
Competition between C–H⋯S and S–H⋯Cl H-bonds in a CHCl3–H2S complex: a combined matrix isolation IR spectroscopic and quantum chemical investigation†
Binod Kumar
Oram
,
Monu
,
Ankita
Kothari
and
Biman
Bandyopadhyay
 *
*
Department of Chemistry, Malaviya National Institute of Technology, Jaipur, J L N Marg, Jaipur-302017, India. E-mail: biman.chy@mnit.ac.in
First published on 2nd December 2024
Abstract
H-bonded complexes between CHCl3 and H2S have been studied in a cold and inert argon matrix using IR spectroscopy. Both molecules were found to act as both a H-bond donor and acceptor, resulting in two different conformers. The more stable one (binding energy 3.25 kcal mol−1) was bound by a C–H⋯S H-bond, while the less stable one (1.90 kcal mol−1) by a S–H⋯Cl H-bond. The H-bonded complex formation has been confirmed by monitoring the spectral changes in νC–H and νS–H fundamental vibrations. The νC–H mode exhibited red shifts by 22.7 cm−1 upon C–H⋯S H-bond formation, while the formation of a S–H⋯Cl H-bond resulted in 24.2 and 25.4 cm−1 red shifts in the νS–H modes. The barrier for conversion of the less stable conformer to the more stable one was found to be 0.6 kcal mol−1. The rigid matrix environment prevented any detectable population transfer that required significant relative movement of the monomeric moieties. Dispersion interaction was found to significantly contribute to the overall stabilization of both conformers, more so for the S–H⋯Cl H-bonded one.
1. Introduction
Hydrogen bonds (H-bonds) involving one or more sulfur atom(s), commonly known as sulfur-centered H-bonds (SCHBs), are known to play significant roles in various biological and chemical processes.1–7 They are known to influence protein structures and their activities.2,7–9 Also, they facilitate the catalytic activities of sulfur-containing enzymes by assisting in substrate recognition.9–11 They have been found to influence crystal packing and biomolecular assembly along with molecular recognition.12–17SCHBs are considered weak compared to the classical H-bond because of S atoms' lower electronegativity (2.58 on the Pauling scale), leading to a weaker polarity of the S–H bonds compared to O–H and N–H bonds (electronegativity of O and N are 3.44 and 3.04, respectively). Dispersion interaction has been shown to play a vital role in stabilizing SCHBs, while electrostatics does the same in classical H-bonds.18,19 However, in some cases, SCHBs have been predicted to exhibit strength comparable to classical H-bonds.20,21
The sulfur atom has been found to act both as an H-bond donor (HBD)22–24 and acceptor (HBA)21,24–35 in SCHBs, both experimentally21,24,27–30,32,35 and theoretically,21,27,28,31,33,34 by various research groups. Recently, Biswal et al. reviewed the potential of the S atom to act as an HBA in binary H-bonded complexes.36 In most of these cases, the SCHBs have an electronegative element as the HBD, while only a few studies on C–H⋯S H-bonds exist.26,29–31 Presence of C–H⋯S H-bonds has been confirmed in various biomolecules as well as in organic and inorganic crystals.15,16,32,37 Ghosh et al. have shown that the C–H⋯S H-bonds possess all the characteristics of H-bonding interaction.26 Later, Fargher et al. discussed in detail its importance in small and large biomolecules.25 Numerous instances of SCHBs with S as the acceptor25–29,31,38 and with a S–H group as the donor23,39–45 are available in the literature. Zhou et al. reported that, between methionine and cysteine, the S–H group in the latter acts as a comparatively better HBD than S atoms in both amino acids as HBA.2 The sulfhydryl group has also been shown to participate in S–H⋯π type H-bond interactions, apart from the σ type SCHBs.17,38,46,47
H2S is the smallest neutral molecule that can form SCHBs as both HBD and HBA, similar to H2O in classical H-bonds. Recently, it has been shown that a H2O–H2S mixed dimer can have two almost isoenergetic conformers, separated only by ∼0.23 kcal mol−1, bound by S–H⋯O and O–H⋯S H-bonds.48 Furthermore, the interactions were found to greatly influence the structures of larger H2O–H2S clusters; O–H⋯S H-bonded conformers preferred cyclic structure, whereas S–H⋯O H-bonded conformers preferred caged structures.48 Therefore, experimental investigation of the competition between two weak SCHBs, with the S atom acting as HBD and HBA, is important to understand how that competition dictates structures of molecular assemblies and, consequently, their properties.
A number of recent experiments are available in the literature that investigate how competition between two intermolecular H-bonds impacts the energetic and structure of binary complexes. Zhao et al. studied the competition between O–H⋯π and O–H⋯Cl interaction between 2,2,2-trifluoromethanol and 3-chloro-2-methyl-1-propene, where the O–H⋯π bound structure was favored by 1 kJ mol−1 over its O–H⋯Cl counterpart.49 Similarly, Mukhopadhyay et al.50 showed that the O–H⋯O bound linear complex between p-fluorophenol and 2,5-dihydrofuran prefers a folded structure with an additional C–H⋯π interaction.50 On the other hand, the intramolecular O–H⋯O H-bond in 1,2-cyclohexanedione is weakened upon the formation of an intermolecular C–H⋯O H-bond with chloroform.51 However, no such studies exist on the competition between two SCHBs, one with S atom as the HBA and the other with a S–H group as the HBD.
Very recently, we have carried out an extensive spectral assignment of the H2S monomer and its clusters up to tetramer in an argon matrix,52 which prompted us to investigate a possible competition between the molecule's capacity to form very weak H-bonds, both as HBD and HBA. We have chosen CHCl3 as the second molecule due to the following two reasons: firstly, H2S binding with the C–H donor of CHCl3 results in one of the weakest known SCHBs with S as the HBA. Secondly, S–H as the HBD and CHCl3 as the HBA would result in an S–H⋯Cl H-bond, an interaction that has not yet been studied experimentally, to the best of our knowledge. In the present work, we have studied the H-bonded complex formation between H2S, CHCl3, and CDCl3 in an argon matrix. Spectral modulations in the νS–H and νC–H/D modes of the two molecules have been monitored to assess how interplay between two types of intermolecular H-bonds dictates the structure, energetic and relative population of the different possible intermolecular complexes. Quantum chemical calculations have been carried out to aid in the interpretation of the experimental findings.
2. Methodology
2.1. Experimental
H2S, CHCl3, and CDCl3 were procured from Sigma Aldrich and were used as such without any further purification. The purity of H2S was 99.5%, and that of CHCl3 was 99.8%, whereas the deuterium content in CDCl3 was 99.8%. Ultra-high purity (N6.0 grade) Argon (Ar) was procured from Messer and was used as the carrier gas to form an inert matrix. All the experiments were carried out with a sample to carrier gas mixing ratio ranging from 1:1000 to 1:10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, while the mixing ratios between the two samples were varied from 1:1 to 1:10.
000, while the mixing ratios between the two samples were varied from 1:1 to 1:10.
The CHCl3–H2S–Ar gas mixtures in the desired mixing ratios were prepared within a custom-made stainless-steel gas mixing chamber. The gas mixing chamber is connected to vacuum pumps to maintain a base pressure of 1 × 10−5 mbar. A digital pressure gauge (Pfeiffer CMR361) was used to monitor the pressure during the preparation of gas mixtures. Subsequently, these gas mixtures were deposited through a nozzle (0.6 mm inner diameter) onto a KBr window mounted on a cold head, maintained at a temperature of 7 K. A closed-cycle helium cryostat (Janis CCS-100/204N) was used to maintain the cold head temperature. The cold head region was evacuated using a turbomolecular pump (Pfeiffer TC400) backed by a rotary vane pump (Pfeiffer Duo 6). The pressure (∼5 × 10−6 mbar) was monitored using a Pirani-Penning gauge (Pfeiffer MPT200). The gas mixtures were deposited for 60 minutes at a controlled flow rate within 3.8 mmol h−1, maintained by a gas dosing valve (Pfeiffer EVN116).
The initially deposited gas mixtures were annealed by gradually increasing the temperature to 35 K to allow diffusion of samples and formation of the intermolecular complexes within the matrix. Subsequently, the matrix was cooled back to 7 K after annealing. All the spectra reported here were recorded using a Bruker Tensor II FTIR spectrophotometer, equipped with a DLaTGS detector and at 1 cm−1 instrument resolution and summed over 500 scans. Further details of the experimental conditions maintained are discussed in our previous work.53,54
2.2. Quantum chemical
Geometry optimizations and frequency calculations of all the studied monomers and association complexes (1:1 and 1:2) have been performed at the ωB97X-D/aug-cc-pV(D+d)Z level. (aug-cc-pV(X+d)Z basis set referred to as aV(X+d)Z hereafter). This particular level of calculation has been shown to provide good geometric and energetic results for intermolecular complexes bound by SCHBs, including C–H⋯S H-bonds.26,48,55 Several other functionals in conjunction with higher basis sets, along with the MP2 method, were also tested. However, none of them predicted usable spectral shifts (Table S1, ESI†) and the ωB97X-D/aV(D+d)Z level was found to provide a significantly better estimation of spectral shifts, in both the νC–H and νS–H regions. The normal mode frequencies calculated at the ωB97X-D/aV(D+d)Z level have been scaled using a scaling factor of 0.963 to match with experimental results. This scaling factor has recently been shown to provide a good match with the experimental frequencies of H2S dimers and oligomers.52 Furthermore, single point energy calculation of all the studied species has been performed at the CCSD(T)-F12/cc-pVTZ-F12 level for geometries optimized at the ωB97X-D/aV(D+d)Z level. We have shown in our earlier works,48,55,56 for multiple systems bound by SCHBs, that the CCSD(T)-F12/cc-pVTZ-F12//ωB97X-D/aV(D+d)Z level provides reliable geometric and energetic values that closely agree with experimental and high level theoretical results.Besides, localized molecular orbital energy decomposition analysis (LMO-EDA)57 has been performed to separate the total interaction energies into various contributing energy components. LMO-EDA calculations have been carried out at the CCSD(T)/aV(Q+d)Z level. Binding energies calculated at this level were found to be appreciably close to both CCSD(T)-F12 values reported herein, CCSD(T)/CBS values for H2S clusters18 and also the experimental binding energy of the H2S dimer.18 According to this methodology, the total interaction energy (EINT) can be written as follows:
| EINT = EES + EEX + EREP + EPOL + EDISP | (1) |
Furthermore, the percentage contribution of all the attractive energy components to the total attractive energy has been calculated using the following equation.
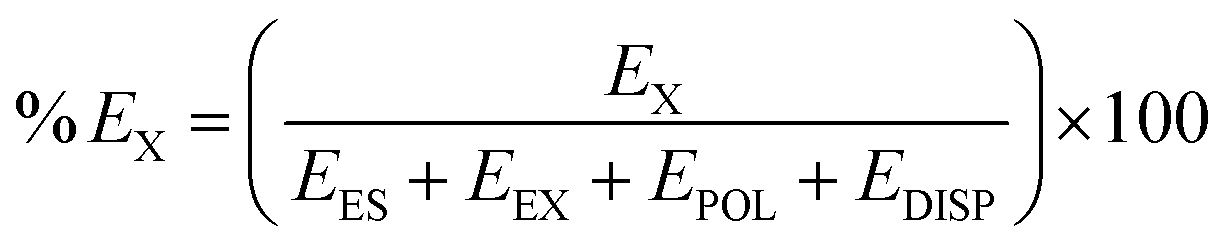 | (2) |
Furthermore, to confirm the formation of H-bonds, atoms in molecules (AIM) and natural bond orbital (NBO) analyses have also been performed. Geometry optimization, frequency calculation, and NBO analysis58 have been performed using the G16 suite of programs.59 AIM analyses60 have been performed using the Multiwfn program package.61 CCSD(T)-F12 calculations have been performed using the ORCA suite of programs.62 LMO-EDA calculations were carried out using the GAMESS program.63
3. Results & discussion
3.1 Experimental results
3.1.1.1 CHCl3. ν C–H transition of monomeric CHCl3 in an Ar matrix is observed at 3053.7 cm−1, in accordance with earlier reported results (Fig. 1A).64,65 Two more bands were observed at 3034.7 and 3038.6 cm−1, red shifted by −19.0 and −13.1 cm−1, respectively, from the monomeric νC–H band. These bands have been attributed in earlier reports to the matrix site, and 1:1 CHCl3–H2O complex, respectively.66,67 Another band at 3045.2 cm−1 was observed in close proximity to a band, reported by Sruthi et al.65 at 3045.7 cm−1 and Pal et al.30 at 3045 cm−1. The band has been assigned to the νC–H band of the CHCl3 dimer, as per the earlier reports.
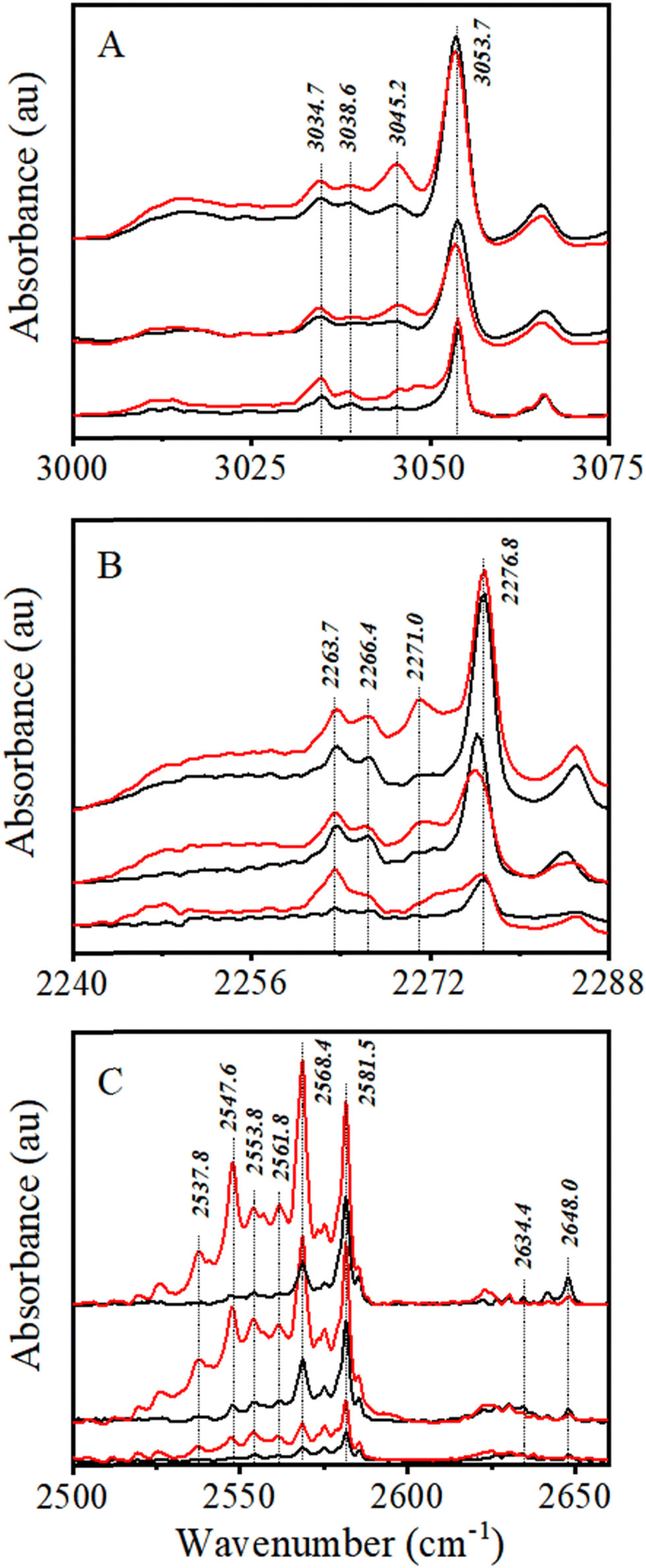 | ||
| Fig. 1 IR spectra of (A) CHCl3, (B) CDCl3, and (C) H2S in the νC–H, νC–D, and νS–H regions at sample to Ar mixing ratios of 1:1000, 1:2000, and 1:5000 from top to bottom. Preannealed and annealed spectra are shown in black and red, respectively. | ||
It is evident from Fig. 1A that only a very marginal amount of (CHCl3)2 was detected at a 1:5000 mixing ratio, while it was more pronounced at 1:2000, especially after annealing. On the other hand, dimer signature is very prominent even in the preannealed spectrum 1:1000 and becomes even stronger after annealing. Although experiments have been carried out at higher CHCl3:Ar mixing proportion, only 1:5000 and 1:2000 CHCl3:Ar mixing ratios have been considered to analyze 1:1 complex formation between CHCl3 and H2S, as any higher order complex formation would be less in these cases.
3.1.1.2 CDCl3. Similarly, νC–D transition of monomeric CDCl3, known to appear at 2276.8 cm−1 in an Ar matrix,65 has been identified accordingly (Fig. 1B). The intensity of the CDCl3νC–D band in the Ar matrix is lower than that of the CHCl3νC–H band, similar to what was observed in nonpolar solutions.68 The CDCl3:Ar mixing ratio was varied from 1:5000 to 1:1000. Similar to CHCl3, a total of three bands at 2284.9, 2271.0, and 2266.4 cm−1, corresponding to the site, homodimer, and CDCl3–H2O complex, respectively, were observed. CDCl3:Ar mixing ratios of 1:5000 and 1:2000 have been considered to analyze the 1:1 complex between CDCl3 and H2S for the same reason as that of CHCl3.
3.1.1.3 H2S. There has been a long-standing ambiguity over assignment of νS–H bands of H2S monomer (ν1 and ν3 modes) in an Ar matrix,69,70 which has been recently addressed.52 Based on that, 2634.4 and 2648.0 cm−1 bands have been assigned to monomer ν1 and ν3 modes, respectively. All the remaining bands appearing within 2550–2650 cm−1 have been assigned to H2S clusters, from dimer to tetramer. Among the most prominent bands, 2581.5 and 2568.4 cm−1 bands were assigned to dimers and trimers, respectively (Fig. 1C).
H2S has a high tendency to form oligomers in a cold, inert matrix.52,71 It has been observed that at no H2S:Ar mixing ratio chosen in this study, the dimer and oligomer bands become less intense than the monomer bands. Similar to CHCl3 and CDCl3, the H2S:Ar mixing ratio has also been fixed at 1:5000 and 1:2000 while studying the 1:1 complex formation.
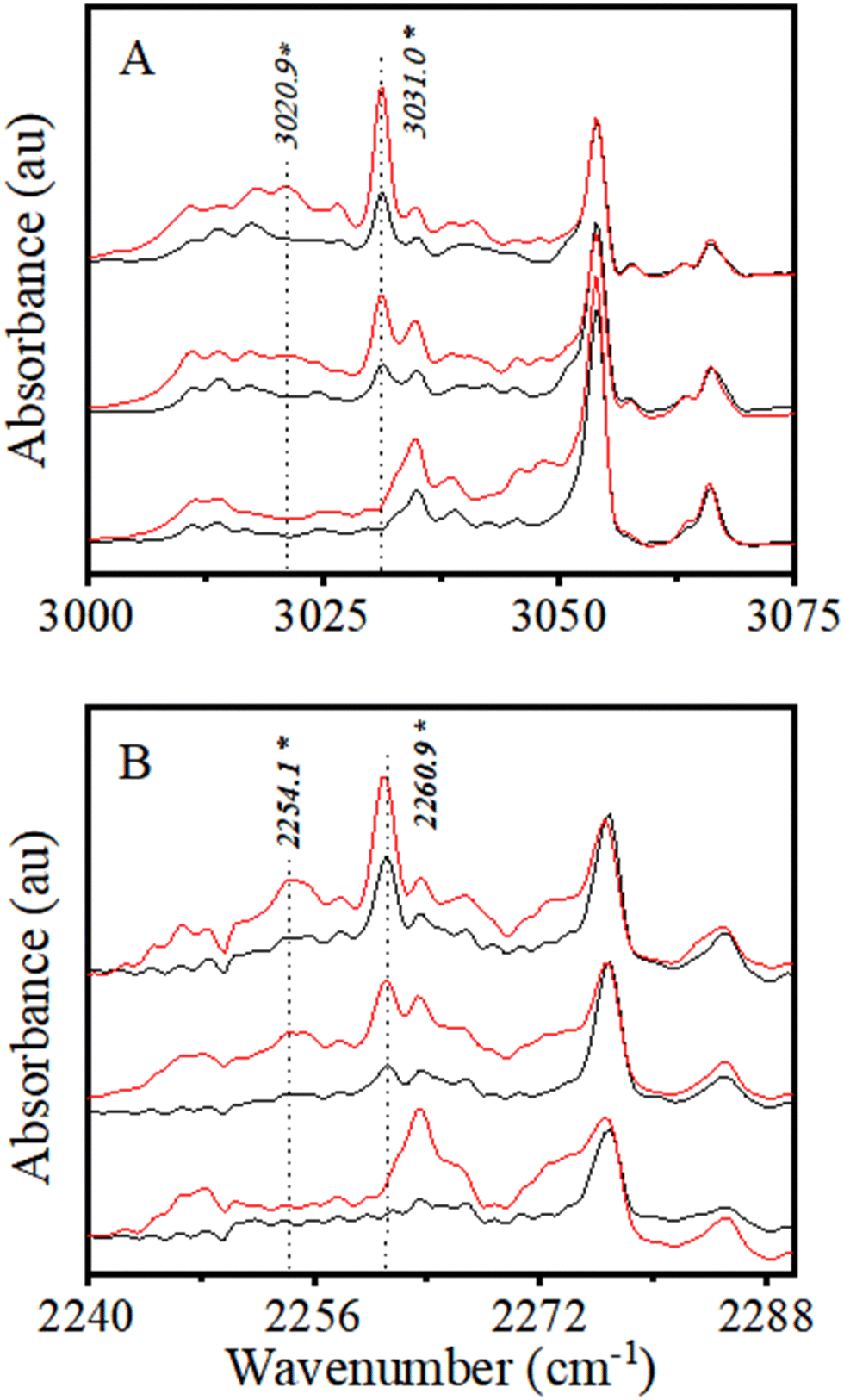 | ||
| Fig. 2 Changes in the νC–H region of CHCl3 (A) and νC–D region of CDCl3, (B) upon addition of H2S. The CHCl3(CDCl3):H2S:Ar mixing ratios in A and B are 1:2:5000; 1:1:5000, and 1:0:5000, both from top to bottom. Preannealed and annealed spectra are shown in black and red, respectively. | ||
| ν C–H/D | ΔνC–H/D | ν S–H | ΔνS–H | |
|---|---|---|---|---|
| CHCl3 | 3053.7 | — | — | — |
| H2S | — | — | 2648.0 | — |
| 2634.4 | ||||
| CHCl3–H2S | 3031.0 | −22.7 | 2622.6 | −25.4 |
| 2610.2 | −24.2 | |||
| CHCl3–(H2S)2 | — | — | 2558.4 | −89.6 |
| (CHCl3)2H2S | 3026.5 | −27.2 | 2599.6 | −48.4 |
| 3020.9 | −32.8 | |||
| CDCl3 | 2276.8 | — | — | — |
| CDCl3–H2S | 2260.9 | −15.9 | 2622.6 | −25.4 |
| 2610.2 | −24.2 | |||
| CDCl3–(H2S)2 | — | — | 2558.4 | −89.6 |
| (CDCl3)2H2S | 2258.1 | −18.7 | 2599.6 | −48.4 |
| 2254.1 | −22.7 |
Finally, it is to be noted that the development of a weak and broad spectral feature was also observed in the presence of H2S on the lower wavenumber side of the 3031.0 cm−1 band, with maxima at ∼3021 cm−1. However, this feature was visible only after annealing. We understand that this feature could have resulted from the formation of mixed clusters larger than a dimer, but it could not be confidently assigned to any specific species. Further experiments were carried out using CDCl3 to ascertain the H-bonded complex formation and support the above spectral assignment.
The above discussion clearly indicates the association complex formation of CHCl3 (and CDCl3) with H2S. Spectral shift in νC–H/D indicates C–H/D to act as HBD and H2S acts as a HBA through the S atom to form a C–H⋯S bound H-bonded complex between CHCl3 (CDCl3) and H2S.
In addition, spectral modulation in the νS–H region of H2S has also been studied due to the complex formation between CHCl3 and CDCl3 with H2S. This will further aid in the characterization of CHCl3 (CDCl3) complexes with H2S.
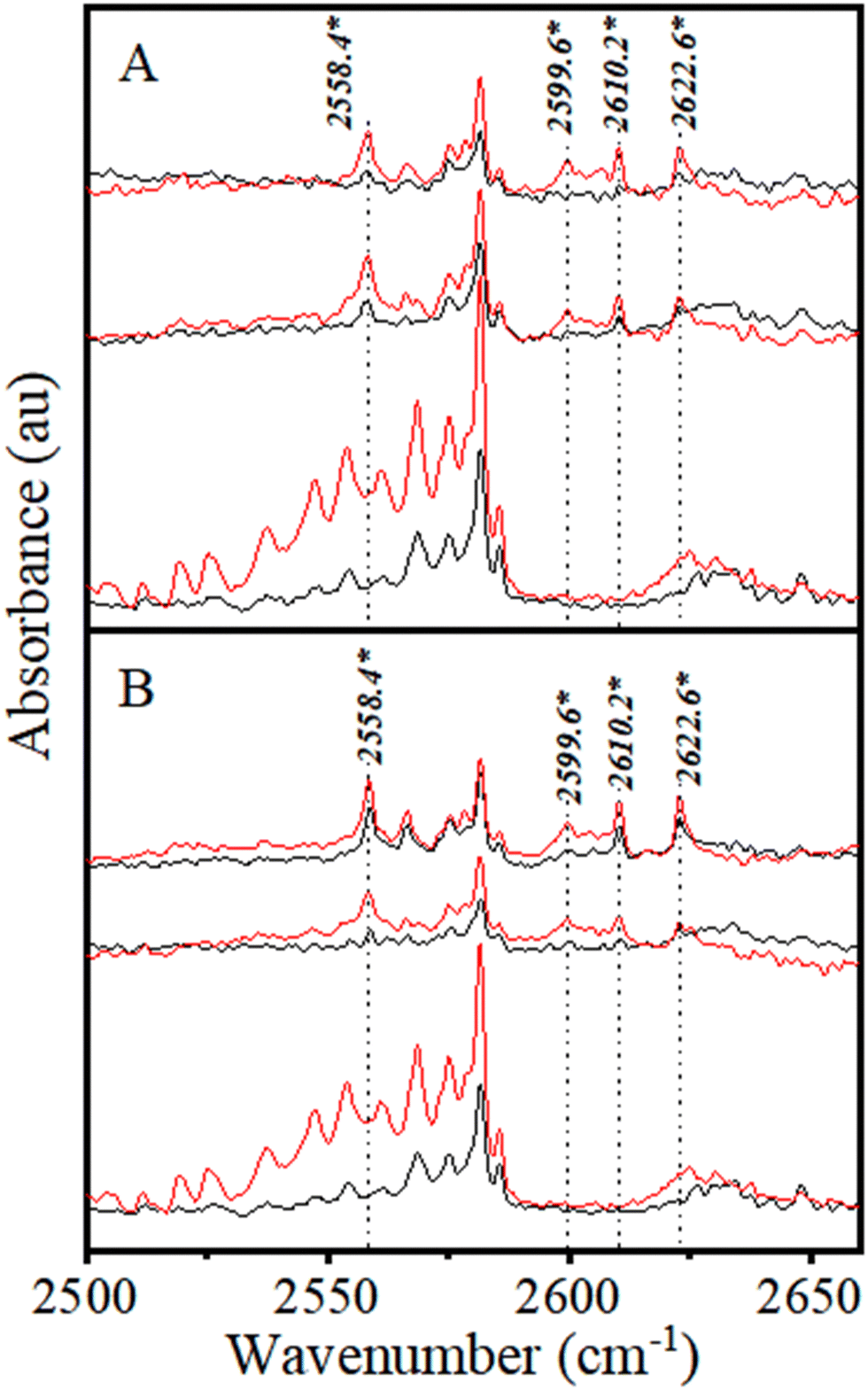 | ||
| Fig. 3 Changes in the νS–H region of H2S upon the addition of CHCl3 (A) and CDCl3 (B). The CHCl3(CDCl3):H2S:Ar mixing ratios in A and B are 2:1:5000; 1:1:5000, and 0:1:5000, both from top to bottom. Preannealed and annealed spectra are shown in black and red, respectively. | ||
From the above discussion, the 2610.2 and 2622.6 cm−1 bands have been assigned to the S–H⋯Cl H-bonded νS–H modes of CHCl3–H2S and CDCl3–H2S complexes (Table 1). It is worth mentioning that the spacing between these two bands (12.4 cm−1) is very close to that between the ν1 and ν3 modes of the H2S monomer (13.6 cm−1), supporting the above assignment. The 2558.4 cm−1 is assigned to CHCl3(H2S)2 and CDCl3(H2S)2 complexes, as its intensity increases with H2S concentration, but not with CHCl3 (and CDCl3) concentration. Consequently, the 2599.6 cm−1 band has been assigned to (CHCl3)2–H2S and (CDCl3)2–H2S complexes as its intensity increases with increasing CHCl3 (and CDCl3) concentration, but not with H2S concentration.
Multiple bands of H2S dimer and oligomers are known to appear within the 2550–2650 cm−1 range of the νS–H region.52 In particular, one band at 2623.0 cm−1, which is known to belong to the cyclic H2S tetramer, is very close to the 2622.6 cm−1 band belonging to the CHCl3–H2S complex.52 Therefore, experiments were conducted at a lower H2S:Ar mixing ratio of 1:10000 to minimize the probability of H2S cluster formation that would help identify and analyse the complex bands. The CHCl3:H2S mixing ratios of 5:1 and 10:1 were used in these experiments to facilitate complex formation (Fig. 4A).
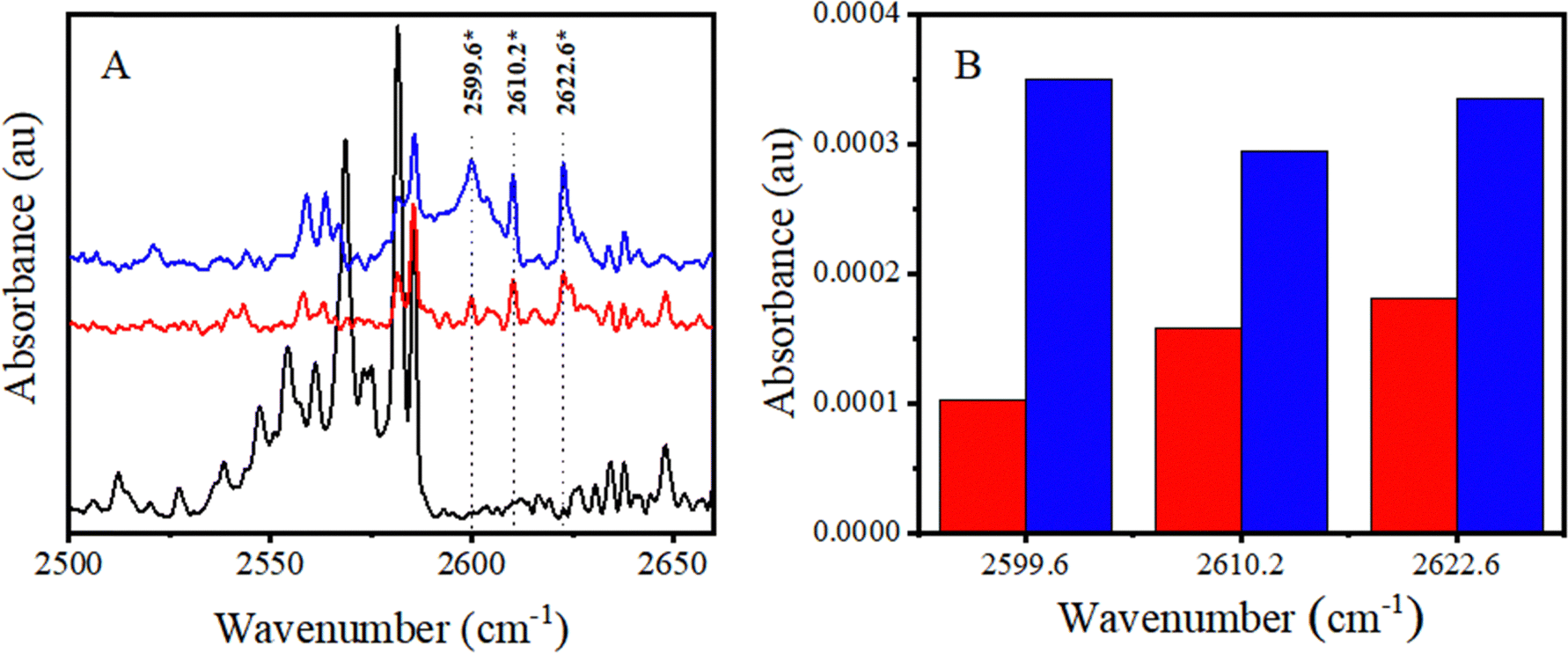 | ||
| Fig. 4 (A) Changes in the νS–H region of H2S upon addition of CHCl3. The CHCl3:H2S:Ar mixing ratios are 0:1:10000 (bottom trace; black); 5:1:10000 (middle trace; red), and 10:1:10000 (top trace; blue). (B) Intensity modulations of νS–H bands upon varying CHCl3 concentration. The CHCl3:H2S:Ar mixing ratios are 5:1:10000 (red), and 10:1:10000 (blue). | ||
No discernible bands are visible in the spectrum of only H2S (bottom trace of Fig. 4A) near any of the three bands that appear upon the addition of CHCl3 at 2599.6, 2610.2, and 2622.6 cm−1 (middle and top traces). Furthermore, it is evident from the plot in Fig. 4B that the intensity of the 2610.2 and 2622.6 cm−1 bands is nearly doubled as the concentration of CHCl3 is doubled. However, the 2599.6 cm−1 band is nearly quadrupled at the same time. This behavior signifies that the 2610.2 and 2622.6 cm−1 bands are due to CHCl3–H2S complex formation, whereas the 2599.6 cm−1 band is due to (CHCl3)2–H2S complex formation. This band can undoubtedly be assigned to a 2:1 complex between CHCl3 and H2S.
Formation of both C–H⋯S and S–H⋯Cl H-bonds is evident from the discussions in the preceding subsections. It is also apparent that both red shifted νC–H and νS–H bands exhibit enhancement in intensities during annealing for every CHCl3(CDCl3):H2S:Ar mixing ratio. This observation indicates enhancement in the population of both C–H⋯S and S–H⋯Cl H-bonded conformers, i.e., conversion of the less stable conformer to a more stable one is not significant upon annealing. Furthermore, the spectra of both the νC–H (νC–D) and νS–H regions have been shown together in Fig. S3–S5 (ESI†) for clear visibility of the relative intensities of the two spectral regions.
3.2 Computational results
Detailed quantum chemical calculations have been performed to support the various experimental observations and understand the spectral behaviors, which are discussed below.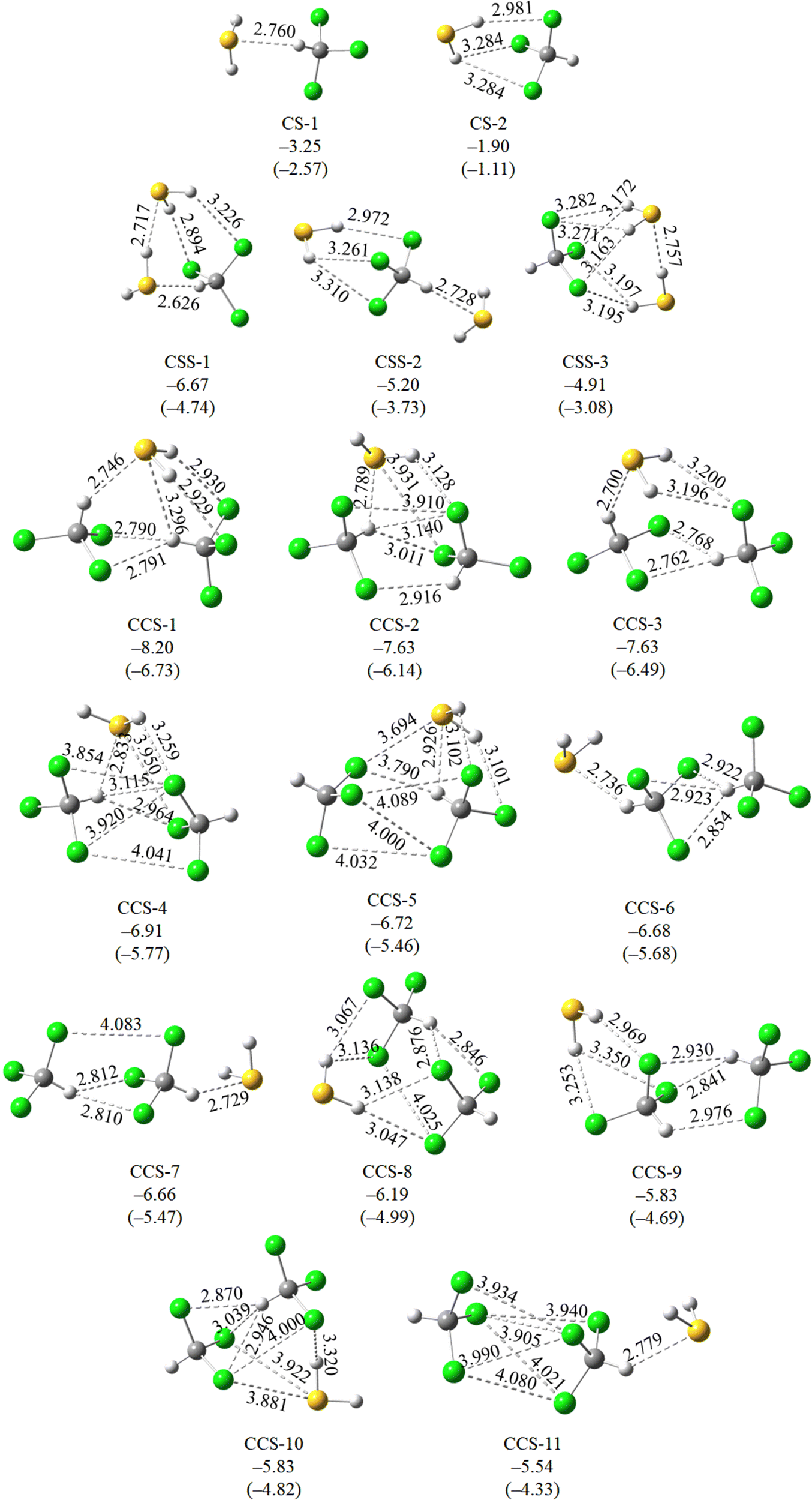 | ||
| Fig. 5 Molecular geometries of all conformers of CHCl3–H2S, CHCl3(H2S)2 and (CHCl3)2H2S optimized at the ωB97X-D/aV(D+d)Z level, along with their binding energetics, and ZPE corrected binding energies (in parenthesis) (in kcal mol−1) calculated at the CCSD(T)-F12/cc-pVTZ-F12 level. | ||
Normal mode frequency calculations predicted 21.2 cm−1 red shift for the νC–H mode of CHCl3 of CS-1, while a 4.4 cm−1 blue shift for CS-2 (Table 2). On the other hand, ν1 and ν3 modes of H2S were predicted to undergo 10.3 and 12.2 cm−1 red shifts, respectively, in CS-1, whereas 16.3 and 17.2 cm−1 red shift in CS-2, in the same order (Table 2). When compared against the experimentally observed spectral shifts, it is evident that the 3031.0 cm−1 band, red shifted by 22.7 cm−1 from the νC–H CHCl3 monomer belongs to CS-1. In contrast, the 2610.2 and 2622.6 cm−1 bands, red shifted by 24.2 and 25.4 cm−1, respectively, from the νS–H bands of H2S monomers belong to CS-2.
| ν C–H | ΔνC–H | ν asymS–H | ΔνasymS–H | ν symS–H | ΔνsymS–H | |
|---|---|---|---|---|---|---|
| Monomer | 3065.4 | 2659.3 | 2644.0 | |||
| CS-1 | 3045.0 | −20.4 | 2649.4 | −9.9 | 2632.3 | −11.7 |
| CS-2 | 3069.6 | +4.2 | 2643.6 | −15.7 | 2627.4 | −16.6 |
| CSS-1 | 3017.3 | −48.1 | 2644.6 | −14.7 | 2622.2 | −21.8 |
| 2641.4 | −17.9 | 2556.8 | −87.2 | |||
| CSS-2 | 3044.7 | −20.7 | 2646.5 | −12.8 | 2630.7 | −13.3 |
| 2646.1 | −13.2 | 2630.6 | −13.4 | |||
| CSS-3 | 3067.2 | +1.8 | 2653.5 | −5.8 | 2637.8 | −6.2 |
| 2644.8 | −14.5 | 2588.5 | −55.5 | |||
| CCS-1 | 3074.2 | +8.8 | 2637.4 | −21.9 | 2623.0 | −21.0 |
| 3050.4 | −15.0 | |||||
| CCS-2 | 3070.5 | +5.1 | 2650.8 | −8.5 | 2634.1 | −9.9 |
| 3060.8 | −4.6 | |||||
| CCS-3 | 3081.4 | +16.0 | 2648.5 | −10.8 | 2634.0 | −10.0 |
| 3042.4 | −22.9 | |||||
| CCS-4 | 3067.2 | +1.8 | 2648.2 | −11.1 | 2631.7 | −12.3 |
| 3056.8 | −8.5 | |||||
| CCS-5 | 3061.1 | −4.3 | 2644.3 | −15.0 | 2628.9 | −15.1 |
| 3067.8 | +2.4 | |||||
| CCS-6 | 3076.2 | +10.8 | 2646.5 | −12.8 | 2630.2 | −13.7 |
| 3045.6 | −19.8 | |||||
| CCS-7 | 3090.5 | +25.1 | 2644.3 | −15.0 | 2628.8 | −15.2 |
| 3046.2 | −19.2 | |||||
| CCS-8 | 3070.8 | +5.4 | 2649.8 | −9.5 | 2635.2 | −8.8 |
| 3072.4 | +7.0 | |||||
| CCS-9 | 3075.6 | +10.2 | 2647.9 | −11.4 | 2631.7 | −12.3 |
| 3071.7 | +6.4 | |||||
| CCS-10 | 3073.9 | +8.6 | 2647.3 | −12.0 | 2631.7 | −12.3 |
| 3069.8 | +4.4 | |||||
| CCS-11 | 3070.3 | +4.9 | 2648.2 | −11.1 | 2630.9 | −13.1 |
| 3052.0 | −13.4 | |||||
Furthermore, different aspects of the C–H⋯S H-bond in the CHCl3–H2S complex are compared against earlier works. Fargher et al.25 extensively examined C–H⋯S interaction in a comprehensive dataset encompassing over 423000 C–H⋯S contacts within more than 86000 structures. Their findings revealed that C–H⋯S contacts exhibit nonlinearity, with the most prevalent H⋯S contact occurring within 3.12–3.25 Å.
The C–H⋯S angle in the CS-1 complex was found to be 137.9° and S⋯H length 2.760 Å, which was notably shorter compared to the range reported by Fargher et al. A prior investigation by Fargher et al. has elucidated that the energies of C–H⋯S interactions typically range between 1–3 kcal mol−1.25 In this context, the −3.25 kcal mol−1 binding energy of the C–H⋯S H-bonded CHCl3–H2S complex aligns well. Given the absence of documentation on S–H⋯Cl H-bonds in the existing literature to the best of our knowledge, our examination is limited to the comparison of only C–H⋯S interaction.
As discussed in the previous section, the intensities of the spectral bands belonging to both CS-1 and CS-2 increase upon annealing. Therefore, conversion of the less stable CS-2 to more stable CS-1 conformer must have been insignificant, if it happened at all. This could be due to kinetically unfavorable interconversion as a result of large barrier height insurmountable at 35 K temperature. A relaxed potential energy scan along ∠Cl–C–S was performed to understand whether the energy barrier was responsible for hindered interconversion. The calculated barrier was ∼0.6 kcal mol−1 (Fig. 6), which was significantly higher than the thermal energy available at the annealing temperature.
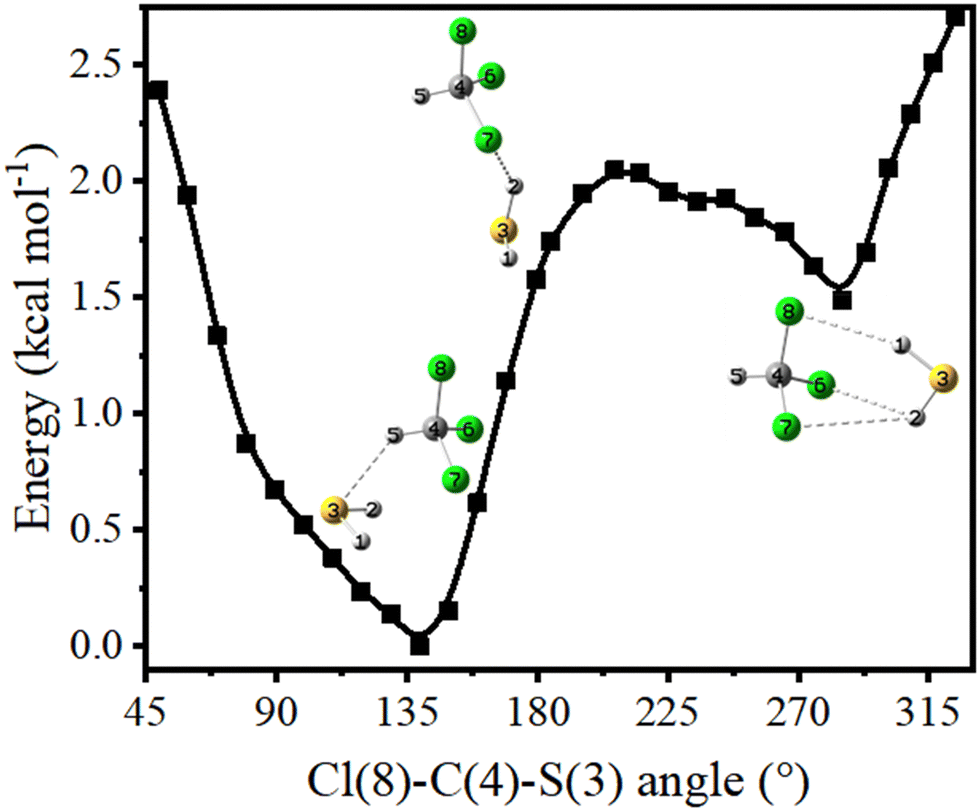 | ||
| Fig. 6 Potential energy landscape for the CHCl3–H2S complex along ∠Cl–C–S. | ||
It is worth mentioning that annealing-induced interconversion between two conformers has been reported for a C–H⋯O H-bonded camphor–CHCl3 complex separated by a very small barrier height of only 0.1–0.2 kcal mol−1, while no such phenomenon could occur for O–H⋯O H-bonded camphor–methanol and camphor–phenol complexes where the barriers were 1.0 and 1.3 kcal mol−1, respectively.72 The barrier in this case is somewhat intermediate of the abovementioned two barriers. Besides, interconversion between the two conformers of H-bonded complexes with camphor required the H-bond donors (i.e., chloroform, methanol, and phenol) to move from one side of the carbonyl oxygen to the other side. On the other hand, here in the CHCl3–H2S complex, H2S needs to rotate by ∼150° and move ∼7.6 Å around CHCl3 for interconversion between CS-1 and CS-2 to happen. A full rotational maneuver by such a larger distance within the rigid, inert matrix is deemed highly improbable. Consequently, annealing experiments resulted in the formation of both CS-1 and CS-2 conformers, with very little or no interconversion between the two.
As discussed in the preceding subsections, intensity modulations of a number of bands in both the νC–H/D and νS–H regions upon concentration variations indicate the presence of higher order complexes. This possibility finds strong support from the self-association of both CHCl3 and H2S, even at the lowest mixing ratios that were evident from experimental spectra in Fig. 1. In order to check that possibility, quantum chemical calculations have also been carried out for CHCl3(H2S)2 and (CHCl3)2H2S.
Normal mode frequency calculations predicted the νS–H mode of the S–H⋯S H-bonded S–H group to undergo 90.6 cm−1 red shifts. When compared against experiments, it is evident that the 2558.4 cm−1 band, red shifted by 89.6 cm−1 from the monomeric ν3 band of H2S belongs to the S–H⋯S H-bonded S–H group of CSS-1. It is worth noting here that the νS–H transition of the S–H⋯S H-bonded S–H group in the H2S dimer appears at 2581.5 cm−1. A comparison of the two transition frequencies indicates that the νS–H transition of the S–H⋯S H-bonded S–H group shows 23.1 cm−1 larger red shift in CHCl3(H2S)2 compared to that in (H2S)2. This observation further supports the energetic and geometrical features of cooperative strengthening of H-bonds in the complex. On the other hand, the νC–H mode of CHCl3 was predicted to show 50.0 cm−1 red shift in CSS-1. As discussed in the previous subsection, experimental spectra showed the appearance of a broad feature near 3020.9 cm−1. This broad feature could be due to the formation of a CHCl3(H2S)2 complex. However, a clear assignment of the νC–H spectral band, corresponding to the CSS-1 conformer could not be achieved.
Normal mode frequency calculations predicted the spectral shift in the νC–H mode of CHCl3 to be within −23.9 cm−1 to +26.1 cm−1, whereas the shift in ν1 and ν3 modes of H2S to be within −8.9 to −22.8 cm−1 (Table 2). However, only one experimental band at 2599.6 cm−1 in the νS–H region could be assigned to the (CHCl3)2H2S complex. Calculation predicted a red shift of −22.8 and −21.8 cm−1 in the ν1 and ν3 modes of H2S in the CCS-1 conformer, where both S–H bonds are H-bonded with the Cl atoms of CHCl3. These calculated spectral shifts are ∼5–6 cm−1 greater than the same in S–H⋯Cl H-bonded binary complex CS-2. The 2599.6 cm−1 band is 10.6 cm−1 more red shifted compared to the experimental band assigned to the C-2 conformer.
Similar to CHCl3(H2S)2, the large difference in binding energies between the two most stable (CHCl3)2H2S conformers indicates that only CCS-1 would have a detectable population in the matrix. The remaining conformers would have negligible population to induce any noticeable spectral sign. Also, the spectral shift in νS–H of CCS-1 is close to that observed experimentally by the 2599.6 cm−1 band. Therefore, the 2599.6 cm−1 band can be assigned to the CCS-1 conformer. However, a clear assignment of the νC–H band corresponding to a particular conformer could not be achieved.
Based on the above discussions it can be stated that certain theoretical results don’t match very closely with the experimental findings, especially the predicted magnitude of spectral shifts due to complex formation with the observed values. This could be due to the fact that the calculation did not consider the influence of matrix environment and there is no robust theoretical model available yet that explicitly considers matrix effects on binding energies and spectral shifts.
The values in Table 3 show that EEX exhibits the highest contribution among the attractive interactions. Upon comparing the percentage contributions of different attractive interactions in CS-1 and CS-2 with those of (H2S)2, it becomes evident that CS-1 demonstrates a marginally higher contribution from EES (25.3%) to that of (H2S)2 (23.9%), while CS-2 exhibits a significantly lower contribution (13.6%), almost half of the above values. The trend is exactly opposite for EDISP; its percentage contribution in CS-2 (33.7%) is more than double that in (H2S)2 (16.4%), while the same in CS-1 (20.7%) is a little higher than in (H2S)2. The contribution of EPOL to overall stabilization is found to be lower, always below 10%, for all three binary complexes. Nevertheless, here also the value for CS-2 (4.4%) is significantly different compared to CS-1 (7.4%) and (H2S)2 (8.9%).
Therefore, in summary, it can be inferred that in terms of the percentage contributions of various interactions the C–H⋯S H-bond in CS-1 is very similar to the S–H⋯S H-bond in (H2S)2. On the other hand, electrostatics plays an appreciably minor role in stabilizing the S–H⋯Cl H-bonded CS-2, which is found to have a significantly higher contribution from dispersion interaction compared to both C–H⋯S and S–H⋯S H-bonds. In order to have a clearer idea about the contribution of dispersion interaction in stabilizing the CHCl3–H2S complex, its value against the total stabilization energies has been compared with earlier investigations of binary complexes featuring SCHBs (Table 4).
| Complex | SCHB | E DISP | E Total |
|
Method | Ref. |
|---|---|---|---|---|---|---|
| H2S–C6H6 | S–H⋯π | −3.56 | −2.85 | 125 | RVS-SCF | 4 |
| H2S–indole | S–H⋯π | −2.95 | −3.34 | 88 | HF/aug-cc-pVDZ | 20 |
| Indole–Me2S | N–H⋯S | −6.57 | −5.75 | 114 | ||
| H2S-3-methylindole | S–H⋯π | −3.02 | −3.26 | 93 | ||
| 3-Methylindole-Me2S | N–H⋯S | −6.69 | −5.66 | 118 | ||
| H2S–indole | S–H⋯π | −7.45 | −5.22 | 143 | RVS-SCF | 38 |
| p-Cresol-diethylsulfide (TT) | O–H⋯S | −4.00 | −7.30 | 55 | HF/aug-cc-pVDZ | 28 |
| p-Cresol–H2S | O–H⋯S | −2.58 | −3.68 | 70 | ||
| H2S–MeOH | S–H⋯O | −2.13 | −1.97 | 108 | MP2/aug-cc-pVDZ | 73 |
| MeOH–H2S | O–H⋯S | −1.70 | −1.61 | 106 | ||
| p-Fluorophenol–H2S | O–H⋯S | −2.24 | −4.12 | 54 | 74 | |
| H2S–H2S | S–H⋯S | −0.78 | −0.87 | 89 | MP2/6-311++G** | 23 |
| H2S–MeOH | S–H⋯O | −2.61 | −1.60 | 61 | ||
| H2S-diethylether (coplanar) | S–H⋯O | −2.84 | −2.47 | 87 | ||
| H2S-diethylether (perpendicular) | S–H⋯O | −2.67 | −3.00 | 113 | ||
| H2S-dibutylether (coplanar) | S–H⋯O | −3.12 | −2.79 | 89 | ||
| H2S-dibutylether (perpendicular) | S–H⋯O | −2.97 | −3.54 | 119 | ||
| H2S-1,4-dioxane (coplanar) | S–H⋯O | −2.63 | −2.12 | 81 | ||
| H2S-1,4-dioxane (perpendicular) | S–H⋯O | −2.35 | −2.39 | 102 | ||
| 1,2,4,5-Tetracyanobenzene-H2S | C–H⋯S | −2.62 | −4.09 | 64 | ωB97XD/cc-pVDZ | 26 |
| 1,2,4,5-Tetracyanobenzene-MeSH | C–H⋯S | −3.20 | −5.15 | 62 | ||
| 1,2,4,5-Tetracyanobenzene-EtSH | C–H⋯S | −5.95 | −5.84 | 102 | ||
| 1,2,4,5-Tetracyanobenzene-Me2S | C–H⋯S | −5.65 | −6.02 | 94 | ||
| 1,2,4,5-Tetracyanobenzene-tetrahydrothiophene | C–H⋯S | −7.03 | −6.93 | 101 | ||
| p-Aminophenol-Me2S | O–H⋯S | −4.73 | −6.20 | 75 | MP2-cp/aug-cc-pVDZ | 75 |
| p-Cresol-Me2S | O–H⋯S | −4.64 | −6.20 | 74 | ||
| Phenol-Me2S | O–H⋯S | −4.56 | −6.24 | 72 | ||
| p-Fluorophenol-Me2S | O–H⋯S | −4.51 | −6.44 | 70 | ||
| cis-β-Naphthol-Me2S | O–H⋯S | −5.04 | −6.62 | 75 | ||
| p-Chlorophenol-Me2S | O–H⋯S | −4.55 | −6.56 | 69 | ||
| p-Cyanophenol-Me2S | O–H⋯S | −4.39 | −6.94 | 64 | ||
| HCONH2-diethyldisulfide | N–H⋯S | −7.15 | −7.27 | 98 | SAPT2+(3)δMP2/aug-cc-pVTZ | 76 |
| HCONH2–Me2S | N–H⋯S | −6.09 | −7.79 | 78 | ||
| CHCl3-dimethylsulfide | C–H⋯S | −6.97 | −4.65 | 149 | SAPT2/aug-cc-pVDZ | 30 |
| CHCl3–H2S (CS-1) | C–H⋯S | −3.24 | −3.25 | 100 | CCSD(T)/aV(Q+d)Z | This work |
| CHCl3–H2S (CS-2) | S–H⋯Cl | −3.04 | −1.90 | 160 |
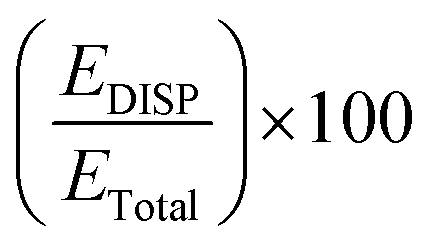
As evident from Table 4, the values of dispersion interaction are either lesser than or similar to the corresponding overall stabilization energies (i.e., entries in column 5 are ≤100%) for most of the studies involving C–H⋯S interactions, except for the CHCl3-dimethylsulfide complex (149%).30 Here also, the value for C–H⋯S H-bonded CS-1 (100%) matched closely with the earlier reports. Notably, there is no such scope of comparison for the S–H⋯Cl H-bonded CS-2 as no prior studies were available in the literature on S–H⋯Cl H-bonded complexes. Nevertheless, the value for CS-2 (160%) is the highest percentage dispersion contribution documented thus far for any binary complexes held by SCHBs. No previous work reports a value greater than 150%. These values provide a quantitative estimation of the contribution of dispersion interaction in total binding energy of the binary complexes bound by SCHBs. The very large value for CS-2 shows that dispersion interaction makes a major contribution towards overall stabilization of the S–H⋯Cl H-bond. When compared against earlier works, the newly reported S–H⋯Cl H-bond is found to have one of the largest dispersion contributions among all the SCHBs reported to date.
To further substantiate the observed H-bond formations in the complex, NBO analysis has been conducted and is discussed below.
Furthermore, AIM analyses have been performed, which shows (3,−1) BCPs at the bond path between molecules (Fig. S6, ESI†). The ρCP and ∇2ρCP values corresponding to the BCP have been provided in Table S3 (ESI†). This further supports the evidence for the formation of both C–H⋯S and S–H⋯Cl H-bonds. Also AIM analysis was found to corroborate the NBO results. The NCI plot clearly shows the attractive regions between the two molecules, which is indicated by the minima observed in the reduced density gradient plot (Fig. S7, ESI†). The reduced density gradient approaches zero, whenever the sign λ2 × ρ approaches zero. These analyses further support the formation of a H-bonded complex in the system.
4. Summary
A H-bonded CHCl3–H2S complex has been investigated in a cold argon matrix using FTIR spectroscopy. A new band at 3031.0 cm−1, red shifted by 22.7 cm−1 from the monomeric νC–H band of CHCl3 was observed due to the formation of a C–H⋯S H-bonded CHCl3–H2S binary complex. Similar spectral features were observed for CDCl3, where the new band at 2260.9 cm−1 was 15.9 cm−1 red shifted from the monomeric νC–D band. On the other hand, two new bands at 2610.2 and 2622.6 cm−1, red shifted 24.2 and 25.4 cm−1 from the monomeric ν1 and ν3 modes of H2S appeared due to a S–H⋯Cl H-bonded conformer of CHCl3–H2S binary complex. Calculations predicted the C–H⋯S H-bonded conformer of the CHCl3–H2S binary complex (CS-1) to be 1.35 kcal mol−1 more stable than its S–H⋯Cl H-bonded counterpart (CS-2), duly supporting the much higher intensity of the νC–H band compared to that of νS–H bands of the binary complexes.No sign of interconversion between the two conformers could be identified experimentally. Subsequently, a relaxed PES scan revealed the energy barrier of converting CS-2 to CS-1 (∼0.6 kcal mol−1) to be too high to overcome at annealing temperature (35 K) and the large relative motions of the two monomers to be improbable in the rigid matrix environment.
Intensity patterns of two more bands at 2558.4 and 2599.6 cm−1 in the νS–H region indicated the presence of larger complexes: the first one belonging to S–H⋯S H-bonded νS–H mode of CHCl3-(H2S)2 and the second one the S–H⋯Cl H-bonded νS–H mode of (CHCl3)2–H2S. Both H-bonds exhibited cooperative strengthening as evident from the 23.1 cm−1 larger spectral shift for the S–H⋯S H-bond in H2S dimer and 10.6 cm−1 higher spectral shift for the S–H⋯Cl H-bond in the CHCl3–H2S binary complex. Both the above bands were assigned to the most stable conformers of CHCl3–(H2S)2 and (CHCl3)2–H2S complexes based on calculated normal mode frequencies.
Energy decomposition analyses reveal dispersion interaction to play an important role in stabilizing SCHBs, which accounted for over one fifth of all the attractive interactions for the C–H⋯S H-bonded complex and over one third for S–H⋯Cl H-bonded one. In fact, the magnitude of dispersion contribution against overall interaction energy of the binary complex has been found to be the greatest for the latter among all the SCHBs reported to date. Additionally, AIM and NBO calculations performed have been found to support the above findings and they were found to corroborate with each other. This study represents the first experimental evidence of an S–H⋯Cl H-bond interaction. Additionally, it offers a comprehensive understanding of the coexistence of two weak SCHBs, namely, C–H⋯S and S–H⋯Cl H-bonds.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
BKO, Monu, and AK acknowledge MNIT Jaipur, CSIR, and CSIR-UGC, respectively, for research fellowships. BB acknowledges SERB-DST, Govt. of India for financial support through sanctioned project (CRG/2020/001601).References
- E. N. Baker and R. E. Hubbard, Prog. Biophys. Mol. Biol., 1984, 44, 97–179 CrossRef CAS PubMed.
- P. Zhou, F. Tian, F. Lv and Z. Shang, Proteins: Struct., Funct., Bioinf., 2009, 76, 151–163 CrossRef CAS.
- L.-H. Xu, Q. Liu, R. Suenram, F. J. Lovas, A. H. Walker, J. Jensen and A. Samuels, J. Mol. Spectrosc., 2004, 228, 243–250 CrossRef CAS.
- D. L. Crittenden, J. Phys. Chem. A, 2009, 113, 1663–1669 CrossRef CAS PubMed.
- T.-a Okamura, Y. Ushijima, Y. Omi and K. Onitsuka, Inorg. Chem., 2013, 52, 381–394 CrossRef CAS PubMed.
- T.-A. Okamura, T. Kaga, S. Yamashita, R. Furuya and K. Onitsuka, J. Org. Chem., 2017, 82, 2187–2192 CrossRef CAS.
- B. W. Beck, Q. Xie and T. Ichiye, Biophys. J., 2001, 81, 601–613 CrossRef CAS PubMed.
- K. Mazmanian, K. Sargsyan, C. D. Grauffel, T. Dudev and C. Lim, J. Phys. Chem. B, 2016, 120, 10288–10296 CrossRef CAS PubMed.
- L. M. Gregoret, S. D. Rader, R. J. Fletterick and F. E. Cohen, Proteins: Struct., Funct., Bioinf., 1991, 9, 99–107 CrossRef CAS PubMed.
- A. D. Pagar, M. D. Patil, D. T. Flood, T. H. Yoo, P. E. Dawson and H. Yun, Chem. Rev., 2021, 121, 6173–6245 CrossRef CAS.
- R. Reddi, K. K. Singarapu, D. Pal and A. Addlagatta, Mol. BioSyst., 2016, 12, 2408–2416 RSC.
- V. S. Minkov and E. V. Boldyreva, J. Phys. Chem. B, 2013, 117, 14247–14260 CrossRef CAS PubMed.
- S. Pathania, R. K. Narang and R. K. Rawal, Eur. J. Med. Chem., 2019, 180, 486–508 CrossRef CAS.
- A. Nangia and G. R. Desiraju, J. Mol. Struct., 1999, 474, 65–79 CrossRef CAS.
- H.-P. Zhou, Y.-M. Zhu, J.-J. Chen, Z.-J. Hu, J.-Y. Wu, Y. Xie, M.-H. Jiang, X.-T. Tao and Y.-P. Tian, Inorg. Chem. Commun., 2006, 9, 90–92 CrossRef CAS.
- J. J. Novoa, M. C. Rovira, C. Rovira, J. Veciana and J. Tarrés, Adv. Mater., 1995, 7, 233 CrossRef CAS.
- R. T. Saragi, M. Juanes, R. Pinacho, J. E. Rubio, J. A. Fernández and A. Lesarri, Symmetry, 2021, 13, 2022 CrossRef CAS.
- S. Sarkar and B. Bandyopadhyay, Phys. Chem. Chem. Phys., 2019, 21, 25439–25448 RSC.
- S. Ghosh, S. Bhattacharyya and S. Wategaonkar, J. Phys. Chem. A, 2015, 119, 10863–10870 CrossRef CAS PubMed.
- H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A, 2009, 113, 12763–12773 CrossRef CAS.
- C. L. Andersen, C. S. Jensen, K. Mackeprang, L. Du, S. Jørgensen and H. G. Kjaergaard, J. Phys. Chem. A, 2014, 118, 11074–11082 CrossRef CAS PubMed.
- A. Paul and R. Thomas, J. Mol. Liq., 2022, 348, 118078 CrossRef CAS.
- A. Bhattacherjee, Y. Matsuda, A. Fujii and S. Wategaonkar, J. Phys. Chem. A, 2015, 119, 1117–1126 CrossRef CAS.
- V. R. Mundlapati, S. Ghosh, A. Bhattacherjee, P. Tiwari and H. S. Biswal, J. Phys. Chem. Lett., 2015, 6, 1385–1389 CrossRef CAS.
- H. A. Fargher, T. J. Sherbow, M. M. Haley, D. W. Johnson and M. D. Pluth, Chem. Soc. Rev., 2022, 51, 1454–1469 RSC.
- S. Ghosh, P. Chopra and S. Wategaonkar, Phys. Chem. Chem. Phys., 2020, 22, 17482–17493 RSC.
- H. S. Biswal, E. Gloaguen, Y. Loquais, B. Tardivel and M. Mons, J. Phys. Chem. Lett., 2012, 3, 755–759 CrossRef CAS PubMed.
- H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A, 2010, 114, 5947–5957 CrossRef CAS.
- E. J. Cocinero, R. Sánchez, S. Blanco, A. Lesarri, J. C. López and J. L. Alonso, Chem. Phys. Lett., 2005, 402, 4–10 CrossRef CAS.
- D. Pal, H. Charaya and S. Chakraborty, ChemPhysChem, 2023, 24, e202300124 CrossRef CAS PubMed.
- M. Domagała and S. J. Grabowski, Chem. Phys., 2010, 367, 1–6 CrossRef.
- L. K. Macreadie, A. J. Edwards, A. S. Chesman and D. R. Turner, Aust. J. Chem., 2014, 67, 1829–1839 CrossRef CAS.
- S. François, M.-M. Rohmer, M. Bénard, A. C. Moreland and T. B. Rauchfuss, J. Am. Chem. Soc., 2000, 122, 12743–12750 CrossRef.
- A. Nowroozi, H. Roohi, H. Hajiabadi, H. Raissi, E. Khalilinia and M. N. Birgan, Comput. Theor. Chem., 2011, 963, 517–524 CrossRef CAS.
- E.-M. Sung and M. D. Harmony, J. Am. Chem. Soc., 1977, 99, 5603–5608 CrossRef CAS.
- H. S. Biswal, S. Bhattacharyya, A. Bhattacherjee and S. Wategaonkar, Int. Rev. Phys. Chem., 2015, 34, 99–160 Search PubMed.
- C. Ma, Q. Zhang, R. Zhang and D. Wang, Chem. – Eur. J., 2006, 12, 420–428 CrossRef PubMed.
- H. S. Biswal and S. Wategaonkar, J. Phys. Chem. A, 2009, 113, 12774–12782 CrossRef CAS PubMed.
- A. Chand, D. K. Sahoo, A. Rana, S. Jena and H. S. Biswal, Acc. Chem. Res., 2020, 53, 1580–1592 CrossRef CAS.
- G. Duan, V. H. Smith Jr and D. F. Weaver, Mol. Phys., 2001, 99, 1689–1699 CrossRef CAS.
- K. Grzechnik, K. Rutkowski and Z. Mielke, J. Mol. Struct., 2012, 1009, 96–102 CrossRef CAS.
- I. A. Lobo, P. A. Robertson, L. Villani, D. J. Wilson and E. G. Robertson, J. Phys. Chem. A, 2018, 122, 7171–7180 CrossRef CAS PubMed.
- M. H. Graneri, D. A. Wild and A. J. McKinley, J. Mol. Spectrosc., 2021, 378, 111440 CrossRef CAS.
- K. K. Mishra, K. Borish, G. Singh, P. Panwaria, S. Metya, M. Madhusudhan and A. Das, J. Phys. Chem. Lett., 2021, 12, 1228–1235 CrossRef CAS.
- M. Solimannejad, M. Gharabaghi and S. Scheiner, J. Chem. Phys., 2011, 134, 024312 CrossRef.
- Y. Jin, W. Li, R. T. Saragi, M. Juanes, C. Pérez, A. Lesarri and G. Feng, Phys. Chem. Chem. Phys., 2023, 25, 12174–12181 RSC.
- E. M. Cabaleiro-Lago, J. Rodríguez-Otero and Á. Peña-Gallego, J. Chem. Phys., 2011, 135, 134310 CrossRef PubMed.
- Monu, B. K. Oram and B. Bandyopadhyay, Comput. Theor. Chem., 2022, 1213, 113740 CrossRef CAS.
- H. Zhao, S. Tang, Q. Zhang and L. Du, RSC Adv., 2017, 7, 22485–22491 RSC.
- D. P. Mukhopadhyay, S. Biswas, A. Chattopadhyay and T. Chakraborty, J. Phys. Chem. A, 2018, 122, 3787–3797 CrossRef CAS PubMed.
- A. K. Samanta, P. Banerjee, B. Bandyopadhyay, P. Pandey and T. Chakraborty, J. Phys. Chem. A, 2017, 121, 6012–6020 CrossRef CAS.
- Monu, B. K. Oram and B. Bandyopadhyay, J. Phys. Chem. A, 2024, 128, 6703–6713 CrossRef CAS.
- B. K. Oram, Monu and B. Bandyopadhyay, Mol. Phys., 2022, 120, e2061387 CrossRef.
- B. K. Oram, Monu and B. Bandyopadhyay, Spectrochim. Acta, Part A, 2020, 230, 118070 CrossRef PubMed.
- Monu, B. K. Oram and B. Bandyopadhyay, Comput. Theor. Chem., 2023, 1225, 114133 CrossRef CAS.
- Monu, B. K. Oram and B. Bandyopadhyay, Phys. Chem. Chem. Phys., 2021, 23, 18044–18057 RSC.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. E. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian, Inc., Wallingford CT, 2016.
- R. F. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen and S. Su, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- S. Oswald and S. Coussan, Low Temp. Phys., 2019, 45, 639–648 CrossRef CAS.
- P. Sruthi, S. Chandra, N. Ramanathan and K. Sundararajan, J. Chem. Phys., 2020, 153, 174305 CrossRef CAS.
- S. Paulson and A. Barnes, J. Mol. Struct., 1982, 80, 151–158 CrossRef CAS.
- F. Ito, J. Chem. Phys., 2012, 137, 014505 CrossRef.
- B. K. Oram, Monu and B. Bandyopadhyay, J. Mol. Struct., 2023, 136749 Search PubMed.
- E. Isoniemi, M. Petterson and L. L. Khriachtchev, J. Phys. Chem. A, 1999, 103, 679–685 CrossRef CAS.
- A. Barnes and J. Howells, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 729–736 RSC.
- S. Y. Tang and C. W. Brown, J. Raman Spectrosc., 1974, 2, 209–215 CrossRef CAS.
- P. Banerjee, P. Pandey and B. Bandyopadhyay, Spectrochim. Acta, Part A, 2019, 209, 186–195 CrossRef CAS PubMed.
- A. Bhattacherjee, Y. Matsuda, A. Fujii and S. Wategaonkar, ChemPhysChem, 2013, 14, 905–914 CrossRef CAS PubMed.
- S. Bhattacharyya and S. Wategaonkar, J. Phys. Chem. A, 2014, 118, 9386–9396 CrossRef CAS PubMed.
- S. Bhattacharyya, S. Ghosh and S. Wategaonkar, Phys. Chem. Chem. Phys., 2021, 23, 5718–5739 RSC.
- T. Lu, J. Zhang, Y. Xu, Z. Wang, G. Feng and Z. Zeng, Spectrochim. Acta, Part A, 2023, 288, 122199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03509g |
| This journal is © the Owner Societies 2025 |