
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mobility of thiolates on Au(111) surfaces†
Daniël R. Duijnstee ab,
Moniek Trompa,
Wesley R. Browne*b and
Aleksandar Staykov*c
ab,
Moniek Trompa,
Wesley R. Browne*b and
Aleksandar Staykov*c
aZernike Institute for Advanced Materials, University of Groningen, The Netherlands
bStratingh Institute for Chemistry, University of Groningen, The Netherlands. E-mail: w.r.browne@rug.nl
cInternational Institute for Carbon Neutral Energy Research, Kyushu University, Japan. E-mail: alex@i2cner.kyushu-u.ac.jp
First published on 12th February 2025
Abstract
Self-assembled monolayers (SAMs), especially those based on thiol containing compounds on gold, are of both practical and fundamental interest. Thiols and thiolates can bind to gold in several ways due to the presence of holes and edges on the surfaces. The variety of binding motifs is increased by the presence of adatoms, i.e. gold atoms present on the surface, that sit between the thiolate and the surface. Although these motifs bind strongly to gold surfaces, they are sufficiently mobile to allow for self-assembly of thiols, either by movement across the surface or by desorption/re-adsorption. The motifs have been investigated primarily in the context of high surface coverage, with some attention given to the mobility of these motifs. Here we focus on the binding in the low-coverage regime, i.e. the initial stage of SAM formation, using theoretical methods. We determine the relative stability of the motifs formed with methane thiolate in the low-coverage regime, and rationalize their relative mobilities. Methane thiolate is used to minimize contributions of intermolecular interactions. Competition between the rates of adsorption, movement, and formation of the motifs can influence the formation of SAMs. In this work we expand the understanding of the early stages of monolayer formation and conclude that the type of motif formed initially depends strongly on the availability of gold adatoms and defects (edges and holes) on the surface at the point of adsorption.
Introduction
Self-assembled monolayers (SAMs) of thiol-containing compounds on gold are of continued interest from a fundamental perspective,1,2 and in applications as diverse as (electro)chemical sensing3,4 and molecule-based electronic junctions.5 Specifically, the application of SAMs in molecular electronics to create new functions is an important milestone in the miniaturization of electronics and in introducing new electronic properties.6 Assembling molecular systems relies on self- or spontaneous assembly from vapor or liquid phase and, in particular, assembly of thiols on gold surfaces has attracted substantial attention. While there are examples of precision positioning at the atomic and molecular level,7 the stability of devices prepared from SAMs of organic compounds is limited by their intrinsically dynamic nature,5 which on the one hand allows the molecules to self-assemble but on the other hand makes it difficult to control their movement after placement at a specific point on the surface.8The bond between sulfur and a binding site on a gold surface is strong with respect to dissociation,9 manifested in the ease with which thiolate-based SAMs form on gold from highly dilute solutions. However, the strength of the bond does not impede significantly the diffusion of molecules over the surface itself,10 nor does it preclude them from reconstructing the surface with this movement: Molina et al. concluded, based on comparison of theoretical results with experimental data,11 that it is energetically favorable, for example, for a Au(111) surface with high methane thiol surface coverage to reconstruct to create adatom-containing thiolate motifs. While the mobility of atoms in metals is, in some cases, a useful property,12 the surface reconstruction caused by gold atom abstraction can be undesirable, for example, in the detailed design of molecular electronic devices and in mechanically controlled break junction (MCBJ) devices. Indeed, MCBJs are a prominent example of a device that is impacted by these effects. MCBJs consist of two metal wires, or a wire and a surface (stm break junction), separated by a few nanometers and are used to probe the conductance of molecules bridging these nanometer gaps. The many reports on such devices4,13–17 present diverse strategies to prepare and use them18 and the effects of the monolayer on the properties of these devices due to reorganization of the gold substrate and other influences are still poorly understood.
Molecular junctions are characterized mostly by conductance traces, a technique that relies on statistical analysis of histograms generated from large numbers of individual measurements,14,19 taking into account the conformational states that the compounds being studied can hold on the surface.20 The results these techniques generate are non-unique: measurements of different molecules or molecular configurations can result in the same conductance trace, meaning one cannot interpret the results of such a technique without additional information. Combining molecular junctions and information provided by spectroscopies or ab initio methods13,15,21,22 can help validate the conclusions reached from conductance measurements, through prediction of the current steps expected for the various motifs23 and/or by narrowing the range of arrangements/orientations of molecules consistent with the data.
Similarly, STM and AFM are employed to obtain detailed information on the atoms in top layers of SAMs that have formed on surfaces, which can be used to extract information regarding the nature of the underlying layers. Due to the limited time resolution and challenges in probing the atoms not exposed on the surface of the SAM, however, it is difficult to draw definitive conclusions with these techniques alone. First principle calculations can help by modeling the various structures of the underlying part of the monolayer forming compound bound directly to the surface, and thus allowing STM/AFM images to be predicted. These can then be correlated with experimental data to support or discount hypotheses.24
Of the various situations that molecules on a surface can be present in, based on earlier theoretical1,11,24–26 and experimental1,24,27–30 studies, three binding motifs are most likely to be dominant (Fig. 1). Furthermore, the surface structure plays an important role in determining relative energetics. For example the ‘staple’ motif, the most commonly investigated of the three, was shown to be lower in energy than the ‘free thiolate’ motif on the Au(111) surface, while the order is reversed on Au(100) and Au(110).31 The thermodynamic stability of these motifs has been compared in several studies, albeit each using different methods, or studying different situations and therefore drawing different conclusions.25,31–33
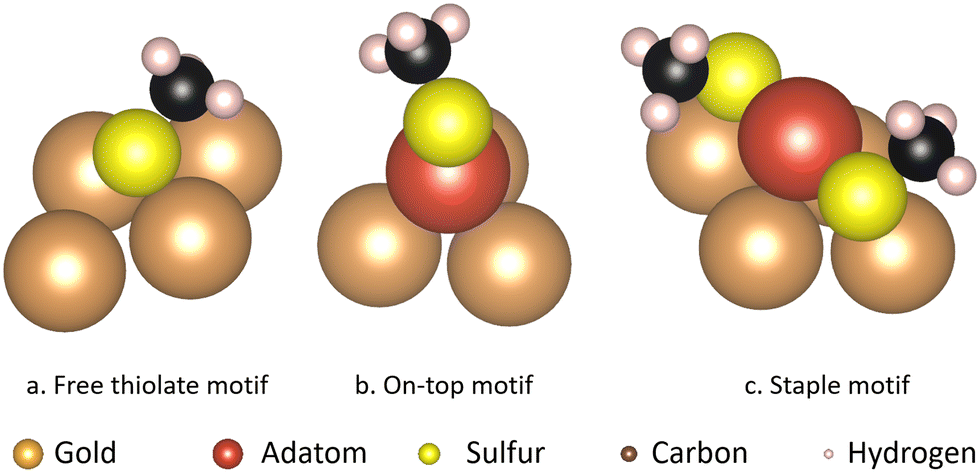 | ||
| Fig. 1 The configuration of atoms in the different motifs and the surface atoms they bind to. (a) Free thiolate motif (b) on-top motif (c) staple motif. The gold atoms are shown in brown, except for the gold adatom that is part of the thiolate motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
One should keep in mind that these theoretical studies cannot easily treat all details of the system at hand. Calculation of a single binding motif in a unit cell does not treat the effects of surface coverage26 and solvent effects,27 both of which have been shown to affect the geometries of molecules binding to a surface.32,33 The initial step in the formation of monolayers, i.e. adsorption, has been investigated both theoretically34 and experimentally.2 Increase and subsequent decrease of potential in potentiometric studies indicate binding of the thiol to the metal, followed by loss of the thiol proton in a two-step adsorption process. This conclusion is in agreement with theoretical studies34 which show that binding of the thiolate is favorable after the atoms in the surface layer of gold relax their positions in order to accommodate its binding.
Furthermore, on pristine Au(111) surfaces the atoms in the top layer are compressed, creating periodic faults known as the (22 × sqrt(3)) herringbone reconstruction.35,36 This reconstruction is of great importance in studies concerning surfaces devoid of any absorbed species, but is lifted upon absorption of strongly interacting species, especially thiolates.37–39
Whereas both the initial adsorption and the properties of the final, full coverage monolayer have been investigated extensively, studies of the behaviour of thiolates in the time between these two situations are scarce. A more complete description of the events between initial absorption, when isolated thiol(ate) species have the space to move around and reconstruct the surface below them,11 and the final crowded monolayer would enhance the utility the information provided by both approaches. As more thiolates attach to the surface, they form a monolayer packed to such an extent that it can influence the chemical properties of attached molecules.40 The motifs in which the molecules move over the surface and the rate of their diffusion have to be investigated in order to describe how quickly a final packing can be achieved in comparison with the time taken for formation of the motifs and surface reconstruction. The type of thiolate-gold motifs that are present in the final monolayer may depend on the rate of adsorption of the thiolate molecules to the surface if formation of a motif is kinetically unfavorable.
Experimentally, mobility has been observed through images in STM where atoms are partially displaced,24 and partially occupied sites in X-ray diffraction data41 indicate that the atoms in question moved during the measurement. These results demonstrate the mobility of thiolate molecules on the surface, but they do not show whether this mobility is due to diffusion or to desorption and subsequent re-adsorption to the surface. Theoretical investigations of the kinetics of diffusion and formation are scarce, with a few studies showing the mobility of the ‘on-top’ motif.26,42 De-en Jiang and coworkers took this a step further, among their other works on binding and mobility of thiolates on gold they compare the mobility of the ‘on-top’ and ‘staple’ motifs43 and the cis–trans conversion of the ‘staple’ motif.44 Although the energetics of the three motifs are reported, the full comparison of their mobilities focussed on in this study has not been reported to date.
In the present study, we focus on the steps between the initial binding of the first thiolate and the formation of highly packed monolayers: the low-coverage regime. The diffusion of bound alkyl thiolates, which can take place during formation, determines which kind of SAMs can form.11 If reconstruction does not occur only the ‘free thiolate’ motif can form; if it does, the other motifs are feasible as well. We work towards the diffusion coefficient for movement of various motifs over a gold surface by analysis of the barriers to their movement, and investigate the viability of existence of the various motifs by analysis of the barriers to formation. Together, these data allow us to estimate the relative rate of diffusion over the gold surface of these motifs.
Methods
The Au(111) surface was selected for the majority of the calculations due to it being the lowest energy, and therefore the most abundant, surface of gold.45 Similarly, for the ridge calculations an edge formed upon meeting of two Au(111) surfaces was used. The alkane thiol studied here is methanethiol, chosen for its small size both to optimize use of computational resources and, more importantly, for the relatively small contributions of interactions between the short alkane chains in different thiols. The latter point allows us to focus on the interaction between methanethiol and the gold surface, and makes the low-coverage calculations a better approximation also for the high-coverage regime.The ab initio calculations were carried out with plane wave density functional theory (DFT) calculations with periodic boundary conditions. These calculations were performed using the Vienna Ab initio Software Package (VASP),46–48 with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional49 applied at the generalized gradient approximation (GGA) level using projector augmented wave (PAW) pseudopotentials.50 All calculations were performed using the DFT-D3 dispersion correction method by Grimme,51 see ESI,† Section S4 for the effect of the dispersion correction on the outcome of the calculations. The configurations of the explicitly treated electrons are 5d106s1 for gold, 3s23p4 for sulfur, and 2s22p2 for carbon.
The energy cutoff for plane waves was set at 400 eV, electron energies were converged to 10−6 eV using Gaussian smearing (Sigma = 0.2) and the configuration of atoms was relaxed to forces below 3 × 10−2 eV Å−1 for each atom. The gold surfaces were constructed from the gold unit cell of which the cell volume, shape and atomic positions were relaxed with a Monkhorst–Pack k-points mesh of 9 × 9 × 9 before constructing the subsequent surfaces.
Slabs were constructed using four layers of gold in the (111) crystallographic direction to approximate the gold surface, see ESI,† Section S6 for the coordinates and cell parameters of all used geometries. The coordinates of the bottom two layers were fixed in order to enforce the effects of a bulk geometry on the top two layers, which were fully relaxed. The edge between the (111) surfaces was approximated by slabs constructed using four layers of gold in the (331) direction, with subsequent removal of part of the top layer of gold atoms in order to create a single edge. On the thickest part of the slab the bottom two layers of atoms were fixed in place, while on the thinnest part of the slab only the bottom layer of gold atoms was fixed in place. All other atoms were fully relaxed. All slab calculations were perfromed with a 3 × 3 × 1 Monkhorst–Pack k-points mesh. A series of higher k-point calculations were done for a sample configuration to confirm that the current choice of k-points is sufficient for the system. (see ESI,† Section S5).
The climbing-image nudged elastic band (CI-NEB) method implemented in VASP transition state theory tools, developed by Henkelman et al.,52 was applied to find the energy of the saddle point of the minimum energy paths of the unit steps of diffusion of the motifs over the surface. Six intermediate images were used between the initial and final states. The spring forces between images were set to 5 eV Å−1 and the configuration of atoms in the intermediate images were relaxed without changing the lattice parameters to forces below 3 × 10−2 eV Å−1 for each atom.
Binding energies of the various motifs to the gold surface were estimated using the following relation:
| Ebind = Eall − (Emotif + Esurf) | (1) |
| pdiff = pall − (pmotif + psurf) | (2) |
COHP was calculated using the LOBSTER software version 5.0.0,53–55 utilizing the wave functions generated from the abovementioned VASP periodic DFT calculations. The LOBSTER calculations were performed with the valence orbitals from the pbeVaspFit201554 set as basis functions for all atom types, and an energy range of +22 to −18 eV around the Fermi level was chosen as all important features were captured within that range.
All single point calculations performed in VASP for the purpose of generating wave functions for LOBSTER were allowed to automatically determine the number of bands used, as the number used always exceeded the number needed to cover the local basis for all atoms in the system.
Results and discussion
The three motifs are considered energetically feasible (vide supra) and are shown both from the top and from a front-facing perspective (Fig. 2). The most favorable binding site (the position of a structure of which the atomic positions are fully relaxed) of the ‘free thiolate’ motif is on a bridge site, displaced partially onto a hollow site. The ‘on-top’ motif binds on a hollow site and the ‘staple’ motif binds with the adatom over a bridge site, with the thiolates binding partially displaced out of a hollow site onto a nearby gold surface atom (Fig. S1, ESI† illustrates the binding sites considered).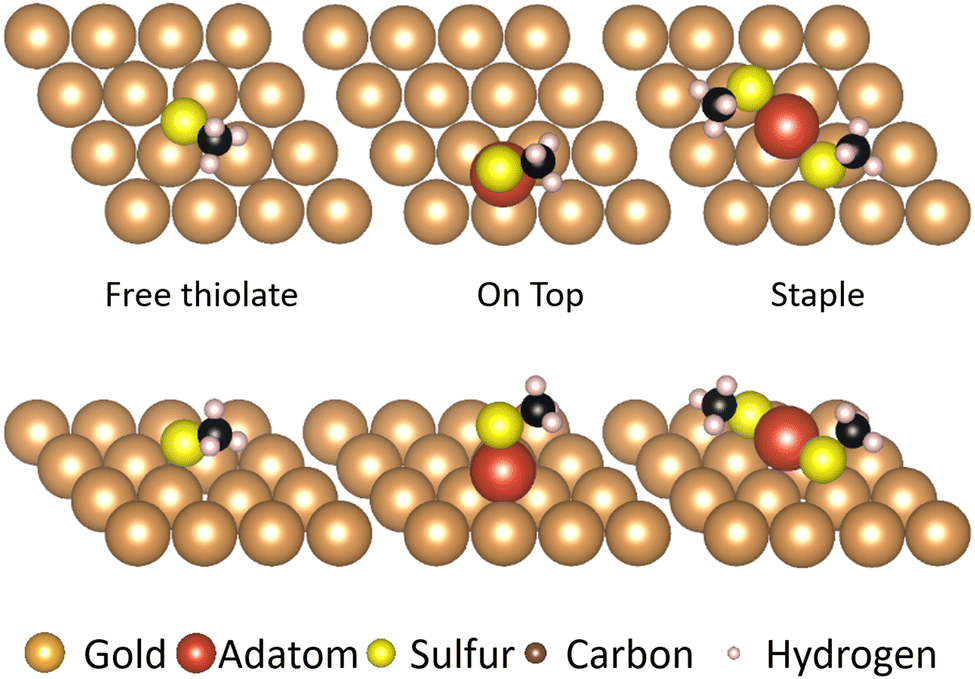 | ||
| Fig. 2 The different forms in which the sulfur can attach to a Au(111) surface. Note that the binding sites for the thiolate vary between bridge site (free thiolate motif), hollow site (on-top motif) and on top of gold atoms (staple motif). The gold atoms are shown in brown, except for the gold adatom that is part of the thiolate motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
Mobility of motifs
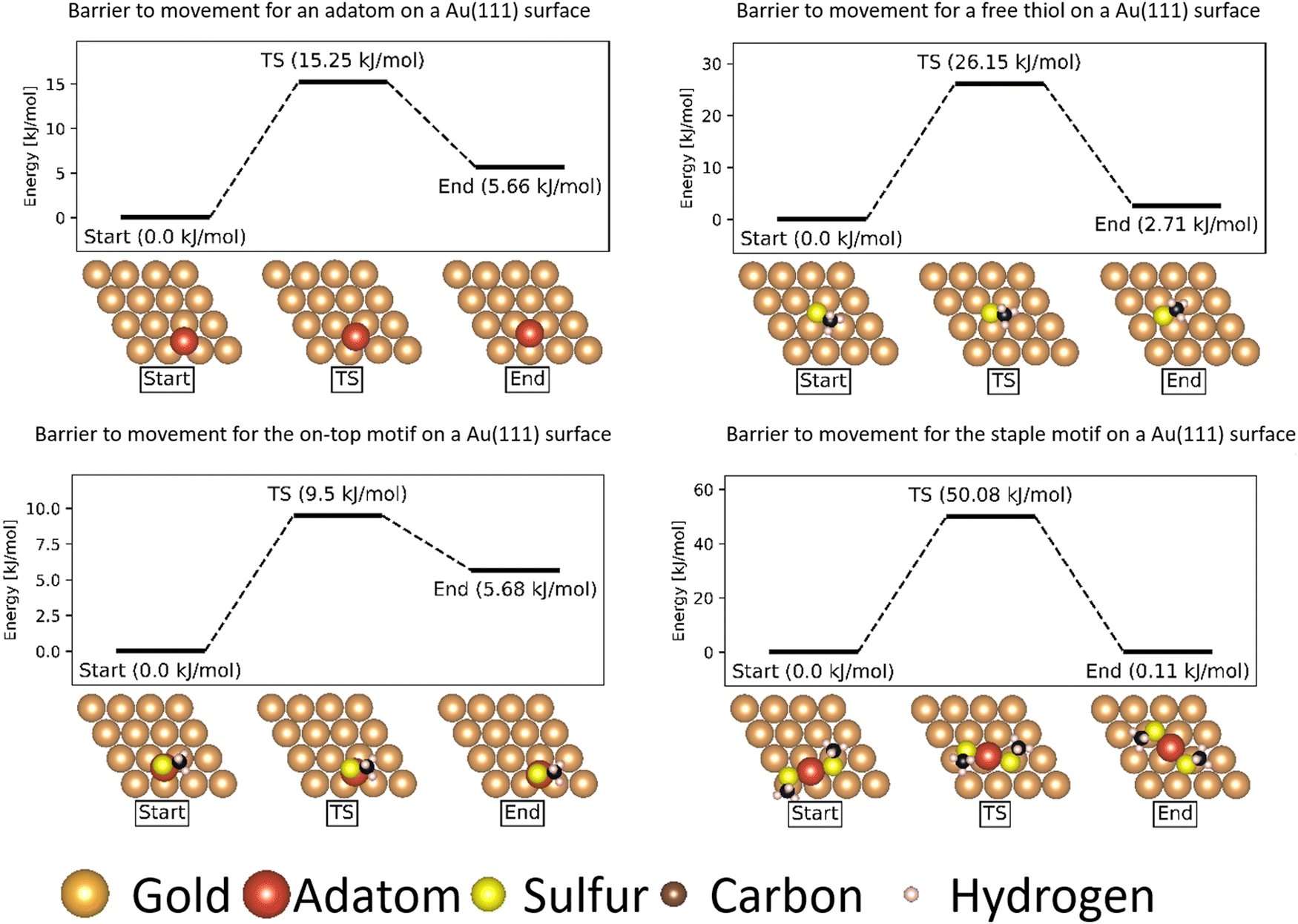 | ||
| Fig. 3 Energy barriers of movement of various motifs on a Au(111) surface. All energies are referenced to the lowest energy state in the movement. The gold atoms are shown in brown, except for the gold adatom that is part of the thiol motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
The movements of the adatom, ‘free thiolate’ motif and ‘on-top’ motifs result in end points that are at different energies than the corresponding starting points. This is caused by the structure of the second layer of the gold surface, which is displaced relative to the top surface such that alternating hollows are situated over another hollow (fcc adsorption site, see ESI,† Section S1) or on top of a gold atom in the second layer (hcp adsorption site), thus creating different interactions with bound molecules depending on which kind of adsorption site they are situated above.
The ‘free thiolate’ motif (Fig. 3), despite binding on a bridge site, is displaced partially onto a hollow site, resulting in a difference in energy depending on the type of hollow it is displaced onto. The ‘staple’ motif has the same energy for its starting and ending positions despite the thiolates being partially present over hollow sites as in the ‘free thiolate’ motif. In the starting situation one thiolate is situated partially over an fcc site while the other is situated over an hcp site. In the final situation the site occupations are reversed, causing the energy differences between binding over the different sites to cancel out and resulting in a similar energy for the initial and final state.
Crystal orbital Hamilton population analysis
Beyond the calculation of the energy barriers, binding energies are often used to help rationalize the data obtained and facilitate intuitive comparison of the various situations. However, binding energies (ESI,† Section S2) alone are an insufficient descriptor of the barriers to movement due to the complexity of the interactions of the motifs with the surface. In a similar situation for a molecular system, one would investigate the orbitals of the system in order to rationalize the properties observed. In this work, this cannot be done directly due to the use of plane waves rather than atomic orbitals as a basis set for the calculation. Electron density difference maps (EDDM) and crystal orbital Hamilton populations (COHP) can be used to gain further understanding on the nature of the system. For those unfamiliar with COHP and/or density of states (DOS) analysis, please refer to ESI,† Section S3.EDDM represent the electron density change (see Methods section) that results from the adsorption of the motif to the gold surface, which allows for visualization of the interactions between the motif and the surface. COHP analysis,56 or more specifically projected COHP (pCOHP)57 allows the use of the localized, chemical reasoning normally done with atomic orbitals that is lost upon use of a plane-wave basis set.
The combination of visual interpretation by EDDM and bonding information from COHP analysis allows us to retrieve not only the chemical bonding of the overall system, but spatial information about the contributions of specific atoms as well. Ultimately, this yields us understanding on the properties of the transition state, and allows us to rationalize our results in a way familiar to the molecular chemist.
The differences in electron density shown by EDDM can be summarized as a decrease in electron density (blue) around the atoms in the motifs and an increase in electron density (yellow) between atoms of the motif and the surface atoms they are bound to. While the increase in electron density is not directly related to the strength of the bonding to the surface, it gives an indication of which surface atoms the motif interacts with. In our COHP analysis, the significantly interacting surface atoms were concluded to be limited to nearest neighbours of the motif, as concluded earlier by Hoffmann et al.58
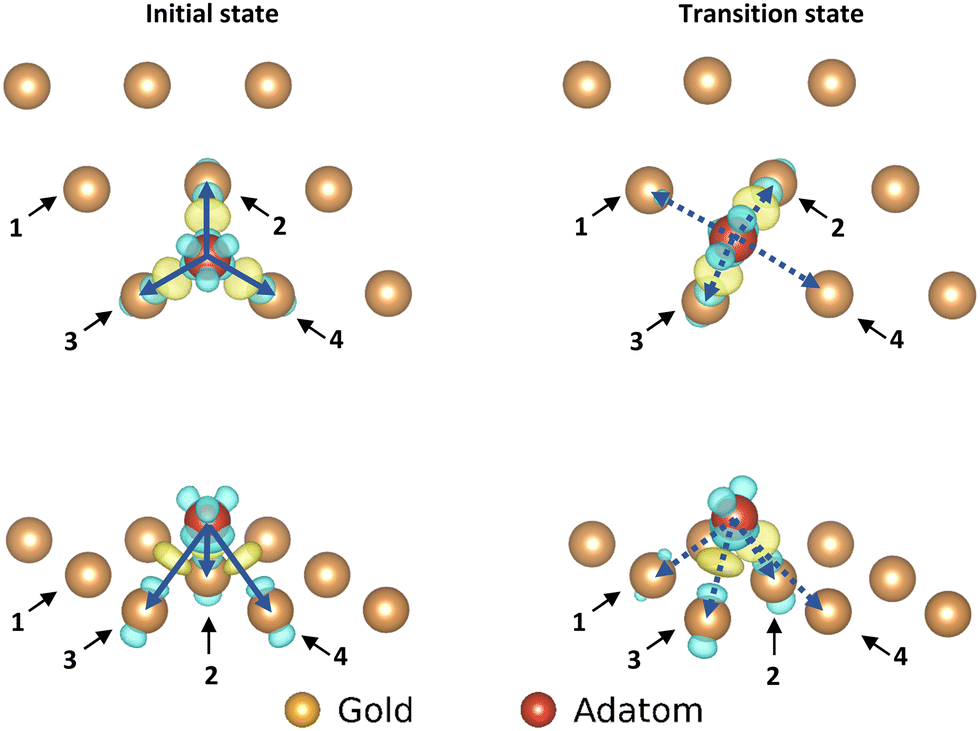 | ||
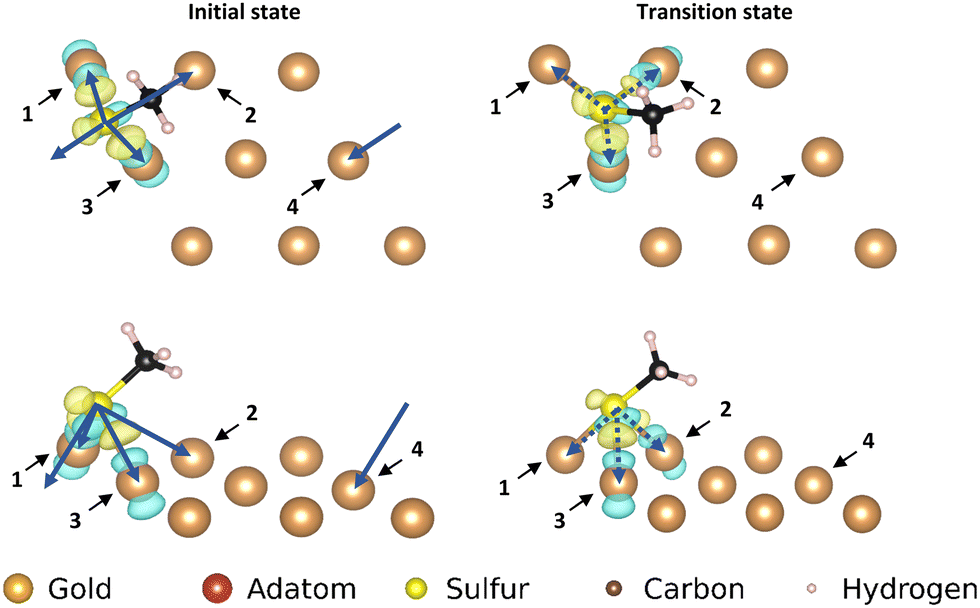
| Fig. 4 Electron density difference map for the adatom; left: top and front view of the initial state of the movement, right: top and front view of the transition state of the movement. Only the top layer of the gold surface is shown for clarity. The arrows correspond with the pairs of atoms used in the corresponding COHP analysis. | ||
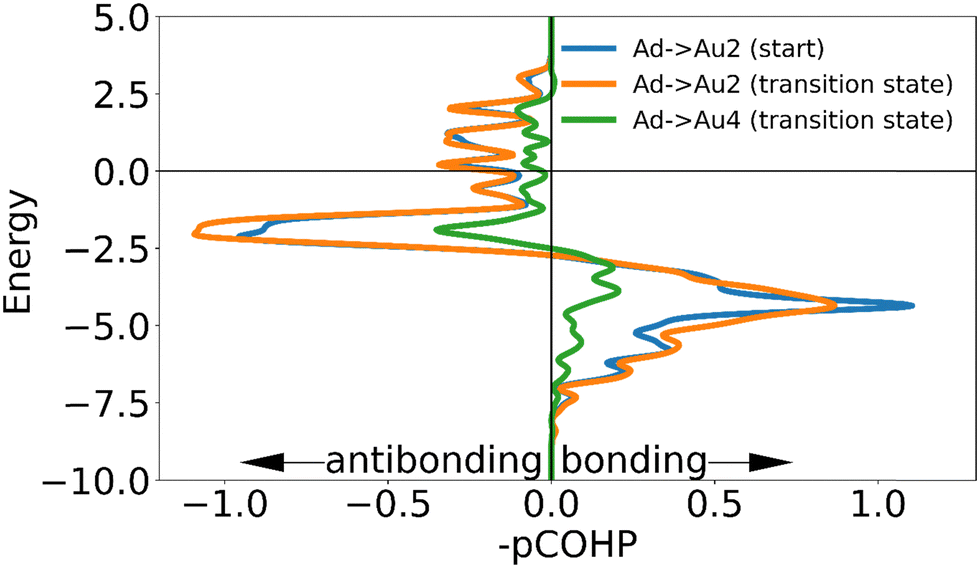 | ||
| Fig. 5 The negative of the projected crystal orbital Hamilton population (–pCOHP) curves calculated for the interactions of one of the surface gold atoms with the gold adatom in the initial and transition state. The x-axis is reversed for ease of analysis for those used to looking at COOP and DOS curves. The positions of the gold surface atoms numbered in the legend are indicated in Fig. 4. Due to the similarity of the –COHP curves of the starting atoms, only one curve is shown here. (see ESI†). | ||
| Initial state | Au1 | Au2 | Au3 | Au4 | Total |
|---|---|---|---|---|---|
| ICOHP with adatom | — | −0.52985 | −0.53355 | −0.58776 | −1.65116 |
| Distance to adatom | — | 2.70862 | 2.71804 | 2.72066 |
| Transition state | Au1 | Au2 | Au3 | Au4 | Total |
|---|---|---|---|---|---|
| ICOHP with adatom | −0.30058 | −0.57073 | −0.57279 | −0.14240 | −1.5865 |
| Distance to adatom | 3.02278 | 2.66909 | 2.66697 | 3.30759 |
In going from the initial state to the transition state the bonds with Au2 and Au3 (which stay close to the adatom during the movement) both gain bonding character and antibonding character, creating a small increase in bonding overall. In contrast the bond with Au4 is reduced by such a degree that the partial bond formation to Au1 does not compensate, and the overall bonding to the surface is weaker.
 | ||
| Fig. 6 Electron density difference map (EDDM) for the ‘free thiolate’ motif; left: top and front view of the initial state of the movement, right: top and front view of the transition state of the movement. Only the top layer of the gold surface is shown for clarity. The arrows correspond with the pairs of atoms used in the corresponding COHP analysis. | ||
| Initial state | Au1 | Au2 | Au3 | Au4 | Total |
|---|---|---|---|---|---|
| ICOHP with sulfur | −1.46516 | −0.03261 | −1.49675 | −0.08524 | −3.07976 |
| Distance to sulfur | 2.44930 | 3.59253 | 2.43687 | 3.16367 |
| Transition state | Au1 | Au2 | Au3 | Au4 | Total |
|---|---|---|---|---|---|
| ICOHP with sulfur | −0.41744 | −0.90250 | −1.50480 | — | −2.82474 |
| Distance to sulfur | 2.76710 | 2.68314 | 2.41260 | — |
The higher energy barrier observed for the movement of a ‘free thiolate’ motif compared to that of the adatom can be rationalized by considering the difference in loss of binding interactions (Fig. 7): the ‘free thiolate’ motif has to break one of two bonds stabilizing the system compared to the adatom, which still has two stabilizing interactions remaining in the transition state. The magnitude of these interactions are apparent from the ICOHP: the ‘free thiolate’ loses more binding interaction in the transition state than the adatom.
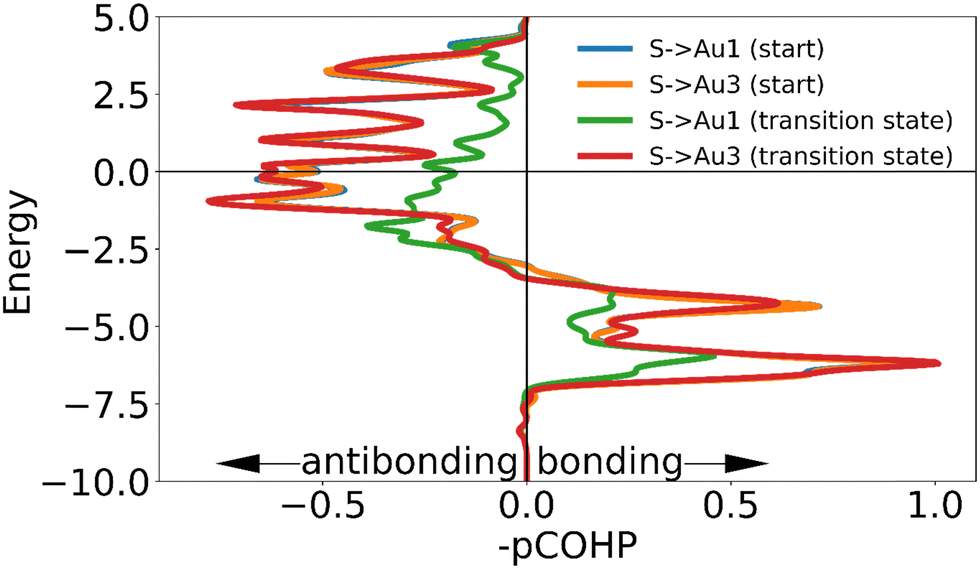 | ||
| Fig. 7 The negative of the projected crystal orbital Hamilton population (–pCOHP) curve calculated for the interactions of one of the surface gold atoms with the sulfur atom of the ‘free thiolate’ motif in the initial and transition state. The x-axis is reversed for ease of analysis for those used to looking at COOP and DOS curves. The positions of the gold surface atoms numbered in the legend are indicated in Fig. 6. Due to the lack of physical significance of some of the –COHP curves in the initial state and the similarity of the –COHP curves of some of the transition state, only two pairs of curves are shown here. (see ESI†). | ||
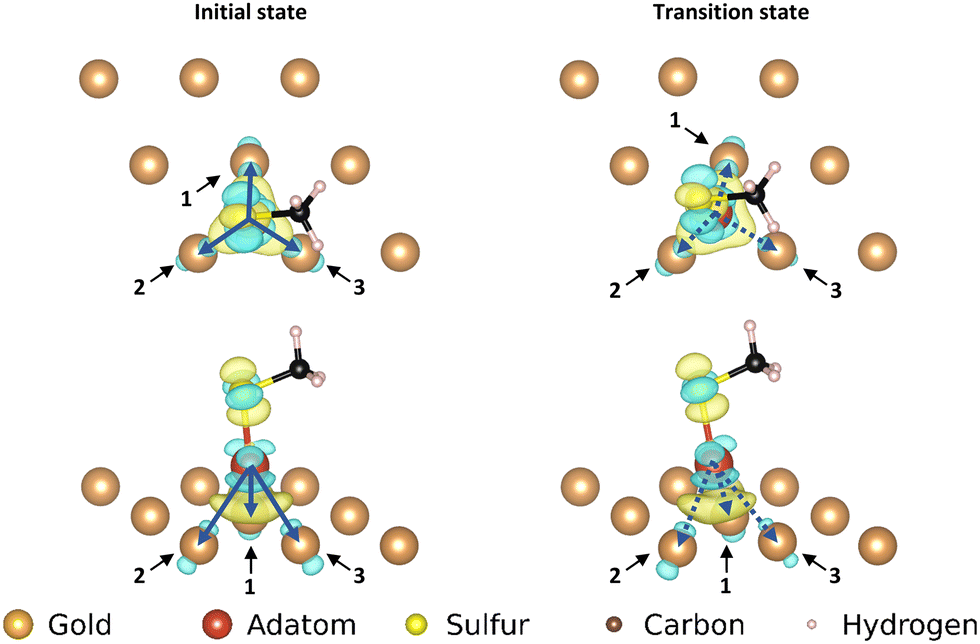 | ||
| Fig. 8 Electron density difference map for the ‘on-top’ motif; left: top and front view of the initial state of the movement, right: top and front view of the transition state of the movement. Only the top layer of the gold surface is shown for clarity. The arrows correspond with the pairs of atoms used in the corresponding COHP analysis. | ||
While the interaction of the sulfur with the adatom causes a larger change of electron density in the EDDM, the COHP curves confirm that this increase in displaced electron density is not directly representative of the strength of the bond to the surface. The COHP curves show that the presence of the sulfur on top of the gold adatom weakens binding interactions between the motif and the surface both in the initial state and the transition state, but that the reduction in binding in the initial state is more severe (Fig. 9, see ESI†). From ICOHP values (Table 3) it can be seen that this stabilizes the transition state relative to the initial state, explaining the lower barrier to movement.
| Initial state | Au1 | Au2 | Au3 | Total |
|---|---|---|---|---|
| ICOHP with adatom | −0.54177 | −0.43695 | −0.47365 | −1.45237 |
| Distance to adatom | 2.72171 | 2.78781 | 2.76623 |
| Transition state | Au1 | Au2 | Au3 | Total |
|---|---|---|---|---|
| ICOHP with adatom | −0.57564 | −0.49700 | −0.33335 | −1.40599 |
| Distance to adatom | 2.69108 | 2.71701 | 2.96812 |
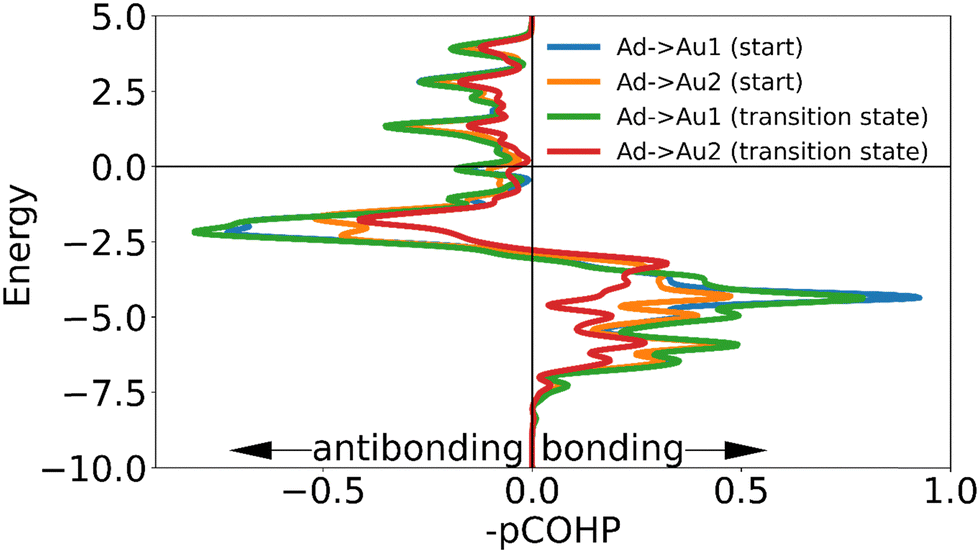 | ||
| Fig. 9 The negative of the projected crystal orbital Hamilton population (–pCOHP) curve calculated for the interactions of the surface gold atoms with the adatom of the ‘on-top’ motif in the initial and transition state. The x-axis is reversed for comparison with COOP and DOS curves. The positions of the gold surface atoms numbered in the legend are indicated in Fig. 8. Due to the similarity of the –COHP curves of some of the initial and transition state, only two pairs of curves are shown here. (see ESI†). | ||
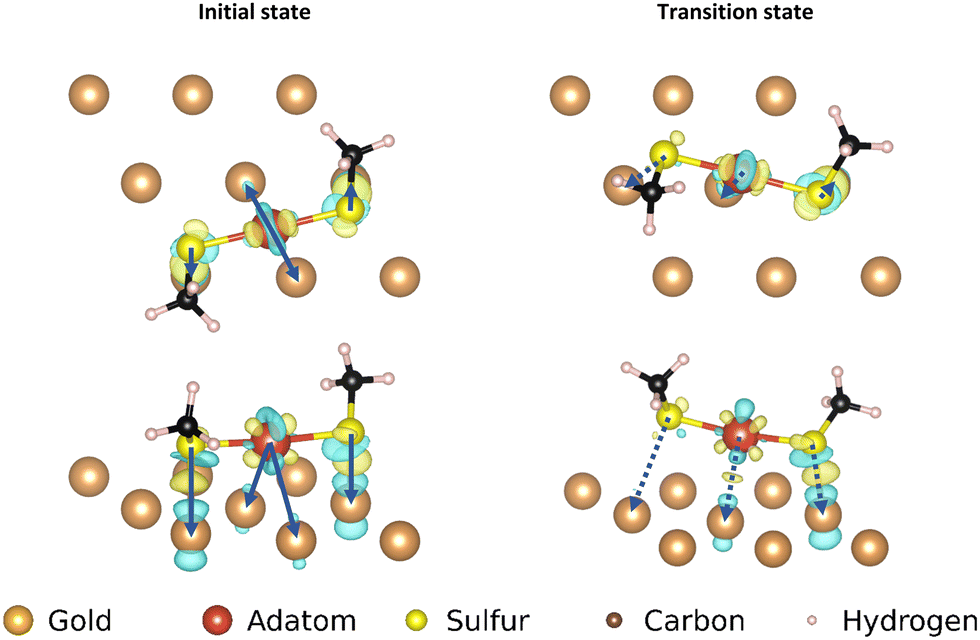 | ||
| Fig. 10 Electron density difference map for the ‘staple’ motif; left: top and front view of the initial state of the movement, right: top and front view of the transition state of the movement. Only the top layer of the gold surface is shown for clarity. The arrows correspond with the pairs of atoms used in the corresponding COHP analysis. Only the interactions with the strongest binding surface atoms for each motif atom are indicated for clarity. | ||
| Atom | Start | TS |
|---|---|---|
| S46 (static) | −1.38223 | −1.4612 |
| S47 (mobile) | −1.3374 | −0.36233 |
| Au37 | −0.52754 | −0.43674 |
| Total | −3.24717 | −2.26027 |
The adatom goes from two approximately equal interactions to one stronger one, which overall creates only a small loss in binding. The high energetic cost of movement can be explained primarily from the changes in interaction of the mobile sulfur atom: in the transition state the linear shape of the motif is retained. As the gold adatom moves directly over a gold atom in the surface the mobile the sulfur atom is much further removed from the surface than in any other transition state. This causes the sulfur atom to lose almost all binding interaction with the surface, without the ability to make a new (partial) bond as is possible in the transition states of the other motifs. The interaction of the static sulfur increases slightly in the transition state, showing that the staple motif acts as a chelating ligand: when the mobile sulfur and the adatom are disattached from the surface, the binding of the static sulfur increases making full dissociation more difficult (Fig. 11).
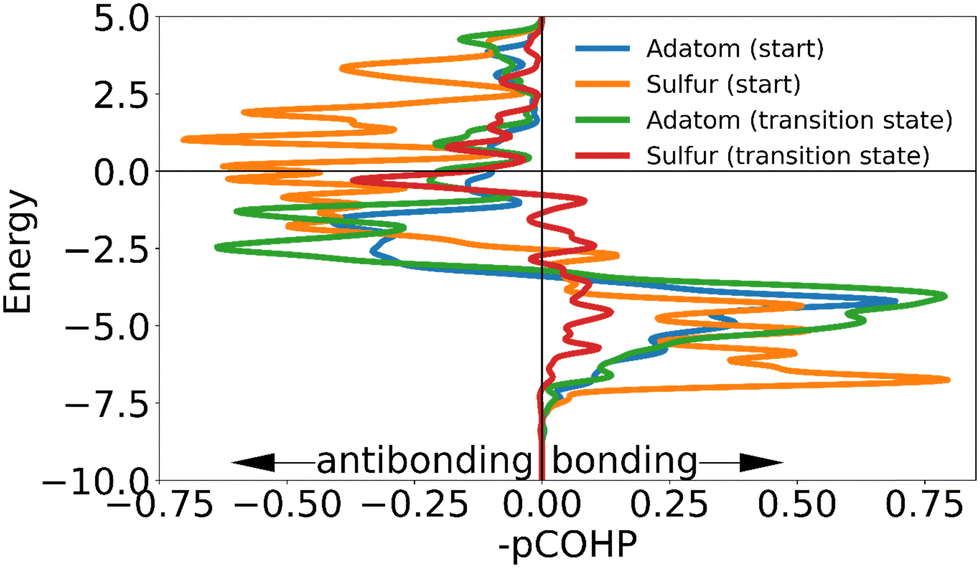 | ||
| Fig. 11 The negative of the projected crystal orbital Hamilton population (–pCOHP) curve calculated for the interactions of the surface gold atoms with the adatom of the ‘staple’ motif in the initial and transition state. The x-axis is reversed for ease of analysis for those used to looking at COOP and DOS curves. Due to the similarity of the –COHP curves of some of the initial and transition state, only some of the curves are shown here. (see ESI†) The transition state curve for sulfur is from the mobile sulfur, the curve for the transition state of the static sulfur is left out for clarity since its shape is very similar to that of the initial state. | ||
Comparison with calculated barriers
As can be seen from Table 5, the integrated COHP values agree with the order of mobility established by the energy barriers: both models indicate a relative ordering of mobility of on-top > adatom > free thiolate > staple. This confirms that despite COHP analysis being a localized model, the quantitative interpretation of COHP curves is representative of the real energy differences in results obtained from plane-wave calculations.| Motif | Initial ICOHP | Transition ICHOP | ICOHP difference | ΔE (kJ mol−1) |
|---|---|---|---|---|
| Ad | −1.65116 | −1.5865 | −0.06466 | 15.25 |
| FT | −3.07976 | −2.82474 | −0.25502 | 26.15 |
| OT | −1.45237 | −1.40599 | −0.04638 | 9.5 |
| St | −3.24717 | −2.26027 | −0.9869 | 50.08 |
The combination of electron density difference maps and crystal orbital Hamilton populations allows us to draw out both visual and quantitative molecular reasoning that was originally lost due to the use of a plane wave basis, and allows for more in-depth analysis of the results than can be done by analysing the energy differences and the DOS.
Formation of motifs
While the barrier to movement of each motif allows us to show their individual mobility, the overall mobility of the system is also largely determined by how much of each motif is present on the surface.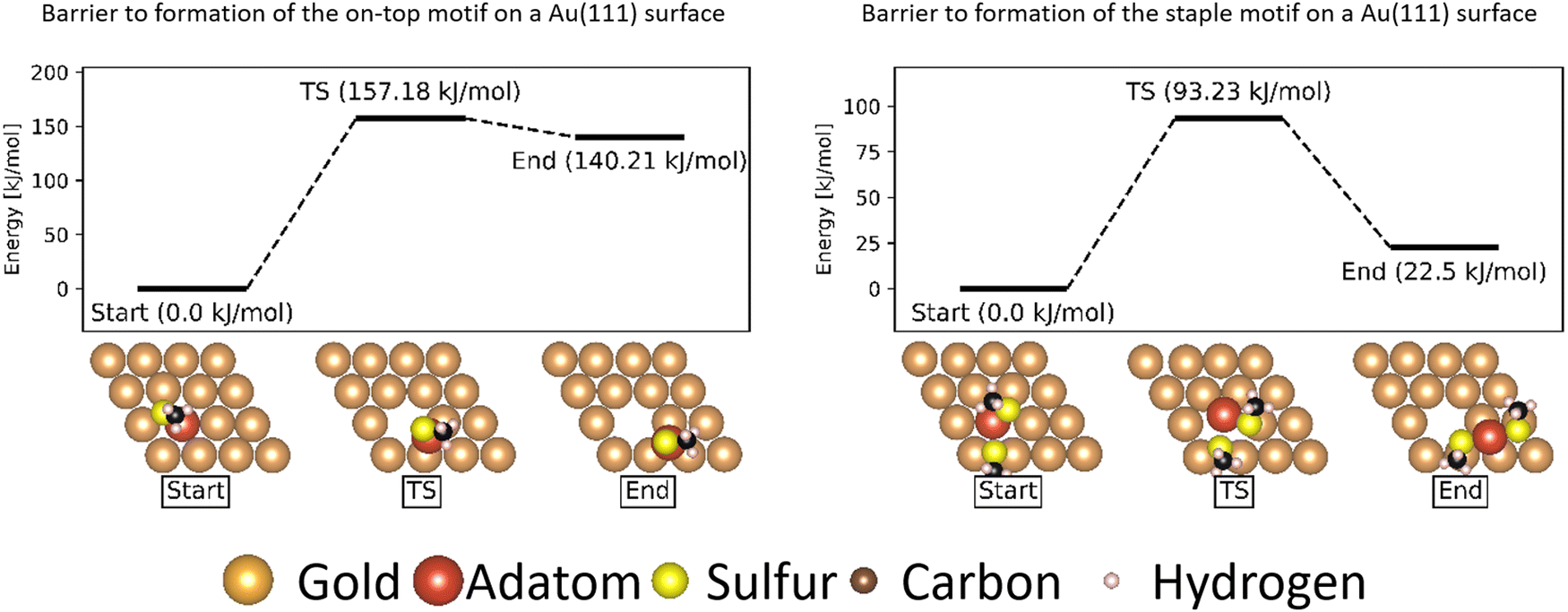 | ||
| Fig. 12 Energy barriers of formation of the on-top and staple motifs on a Au(111) surface. All energies are referenced to the starting state in the formation. The gold atoms are shown in brown, except for the gold adatom that is part of the thiolate motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
In the case of atomically flat surfaces, immediately after monolayer formation the only motif that should be present is the ‘free thiolate’ motif. The significantly higher energy barrier calculated indicates that both the ‘on-top’ motif and the ‘staple’ motif do not spontaneously form on such a surface, and thus would not contribute significantly to the mobility of thiolate motifs in an experiment starting from a flat surface.
Instead, it is more likely that the adatoms necessary to form the motifs are released from disuniformities on the surface such as terraces and ridges between surfaces. In a study on the growth of Au(111) surfaces Ali et al. show that generation of an adatom from a step-edge is thermodynamically favourable, and has a much lower energy barrier (13.5 kJ mol−1) than what is calculated for the extraction of an adatom from a flat surface.59
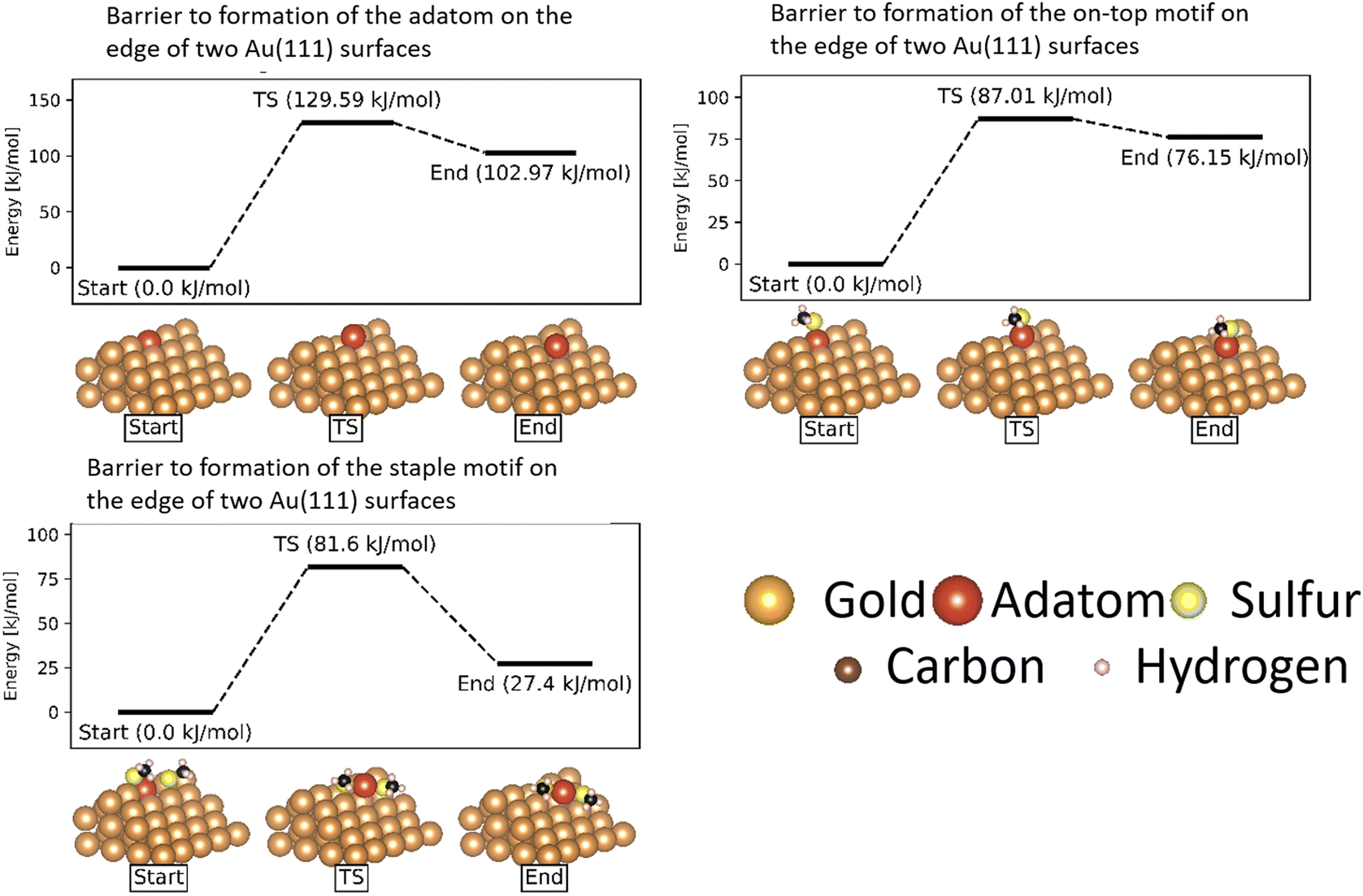 | ||
| Fig. 13 Energy barriers of formation of the on-top and staple motifs on the ridge between two Au(111) surfaces. All energies are referenced to the starting state in the formation. The gold atoms are shown in brown, except for the gold adatom that is part of the thiolate motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
Thermodynamic stability comparison
While comparisons of the thermodynamic stability of the motifs have been made in earlier studies through the use of absorption or binding energies to facilitate comparison,31–33 while others used the combination of energies from different calculations in order to compare the various motifs, which include different numbers of atoms.25 As such, energies like those of gold bulk or surface atoms needed to be added or subtracted from results in order to achieve a meaningful quantity. While these methods are the only way to achieve such a comparison for high-coverage systems, in low coverage systems it is possible to use large unit cells containing all the necessary components of the various motifs separated spatially. This allows for a more reliable comparison of the energies of the various motifs, as the energies of the gold bulk and surface atoms utilized in the aforementioned studies do not fully reflect the properties of the systems they were applied to, nor do binding or absorption energies reflect the full thermodynamics of the system at hand.Fig. 14 shows the larger surfaces used to achieve this comparison. The comparison can be divided into two parts depending on where the adatom utilized for the motif comes from: in the top row an adatom is already present on the gold surface (named here ‘full surface’ in this text, see Methods), while in the bottom row a vacancy is created in order to supply the motifs with the necessary adatom (named here ‘surface with vacancy’). As can be seen in Table 6 this makes a difference as to which motifs are energetically feasible:
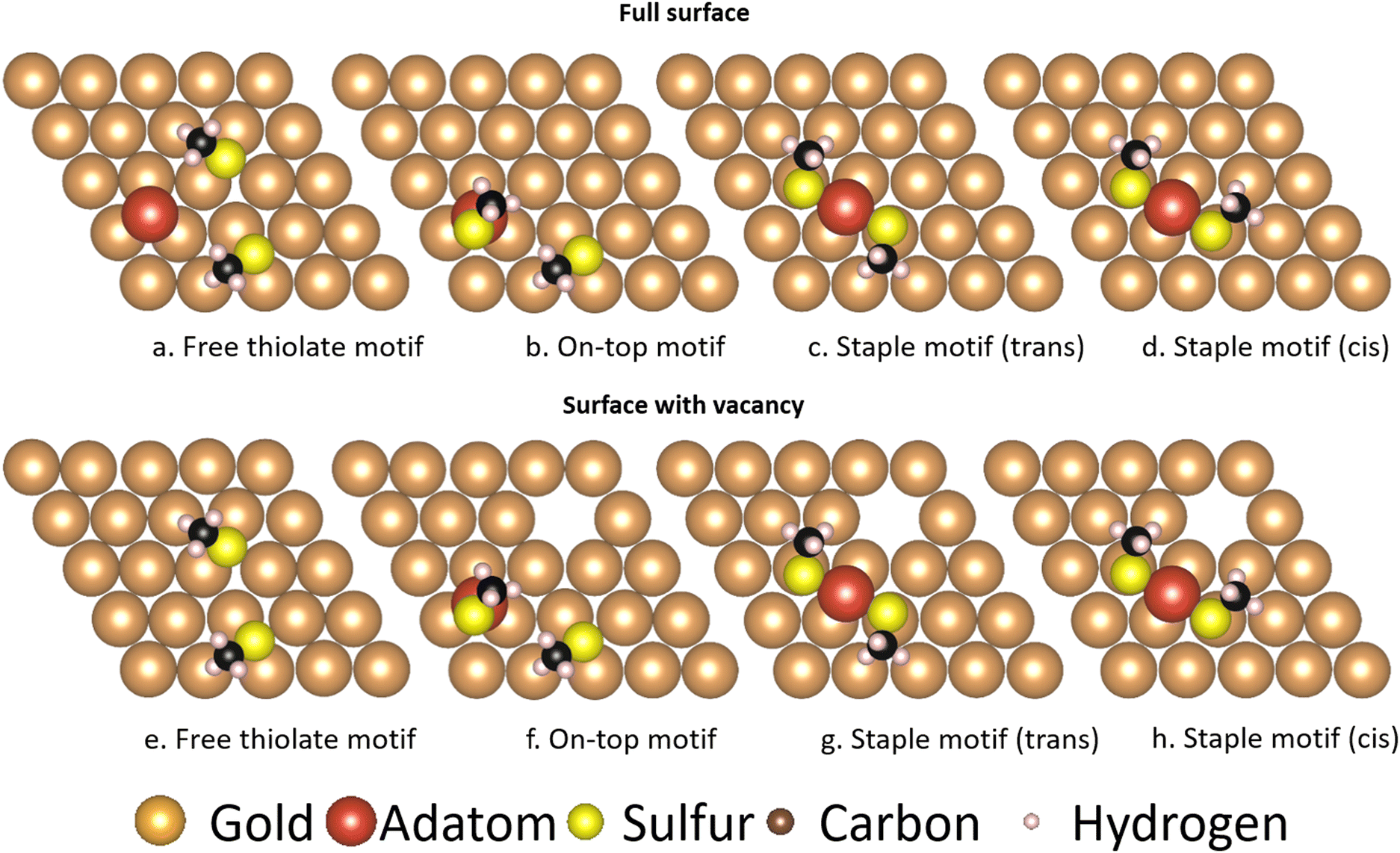 | ||
| Fig. 14 Motifs calculated on larger surfaces of gold in order to make an accurate energy comparison. The top row contains enough gold atoms to generate the motifs without creating vacancies in the gold surface. The surfaces in the bottom row are built with one gold atom less in order to investigate the effect of a vacancy being created upon generation of the adatom containing motifs. The gold atoms are shown in brown, except for the gold adatom that is part of the thiolate motifs which is shown in red. The sulfur atoms are shown in yellow, the carbon atoms in black and the hydrogen atoms in pink. | ||
| Relative energy [kJ mol−1] | Free thiolate | On-top | Staple | Staple cis |
|---|---|---|---|---|
| Full surface | 0 | 8.85 | −102.18 | −103.71 |
| With vacancies | 0 | 144.22 | 33.21 | 30.88 |
In the full surfaces, the ‘staple’ motifs are energetically favored, and given enough time the system should mainly consist of them. In the surfaces with vacancies, the ‘on-top’ motif is energetically inaccessible, and similarly the ‘staple’ motifs will constitute only a small fraction of the motifs on the surface. The results of the full surface, where adatoms are freely available, are in agreement with the thermodynamic comparison by Grönbeck et al.,25 and are in qualitative agreement with the binding energies found by Hu et al.31 and the adsorption energies found by Löfgren et al.33 A study by Ferrighi et al. comes to different results using the M06-L functional, where the adsorption energy of the ‘free thiolate’ motif is slightly larger than that of the ‘staple’ motif.32 The results of the surface with vacancies show that introducing a vacancy in the surface to generate the motifs is costly in energy, and will therefore change the ratio of motifs present in experiments conducted on atomically flat surfaces.
Conclusion
In this work we investigated the mobility of various motifs over the Au(111) surface, as well as their formation from the (111) and (331) surfaces, through use of periodic density functional theory. The same computational methods were used to investigate the relative thermodynamic stability of these motifs. The combination of these results allow us to improve the understanding on how the early stages of monolayer formation occur.Comparison of the relative thermodynamic stability of the motifs indicates that the distribution of thiolates over the possible motifs depends strongly on the roughness of the initial gold surface. On a roughened surface (where adatoms are present) at equilibrium, the thiolates are mostly present in the form of the ‘staple’ motif. On an atomically flat surface (where adatoms are not present), the thiolates are mainly present as the ‘free thiolate’ motif, with small amounts of ‘staple’ motif statistically present. The ‘on-top’ motif was found to represent only a minor fraction of the thiolates on the surface in either situation.
In experimental studies a pristine Au(111) surface will rearrange to the ‘herringbone’ reconstructed surface, this introduces alternative points to keep into account when analysing these results: the ‘herringbone’ reconstructed surface contains 23 atoms per 22 top layer atoms in a fully relaxed state. This means that binding of the first thiolate moiety per reconstructed unit cell will liberate the excess atom instead of creating a vacancy, thus creating an energetic situation more similar to the ‘full’ surface than the surface with vacancies. Continuing onwards from this point, the newly reformed Au(111) unit cell will contain no more excess atoms and further extraction of adatoms will incur the abovementioned energetic cost.
Despite the ‘staple’ motifs being somewhat thermally accessible the investigations on the formation of the motifs, however, indicate that the barrier to formation is much higher than the energy available at room temperature. If adatoms are absent initially, the surface initially should not contain a significant amount of the ‘staple’ motif. Rather, it should consist mostly of the kinetically accessible ‘free thiolate’ motif that forms initially upon binding to the surface, and will include a few ‘staple’ motif structures obtained by liberating the excess surface atom present in the ‘herringbone’ reconstruction.
The present study is focused on chemistry at the solid/vacuum (/sparse gas) interface, and thus cannot be directly compared to experimental studies at the solid/liquid interface. Extrapolation of the results requires consideration of the effects of additional processes that will be possible at the solid/liquid interface such as competitive absorption, alteration of barrier heights due to stabilization by the solvent, etc.
In conclusion, if adatoms are initially present, the ‘staple’ motif should constitute most of the thiolates on the surface, resulting in a low average mobility. On a surface that is devoid of initially present gold adatoms the combination of the barrier heights for the motifs and the probability of their formation, the mobility of thiolate containing species should be higher. In this regime, most of the thiolates should be present as the ‘free thiolate’ motif, which has greater mobility than the ‘staple’ motif.
With time, adatoms liberated from features on the surface such as step-edges59 will encounter the ‘free thiolate’ motifs present on the surface, and form ‘staple’ motif upon encountering another bound thiolate molecule, resulting in a similarly low mobility as described above. In this study we predict a low average mobility of thiolate containing motifs on the surface, which matches the experimental observations concerning the length of the process of formation of a full monolayer.1 Our study provides insight into the initial step of the monolayer formation, and rationalizes the relative mobilities of the motifs present on the surface.
Data availability
All coordinates for optimised calculations are provided as ESI.† COHP data is stored on the Habrok cluster of the University of Groningen and is available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Hábrák high performance computing cluster and the University of Groningen for funding (DRD).References
- R. V. Mom, S. T. A. G. Melissen, P. Sautet, J. W. M. Frenken, S. N. Steinmann and I. M. N. Groot, The Pressure Gap for Thiols: Methanethiol Self-Assembly on Au(111) from Vacuum to 1 bar, J. Phys. Chem. C, 2019, 123, 12382–12389 CrossRef CAS.
- M. Cohen-Atiya and D. Mandler, Studying thiol adsorption on Au, Ag and Hg surfaces by potentiometric measurements, J. Electroanal. Chem., 2003, 550–551, 267–276 CrossRef CAS.
- D. Mandler and S. Kraus-Ophir, Self-assembled monolayers (SAMs) for electrochemical sensing, J. Solid State Electrochem., 2011, 15, 1535–1558 CrossRef CAS.
- H. Atesci, V. Kaliginedi, J. A. Celis Gil, H. Ozawa, J. M. Thijssen, P. Broekmann, M. Haga and S. J. Van Der Molen, Humidity-controlled rectification switching in ruthenium-complex molecular junctions, Nat. Nanotechnol., 2018, 13, 117–121 CrossRef CAS PubMed.
- L. Newton, T. Slater, N. Clark and A. Vijayaraghavan, Self assembled monolayers (SAMs) on metallic surfaces (gold and graphene) for electronic applications, J. Mater. Chem. C, 2013, 1, 376–393 RSC.
- A. J. Bergren, L. Zeer-Wanklyn, M. Semple, N. Pekas, B. Szeto and R. L. McCreery, Musical molecules: the molecular junction as an active component in audio distortion circuits, J. Phys.: Condens. Matter, 2016, 28, 094011 CrossRef PubMed.
- T. A. Jung, R. R. Schlittler, J. K. Gimzewski, H. Tang and C. Joachim, Controlled Room-Temperature Positioning of Individual Molecules: Molecular Flexure and Motion, Science, 1996, 271, 181–184 CrossRef CAS.
- R. L. Carroll and C. B. Gorman, The Genesis of Molecular Electronics, Angew. Chem., Int. Ed., 2002, 41, 4378–4400 CrossRef PubMed.
- F. Davis and S. P. J. Higson, Structured thin films as functional components within biosensors, Biosens. Bioelectron., 2005, 21, 1–20 CrossRef CAS PubMed.
- N. J. Tao, Electron transport in molecular junctions, Nat. Nanotechnol., 2006, 1, 173–181 CrossRef CAS PubMed.
- L. M. Molina and B. Hammer, Theoretical study of thiol-induced reconstructions on the Au(111) surface, Chem. Phys. Lett., 2002, 360, 264–271 CrossRef CAS.
- A. Hamdan, X. Glad and M. S. Cha, Synthesis of Copper and Copper Oxide Nanomaterials by Pulsed Electric Field in Water with Various Electrical Conductivities, Nanomaterials, 2020, 10, 1347 CrossRef CAS PubMed.
- N. Xin, C. Hu, H. Al Sabea, M. Zhang, C. Zhou, L. Meng, C. Jia, Y. Gong, Y. Li, G. Ke, X. He, P. Selvanathan, L. Norel, M. A. Ratner, Z. Liu, S. Xiao, S. Rigaut, H. Guo and X. Guo, Tunable Symmetry-Breaking-Induced Dual Functions in Stable and Photoswitched Single-Molecule Junctions, J. Am. Chem. Soc., 2021, 143, 20811–20817 CrossRef CAS PubMed.
- D. Stefani, K. J. Weiland, M. Skripnik, C. Hsu, M. L. Perrin, M. Mayor, F. Pauly and H. S. J. Van Der Zant, Large Conductance Variations in a Mechanosensitive Single-Molecule Junction, Nano Lett., 2018, 18, 5981–5988 CrossRef CAS PubMed.
- L. Meng, N. Xin, C. Hu, J. Wang, B. Gui, J. Shi, C. Wang, C. Shen, G. Zhang, H. Guo, S. Meng and X. Guo, Side-group chemical gating via reversible optical and electric control in a single molecule transistor, Nat. Commun., 2019, 10, 1450 CrossRef PubMed.
- I. Díez-Pérez, J. Hihath, Y. Lee, L. Yu, L. Adamska, M. A. Kozhushner, I. I. Oleynik and N. Tao, Rectification and stability of a single molecular diode with controlled orientation, Nat. Chem., 2009, 1, 635–641 CrossRef PubMed.
- E. Lörtscher, B. Gotsmann, Y. Lee, L. Yu, C. Rettner and H. Riel, Transport Properties of a Single-Molecule Diode, ACS Nano, 2012, 6, 4931–4939 CrossRef PubMed.
- R. L. McCreery and A. J. Bergren, Progress with Molecular Electronic Junctions: Meeting Experimental Challenges in Design and Fabrication, Adv. Mater., 2009, 21, 4303–4322 CrossRef CAS PubMed.
- E. H. Huisman, M. L. Trouwborst, F. L. Bakker, B. J. Van Wees and S. J. Van Der Molen, The mechanical response of lithographically defined break junctions, J. Appl. Phys., 2011, 109, 104305 CrossRef.
- Y. Tsuji, A. Staykov and K. Yoshizawa, Orbital Views of Molecular Conductance Perturbed by Anchor Units, J. Am. Chem. Soc., 2011, 133, 5955–5965 CrossRef CAS PubMed.
- J. D. Steen, A. Volker, D. R. Duijnstee, A. S. Sardjan and W. R. Browne, pH-Induced Changes in the SERS Spectrum of Thiophenol at Gold Electrodes during Cyclic Voltammetry, J. Phys. Chem. C, 2022, 126, 7680–7687 CrossRef CAS PubMed.
- D. Dulić, S. J. Van Der Molen, T. Kudernac, H. T. Jonkman, J. J. D. De Jong, T. N. Bowden, J. Van Esch, B. L. Feringa and B. J. Van Wees, One-Way Optoelectronic Switching of Photochromic Molecules on Gold, Phys. Rev. Lett., 2003, 91, 207402 CrossRef PubMed.
- M. S. Inkpen, Z. Liu, H. Li, L. M. Campos, J. B. Neaton and L. Venkataraman, Non-chemisorbed gold–sulfur binding prevails in self-assembled monolayers, Nat. Chem., 2019, 11, 351–358 CrossRef CAS PubMed.
- J. Gao, H. Lin, X. Qin, X. Zhang, H. Ding, Y. Wang, M. Rokni Fard, D. Kaya, G. Zhu, Q. Li, Y. Li, M. Pan and Q. Guo, Probing Phase Evolutions of Au-Methyl-Propyl-Thiolate Self-Assembled Monolayers on Au(111) at the Molecular Level, J. Phys. Chem. B, 2018, 122, 6666–6672 CrossRef CAS PubMed.
- H. Grönbeck, H. Häkkinen and R. L. Whetten, Gold−Thiolate Complexes Form a Unique c (4 × 2) Structure on Au(111), J. Phys. Chem. C, 2008, 112, 15940–15942 CrossRef.
- F. P. Cometto, P. Paredes-Olivera, V. A. Macagno and E. M. Patrito, Density Functional Theory Study of the Adsorption of Alkanethiols on Cu(111), Ag(111), and Au(111) in the Low and High Coverage Regimes, J. Phys. Chem. B, 2005, 109, 21737–21748 CrossRef CAS PubMed.
- N. Camillone, T. Y. B. Leung, P. Schwartz, P. Eisenberger and G. Scoles, Chain Length Dependence of the Striped Phases of Alkanethiol Monolayers Self-Assembled on Au(111): An Atomic Beam Diffraction Study, Langmuir, 1996, 12, 2737–2746 CrossRef CAS.
- T. Bürgi, Properties of the gold–sulphur interface: from self-assembled monolayers to clusters, Nanoscale, 2015, 7, 15553–15567 RSC.
- O. Voznyy, J. J. Dubowski, J. T. Yates and P. Maksymovych, The Role of Gold Adatoms and Stereochemistry in Self-Assembly of Methylthiolate on Au(111), J. Am. Chem. Soc., 2009, 131, 12989–12993 CrossRef CAS PubMed.
- A. Chaudhuri, M. Odelius, R. G. Jones, T.-L. Lee, B. Detlefs and D. P. Woodruff, The structure of the Au(111)/methylthiolate interface: New insights from near-edge X-ray absorption spectroscopy and X-ray standing waves, J. Chem. Phys., 2009, 130, 124708 CrossRef CAS PubMed.
- G. Hu, R. Jin and D. Jiang, Beyond the staple motif: a new order at the thiolate–gold interface, Nanoscale, 2016, 8, 20103–20110 RSC.
- L. Ferrighi, Y. Pan, H. Grönbeck and B. Hammer, Study of Alkylthiolate Self-assembled Monolayers on Au(111) Using a Semilocal meta-GGA Density Functional, J. Phys. Chem. C, 2012, 116, 7374–7379 CrossRef CAS.
- J. Löfgren, H. Grönbeck, K. Moth-Poulsen and P. Erhart, Understanding the Phase Diagram of Self-Assembled Monolayers of Alkanethiolates on Gold, J. Phys. Chem. C, 2016, 120, 12059–12067 CrossRef PubMed.
- P. G. Lustemberg, M. L. Martiarena, A. E. Martínez and H. F. Busnengo, The Reaction Pathways for HSCH 3 Adsorption on Au(111): A Density Functional Theory Study, Langmuir, 2008, 24, 3274–3279 CrossRef CAS PubMed.
- P. Li and F. Ding, Origin of the herringbone reconstruction of Au(111) surface at the atomic scale, Sci. Adv., 2022, 8, eabq2900 CrossRef CAS PubMed.
- F. Hanke and J. Björk, Structure and local reactivity of the Au(111) surface reconstruction, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235422 CrossRef.
- A. D. Jewell, S. J. Kyran, D. Rabinovich and E. C. H. Sykes, Effect of Head-Group Chemistry on Surface-Mediated Molecular Self-Assembly, Chem. – Eur. J., 2012, 18, 7169–7178 CrossRef CAS PubMed.
- F.-S. Li, W. Zhou and Q. Guo, Uncovering the hidden gold atoms in a self-assembled monolayer of alkanethiol molecules on Au(111), Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 113412 CrossRef.
- N. Preetha Genesh, D. Cui, D. Dettmann, O. MacLean, T. K. Johal, A. V. Lunchev, A. C. Grimsdale and F. Rosei, Selective Self-Assembly and Modification of Herringbone Reconstructions at a Solid–Liquid Interface of Au(111), J. Phys. Chem. Lett., 2023, 14, 3057–3062 CrossRef CAS PubMed.
- O. Ivashenko, H. Logtenberg, J. Areephong, A. C. Coleman, P. V. Wesenhagen, E. M. Geertsema, N. Heureux, B. L. Feringa, P. Rudolf and W. R. Browne, Remarkable Stability of High Energy Conformers in Self-Assembled Monolayers of a Bistable Electro- and Photoswitchable Overcrowded Alkene, J. Phys. Chem. C, 2011, 115, 22965–22975 CrossRef CAS.
- A. Cossaro, R. Mazzarello, R. Rousseau, L. Casalis, A. Verdini, A. Kohlmeyer, L. Floreano, S. Scandolo, A. Morgante, M. L. Klein and G. Scoles, X-ray Diffraction and Computation Yield the Structure of Alkanethiols on Gold(111), Science, 2008, 321, 943–946 CrossRef CAS PubMed.
- Ö. Kap, N. Kabanov, M. Tsvetanova, C. Varlikli, A. L. Klavsyuk, H. J. W. Zandvliet and K. Sotthewes, Structural Stability of Physisorbed Air-Oxidized Decanethiols on Au(111), J. Phys. Chem. C, 2020, 124, 11977–11984 CrossRef.
- D. Jiang and S. Dai, Diffusion of the Linear CH 3 S−Au−SCH 3 Complex on Au(111) from First Principles, J. Phys. Chem. C, 2009, 113, 3763–3766 CrossRef CAS.
- D. Jiang and S. Dai, Cis–trans conversion of the CH3S–Au–SCH3 complex on Au(111), Phys. Chem. Chem. Phys., 2009, 11, 8601 RSC.
- N. A. Kautz and S. A. Kandel, Alkanethiol/Au(111) Self-Assembled Monolayers Contain Gold Adatoms: Scanning Tunneling Microscopy before and after Reaction with Atomic Hydrogen, J. Am. Chem. Soc., 2008, 130, 6908–6909 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Bloechl, Crystal orbital Hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal Orbital Hamilton Population (COHP) Analysis As Projected from Plane-Wave Basis Sets, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- M. Tachibana, K. Yoshizawa, A. Ogawa, H. Fujimoto and R. Hoffmann, Sulfur−Gold Orbital Interactions which Determine the Structure of Alkanethiolate/Au(111) Self-Assembled Monolayer Systems, J. Phys. Chem. B, 2002, 106, 12727–12736 CrossRef CAS.
- A. Ali and H. Jónsson, Mechanism of Interlayer Transport on a Growing Au(111) Surface: 2D vs. 3D Growth, Surf. Interfaces, 2022, 31, 101944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03709j |
| This journal is © the Owner Societies 2025 |