
Theoretical insights into the vibrational spectra and chemical bonding of Ln(III) complexes with a tripodal N4O3 ligand along the lanthanide series†
Francielle C.
Machado
 a,
Mateus
Quintano
*ab,
Carlos V.
Santos-Jr
c,
Albano N.
Carneiro Neto
d,
Elfi
Kraka
b,
Ricardo L.
Longo
*a and
Renaldo T.
Moura Jr.
*be
a,
Mateus
Quintano
*ab,
Carlos V.
Santos-Jr
c,
Albano N.
Carneiro Neto
d,
Elfi
Kraka
b,
Ricardo L.
Longo
*a and
Renaldo T.
Moura Jr.
*be
aDepartamento de Química Fundamental, Universidade Federal de Pernambuco, Cidade Universitária, 50740-560, Recife, PE, Brazil. E-mail: mateusmquintano@gmail.com; ricardo.longo@ufpe.br
bComputational and Theoretical Chemistry Group (CATCO), Department of Chemistry, Southern Methodist University, Dallas, TX 75275, USA. E-mail: renaldotmjr@gmail.com
cDepartment of Chemistry, Federal University of Paraíba, João Pessoa, PB 58033-455, Brazil
dPhysics Department and CICECO – Aveiro Institute of Materials University of Aveiro, Aveiro 3810-193, Portugal
eDepartment of Chemistry and Physics, Center of Agrarian Sciences, Federal University of Paraíba, Areia, PB 58397-000, Brazil
First published on 11th November 2024
Abstract
This study provides new theoretical insights into the vibrational spectra of Ln(III) complexes, along the lanthanide series by utilizing the LModeAGen protocol and integrating cutting-edge topological ideas. It provides a quantitative interpretation of the vibrational spectra of [Ln(trensal)] complexes at the B3LYP/MWBn(Ln)/6-311++G** (n from 46 (La) to 60 (Lu)) level using the characterization of normal modes from the local vibrational mode theory. This involves decomposing normal vibrational modes related to the complex formation, distortions in the coordination sphere, and C–H vibrations into local mode contributions, offering particularly promising results aimed at the design of highly luminescent lanthanide complexes. This study also delivers key theoretical insights into the chemical bonding of the coordination sphere of [Ln(trensal)] by combining the local vibrational mode theory and the bond overlap model to achieve relationships between bond properties, including those of the Badger-type. Altogether, we present the theoretical framework necessary to quantitatively interpret the vibrational spectra of [Ln(trensal)] complexes along the lanthanide series and gain a better understanding of the lanthanide-ligand chemical bonds, thereby enhancing our understanding of their chemistry and guiding future design efforts.
Introduction
Rare earth elements, 15 of which belong to the lanthanide (La–Lu) series, are present in moderate amounts in the lithosphere and have special attributes that make them useful in catalysts, lasers, magnets, batteries, and even biological applications.1–5 From a chemistry perspective, a better understanding of chemical bonding in lanthanide-containing compounds should facilitate these applications.6 To this end, we have employed the local vibrational mode (LVM) theory, originally proposed by Konkoli and Cremer,7 and the bond overlap model (BOM)8–10 for investigating Ln(III) ions in complexes with the tripodal trensal ligand11 (see Fig. 1a) along the La–Lu series. This investigation of the entire series is expected to contribute to the fundamental chemistry of Pm(III), which is quite unknown.12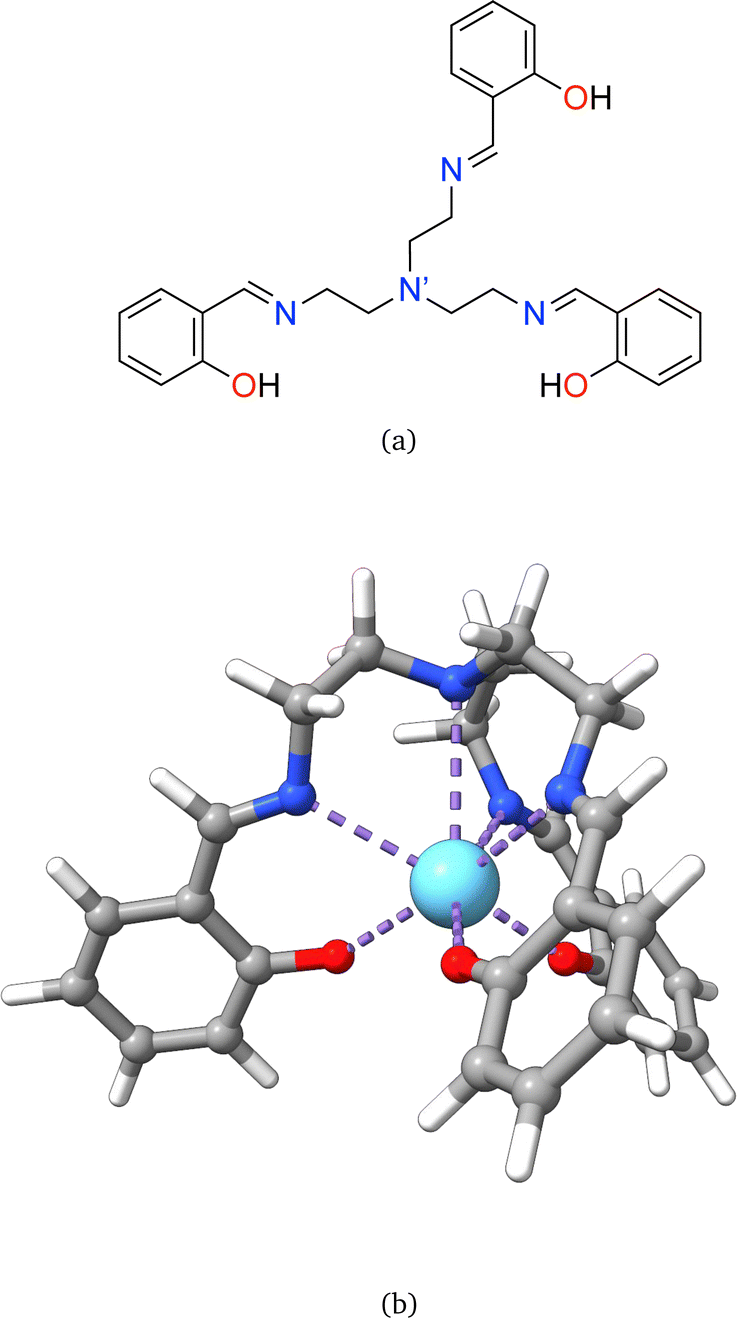 | ||
| Fig. 1 (a) Schematic 2D representation of the neutral form of the unbound trensal ligand (H3 trensal), and (b) the 3D visualization of trensal bound to a trivalent lanthanide ion (oxygen, nitrogen, lanthanide, carbon, and hydrogen are depicted in red, blue, light blue, grey, and white, respectively). | ||
Trivalent lanthanide ions, Ln(III), possess a unique electronic structure, which distinguishes them from other elements as their 4f valence orbitals are shielded by the filled 5s and 5p orbitals, isolating the 4f electrons from external chemical influences.5,13 The trensal ligand coordinates Ln(III) in the trianionic heptadentate (N4O3) form.14–18 Due to the C3 point symmetry, the coordination sphere of the resulting complex, [Ln(trensal)] (see Fig. 1b), can be divided into three distinct families: the Ln–O, Ln–N, and Ln–N′ fragments.15 N′ denotes the apical nitrogen atom (within the C3 axis), which is non-equivalent to the other three nitrogen atoms and is expected to be unbound in complexes of first-row transition metals due to a decrease in denticity to six.16,18
The formation of [Ln(trensal)] is experimentally confirmed by a strong infrared (IR) band attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch.14,15,19–23 Consequently, the sensitivity of IR spectroscopy to lanthanide coordination can be linked to specific normal vibrational modes within the fingerprint region of the vibrational spectra. However, the high delocalization of normal vibrational modes makes it difficult to pinpoint specific bond stretching contributions, as extensively emphasized in the literature by some of us in the context of various chemical systems.8,24–30 In other words, assigning the strong IR characteristic band typical of [Ln(trensal)] solely to the imine CN stretching vibration, without quantitative spectral analysis, is incomplete due to the delocalization conundrum.
N stretch.14,15,19–23 Consequently, the sensitivity of IR spectroscopy to lanthanide coordination can be linked to specific normal vibrational modes within the fingerprint region of the vibrational spectra. However, the high delocalization of normal vibrational modes makes it difficult to pinpoint specific bond stretching contributions, as extensively emphasized in the literature by some of us in the context of various chemical systems.8,24–30 In other words, assigning the strong IR characteristic band typical of [Ln(trensal)] solely to the imine CN stretching vibration, without quantitative spectral analysis, is incomplete due to the delocalization conundrum.
Delocalization also hinders the study of Badger-type relationships,31,32 as the correlation with bond lengths requires breaking down polyatomic molecules into quasi-diatomic molecules31 (or recently referred to as “diatomics within molecules”33) to obtain local mode properties such as frequencies or force constants. In this respect, relationships between bond properties can enhance our understanding of chemical bonding, especially when different quantum chemistry methods can be employed for analysis. Our combined protocol, which integrates LVM with BOM, exemplifies this approach. Often, one bond property is easily obtained experimentally, while another is more challenging to assess. For example, measuring or calculating the vibrational spectrum of a compound is typically more practical than performing a detailed structural analysis.31 In cases like that of Pm, where experimental data on chemical bonding is unavailable,12 vibrational spectroscopy is expected to become an invaluable tool for gaining insights into bond properties.
Furthermore, recent interest in designing luminescent lanthanide complexes has focused on identifying the normal vibrational modes, calculated at the density functional theory34 (DFT) level in the gas phase, most strongly coupled to the electronic states, which for [Yb(trensal)] are mainly within the frequency range of 0–500 cm−1 and simultaneously distort the first coordination sphere while breaking the C3 symmetry of the molecule.35 Moreover, the design should take into consideration the character of the vibrational modes that lead to non-radiative deactivation of excited states.13 To take a case in point, the C–H vibrations exhibiting significant anharmonicity in [Eu(trensal)], for instance, can give rise to the undesired dissipation pathway of the excitation energy.36 As a result, the normal mode decomposition of the C–H related region of the IR spectrum is germane to our approach as well.
So far, a theoretical framework that aids in the elucidation of the vibrational spectra of [Ln(trensal)] complexes along the lanthanide series has been lacking and is of paramount interest for gaining a better understanding of the lanthanide chemistry involved. This clearly calls for new ideas to facilitate the quantitative spectral analysis via normal mode decomposition. In this work, we have addressed this need by utilizing the characterization of normal mode (CNM) procedure,25,37,38 an integral part of the LVM theory, as a key tool.
It is worth mentioning as a motivation that the recent application of local mode analysis in lanthanide spectroscopy has provided new insights into the degree of normal mode delocalization and the bonding behavior within the coordination sphere of lanthanide complexes.8 To the best of our knowledge, the present study presents for the first time a complete characterization of the [Ln(trensal)] complexes along the La–Lu series by the combined computational approach herein proposed, aiming to give:
(i) A quantitative interpretation via CNM spanning the gas-phase DFT-computed IR spectra to elucidate the normal vibrational modes associated with peaks linked to distortion within the first coordination sphere with disruption of the C3 symmetry, complex formation, and C–H vibrations.
(ii) A comparative analysis involving the local mode force constant, the ligand effective polarizability from BOM, bond distances, and ionic radii to gain a better understanding of chemical bonding within the coordination sphere.
(iii) A pairwise comparison between [Er(trensal)] and [Fe(trensal)] to probe the change in bonding of the trensal ligand by the apical nitrogen using the local mode force constant.
Methodology
Normal mode analysis
According to the GF method,39,40 the Lagrangian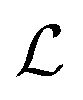 for a polyatomic molecule composed of N atoms undergoing harmonic vibrations can be conveniently expressed using the following matrix formulation:
for a polyatomic molecule composed of N atoms undergoing harmonic vibrations can be conveniently expressed using the following matrix formulation: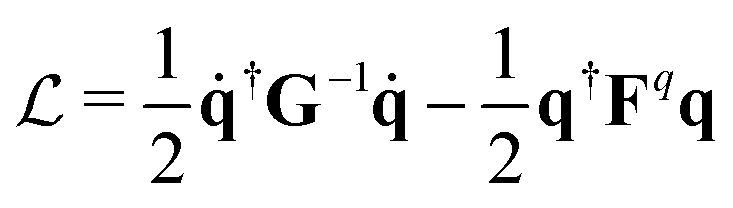 | (1) |
 | (2) |
![[D with combining tilde]](https://www.rsc.org/images/entities/b_char_0044_0303.gif) . The internal coordinate vector q can be defined as the result of a linear transformation applied to the normal coordinate vector Q, expressed as:
. The internal coordinate vector q can be defined as the result of a linear transformation applied to the normal coordinate vector Q, expressed as:| q = Q | (3) |
![[d with combining tilde]](https://www.rsc.org/images/entities/char_0064_0303.gif) μ (μ = 1,…,Nvib) in internal coordinates constitute the columns of the matrix .
μ (μ = 1,…,Nvib) in internal coordinates constitute the columns of the matrix .
The vibrational secular equation in internal coordinates is expressed as:
| Fq = G−1Λ | (4) |
| †G−1 = I | (5) |
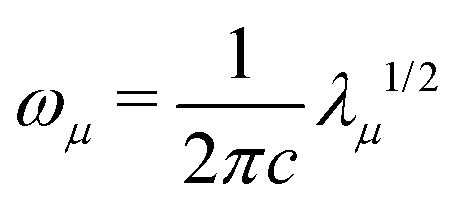 | (6) |
The Wilson B matrix, represented as a Nvib × 3N rectangular matrix, incorporates the first derivatives of the internal coordinates qn (n = 1,…,Nvib) with respect to the Cartesian coordinates xi (i = 1,…,3N) as its elements Bni. This matrix serves to establish the connection between internal and Cartesian coordinates:39
| q = Bx | (7) |
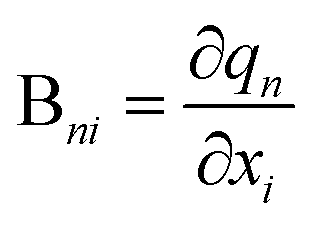 | (8) |
| C = M−1B†G−1 | (9) |
Renormalization of , via the diagonal matrix MR of reduced mass elements mRμ, in conformity with
| D = (MR)1/2 | (10) |
| D†G−1D = MR | (11) |
| D†FqD = K | (12) |
| Fq = C†FxC | (13) |
| L = CD | (14) |
Multiplying eqn (3) from the left by †G−1, taking into account eqn (5), yields:
| Q = †G−1q | (15) |
In other words, normal coordinates are linear combinations of internal coordinates. This results in normal vibrational modes typically spread out across the molecule, thereby restricting the utility of normal mode frequencies and force constants as measures of bond strength. To address this, local vibrational modes and their corresponding frequencies and force constants are necessary. The delocalization also renders it impractical to track subtle structural changes and variations in the molecule's surroundings without a method for normal mode decomposition. In this regard, the CNM procedure emerges as a novel and powerful tool as an integral part of the LVM theory. It offers a quantitative approach to decomposing normal vibrational modes into local mode contributions. As a result, it facilitates the quantitative analysis of vibrational spectra, offering insights into the molecular fragments involved in a novel and innovative manner.
Local mode analysis
The local mode vectors an associated with the internal coordinates qn can be calculated utilizing the normal vibrational modes dn and the diagonal force constant matrix K, as demonstrated by Konkoli and Cremer in their seminal paper on the local vibrational mode theory:7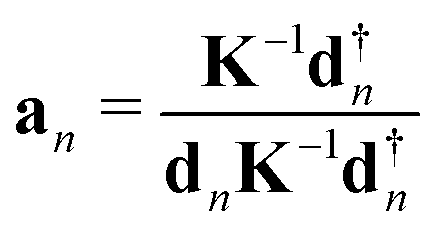 | (16) |
| axn = Lan | (17) |
The local mode force constant kan can be calculated as:
| kan = a†nKan = (dnK−1d†n)−1 | (18) |
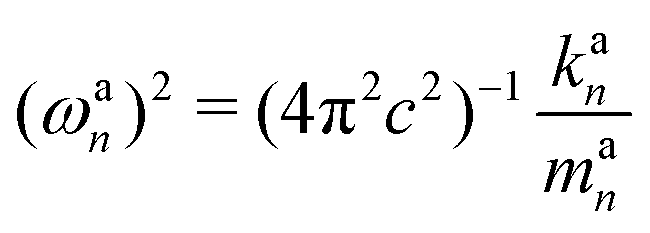 | (19) |
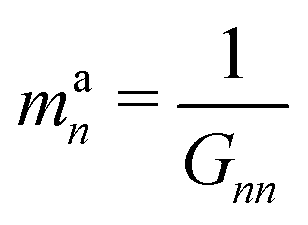 | (20) |
Each normal vibrational mode, including those of significant interest corresponding to peaks in the vibrational spectrum, can be decomposed into the percentage contributions of non-redundant local vibrational modes from a complete set using the characterization of normal mode procedure.26,30 This is based on an adiabatic connection scheme (ACS).42 The degree of overlap, represented by Snμ,25,37,38 is defined as:
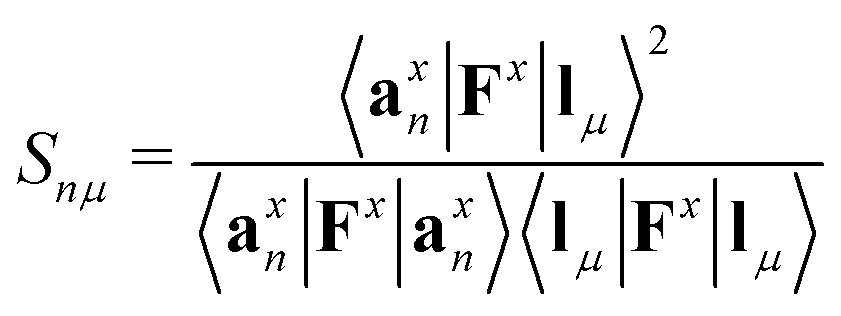 | (21) |
 | (22) |
A CNM plot illustrates normal mode decompositions, with the local mode contributions C%nμ shown as distinct components forming the bars that represent the normal modes in a diagram. The normal frequencies corresponding to the decomposed normal modes are displayed on the plot's x-axis. For further details on the LVM theory, the reader is referred to two comprehensive feature articles.25,38
The following set of formulas defines the number of bond (Nb), angle (Na), and dihedral (Nd) parameters necessary to form a topologically complete and non-redundant set of local vibrational modes for CNM30 and implemented in LModeAGen:26
| Nb = N + f − 2 | (23) |
| Na = N − f − 1 | (24) |
| Nd = N − 3 | (25) |
From the perspective of graph theory, f represents the number of faces of the molecular graph, whether it is of tree, cycle, or polyhedral type.30
Given both the significant delocalization observed in the normal vibrational modes associated with the infrared-active peaks and the dimension of the vibrational space of the Ln-containing complexes along the La–Lu series, it is recommended to perform the CNM analysis using local mode families. For the C–H family, this involves merging all the percentage contributions of C–H local modes, 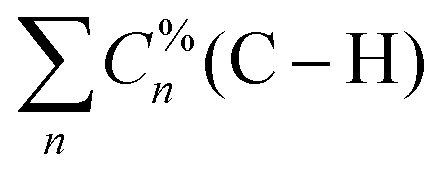 , to form the C–H entry in Fig. 6. A similar rationale applies to other local mode families. For visualization purposes, Σσ comprises the summation of all σ local mode contributions below the threshold of 5%.
, to form the C–H entry in Fig. 6. A similar rationale applies to other local mode families. For visualization purposes, Σσ comprises the summation of all σ local mode contributions below the threshold of 5%.
Ligand effective polarizability
The bond overlap model (BOM)9,10 explains the dynamic coupling (DC) mechanism43–45via overlap and ligand effective polarizabilities (α = α′ + αOP), with αOP and α′ denoting bond overlap polarization and ligand polarizability contributions to DC, respectively.9,10 Improved effective ligand polarizabilities (α′) are obtained by decomposing the molecular polarizability for the entire complex into localized molecular orbitals (LMOs) contributions, which are relevant for understanding ligand interactions with Ln(III).8 It should be mentioned that BOM has recently been implemented for multiconfigurational wave functions.46 For up-to-date applications of BOM, readers are referred to the recent literature.47–53Computational details
The complexes formed with the trensal ligand14–17 binding to Ln(III) ions from lanthanum to lutetium were investigated. Very little has been found in the literature on Pm, which highlights the importance of this investigation encompassing the entire series. Equilibrium geometries, Hessian matrices, and associated normal vibrational modes, as well as finite field calculations, employed the B3LYP54,55 functional in combination with the 6-311++G** basis set56,57 to describe all ligand atoms as well as a pseudopotential scheme (MWBn, n varies with atomic number)58 and valence electrons basis set for Ln(III) ions utilizing Gaussian16.59 For [Fe(trensal)], the results were obtained at the UB3LYP/6-311++G** level. Multiwfn60 was employed for obtaining the localized molecular orbital (LMO), while ChemBOS8,47,61 decomposed the molecular polarizability into LMO contributions. The automatic generation of complete and non-redundant sets of local mode parameters was accomplished through the newly developed LModeAGen protocol,26,30 with local mode analysis being conducted using the standalone LModeA package.62 All the geometries were visualized using UCSF ChimeraX,63 and their Cartesian coordinates are provided in the ESI.† The values of trivalent ionic radii used for comparison were taken from the literature.64 Additionally, in the ESI,† Fig. S3 corroborates the soundness of our model on a comparative basis with the PBE065,66 functional.Results
CNM analysis of selected normal modes
This study aims at assessing the topologically complete and non-redundant set of local vibrational modes30 for the CNM analysis of the gas-phase DFT-computed vibrational spectra of [Ln(trensal)] complexes at the B3LYP/MWBn(Ln)/6-311++G** level, as implemented in LModeAGen.26 The molecular graph associated with [Ln(trensal)], featuring f = 10, exhibits Nb = 70, Na = 51, and Nd = 59, with Nvib = Nb + Na + Nd = 180. Fig. 2 shows three out of the ten faces that constitute the associated molecular graph, with the infinite face accounting for the tenth one.30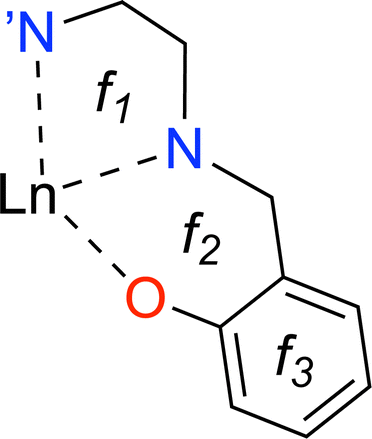 | ||
| Fig. 2 A 2D representation of one-third of [Ln(trensal)], depicting three out of the ten faces that compose the associated molecular graph. | ||
Fig. 3 shows the gas-phase DFT-computed IR spectrum of [Eu(trensal)], which, albeit slightly blue-shifted, is in considerable agreement with the experimental data extracted from the literature.36 On average, the IR spectra obtained along the La–Lu series exhibit similar patterns, justifying the selection of a single spectrum for discussion. The peaks identified in the spectrum due to distortion within the first coordination sphere with disruption of the C3 symmetry (p1), complex formation (p2), and C–H vibrations (p3) are illustrated in Fig. 3.
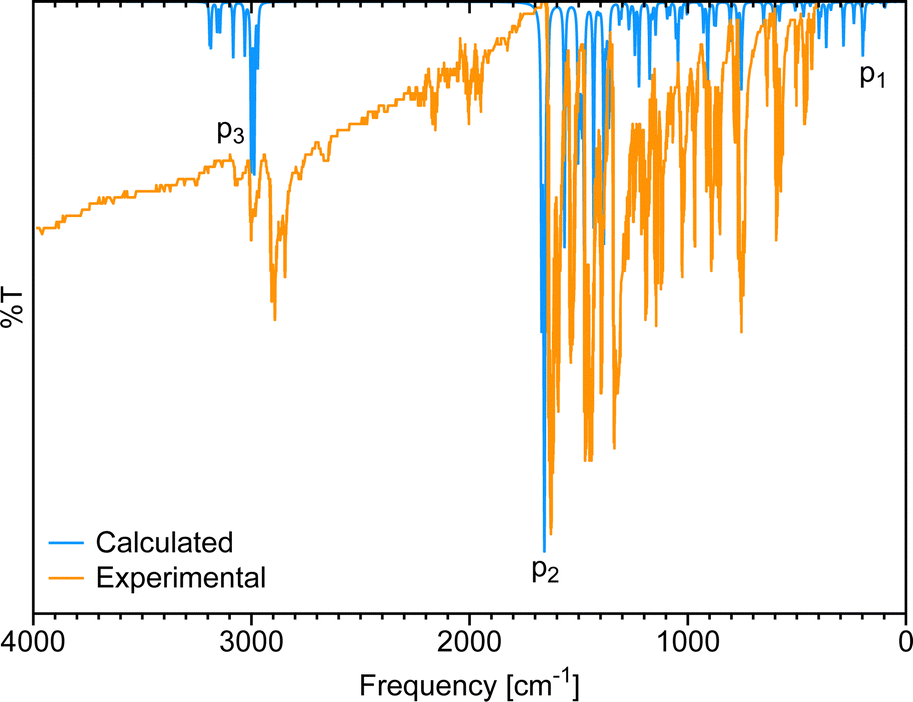 | ||
| Fig. 3 Comparison of the calculated IR spectrum at the B3LYP/MWB52/6-311++G** level of theory with the experimental ATR-IR spectrum36 of [Eu(trensal)]. | ||
Distortion within the first coordination sphere (p1)
As mentioned in the literature, characterizing gas-phase DFT-computed normal vibrational modes strongly coupled to electronic states can be instrumental in designing luminescent lanthanide complexes.35 For instance, in [Yb(trensal)], these modes fall predominantly within 0–500 cm−1, distorting the first coordination sphere and breaking the C3 point symmetry.35 We selected the peak of highest intensity in the frequency range of 0–500 cm−1 (p1) to perform the CNM analysis on the corresponding normal modes along the La–Lu series.Fig. 4 presents the CNM analysis for the degenerate normal vibrational modes associated with p1, spanning the frequency range of 196–200 cm−1 along the lanthanide series. Consistent with the literature, Fig. 4 agrees with the previous report that these normal modes, which distort the first coordination sphere in [Yb(trensal)], are of E-symmetry.35 Most striking is the substantial difference in the patterns of these normal modes of E-symmetry, as shown in Fig. 4.
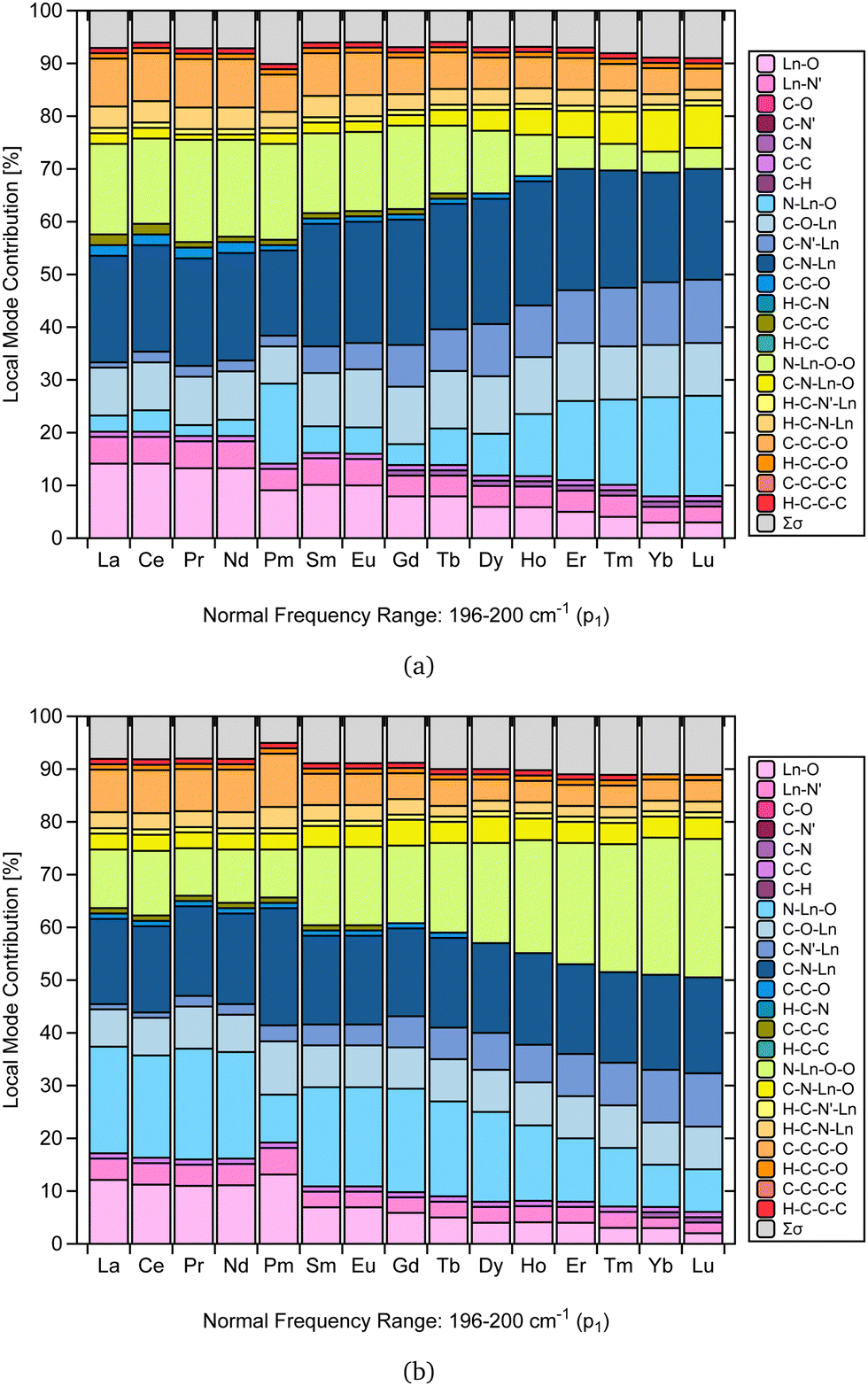 | ||
| Fig. 4 CNM analysis of the degenerate E-symmetry normal vibrational modes (a) and (b) associated with p1 along the La–Lu series at the B3LYP/MWBn(Ln)/6-311++G** level of theory. | ||
By comparing Fig. 4a and b, we can infer that the breaking of the C3 symmetry occurs through motions with varying degrees of distortion in the first coordination sphere along the lanthanide series. Also, the local mode contributions associated with the molecular fragments N–Ln–O–O, N–Ln–O, C–O–Ln, C–N'–Ln, and C–N–Ln exhibit opposite trends. In Fig. 4a, the percentage values of N–Ln–O–O show an overall decrease, while those of N–Ln–O show an increase. However, this pattern is reversed in Fig. 4b. For C–O–Ln, C–N′–Ln, and C–N–Ln, the ascending trend observed in Fig. 4a shifts to a descending trend in Fig. 4b.
Importantly, the CNM analysis was successful in identifying and quantifying the contributions of the molecular fragments involved in the motion described by the normal vibrational modes that distort the first coordination sphere. Fig. 4 is a particularly remarkable outcome in this regard, raising the possibility that CNM can quantitatively aid in the guided design suggested in the literature.35 This finding is reassuring, as it opens up future research endeavors focused on different ligands and substituent effects.
Vibrations related to the complex formation (p2)
Another relevant aspect of this investigation is the critical examination of the assignment of the strong IR characteristic band typical of Ln(trensal), which is an experimental evidence for the formation of the complex.14,15,19–23 In this context, much of the assignments up to now has been qualitative in nature. The main challenge faced by qualitative assignments is the lack of a quantitative spectral analysis that assesses the composition of the characteristic normal vibrational modes.Fig. 5 makes a considerable contribution to the characterization of [Ln(trensal)] by demonstrating, based on a strong theoretical framework, that the fingerprint normal vibrational modes indicative of [Ln(trensal)] formation in the frequency range of 1656–1658 cm−1 along the lanthanide series are composed not only of C–N contributions but also of C–C and H–C–N ones. In future investigations, we recommend using CNM to interpret vibrational spectra for the assignment of peaks that indicate the formation of Ln-containing complexes.
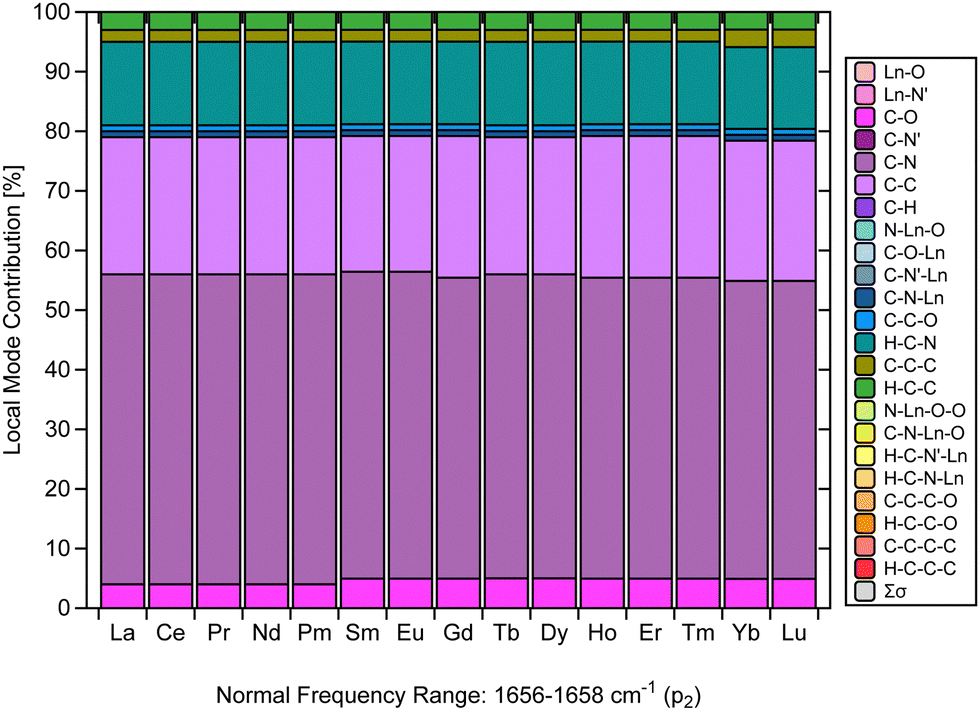 | ||
| Fig. 5 CNM analysis of the normal vibrational mode associated with p2 along the La–Lu series at the B3LYP/MWBn(Ln)/6-311++G** level of theory. | ||
C–H vibrations (p3)
Another finding that stands out from the results is the CNM analysis depicted in Fig. 6, which confirms that the C–H stretching vibrations in [Ln(trensal)] are nicely decoupled from other stretching, bending, or torsional vibrations.31 The normal vibrational modes within 2984–2992 cm−1 along the La–Lu series are fully localized with respect to the C–H family. It is crucial to consider vibrational modes leading to non-radiative deactivation of excited states, as seen in [Eu(trensal)], where C–H high energy oscillators lead to undesired energy dissipation and luminescence quenching.36 Because local vibrational modes are related to the local modes of overtone spectroscopy,31,67 further studies are needed and highly recommended to test the monitoring of C–H stretching vibrations in [Ln(trensal)] (and other complexes as well) via local mode analysis. This approach can conceivably be hypothesized to be relevant to strategies, such as deuteration or fluorination, for reducing undesired relaxation pathways aimed at the design of highly luminescent lanthanide complexes.13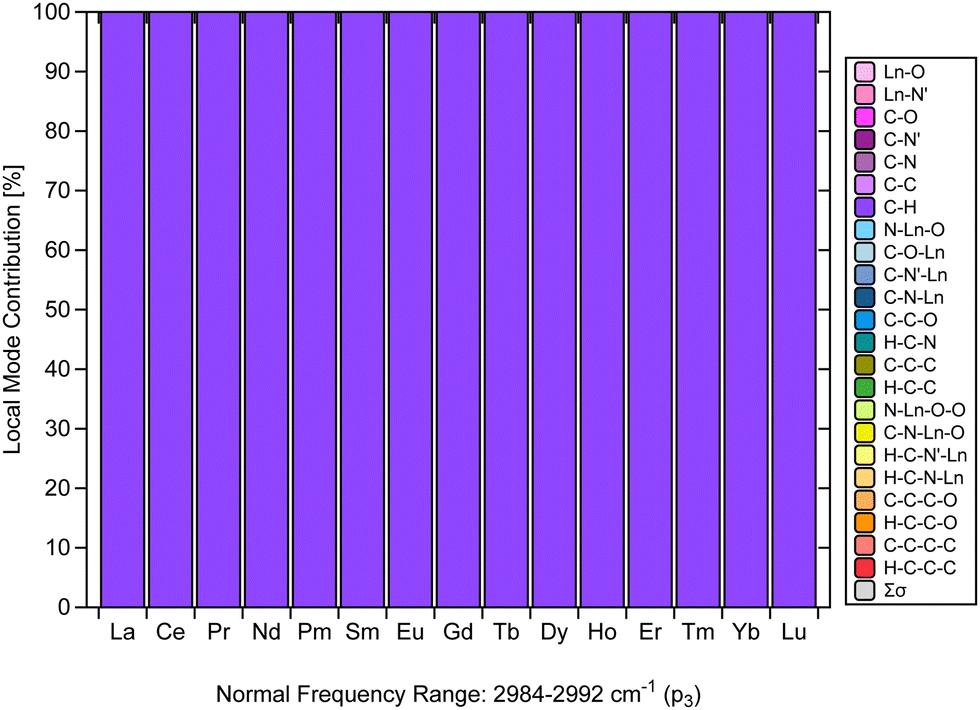 | ||
| Fig. 6 CNM analysis of the normal vibrational mode associated with p3 along the La–Lu series at the B3LYP/MWBn(Ln)/6-311++G** level of theory. | ||
Relationships between bond properties
The C3 point symmetry results in the equivalence between the oxygen atoms and the nitrogen atoms, except for the apical nitrogen within the coordination sphere of [Ln(trensal)], explaining the three families of lanthanide-ligand bonds depicted in Fig. 7.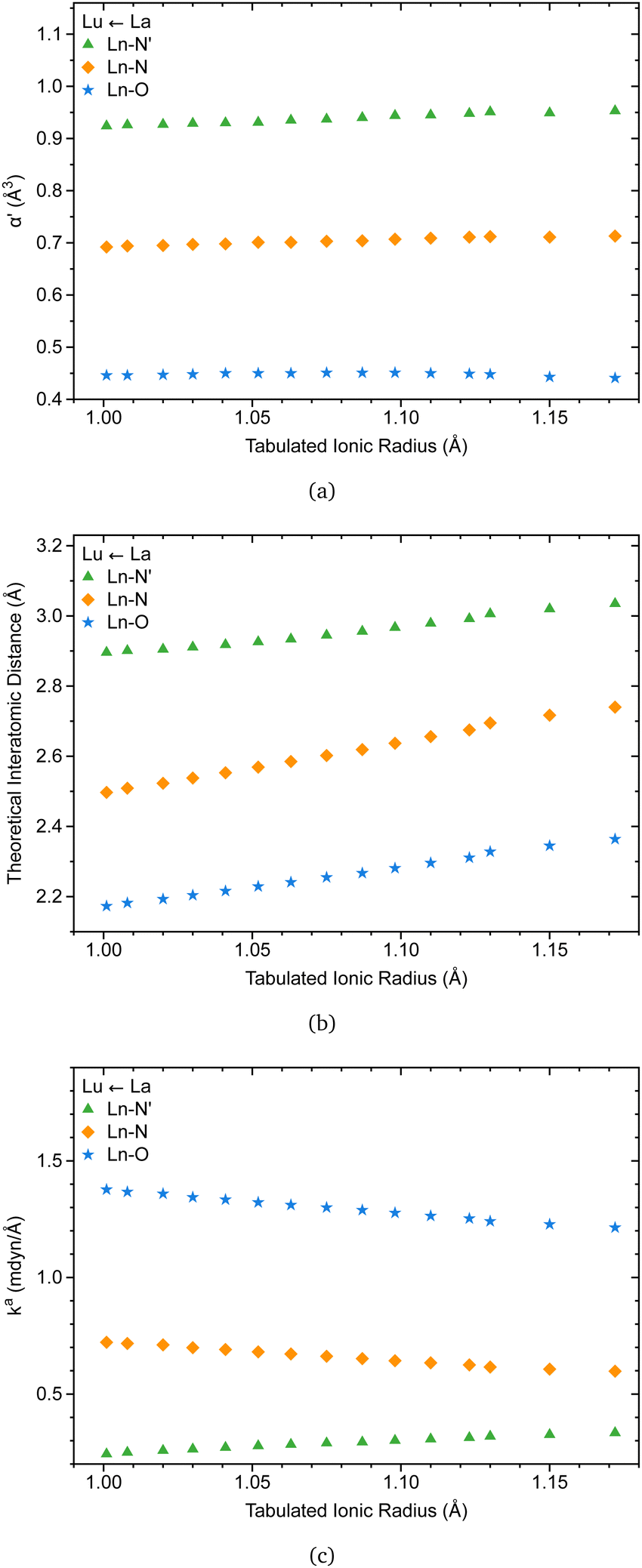 | ||
| Fig. 7 Ln(III)–L effective polarizability (a), interatomic distance (b), and local force constant (c) vs. Ln(III) ionic radius along the lanthanide series (Lu ← La). The results were obtained at the B3LYP/MWBn(Ln)/6-311++G** level of theory. | ||
Recently, there has been renewed interest in Badger-type relationships, with implications for pairs of atoms in the periodic table, including lanthanides.33 In this context, the knowledge from the literature that the coordination number of [Ln(trensal)] remains consistent throughout the lanthanide series is particularly relevant, as it enables relationships between ionic radii and interatomic distances within the coordination sphere.15 It may be the case therefore that Fig. 7a supports this idea, as the ligand polarizabilities remain practically unchanged throughout the series, reflecting the consistency of polarization of the chemical environment around the Ln(III) ions.
Besides, Fig. 7b is in accord with a previous study indicating that interatomic distances within the coordination sphere decrease as the ionic radii decrease along the series.15 However, caution must be applied, as interatomic distances alone cannot provide a reliable description of bond strength. This is where the local force constant plays a crucial role. The relationship between the local force constant and ionic radius in Fig. 7c suggests that the intrinsic strength of Ln–O and Ln–N bonds follows an inverse correlation with the ionic radius. Closer inspection of Fig. 7c raises the possibility that Ln–N′ bonds follow the opposite trend, prompting an investigation into the relationship between the local force constant and interatomic distance.
Fig. 8a and b indicate that Ln–O and Ln–N bonds adhere to the generalized Badger's rule along the lanthanide series; namely, the shorter the interatomic distance, the stronger the interaction. What stands out is Fig. 8c, where Ln–N′ follows the opposite trend. In accordance with the present results, previous studies have demonstrated that a shorter bond is not necessarily a stronger bond.29,68–74 This intriguing finding may be attributed to the specific nature of the bond involving the apical nitrogen.
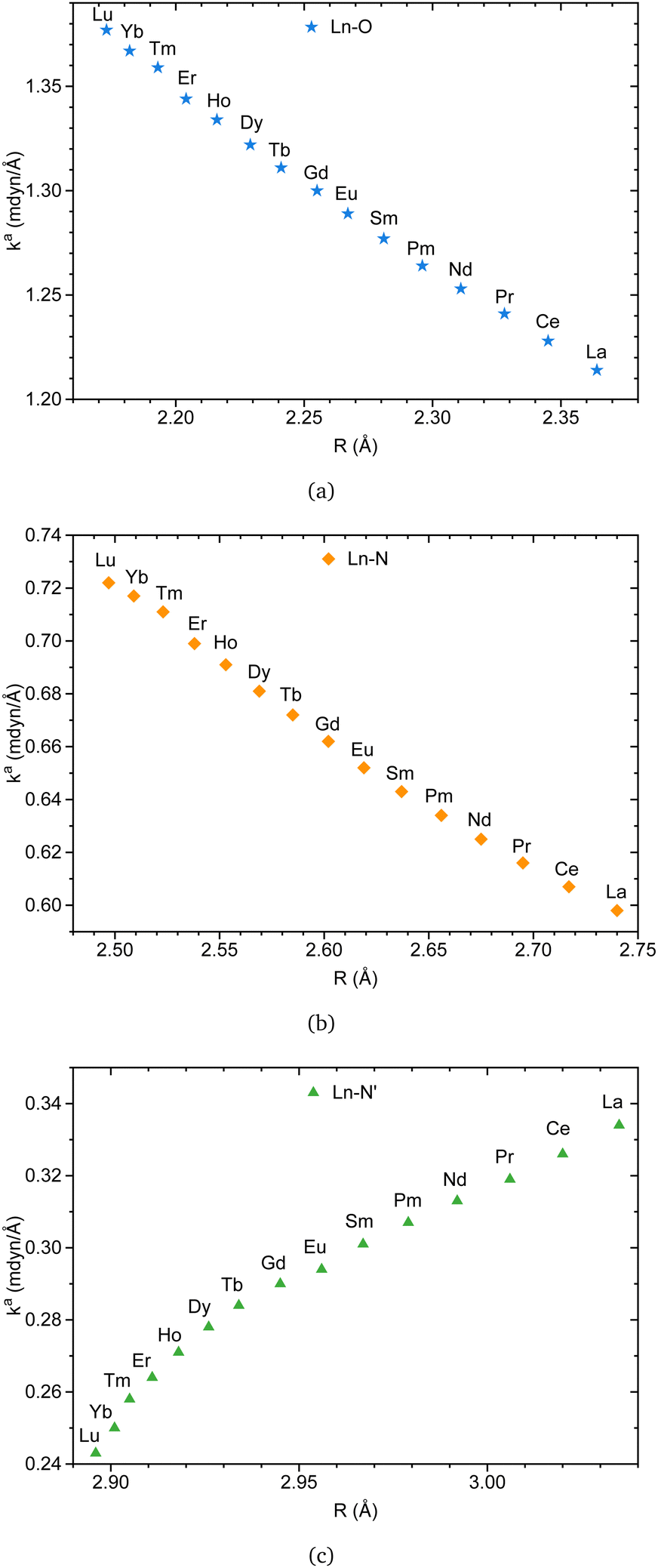 | ||
| Fig. 8 Ln(III)–L local force constant vs. interatomic distance for Ln–O (a), Ln–N (b), and Ln–N′ (c) bonds. The results were obtained at the B3LYP/MWBn(Ln)/6-311++G** level of theory. | ||
In Fig. S1 in the ESI,† experimental interatomic distances for the structural parameters within the coordination sphere of [Ln(trensal)], taken from the literature,18 are correlated with the corresponding B3LYP/MWBn(Ln)/6-311++G** values throughout the series. Because experimental data on [La(trensal)] and [Pm(trensal)] are not reported in the literature from which we took the structural parameters for comparison,18 they are omitted from Fig. S1 (ESI†). There is a linear relationship between the experimental and theoretical quantities for the Ln–O, Ln–N, and Ln–N′ bonds (correlation coefficient, r2 > 0.9), which reassures that our calculated gas-phase structures are consistent with crystallographic structural characterization.16–18 Fig. S2 (ESI†) offers insights into [Gd(trensal)], demonstrating that our calculations produced interatomic distances more closely related to the atomic radii.64
To date, research has tended to focus on the Ln–N′ interatomic distance rather than the intrinsic strength to assess the change in denticity for the complexation of trensal to the smaller trivalent transition-metal ions, leading to a distorted octahedron.16,18 In light of local vibrational mode theory, the local force constant can contribute to a better understanding of the bonding involving the apical nitrogen. For the sake of computational convenience, [Fe(trensal)] has been selected to probe the hexadentate coordination expected for first-row transition metals. What can be clearly seen in Table 1 is the manifestation of the three-fold symmetry relating the equivalent nitrogen and oxygen atoms in [Er(trensal)], with a marked difference in both interatomic distance and local force constant for the Er–N′ bond.
| [M(trensal)] | ||||
|---|---|---|---|---|
| M = Er(III)a | M = Fe(III)b | |||
| M–L | R (Å) | k a (mdyn Å−1) | R (Å) | k a (mdyn Å−1) |
| a At the B3LYP/MWB57(Er)/6-311++G** level of theory. b At the UB3LYP/6-311++G** level of theory with a spin multiplicity of a doublet. | ||||
| M1–O3 | 2.204 | 1.344 | 1.913 | 1.570 |
| M1–O5 | 2.204 | 1.344 | 1.921 | 1.725 |
| M1–O6 | 2.204 | 1.344 | 1.898 | 1.725 |
| M1–N′2 | 2.911 | 0.264 | 3.607 | 0.265 |
| M1–N4 | 2.538 | 0.699 | 2.034 | 1.321 |
| M1–N7 | 2.538 | 0.699 | 2.036 | 1.179 |
| M1–N19 | 2.538 | 0.699 | 2.006 | 1.422 |
The [Er(trensal)] complex was selected because it presents the local force constant value for the Er–N′ bond that closely matches that of the Fe–N′ bond in [Fe(trensal)]. What is striking in Table 1 is the dramatic difference in interatomic distance between Er–N′ and Fe–N′, despite their corresponding local force constant values being practically equivalent. Once again, this emphasizes that interatomic distance is not always a reliable descriptor of bond strength. These results therefore need to be interpreted with caution. Despite the similarity in intrinsic strength between Er–N′ and Fe–N′, the three equivalent Er–N bonds are more than twice as strong as Er–N′. For [Fe(trensal)], the weakest of the Fe–N bonds in the distorted octahedron is more than four times as strong as Fe–N′. This discrepancy in intrinsic strength could be attributed to the apical nitrogen not being bound in [Fe(trensal)], offering an alternative explanation for this change in denticity for the first time.
Conclusions
In this investigation, our primary objective was to utilize the characterization of normal mode (CNM) procedure from local vibrational mode theory to assess the normal vibrational modes that characterize the formation of the complex, the distortions in the first coordination sphere, and C–H vibrations of Ln(III) complexes, specifically [Ln(trensal)], along the lanthanide series. Our secondary aim was to combine the bond overlap model with local vibrational mode theory to analyze the chemical bonding within the coordination sphere of [Ln(trensal)]. The following key contributions should be emphasized:• We have characterized the E-symmetry normal vibrational modes that distort the first coordination sphere in [Ln(trensal)], revealing the varying degrees of such distortions along the La–Lu series for the first time. This discovery is promising as it opens avenues for researches on different ligands and substituent effects, suggesting that CNM can quantitatively support guided design as proposed in the literature.35
• We have critically examined the assignment of the strong infrared band of [Ln(trensal)], which has been used experimentally as evidence for the formation of the complex.14,15,19–23 We recommend using CNM to interpret vibrational spectra for assigning peaks indicative of Ln-containing complex formation in future studies.
• We have confirmed that the C–H stretching vibrations in [Ln(trensal)] are effectively decoupled from other stretching, bending, or torsional vibrations. This finding motivates further CNM studies aimed at minimizing undesired relaxation pathways.
• We have demonstrated that a shorter Ln–N′ bond does not indicate a stronger bond, which may be due to the unique features of the bond involving the apical nitrogen.
• For the first time, we have provided an alternative explanation based on the local force constant for the change in denticity of the trensal ligand when complexed to Fe(III).
Altogether, these findings enhance our understanding of the vibrational spectroscopy and chemical bonding of lanthanide complexes, offering valuable insights for future research.
Data availability
The data supporting this article have been included as part of the ESI.† Also, the codes utilized in this manuscript can be downloaded and accessed at http://www.gpqtc.chembos.website/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FCM thanks CAPES for the Master's scholarship as well as UFPE. MQ, RTMJr, and EK thank the National Science Foundation, Grant CHE 2102461, and SMU's Center for Research Computing for providing generous computational resources. MQ thanks SMU for the postdoctoral fellowship (08/2023–04/2024) and FACEPE for the postdoctoral fellowship BFP-0039-1.06/24 (09/2024–09/2025). RLL thanks CNPq for the PQ-fellowship (Proc. 308000/2022-6). RTMJr thanks the Brazilian National Council for Scientific and Technological Development - CNPq, grant numbers 406483/2023-0, and 310988/2023-3.References
- R. A. Rocha, K. Alexandrov and C. Scott, Microb. Biotechnol., 2024, 17, e14503 CrossRef PubMed.
- S.-L. Liu, H.-R. Fan, X. Liu, J. Meng, A. R. Butcher, L. Yann, K.-F. Yang and X.-C. Li, Ore Geol. Rev., 2023, 157, 105428 CrossRef.
- D. Gielen and M. Lyons ( 2022), Critical materials for the energy transition: Rare earth elements, International Renewable Energy Agency, Abu Dhabi Search PubMed.
- J.-C. G. Bünzli, Coord. Chem. Rev., 2015, 293–294, 19–47 CrossRef.
- S. Cotton, Lanthanide and Actinide Chemistry, John Wiley & Sons, 2006 Search PubMed.
- T. Vitova, P. Roesky and S. Dehnen, Commun. Chem., 2022, 5, 12 CrossRef CAS PubMed.
- Z. Konkoli and D. Cremer, Int. J. Quantum Chem., 1998, 67, 1–9 CrossRef CAS.
- R. T. Moura Jr., M. Quintano, C. V. Santos-Jr, V. A. Albuquerque, E. C. Aguiar, E. Kraka and A. N. Carneiro Neto, Opt. Mater., 2022, 16, 100216-1–100216-15 Search PubMed.
- A. N. Carneiro Neto, R. T. Moura Jr., E. C. Aguiar, C. V. Santos and M. A. F. L. B. de Medeiros, J. Lumin., 2018, 201, 451–459 CrossRef CAS.
- R. T. Moura Jr., A. N. Carneiro Neto, R. L. Longo and O. L. Malta, J. Lumin., 2016, 170, 420–430 CrossRef.
- N. Gündüz, T. Gündüz, M. Hursthouse, H. G. Parkes, L. S. Shaw (née Gözen), R. A. Shaw and M. Tüzün, J. Chem. Soc. Perkin Trans. II, 1985, 899–902 RSC.
- D. M. Driscoll, F. D. White, S. Pramanik, J. D. Einkauf, B. Ravel, D. Bykov, S. Roy, R. T. Mayes, L. H. Delmau, S. K. Cary, T. Dyke, A. Miller, M. Silveira, S. M. VanCleve, S. M. Davern, S. Jansone-Popova, I. Popovs and A. S. Ivanov, Nature, 2024, 629, 819–823 CrossRef CAS.
- E. Kreidt, C. Kruck and M. Seitz, Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 2018, vol. 53, pp. 35–79 Search PubMed.
- M. Kanesato, T. Yokoyama, O. Itabashi, T. M. Suzuki and M. Shiro, Bull. Chem. Soc. Jpn., 1996, 69, 1297–1302 CrossRef CAS.
- M. Kanesato and T. Yokoyama, Chem. Lett., 1999, 137–138 CrossRef CAS.
- P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Aust. J. Chem., 2000, 53, 229–231 CrossRef CAS.
- B. M. Flanagan, P. V. Bernhardt, E. R. Krausz, S. R. Lüthi and M. J. Riley, Inorg. Chem., 2001, 40, 5401–5407 CrossRef CAS PubMed.
- B. M. Flanagan, P. V. Bernhardt, E. R. Krausz, S. R. Lüthi and M. J. Riley, Inorg. Chem., 2002, 20, 5024–5033 CrossRef PubMed.
- S. Salehzadeh, S. M. Nouri, H. Keypour and M. Bagherzadeh, Polyhedron, 2005, 24, 1478–1486 CrossRef CAS.
- C. D. Buch, D. Mitcov and S. Piligkos, Dalton Trans., 2020, 49, 13557–13565 RSC.
- C. D. Buch, S. H. Hansen, C. M. Tram, D. Mitcov and S. Piligkos, Inorg. Chem., 2020, 59, 16328–16340 CrossRef CAS PubMed.
- C. D. Buch, S. H. Hansen, D. Mitcov, C. M. Tram, G. S. Nichol, E. K. Brechin and S. Piligkos, Chem. Sci., 2021, 12, 6983–6991 RSC.
- Y. Zhou, C. D. Buch, S. H. Hansen and S. Piligkos, Dalton Trans., 2023, 52, 8792–8799 RSC.
- M. Quintano and E. Kraka, Chem. Phys. Lett., 2022, 803, 139746-1–139746-7 CrossRef.
- E. Kraka, M. Quintano, H. W. L. Force, J. J. Antonio and M. Freindorf, J. Phys. Chem. A, 2022, 126, 8781–8900 CrossRef CAS.
- R. T. Moura, Jr., M. Quintano, J. J. Antonio, M. Freindorf and E. Kraka, J. Phys. Chem. A, 2022, 126, 9313–9331 CrossRef.
- M. Quintano, A. A. A. Delgado, R. T. Moura Jr, M. Freindorf and E. Kraka, Electron Struct., 2022, 4, 044005-1–044005-17 Search PubMed.
- M. Quintano, R. T. Moura Jr. and E. Kraka, Chem. Phys. Lett., 2023, 826, 140654-1–140654-7 CrossRef.
- M. Quintano, R. T. Moura Jr. and E. Kraka, J. Mol. Model., 2024, 30, 102 CrossRef CAS PubMed.
- M. Quintano, R. T. Moura Jr. and E. Kraka, Chem. Phys. Lett., 2024, 849, 141416 CrossRef CAS.
- E. Kraka, J. A. Larsson and D. Cremer, Computational Spectroscopy, Wiley, New York, 2010, pp. 105–149 Search PubMed.
- A. Madushanka, R. T. Moura, Jr., N. Verma and E. Kraka, Int. J. Mol. Sci., 2023, 24, 6311 CrossRef CAS.
- D. L. Crittenden, AIP Adv., 2023, 13, 115323 CrossRef CAS.
- R. G. Parr and Y. Weitao, Density-Functional Theory of Atoms and Molecules, Oxford University Press, 1994 Search PubMed.
- J. G. C. Kragskow, J. Marbey, C. D. Buch, J. Nehrkorn, M. Ozerov, S. Piligkos, S. Hill and N. F. Chilton, Nat. Commun., 2022, 13, 825 CrossRef CAS PubMed.
- S. K. Kuppusamy, E. Vasilenko, W. Li, J. Hessenauer, C. Ioannou, O. Fuhr, D. Hunger and M. Ruben, J. Phys. Chem. C, 2023, 127, 10670–10679 CrossRef CAS.
- Z. Konkoli and D. Cremer, Int. J. Quantum Chem., 1998, 67, 29–40 CrossRef CAS.
- E. Kraka, W. Zou and Y. Tao, WIREs: Comput. Mol. Sci., 2020, 10, 1480 Search PubMed.
- E. B. Wilson, J. C. Decius and P. C. Cross, Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra, McGraw-Hill, New York, 1955 Search PubMed.
- E. B. Wilson, J. Chem. Phys., 1941, 9, 76–84 CrossRef CAS.
- W. Zou and D. Cremer, Chem. Eur. J., 2016, 22, 4087–4097 CrossRef CAS.
- W. Zou, R. Kalescky, E. Kraka and D. Cremer, J. Chem. Phys., 2012, 137, 084114 CrossRef.
- C. K. Jørgensen and B. R. Judd, Mol. Phys., 1964, 8, 281–290 CrossRef.
- S. F. Mason, R. D. Peacock and B. Stewart, Mol. Phys., 1975, 30, 1829–1841 CrossRef CAS.
- B. R. Judd, J. Chem. Phys., 1979, 70, 4830–4833 CrossRef CAS.
- C. Santos-Jr and R. Moura Jr., ChemRxiv, 2023, 1–21 Search PubMed.
- R. T. Moura Jr., A. N. C. Neto, O. L. Malta and R. L. Longo, J. Mol. Model., 2020, 26, 301 CrossRef PubMed.
- C. V. Santos-Jr, E. M. Lima and R. T. Moura Jr., Comput. Theor. Chem., 2021, 1206, 113457 CrossRef CAS.
- C. V. Santos-Jr, M. A. F. de Souza, E. Kraka and R. T. Moura Jr., Chem. Phys. Lett., 2022, 787, 139282 CrossRef CAS.
- C. V. Santos, S. A. Monteiro, A. S. C. Soares, I. C. A. Souto and R. T. Moura Jr., J. Phys. Chem. A, 2023, 127, 7997–8014 CrossRef.
- C. V. Santos-Jr, G. M. B. Da Silva, R. P. Dias, R. T. Moura Jr. and J. C. S. Da Silva, Adv. Theory Simul., 2024, 7(5), 2301148 CrossRef CAS.
- W. G. Barbosa, C. V. Santos-Jr, R. B. Andrade, J. R. Lucena Jr and R. T. Moura Jr, J. Mol. Model., 2024, 30, 139 CrossRef CAS.
- W. Thor, A. N. Carneiro Neto, R. T. Moura Jr., K.-L. Wong and P. A. Tanner, Coord. Chem. Rev., 2024, 517, 215927 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1989, 90, 1730–1734 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsujiet al., Gaussian 16 Revision C.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- C. V. Santos-Jr, E. M. Lima and R. T. Moura Jr., Comput. Theor. Chem., 2021, 1206, 113457 CrossRef CAS.
- W. Zou, R. Moura Jr., M. Quintano, F. Bodo, Y. Tao, M. Freindorf, M. Z. Makoś, N. Verma, D. Cremer and E. Kraka, LModeA2023, Computational and Theoretical Chemistry Group (CATCO), Southern Methodist University, Dallas, TX, USA, 2023 Search PubMed.
- E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, e4792 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., A, 1976, 32, 751–767 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- V. Vetere, C. Adamo and P. Maldivi, Chem. Phys. Lett., 2000, 325, 99–105 CrossRef CAS.
- D. Cremer and E. Kraka, Curr. Org. Chem., 2010, 14, 1524–1560 CrossRef CAS.
- E. Kraka, D. Setiawan and D. Cremer, J. Comput. Chem., 2015, 37, 130–142 CrossRef.
- D. Setiawan, E. Kraka and D. Cremer, J. Phys. Chem. A, 2015, 119, 9541–9556 CrossRef CAS.
- E. Kraka and D. Cremer, Rev. Proc. Quim., 2012, 39–42 Search PubMed.
- D. Cremer, A. Wu, J. A. Larsson and E. Kraka, J. Mol. Model., 2000, 6, 396–412 CrossRef CAS.
- M. Kaupp, B. Metz and H. Stoll, Angew. Chem., Int. Ed., 2000, 39, 4607–4609 CrossRef CAS.
- M. Kaupp and S. Riedel, Inorg. Chim. Acta, 2014, 357, 1865–1872 CrossRef.
- B. A. Lindquist and T. H. Dunning, Jr., J. Phys. Chem. Lett., 2013, 4, 3139–3143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03677h |
| This journal is © the Owner Societies 2025 |