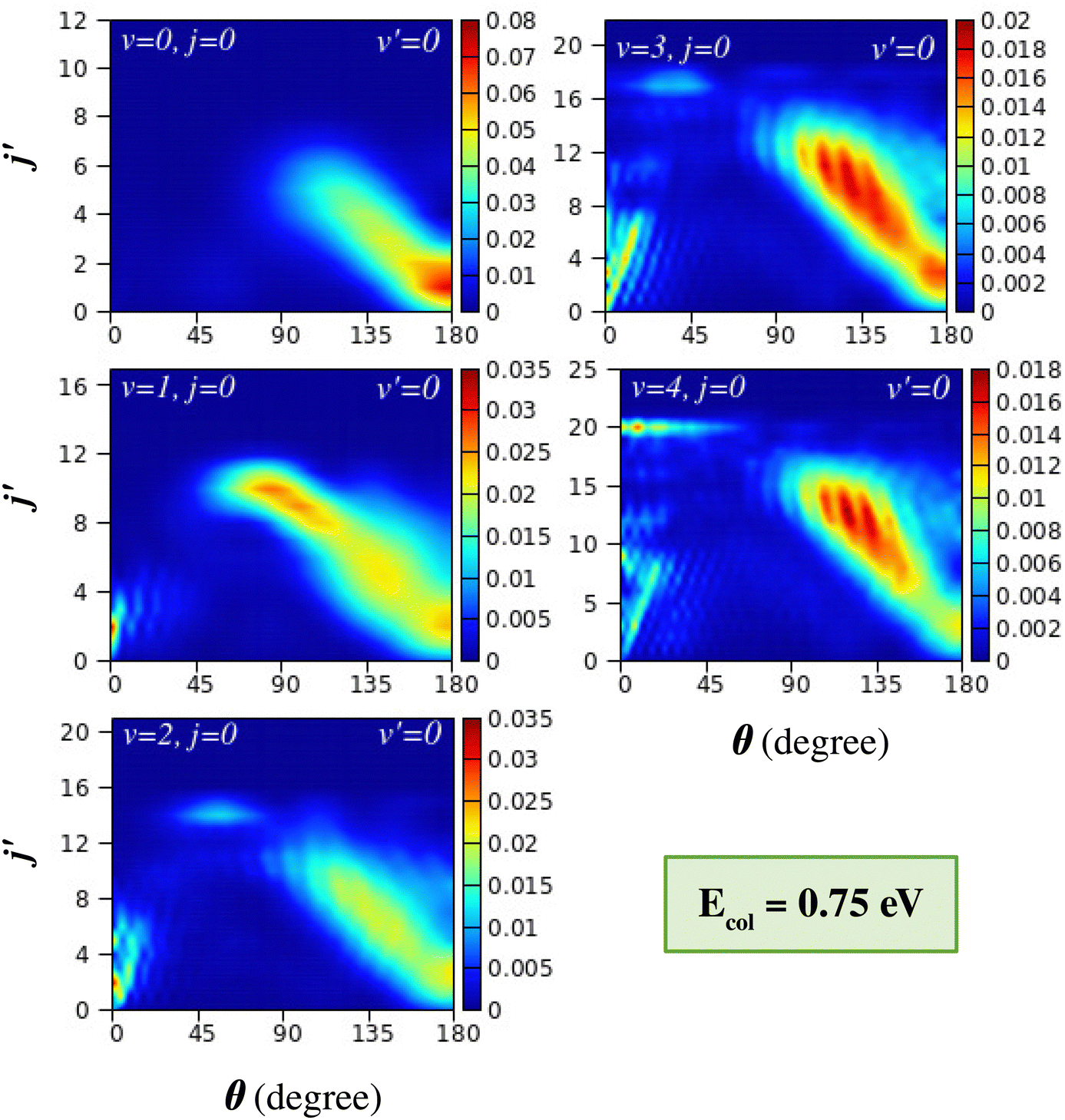 | ||
| Fig. 7 Same as in Fig. 6, but at Ecol = 0.75 eV. | ||
It can be seen from Fig. 6 that for reagent H2 (v = 0, j = 0) the DCSs are dominated by backward scattering with the peak of the angular distribution, θpeak, close to 180°. Moreover, the products are formed at the lower rotational levels, within j′ = 4. This behaviour, that is, backward scattering with low rotational excitation of the product diatom, supports the conventional rebound or direct recoil mechanism, where near head-on collisions between the reagent atom and diatom with very low impact parameters (b or J ≈ 0) result in backward-scattered products with very low rotational excitation. Such mechanisms that are dominant at low collision energy access the collinear transition-state in accordance with the MEP of the H + H2 reaction. It is to be noted here that the collision energy of 0.5 eV is very close to the threshold energy of the (v = 0, j = 0) vibrationally adiabatic potential.
With successive vibrational excitations of the reagent diatom, the backward scattering found in H2 (v = 0, j = 0) gradually changes to sideways scattering, but remains in the backward hemisphere. These sideways scattered products are now rotationally excited and the rotational excitation increases with increasing reagent vibrational excitation. For the vibrationally excited reagent H2 (v = 3–4, j = 0), a relatively large range of product rotational levels are populated and the peak value of the angular distribution decreases as the j′ quantum number increases. Such a phenomenon is well known in H + H2 reaction dynamics67,76,77 and has been termed “negative j′–θ correlation”.78 It is believed that the rotationally excited products that scatter into the sideways direction of the backward hemisphere result from glancing collisions.76,77 This means that the head-on or near zero-impact-parameter collisions lead to rotationally cold products in the extreme backward direction, and as the impact parameter increases, the collisions become more glancing, which leads to rotationally excited products in the sideways direction. This phenomenon has been found in a crossed molecular beam experiment on the H + D2 (v = 0, j) → HD (v′, j′) + D reaction67,76–78,116 and recent experimental work on the H + HD (v = 0, j = 0) → H2 (v′, j′) + H reaction117 in various collision energy ranges.
The transition from conventional rebound to glancing collision mechanism generally occurs when the collision energy between the reagents increases, as is well established in the literature for the case of the (v = 0, j = 0) level of the reagent diatom.76,77,116 This can be understood in the present case by comparing the DCSs of the top-left panels of Fig. 6 and 7, where the latter figure displays the j′-resolved DCSs at a slightly higher collision energy of 0.75 eV. At this collision energy, it can be seen that, in addition to the rebound mechanism for the low j′ levels, the glancing collision mechanism also appears for higher j′ states, showcasing the “negative j′–θ correlation” for H2 (v = 0, j = 0). The gradual transition of the mechanism is due to the fact that with an increase in the collision energy, the colliding partners can access the region of high impact parameters and hence can undergo glancing collisions. However, in the present study, it is found that the transition of the mechanism from rebound to glancing can also occur when increasing the vibrational energy of the reagent diatom at the same collision energy (see Fig. 6). This is consistent with the fact that the reaction is dominated by higher partial waves in the case of vibrationally excited reagents as compared to the case of H2 (v = 0, j = 0) at the same value of collision energy, which has already been discussed in Section III A (see Fig. 3).
In addition to the backward scattering and sideways scattering in the backward hemisphere, forward scattering also appears with successive vibrational excitation of the reagent diatom (see Fig. 6 and 7). Although no forward scattering is seen in the case of reagent H2 (v = 0, j = 0) at Ecol = 0.5 and 0.75 eV, products scatter to the forward direction at higher energies (see Fig. 1(a) and Fig. S5(a), ESI†). The forward scattering is seen for low j′ product states and becomes more prominent with increasing reagent vibrational excitation at the same value of collision energy. The lowest j′ product states peak in the extreme forward direction (θ ≃ 0°), and with increasing j′ the peak of the forward angular distribution gradually shifts towards the sideways direction but remains in the forward hemisphere. This results in a situation where the θpeak value increases with increasing j′ quantum number in the forward-scattering region. This phenomenon can be designated as “positive j′–θ correlation” in the forward scattering. This effect can be clearly seen for reagent H2 (v = 3–4, j = 0) at Ecol = 0.5 eV (see Fig. 6) and for H2 (v = 2–4, j = 0) at Ecol = 0.75 eV (see Fig. 7). In contrast to the “negative j′–θ correlation” in the backward hemisphere, a relatively low number of product rotational states are populated in the forward scattering, which is why it does not extend to the extreme sideways region. Moreover, in contrast to the broad angular distribution in the backward hemisphere, which originates from a broad range of impact parameters, the forward-scattering angular distributions are narrow and dominated by angular oscillations.
As we see from the above discussion, the “positive j′–θ correlation” is seen with increasing reagent vibrational energy at a specific collision energy. However, it would be very interesting to investigate if the same correlation appears for low vibrational levels of reagents at relatively higher collision energies. To examine this, the j′-resolved DCSs of the H + H2 (v = 0–1, j = 0) → H2 (v′ = 0–1, j′) + H reaction at Ecol = 1.25 eV are shown in Fig. S12 (ESI†). It can be seen from this figure that “positive j′–θ correlation” in forward scattering also exists for low vibrational levels of the reagent but at higher collision energies. Similar to the characteristics of the correlation found for highly vibrationally excited reagents (see Fig. 6 and 7), a smaller number of product rotational levels are populated in this case with similar narrow forward-scattering angular distributions.
Previous investigations of forward scattering in the hydrogen exchange reaction and its isotopic variants8,13–16,35,36,65,69–75,107,118–121 mostly focused on the search for the resonance feature,8,69,70,107,118–121 its relation with the QBSs13–16 and time-delayed mechanism,13,71,72 and the analysis of angular oscillations65 originating from the nearside–farside interference.35,36,73–75 We note that, to the best of our knowledge, the phenomenon of “positive j′–θ correlation” in the forward scattering of the H + H2 exchange reaction is reported here for the first time. A search for this phenomenon is also carried out for higher v′ product states by inspecting a number of j′-resolved state-to-state DCSs at different collision energies and for different reagent vibrational levels. It is found that such “positive j′–θ correlation” in higher v′ product states is not very distinct. For some v′ levels it is obscure and for some it is not clear (the results are not shown here for brevity). However, it can be anticipated that this correlation may be seen in higher v′ product states at a sufficiently high value of collision energy when the v′ levels are significantly populated.
E. Insights into the reaction mechanism
Upon examining different types of j′–θ correlation in the backward and forward scattering of the hydrogen exchange reaction, we are now in a position to elucidate more details to better understand the reaction mechanism as a consequence of these correlations. The correlation among the impact parameters b, j′ and θpeak for the glancing collision mechanism has been qualitatively explained by the line-of-centers nearly elastic specular scattering (LOCNESS) model.76,77 This model predicts a linear correlation between the cosine of the most probable scattering angle and the square of the reduced impact parameter (b2/bmax2, bmax being the maximum impact parameter) by considering the colliding entities as hard-spheres (see ref. 76 for more details). The impact parameter is directly proportional to the orbital angular momentum, which is equal to the total angular momentum J for the j = 0 case. Moreover, according to the kinematic constraints, the rotational angular momentum of the product diatom has an approximately linear relation (directly proportional) with the initial orbital angular momentum for collinearly dominated reactions, such as H + H2, with vibrationally and/or rotationally cold reagents.122 Hence, for this reaction, J and j′ (or more precisely b and j′) possess an approximately linear relation. Thus, according to the LOCNESS model, cosθpeak and j′2 should have an approximately linear correlation between them. This, in fact, has already been shown in the case of the other isotopic variants of this reaction where the reagent diatom is in its ground ro-vibrational level.67,76,77,117
In order to understand what emerged in the present scenario and in a similar spirit, the correlation among θpeak, j′ and J in the case of the glancing collision mechanism (in the backward hemisphere) at Ecol = 0.75 eV for H2 (v = 3, j = 0) and product v′ = 0 is examined here. More precisely, the correlations between j′ and θpeak, and between j′ and Jpeak are considered. The Jpeak denotes the most dominant peak value of J in the state-to-state opacity function. The Jpeak and θpeak values are extracted from the respective opacity function and state-to-state DCSs for different values of j′. The correlation between cosθpeak and j′2, and that between j′ and Jpeak are plotted in Fig. 8(a) and (b), respectively. It can be seen that the cosine of the θpeak values and j′2 have an approximately linear correlation. This observation is in line with the LOCNESS model. Moreover, the Jpeak and j′ quantum numbers also have an approximately linear correlation (see Fig. 8(b)), suggesting the lower j′ products are formed from low J and higher j′ products from high J collisions, as per the prediction of the LOCNESS model.
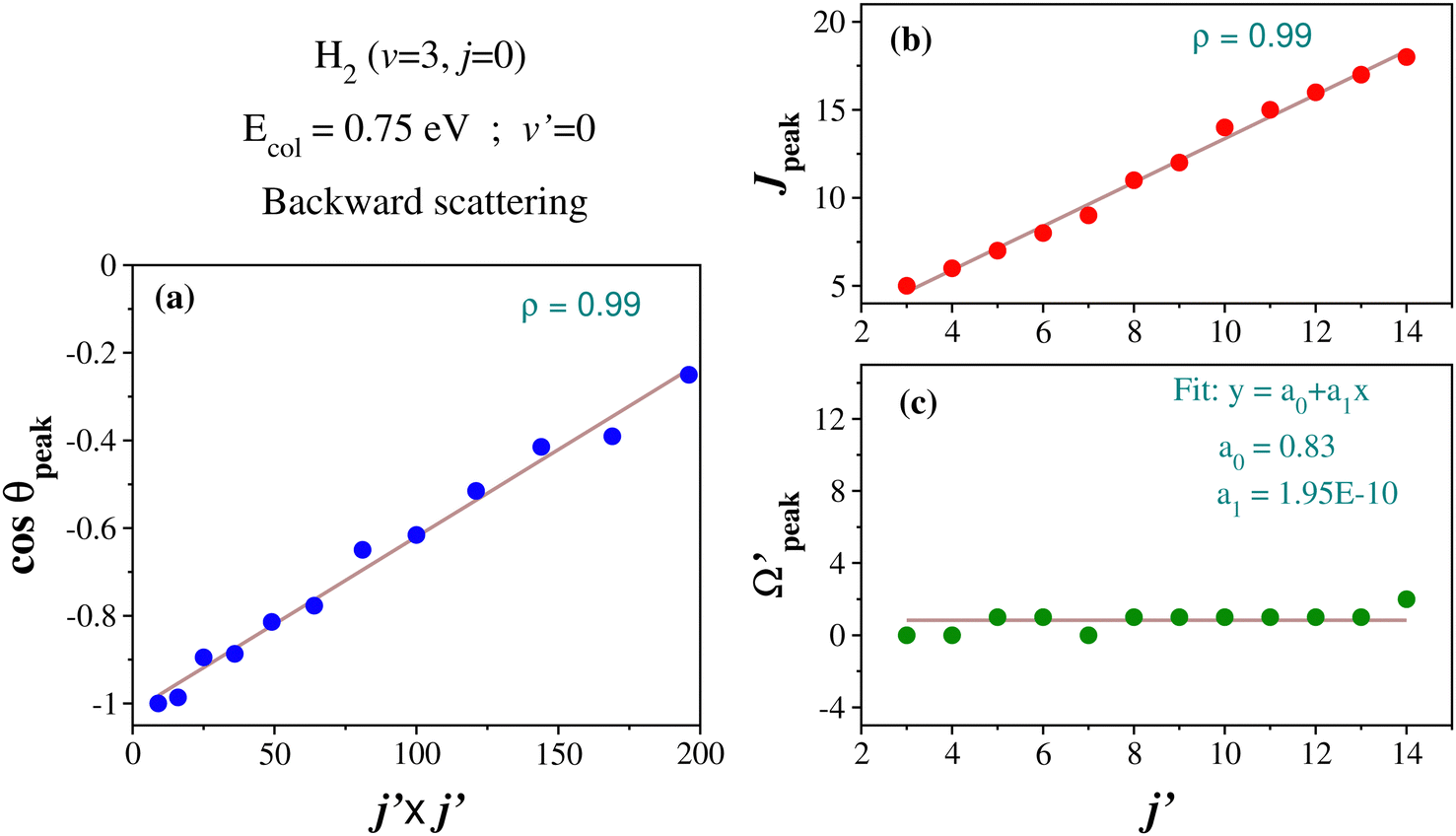 | ||
Fig. 8 Correlation between the cosine of the peak value of the scattering angle θpeakand the square of the rotational quantum number j′ (panel a), between Jpeak and j′ (panel b), and between 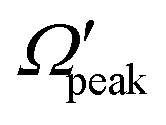 and j′ (panel c) in the backward scattering of the H + H2 (v = 3, j = 0) → H2 (v′ = 0, j′) + H reaction at Ecol = 0.75 eV. The solid lines are least-squares fits to the corresponding data using a linear equation of the type y = a0 + a1x. The Pearson correlation coefficients (ρ) are mentioned inside the corresponding panels to show the quality of the fits. and j′ (panel c) in the backward scattering of the H + H2 (v = 3, j = 0) → H2 (v′ = 0, j′) + H reaction at Ecol = 0.75 eV. The solid lines are least-squares fits to the corresponding data using a linear equation of the type y = a0 + a1x. The Pearson correlation coefficients (ρ) are mentioned inside the corresponding panels to show the quality of the fits. | ||
In addition to the cosθpeak–j′2 and Jpeak–j′ correlation, the relationship between the product rotational angular momentum and its most probable helicity state (represented by the projection quantum number, 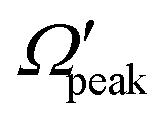 ) is also evaluated for each j′ level. The
) is also evaluated for each j′ level. The 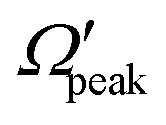 values are extracted by plotting the state-to-state DCSs as a function of Ω′ at the corresponding θpeak values of each j′, followed by the selection of the most probable Ω′ quantum number. The correlation between the
values are extracted by plotting the state-to-state DCSs as a function of Ω′ at the corresponding θpeak values of each j′, followed by the selection of the most probable Ω′ quantum number. The correlation between the  and j′ is shown in panel (c) of Fig. 8. It can be seen that unlike the θpeak and Jpeak, the
and j′ is shown in panel (c) of Fig. 8. It can be seen that unlike the θpeak and Jpeak, the 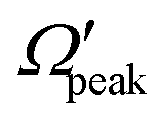 values are almost independent of the corresponding j′ quantum number and tend to be in the lowest possible helicity states. This happens even for the highly rotationally excited product states, e.g.,
values are almost independent of the corresponding j′ quantum number and tend to be in the lowest possible helicity states. This happens even for the highly rotationally excited product states, e.g.,  for j′ = 13. This indicates that the rotational angular momentum vector of the product diatom preferentially remains perpendicular to the products’ recoil direction, suggesting an almost “coplanar detachment mechanism”. This finding is also in accordance with the kinematic constraint predictions for the collinearly dominated encounters in the H + H2 reaction, as described in ref. 122. Similar types of correlation among θpeak, Jpeak,
for j′ = 13. This indicates that the rotational angular momentum vector of the product diatom preferentially remains perpendicular to the products’ recoil direction, suggesting an almost “coplanar detachment mechanism”. This finding is also in accordance with the kinematic constraint predictions for the collinearly dominated encounters in the H + H2 reaction, as described in ref. 122. Similar types of correlation among θpeak, Jpeak, 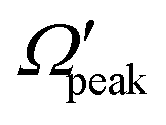 and j′ have also been found for reagent H2 (v = 4, j = 0) and product v′ = 0, and are shown in Fig. S13 and S14 (ESI†) at Ecol = 0.5 and 0.75 eV, respectively (see panels (a)–(c)).
and j′ have also been found for reagent H2 (v = 4, j = 0) and product v′ = 0, and are shown in Fig. S13 and S14 (ESI†) at Ecol = 0.5 and 0.75 eV, respectively (see panels (a)–(c)).
It is surprising to notice that the present behaviour of the rotationally excited product in the sideways direction for vibrationally excited reagents has a remarkable resemblance to the prediction of the LOCNESS model, even though the underlying presumption of the kinematic constraints of ref. 122 is applicable only for vibrationally and/or rotationally cold reagents. This resemblance between the present numerically exact quantum scattering results and the prediction of the above model suggests that the dynamics of the sideways scattered products from the vibrationally excited reagent can be understood with the help of the LOCNESS model. Therefore, the scattering of these products in the backward hemisphere follows a similar glancing collision mechanism to that found in the case of the ground ro-vibrational level reagent.
Similar to the analysis done in the case of the glancing collision mechanism (see Fig. 8), the correlations among θpeak, j′, Jpeak and 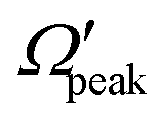 are also examined here for the “positive j′–θ correlation” in the forward scattering of reagent H2 (v = 3, j = 0) and product v′ = 0 at Ecol = 0.75 eV. The results are plotted in Fig. 9. It is to be noted that instead of the cosine of the θpeak, the value of the θpeak is plotted against the j′ quantum number. The Jpeak values are extracted here from the J-dependent partial DCSs, instead of the state-to-state opacity functions, by plotting them against J for the corresponding θpeak for each j′. The θpeak and
are also examined here for the “positive j′–θ correlation” in the forward scattering of reagent H2 (v = 3, j = 0) and product v′ = 0 at Ecol = 0.75 eV. The results are plotted in Fig. 9. It is to be noted that instead of the cosine of the θpeak, the value of the θpeak is plotted against the j′ quantum number. The Jpeak values are extracted here from the J-dependent partial DCSs, instead of the state-to-state opacity functions, by plotting them against J for the corresponding θpeak for each j′. The θpeak and 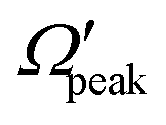 values are extracted in a similar fashion, as that is done in the analysis of the glancing collision mechanism. It can be seen from Fig. 9(a) that the θpeak values and the j′ quantum number possess a linear correlation, which is qualitatively similar to what is seen in the “negative j′–θ correlation” in the backward hemisphere. This suggests that the rotationally excited products going to the forward hemisphere tend to scatter into the sideways region, but not too far from the extreme forward direction. In contrast to Fig. 8(b), the correlation between Jpeak and j′ here is not linear, as can be seen from panel (b) of Fig. 9. Rather, the Jpeak values are almost independent of the j′ quantum number and acquire the highest possible values. This suggests that unlike the glancing collision mechanism, both the rotationally excited and rotationally cold forward-scattered products are formed from a similar type of high-impact-parameter collision.
values are extracted in a similar fashion, as that is done in the analysis of the glancing collision mechanism. It can be seen from Fig. 9(a) that the θpeak values and the j′ quantum number possess a linear correlation, which is qualitatively similar to what is seen in the “negative j′–θ correlation” in the backward hemisphere. This suggests that the rotationally excited products going to the forward hemisphere tend to scatter into the sideways region, but not too far from the extreme forward direction. In contrast to Fig. 8(b), the correlation between Jpeak and j′ here is not linear, as can be seen from panel (b) of Fig. 9. Rather, the Jpeak values are almost independent of the j′ quantum number and acquire the highest possible values. This suggests that unlike the glancing collision mechanism, both the rotationally excited and rotationally cold forward-scattered products are formed from a similar type of high-impact-parameter collision.
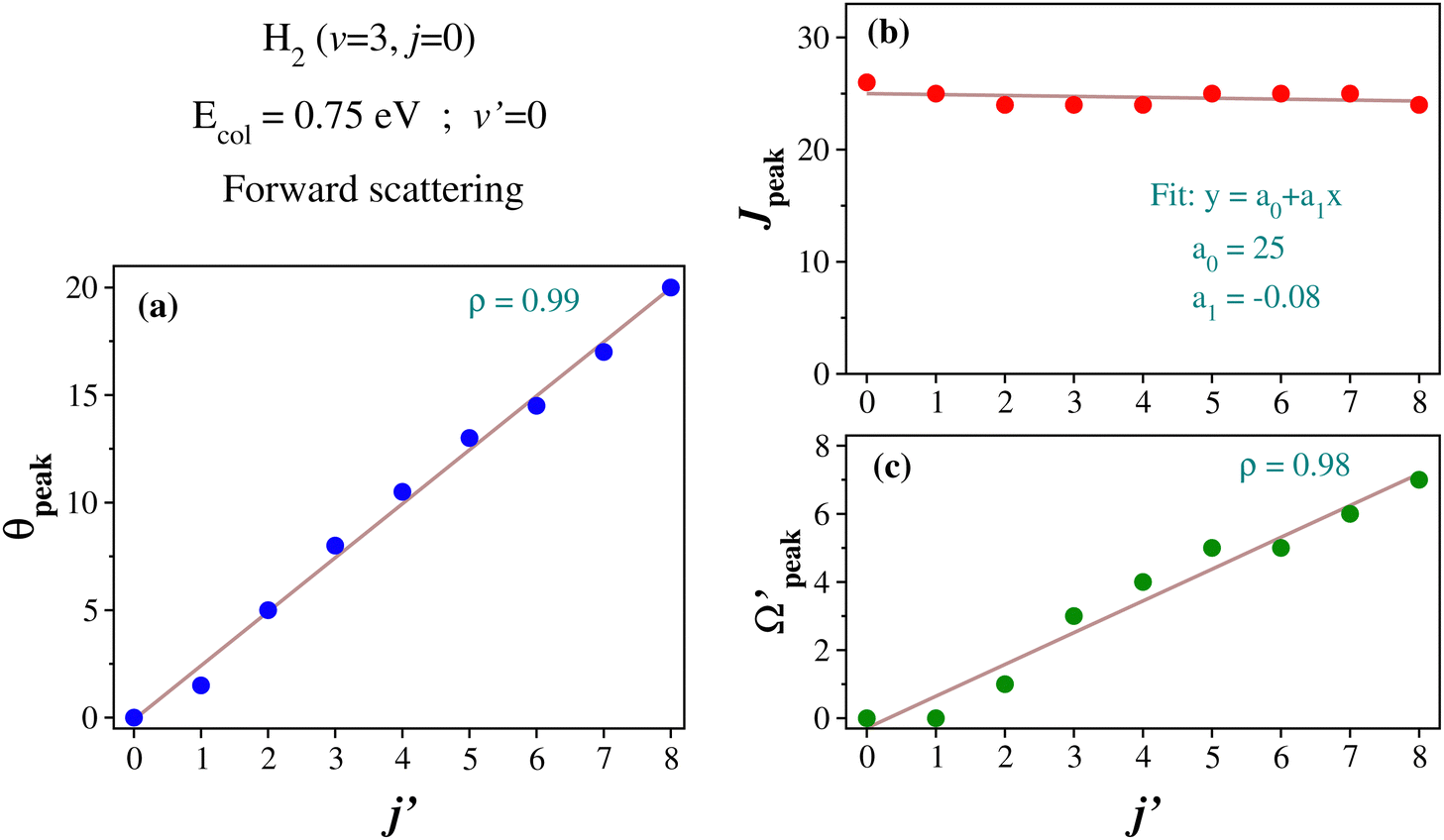 | ||
Fig. 9 Correlation between the peak value of the scattering angle θpeak and the rotational quantum number j′ (panel a), between Jpeak and j′ (panel b), and between  and j′ (panel c) in the forward scattering of the H + H2 (v = 3, j = 0) → H2 (v′ = 0, j′) + H reaction at Ecol = 0.75 eV. The solid lines are least-squares fits to the corresponding data using a linear equation of the type y = a0 + a1x. The Pearson correlation coefficients (ρ) are mentioned inside the corresponding panels to show the quality of the fit. and j′ (panel c) in the forward scattering of the H + H2 (v = 3, j = 0) → H2 (v′ = 0, j′) + H reaction at Ecol = 0.75 eV. The solid lines are least-squares fits to the corresponding data using a linear equation of the type y = a0 + a1x. The Pearson correlation coefficients (ρ) are mentioned inside the corresponding panels to show the quality of the fit. | ||
In order to gain further understanding, the correlation between the 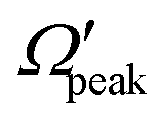 and j′ is plotted in panel (c) of Fig. 9. It can be seen that the
and j′ is plotted in panel (c) of Fig. 9. It can be seen that the 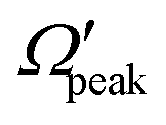 values have an almost linear relation with the j′ quantum number and tend to possess the highest possible helicity quantum number. This indicates that the most dominant contribution to the “positive j′–θ correlation” comes from those helicity states whose quantum numbers lie close to the value of the corresponding j′ quantum number. In such cases, the rotational angular momentum vector of the product diatom preferentially remains parallel to the products’ recoil direction while departing from the triatomic complex. This suggests a “non-coplanar detachment mechanism” for the forward-scattered products that are rotationally excited.
values have an almost linear relation with the j′ quantum number and tend to possess the highest possible helicity quantum number. This indicates that the most dominant contribution to the “positive j′–θ correlation” comes from those helicity states whose quantum numbers lie close to the value of the corresponding j′ quantum number. In such cases, the rotational angular momentum vector of the product diatom preferentially remains parallel to the products’ recoil direction while departing from the triatomic complex. This suggests a “non-coplanar detachment mechanism” for the forward-scattered products that are rotationally excited.
Similar conclusions can be drawn from the correlations among θpeak, Jpeak, 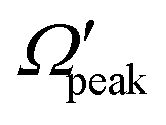 and j′ in the forward scattering of the v′ = 0 level product from reagent H2 (v = 4, j = 0). The results of the latter are shown in panels (d)–(f) of Fig. S13 and S14 (ESI†) at Ecol = 0.5 and 0.75 eV, respectively. Moreover, such correlations in the case of reagent H2 (v = 0, j = 0) are shown in Fig. S15 (ESI†) for the v′ = 0 product state at a relatively high collision energy of 1.25 eV. The “positive j′–θ correlation” and non-coplanar detachment mechanism are also seen, even in the forward scattering of the low vibrational level reagent H2 (v = 0, j = 0) at higher collision energies.
and j′ in the forward scattering of the v′ = 0 level product from reagent H2 (v = 4, j = 0). The results of the latter are shown in panels (d)–(f) of Fig. S13 and S14 (ESI†) at Ecol = 0.5 and 0.75 eV, respectively. Moreover, such correlations in the case of reagent H2 (v = 0, j = 0) are shown in Fig. S15 (ESI†) for the v′ = 0 product state at a relatively high collision energy of 1.25 eV. The “positive j′–θ correlation” and non-coplanar detachment mechanism are also seen, even in the forward scattering of the low vibrational level reagent H2 (v = 0, j = 0) at higher collision energies.
Therefore, it is the helicity state of the product rotational angular momentum that is responsible for the “positive j′–θ correlation” observed in the forward scattering. This is in contrast to the “negative j′–θ correlation” in the backward hemisphere where the orbital angular momentum or the impact parameter is responsible. Moreover, the underlying mechanisms of the two j′–θ correlations are in contrast with each other. One undergoes a “coplanar detachment mechanism” with glancing collisions (negative j′–θ) to produce backward-scattered products, whereas the other undergoes a “non-coplanar detachment” mechanism involving high-impact-parameter collisions (positive j′–θ) to produce forward-scattered products. These two contrasting scenarios are noteworthy and shown in Fig. 10 in terms of a schematic diagram.
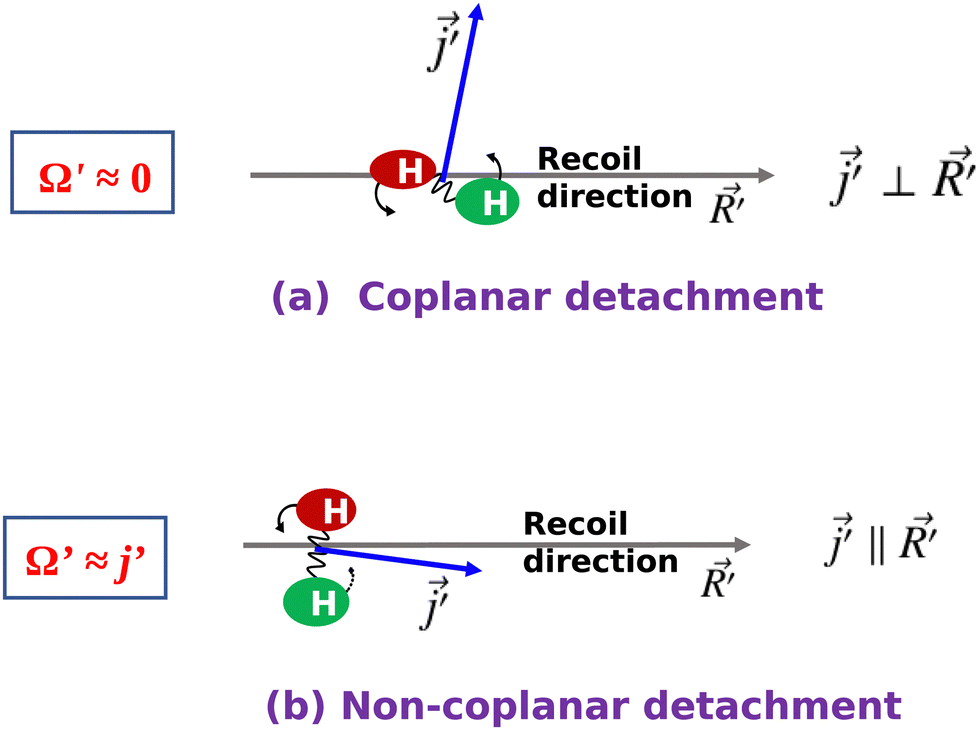 | ||
| Fig. 10 Schematic diagrams depicting the two contrasting mechanisms in the “negative j–θ” in backward scattering and in the “positive j′–θ” in forward scattering for the H + H2 → H2 + H reaction. | ||
IV. Summary and outlook
The effect of reagent vibration on the scattering mechanism of the hydrogen exchange reaction is studied in detail. A TDQM approach is employed both on the single lower adiabatic PES and a coupled two-state diabatic surface developed by us. Initial state-selected total and product state-resolved DCSs are computed and used to underlie the scattering mechanism. Vibrational excitation of the reagent diatom is found to enhance the forward scattering of the products and makes the backward scattering less prominent. The magnitude of forward scattering increases with increasing collision energy, whereas the backward scattering decreases. The analysis of the J-dependent partial DCSs revealed that the forward scattering mainly emerges from the higher partial waves and backward scattering from the lower partial waves. In the case of vibrationally excited reagents, an opposite behaviour between the forward- and backward-scattered products is discovered in the product vibrational energy disposal at higher collision energies. Enhancement of collision energy reduces the vibrational excitation of the backward-scattered products to a significant extent, but not in the case of the forward-scattered products. The reduced amount of product vibrational energy mostly flows into the product rotation of the backward-scattered products. The effect of reagent vibration on the j′-resolved state-to-state DCSs is also examined. Two different mechanisms corresponding to two contrasting phenomena emerged. One is the “negative j′–θ correlation” in the backward scattering and the other one is the “positive j′–θ correlation” in the forward scattering. The underlying mechanisms of these two phenomena are different; the former one is due to a “coplanar detachment mechanism” where the product diatom rotational angular momentum vector preferentially lies perpendicular to the recoil direction, whereas the latter one is due to a “non-coplanar detachment mechanism” where the product diatom rotational angular momentum vector preferentially lies parallel to the recoil direction.We note that the two mechanisms mentioned above are elucidated in a straightforward manner from the scattering properties of the most probable Ω′ states of the product diatom and hence do not consider the contribution of other Ω′ states. Thus, in the present circumstances, the two mechanisms can be regarded as the most dominant ones. To understand the role of other non-dominant Ω′ states, a proper stereo-dynamical investigation of product rotational angular momentum polarization (for example, k − k′ − j′) is needed. In particular, the role of so-called non-dominant Ω′ states might become interesting in the forward scattering which is dominated by angular oscillations originating from nearside–farside quantum interference. Furthermore, we reiterate that since the primary aim of this work is to unravel new mechanistic insights only for the reactive process, the three H atoms are considered as distinguishable. This means that the contributions from inelastic processes, which would be physically identical to the reactive process for the H + H2 → H2 + H reaction, are not considered here explicitly. Nonetheless, we envisage that the underlying scattering mechanisms of inelastic scattering are expected to be different from the reactive ones.
Author contributions
Jayakrushna Sahoo: methodology (lead), investigation (lead), software (lead), data curation (lead), formal analysis (lead), writing – original draft (lead), visualisation (lead). Sugata Goswami: investigation (supporting), data curation (supporting), writing – original draft (supporting). S. Mahapatra: conceptualisation (lead), resources (lead), writing – review & editing (lead), supervision (lead), project administration (lead), funding acquisition (lead).Data availability
Data for this article, including those in the ESI,† are available on Github at https://github.com/JaySahoo/HH2_dcs2.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study is financially supported in part by a grant (No. CRG/2022/001425) from the Science and Engineering Research Board, Department of Science and Technology, Government of India. The computational facilities provided by the Translational Research Facility (UPE-II of UGC) and the Centre for Modelling, Simulation and Design (CMSD) at the University of Hyderabad are gratefully acknowledged.References
- F. London, Z. Elektrochem., 1929, 35, 552–555 CrossRef CAS.
- H. Eyring and M. Polanyi, Z. Phys. Chem., 2013, 227, 1221–1245 CAS.
- H. Eyring and S. H. Lin, in Potential Energy Surfaces, ed. H. Eyring, D. Henderson and W. Jost, Academic Press, New York, 1974, vol. VIA, ch. 3, pp. 121–186 Search PubMed.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- R. D. Levine and S.-F. Wu, Chem. Phys. Lett., 1971, 11, 557–561 CrossRef.
- G. C. Schatz and A. Kuppermann, Phys. Rev. Lett., 1975, 35, 1266–1269 CrossRef CAS.
- W. H. Miller and J. Z. H. Zhang, J. Phys. Chem., 1991, 95, 12–19 CrossRef CAS.
- F. Fernández-Alonso and R. N. Zare, Annu. Rev. Phys. Chem., 2002, 53, 67–99 CrossRef PubMed.
- D. C. Chatfield, R. S. Friedman, D. G. Truhlar, B. C. Garrett and D. W. Schwenke, J. Am. Chem. Soc., 1991, 113, 486–494 CrossRef CAS.
- D. C. Chatfield, R. S. Friedman, D. G. Truhlar and D. W. Schwenke, Faraday Discuss. Chem. Soc., 1991, 91, 289–304 RSC.
- D. C. Chatfield, R. S. Friedman, D. W. Schwenke and D. G. Truhlar, J. Phys. Chem., 1992, 96, 2414–2421 CrossRef CAS.
- S. A. Harich, D. Dai, C. C. Wang, X. Yang, S. D. Chao and R. T. Skodje, Nature, 2002, 419, 281–284 CrossRef CAS.
- D. Dai, C. C. Wang, S. A. Harich, X. Wang, X. Yang, S. D. Chao and R. T. Skodje, Science, 2003, 300, 1730–1734 CrossRef CAS PubMed.
- R. T. Skodje and X. Yang, Int. Rev. Phys. Chem., 2004, 23, 253–287 Search PubMed.
- X. Yang, Annu. Rev. Phys. Chem., 2007, 58, 433–459 CrossRef CAS PubMed.
- S. Mahapatra, H. Köppel and L. S. Cederbaum, J. Phys. Chem. A, 2001, 105, 2321–2329 CrossRef CAS.
- R. F. Lu, T. S. Chu, Y. Zhang, K. L. Han, A. J. C. Varandas and J. Z. H. Zhang, J. Chem. Phys., 2006, 125, 133108 CrossRef PubMed.
- B. Jayachander Rao, R. Padmanaban and S. Mahapatra, Chem. Phys., 2007, 333, 135–147 CrossRef.
- F. Bouakline, S. C. Althorpe and D. P. Ruiz, J. Chem. Phys., 2008, 128, 124322 CrossRef.
- B. K. Kendrick, J. Chem. Phys., 2018, 148, 044116 CrossRef PubMed.
- J. Sahoo and S. Mahapatra, Phys. Chem. Chem. Phys., 2023, 25, 28309–28325 RSC.
- A. Kuppermann and Y. M. Wu, Chem. Phys. Lett., 1993, 205, 577–586 CrossRef CAS.
- B. K. Kendrick, J. Chem. Phys., 2000, 112, 5679–5704 CrossRef CAS.
- B. K. Kendrick, J. Phys. Chem. A, 2003, 107, 6739–6756 CrossRef CAS.
- J. C. Juanes-Marcos and S. C. Althorpe, J. Chem. Phys., 2005, 122, 204324 CrossRef.
- J. C. Juanes-Marcos, S. C. Althorpe and E. Wrede, Science, 2005, 309, 1227–1230 CrossRef CAS PubMed.
- J. C. Juanes-Marcos, S. C. Althorpe and E. Wrede, J. Chem. Phys., 2007, 126, 044317 CrossRef.
- F. Bouakline, S. C. Althorpe, P. Larregaray and L. Bonnet, Mol. Phys., 2010, 108, 969–980 CrossRef CAS.
- D. Yuan, Y. Guan, W. Chen, H. Zhao, S. Yu, C. Luo, Y. Tan, T. Xie, X. wang, Z. Sun, D. H. Zhang and X. Yang, Science, 2018, 362, 1289–1293 CrossRef CAS.
- D. Yuan, Y. Huang, W. Chen, H. Zhao, S. Yu, C. Luo, Y. Tan, S. Wang, X. Wang, Z. Sun and X. Yang, Nat. Commun., 2020, 11, 3640 CrossRef CAS PubMed.
- Y. Xie, H. Zhao, Y. Wang, T. Wang, X. Xu, C. Xiao, Z. Sun, D. H. Zhang and X. Yang, Science, 2020, 368, 767–771 CrossRef CAS PubMed.
- B. Zhou, B. Yang, N. Balakrishnan, B. K. Kendrick and P. C. Stancil, J. Phys. Chem. Lett., 2020, 11, 4970–4975 CrossRef CAS.
- Y. Bai, B. Buren, Z. Yang, B. Zhou and M. Chen, J. Comput. Chem., 2021, 42, 2334–2340 CrossRef CAS.
- C. Xiahou and J. N. L. Connor, Phys. Chem. Chem. Phys., 2021, 23, 13349–13369 RSC.
- C. Xiahou and J. N. L. Connor, J. Phys. Chem. A, 2021, 125, 8734–8750 CrossRef CAS PubMed.
- B. Jayachander Rao and S. Mahapatra, Ind. J. Phys., 2007, 81, 1003–1021 Search PubMed.
- S. Ghosal, B. J. Rao and S. Mahapatra, J. Chem. Sci., 2007, 119, 401–407 CrossRef CAS.
- B. Jayachander Rao and S. Mahapatra, J. Chem. Sci., 2009, 121, 789–795 CrossRef.
- T. R. Rao, B. J. Rao and S. Mahapatra, Chem. Phys., 2009, 365, 129–137 CrossRef.
- T.-S. Chu, K.-L. Han, M. Hankel, G. G. Balint-Kurti, A. Kuppermann and R. Abrol, J. Chem. Phys., 2009, 130, 144301 CrossRef PubMed.
- B. K. Kendrick, Chem. Phys., 2018, 515, 387–399 CrossRef CAS.
- B. K. Kendrick, J. Phys. Chem. A, 2019, 123, 9919–9933 CrossRef CAS.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, Phys. Rev. Lett., 2015, 115, 153201 CrossRef CAS PubMed.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, J. Chem. Phys., 2016, 145, 164303 CrossRef CAS.
- J. Hazra, B. K. Kendrick and N. Balakrishnan, J. Phys. B: At., Mol. Opt. Phys., 2016, 49, 194004 CrossRef.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, New J. Phys., 2016, 18, 123020 CrossRef.
- B. K. Kendrick, J. Hazra and N. Balakrishnan, J. Chem. Phys., 2017, 147, 074302 CrossRef.
- Y. Sun and A. Dalgarno, Astrophys. J., 1994, 427, 1053–1056 CrossRef CAS.
- F. Lique, P. Honvault and A. Faure, J. Chem. Phys., 2012, 137, 154303 CrossRef PubMed.
- F. Lique, Mon. Not. R. Astron. Soc., 2015, 453, 810–818 CrossRef CAS.
- D. Bossion, S. Ndengué, H.-D. Meyer, F. Gatti and Y. Scribano, J. Chem. Phys., 2020, 153, 081102 CrossRef CAS.
- B. Zhou, B. Yang, N. Balakrishnan, B. K. Kendrick, M. Chen and P. C. Stancil, Mon. Not. R. Astron. Soc., 2021, 507, 6012–6019 CrossRef CAS.
- R. N. Porter and M. Karplus, J. Chem. Phys., 1964, 40, 1105–1115 CrossRef CAS.
- D. G. Truhlar and C. J. Horowitz, J. Chem. Phys., 1978, 68, 2466–2476 CrossRef CAS.
- D. G. Truhlar and C. J. Horowitz, J. Chem. Phys., 1979, 71, 1514 CrossRef.
- A. J. C. Varandas, F. B. Brown, C. A. Mead, D. G. Truhlar and N. C. Blais, J. Chem. Phys., 1987, 86, 6258–6269 CrossRef CAS.
- A. I. Boothroyd, W. J. Keogh, P. G. Martin and M. R. Peterson, J. Chem. Phys., 1991, 95, 4343–4359 CrossRef CAS.
- A. I. Boothroyd, W. J. Keogh, P. G. Martin and M. R. Peterson, J. Chem. Phys., 1996, 104, 7139–7152 CrossRef CAS.
- Y.-S. M. Wu, A. Kuppermann and J. B. Anderson, Phys. Chem. Chem. Phys., 1999, 1, 929–937 RSC.
- S. L. Mielke, B. C. Garrett and K. A. Peterson, J. Chem. Phys., 2002, 116, 4142–4161 CrossRef CAS.
- R. Abrol and A. Kuppermann, J. Chem. Phys., 2002, 116, 1035–1062 CrossRef CAS.
- F. J. Aoiz, L. Banares and V. J. Herrero, Int. Rev. Phys. Chem., 2005, 24, 119–190 Search PubMed.
- M. Qiu, L. Che, Z. Ren, D. Dai, X. Wang and X. Yang, Rev. Sci. Instrum., 2005, 76, 083107 CrossRef.
- D. Yuan, S. Yu, W. Chen, J. Sang, C. Luo, T. Wang, X. Xu, P. Casavecchia, X. Wang, Z. Sun, D. H. Zhang and X. Yang, Nat. Chem., 2018, 10, 653–658 CrossRef CAS PubMed.
- P. G. Jambrina, D. Herráez-Aguilar, F. J. Aoiz, M. Sneha, J. Jankunas and R. N. Zare, Nat. Chem., 2015, 7, 661–667 CrossRef CAS PubMed.
- D. Yuan, W. Chen, C. Luo, Y. Tan, S. Li, Y. Huang, Z. Sun, X. Yang and X. Wang, J. Phys. Chem. Lett., 2020, 11, 1222–1227 CrossRef CAS.
- J. Jankunas, M. Sneha, R. N. Zare, F. Bouakline, S. C. Althorpe, D. Herráez-Aguilar and F. J. Aoiz, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15–20 CrossRef CAS PubMed.
- F. J. Aoiz, V. J. Herrero and V. S. Rábanos, J. Chem. Phys., 1992, 97, 7423–7436 CrossRef CAS.
- F. J. Aoiz, L. Banares, M. J. D’Mello, V. J. Herrero, V. S. Rábanos, L. Schnieder and R. E. Wyatt, J. Chem. Phys., 1994, 101, 5781–5791 CrossRef CAS.
- S. C. Althorpe, F. Fernández-Alonso, B. D. Bean, J. D. Ayers, A. E. Pomerantz, R. N. Zare and E. Wrede, Nature, 2002, 416, 67–70 CrossRef CAS.
- S. J. Greaves, D. Murdock and E. Wrede, J. Chem. Phys., 2008, 128, 164307 CrossRef.
- D. Sokolovski, Chem. Phys. Lett., 2003, 370, 805–812 CrossRef CAS.
- J. N. L. Connor, Phys. Chem. Chem. Phys., 2004, 6, 377–390 RSC.
- C. Xiahou and J. N. L. Connor, Mol. Phys., 2006, 104, 159–175 CrossRef CAS.
- F. Fernández-Alonso, B. D. Bean and R. N. Zare, J. Chem. Phys., 1999, 111, 1035–1042 CrossRef.
- F. Fernández-Alonso, B. D. Bean and R. N. Zare, J. Chem. Phys., 1999, 111, 2490–2498 CrossRef.
- M. Sneha, H. Gao, R. N. Zare, P. G. Jambrina, M. Menéndez and F. J. Aoiz, J. Chem. Phys., 2016, 145, 024308 CrossRef PubMed.
- J. Jankunas, R. N. Zare, F. Bouakline, S. C. Althorpe, D. Herráez-Aguilar and F. J. Aoiz, Science, 2012, 336, 1687–1690 CrossRef CAS PubMed.
- T. C. Allison, R. S. Friedman, D. J. Kaufman and D. G. Truhlar, Chem. Phys. Lett., 2000, 327, 439–445 CrossRef CAS.
- W. E. Perreault, N. Mukherjee and R. N. Zare, J. Chem. Phys., 2016, 145, 154203 CrossRef PubMed.
- N. Mukherjee, W. E. Perreault and R. N. Zare, J. Phys. B: At., Mol. Opt. Phys., 2017, 50, 144005 CrossRef.
- W. E. Perreault, N. Mukherjee and R. N. Zare, J. Chem. Phys., 2019, 150, 234201 CrossRef.
- W. E. Perreault, H. Zhou, N. Mukherjee and R. N. Zare, Phys. Rev. Lett., 2020, 124, 163202 CrossRef CAS PubMed.
- Y. Wang, W. Wang, Y. Xie, T. Wang and X. Yang, Rev. Sci. Instrum., 2020, 91, 053001 CrossRef CAS.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 138, 234104 CrossRef.
- G. D. Barg, H. R. Mayne and J. P. Toennis, J. Chem. Phys., 1981, 74, 1017–1025 CrossRef CAS.
- S. Goswami, J. Sahoo, S. K. Paul, T. R. Rao and S. Mahapatra, J. Phys. Chem. A, 2020, 124, 9343–9359 CrossRef CAS.
- M. Hankel, S. C. Smith, R. J. Allan, S. K. Gray and G. G. Balint-Kurti, J. Chem. Phys., 2006, 125, 164303 CrossRef.
- M. Hankel, S. C. Smith, S. K. Gray and G. G. Balint-Kurti, Comput. Phys. Commun., 2008, 179, 569–578 CrossRef CAS.
- S. K. Gray and G. G. Balint-Kurti, J. Chem. Phys., 1998, 108, 950–962 CrossRef CAS.
- J. Huang and D. H. Zhang, J. Chem. Phys., 2020, 153, 141102 CrossRef CAS.
- H. Yang, K. L. Han, G. C. Schatz, S. C. Smith and M. Hankel, Phys. Chem. Chem. Phys., 2010, 12, 12711–12718 RSC.
- M. Hankel, S. C. Smith and A. J. C. Varandas, Phys. Scr., 2011, 84, 028102 CrossRef.
- Z. Shen, H. Ma, C. Zhang, M. Fu, Y. Wu, W. Bian and J. Cao, Nat. Commun., 2017, 8, 14094 CrossRef CAS PubMed.
- S. Goswami, J. Sahoo, T. R. Rao, B. Bussery-Honvault, P. Honvault and S. Mahapatra, Eur. Phys. J. D, 2018, 72, 225 CrossRef.
- Y. Wu, J. Cao, H. Ma, C. Zhang, W. Bian, D. Nunez-Reyes and K. M. Hickson, Sci. Adv., 2019, 5, eaaw0446 CrossRef CAS.
- Y. Wu, J. Cao and W. Bian, J. Phys. Chem. A, 2020, 124, 801–809 CrossRef CAS.
- J. Cao, Y. Wu, H. Ma, Z. Shen and W. Bian, Phys. Chem. Chem. Phys., 2021, 23, 6141–6153 RSC.
- J. Sahoo, A. M. S. Rawat and S. Mahapatra, J. Phys. Chem. A, 2021, 125, 3387–3397 CrossRef CAS.
- J. Sahoo, A. M. S. Rawat and S. Mahapatra, Phys. Chem. Chem. Phys., 2021, 23, 27327–27339 RSC.
- G. G. Balint-Kurti, Int. Rev. Phys. Chem., 2008, 27, 507–539 Search PubMed.
- W. Gropp, E. Lusk and A. Skjellum, Using MPI: Portable Parallel Programming with the Message Passing Interface, MIT Press, Cambridge, MA, 2nd edn, 1999 Search PubMed.
- Message Passing Interface, https://www.mcs.anl.gov/research/projects/mpi/, accessed February 22, 2021.
- A. R. Edmonds, Angular Momentum in Quantum Chemistry, Princeton University Press, Princeton, 1960 Search PubMed.
- R. N. Zare, Angular Momentum, Willey, New York, 1988 Search PubMed.
- J. Z. H. Zhang and W. H. Miller, J. Chem. Phys., 1989, 91, 1528–1547 CrossRef CAS.
- P. G. Jambrina, M. Menéndez and F. J. Aoiz, Chem. Sci., 2018, 9, 4837–4850 RSC.
- P. G. Jambrina, M. Menéndez, A. Zanchet, E. García and F. J. Aoiz, Phys. Chem. Chem. Phys., 2019, 21, 14012–14022 RSC.
- P. G. Jambrina, L. González-Sánchez, J. Aldegunde, V. Sáez-Rábanos and F. J. Aoiz, J. Phys. Chem. A, 2019, 123, 9079–9088 CrossRef CAS PubMed.
- S. Mahapatra and H. Köppel, Phys. Rev. Lett., 1998, 81, 3116–3119 CrossRef CAS.
- A. Thiel and H. Köppel, J. Chem. Phys., 1999, 110, 9371–9383 CrossRef CAS.
- H. Köppel, S. Mahapatra and A. Thiel, in Theoretical study of the Rydberg emission spectrum of triatomic Hydrogen, ed. G. Bevilacqua, L. Martinelli and N. Terzi, World Scientific, Singapore, 1999, pp. 327–334 Search PubMed.
- T. C. Thompson and C. A. Mead, J. Chem. Phys., 1985, 82, 2408–2417 CrossRef CAS.
- J. Zhao, D.-G. Yue, L.-L. Zhang, S. Gao, Z.-B. Liu and Q.-T. Meng, Chin. Phys. B, 2021, 30, 073102 CrossRef CAS.
- E. Wrede, L. Schnieder, K. H. Welge, F. J. Aoiz, L. Banares, J. F. Castillo, B. Martínez-Haya and V. J. Herrero, J. Chem. Phys., 1999, 110, 9971–9981 CrossRef CAS.
- C. Luo, Y. Tan, S. Li, Z. Lu, Y. Shu, W. Chen, D. Yuan, X. Yang and X. Wang, J. Phys. Chem. A, 2022, 126, 4444–4450 CrossRef CAS PubMed.
- J. Z. Zhang and W. H. Miller, Chem. Phys. Lett., 1989, 159, 130–133 CrossRef CAS.
- F. Fernandez-Alonso, B. D. Bean, J. D. Ayers, A. E. Pomerantz, R. N. Zare, L. Banares and F. J. Aoiz, Angew. Chem., Int. Ed., 2000, 39, 2748–2752 CrossRef CAS.
- F. J. Aoiz, L. Banares, J. F. Castillo and D. Sokolovski, J. Chem. Phys., 2002, 117, 2546–2556 CrossRef CAS.
- B. K. Kendrick, J. Chem. Phys., 2001, 114, 8796–8819 CrossRef CAS.
- I. Schechter, R. D. Levine and R. G. Gordon, J. Phys. Chem., 1991, 95, 8201–8205 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03433c |
| ‡ Present address: Laboratoire Univers et Particules de Montpellier, Université de Montpellier, UMR-CNRS 5299, 34095 Montpellier Cedex, France. |
| § Present address: Department of Chemistry, Medi-Caps University, A.B. Road, Pigdamber, Indore – 453 331 (M.P.), India. |
| This journal is © the Owner Societies 2025 |