
Evidence of strong O–H⋯C interactions involving apical pyramidane carbon atoms as hydrogen atom acceptors†
Ivana S.
Veljković
 a,
Miroslavka
Malinić
b and
Dušan Ž.
Veljković
*b
a,
Miroslavka
Malinić
b and
Dušan Ž.
Veljković
*b
aUniversity of Belgrade – Institute of Chemistry, Technology and Metallurgy – National Institute of the Republic of Serbia, Belgrade, Serbia
bUniversity of Belgrade – Faculty of Chemistry, Studentski trg 12-16, Belgrade, Serbia. E-mail: vdusan@chem.bg.ac.rs
First published on 27th December 2024
Abstract
Using high-level quantum chemical calculations, we predicted a strong O–H⋯C interaction between the apical carbon atoms of pyramidane and its derivatives and water molecules. Analysis of calculated electrostatic potential maps showed that there are areas of strong negative potential above apical carbon atoms in all studied structures. The results of quantum chemical calculations showed that the O–H⋯C interaction between the hydrogen atom of water and the apical carbon atom of pyramidane derivatives with four –CH3 substituents is unexpectedly strong, ΔECCSD(T)/CBS = −7.43 kcal mol−1. The strong hydrogen bonds were also predicted in the case of unsubstituted pyramidane (ΔECCSD(T)/CBS = −6.41 kcal mol−1) and pyramidane with four –OH substituents (ΔECCSD(T)/CBS = −5.87 kcal mol−1). Although there are not many crystal structures of pyramidane-like molecules, we extracted examples of pyramidal-shaped molecules with apical carbon atoms from the Cambridge Structural Database and analyzed their hydrogen-bonding patterns. Analysis of crystal structures confirmed the existence of short non-covalent contacts between apical carbon atoms and neighboring hydrogen atoms.
Introduction
Hydrogen bonds play a crucial role in numerous chemical, physical, and biological phenomena and processes.1–9 According to an IUPAC definition, a hydrogen bond (depicted as D–H⋯A) is formed between a hydrogen atom bonded to an electronegative atom (donor atom, D) and another electron-rich atom (acceptor atom, A).10 In more general terms, the acceptor does not have to be an electron-rich atom but could be any species with increased electron density (including aromatic systems, π-bonds, radicals, etc.). Along these lines, in addition to usual hydrogen atom acceptors like O, N, or F atoms, there is a possibility that other atoms may play that role, too. This was discussed in detail in a very recent review on specific types of hydrogen bonds.11 One of the unexpected candidates for the role of hydrogen atom acceptor is the carbon atom.11–14 Although electron-rich carbon atoms are not usual in molecules, there are indeed examples of organic and organometallic compounds in which an increased electron density is detected around the C atom. An early example of a non-covalent carbon-hydrogen contact was noticed in the crystal structure of the carbene complex.12 The C–H⋯C bond between two carbene moieties was analysed in terms of geometry, and the analysis showed that the C–H⋯C angle is almost linear (172.5°), which is the characteristic of hydrogen bonds. Also, the distance between interacting C and H atoms (2.02 Å) was shorter than the sum of their van der Waals radii, indicating the existence of a hydrogen bond. In the very recent study performed by S. Grabowski, C–H+⋯C hydrogen bonds between carbon atoms of N-heterocyclic carbenes (NHC) and a proton of a hydrogen-containing cation were detected in crystal structures extracted from the Cambridge Structural Database (CSD).14 The results of density functional theory (DFT) calculations performed at the BP86-D4/TZ2P level showed that these interactions may be quite strong, as expected for charged species like cations.14Another theoretical study showed that carbenes may also be involved in X–H⋯C interactions with neutral species.13Ab initio calculations performed at the MP2/aug-cc-pVTZ level showed that carbenes and carbodiphosphoranes may be involved in X–H⋯C interactions (X = O, C) in which divalent carbon atoms act as hydrogen atom acceptors.13 The results of quantum chemical calculations indicate that a strong O–H⋯C interaction may be formed between imidazole-2-ylidene and a water molecule (ΔE = −9.84 kcal mol−1). These unusual properties of carbon atoms were explained by the presence of lone pairs on C atoms in these specific systems. Examples of unusual X–H⋯C interactions involving carbon atoms of a methyl group as hydrogen atom acceptors were recognized in crystal structures, too.15
Pyramidane (tetracyclo[2.1.0.013,2.035]pentane) is a hydrocarbon molecule with a pyramidal-shape structure (Fig. 1).16 Due to its unusual bonding characteristics, possible application as a high-energy material precursor, and the fact that it has never been synthesized, the pyramidane molecule has attracted significant attention from both theoretical and synthetic chemists.16–22 However, many heteroatom pyramidane analogs were synthesized and structurally characterized, including both charged and neutral chemical species.17–20
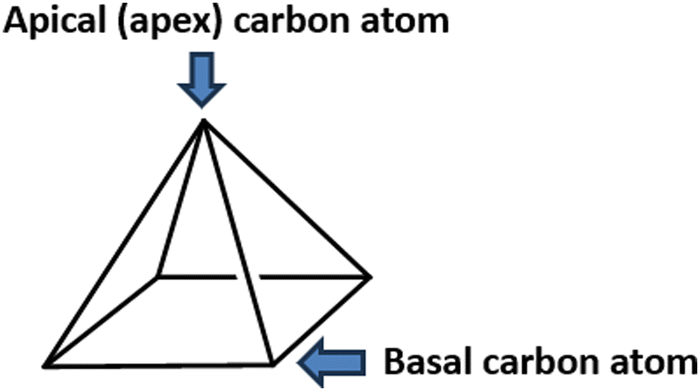 | ||
| Fig. 1 The structure of a pyramidane molecule. The carbon atom on the top of the pyramidal structure is designated as an “apical” or an “apex” carbon atom, while other carbon atoms are designated as “basal” carbon atoms. | ||
Combined experimental and theoretical studies indicate the presence of non-classical bonding in the pyramidane molecule and its derivatives, with a high degree of ionicity of the bonds between apical and basal carbon atoms.21 The results of DFT calculations show that an electron pair with a very high s character exists at the apex of the pyramidal structure.21 These results indicate that the apical carbon atoms in the pyramidane molecule and its derivatives may act as hydrogen atom acceptors in hydrogen bonds. Furthermore, calculations performed within the same study showed that the nature of substituents on basal carbon atoms significantly affects the structure and bonding in pyramidal molecules, which may also have consequences in terms of non-covalent bonding patterns.
Although covalent bonding in pyramidane and many of its derivatives was extensively studied in the past, a systematic study of non-covalent interactions involving these molecules has not been performed yet. On the other hand, it is known that in the case of some strained high-energy molecules, hydrogen bonds play very important roles in terms of their detonation properties.22 DFT calculations performed on a series of nitrocyclohydrocarbon explosives showed that hydrogen bonding affects the sensitivity of these molecules towards detonation.22
In this study, we have performed high-level ab initio calculations to predict and characterize possible O–H⋯C interactions involving the apical carbon atom of pyramidane and substituted pyramidane molecules as hydrogen atom acceptors. We also studied the influence of substituents attached to base carbon atoms on the properties of hydrogen bonds involving apical carbon atoms.
Methodology
All geometry optimizations, vibrational spectra, wave function files, and interaction energy calculations performed within this study were performed using the Gaussian0923 software package and the MP2 method24 combined with the aug-cc-pVTZ basis set.25a Interaction energies were calculated using the full counterpoise correction method to account for the basis set superposition error (BSSE). For the most stable geometries, interaction energies were re-calculated using a very accurate CCSD(T)/CBS26,27 level of theory by applying the double-triple (D, T) extrapolation scheme proposed by Helgaker and coworkers.28 Energies of O–H⋯C interactions were calculated for model systems consisting of pyramidane and its derivatives and water molecules. Studied interactions involved the apical C atom of pyramidane (and its derivatives) and the hydrogen atom of a water molecule. Molecular electrostatic potentials (MEPs) were calculated and visualized using the Wave Function Analysis – Surface Analysis Suite (WFA–SAS) software.29 Energy decomposition analysis (EDA) was done using the PSI4 software and the SAPT2/aug-cc-pVTZ level of theory.30,31 All structures were visualized using Avogadro and Mercury programs.32,33 The Cambridge Structural Database (CSD)34 was searched using the ConQuest35 software to find all square-pyramidal structures in which an apical carbon atom interacts with a hydrogen atom. The CSD was searched for all square-pyramidal structures with a carbon atom at the apex that is involved in a non-covalent contact with a hydrogen atom of another molecule. The distance between interacting C and H atoms was ≤ 2.9 Å (the sum of van der Waals radii for C and H), while the hydrogen atom positions were normalized.36 The R factor was chosen to be < 0.05, and only non-disordered structures with 3D coordinates determined were considered for analysis. Hirshfeld maps were calculated using the Crystal Explorer software.37 Hirshfeld surfaces for ZUQRUY and VIZMUN structures were calculated and mapped over dnorm. Non-covalent index (NCI) analysis was performed using Multiwfn and VMD programs at the MP2/aug-cc-pVTZ level, while the bond critical points were calculated and visualized in the Multiwfn program.38–40 Natural bond orbital (NBO) analysis was performed at the MP2/cc-pVTZ level.25b,41,42Results and discussion
Geometry optimizations and molecular electrostatic potential calculations
The optimized structures of pyramidane and its derivatives (Tables S1–S6, ESI†) are given in Fig. 2.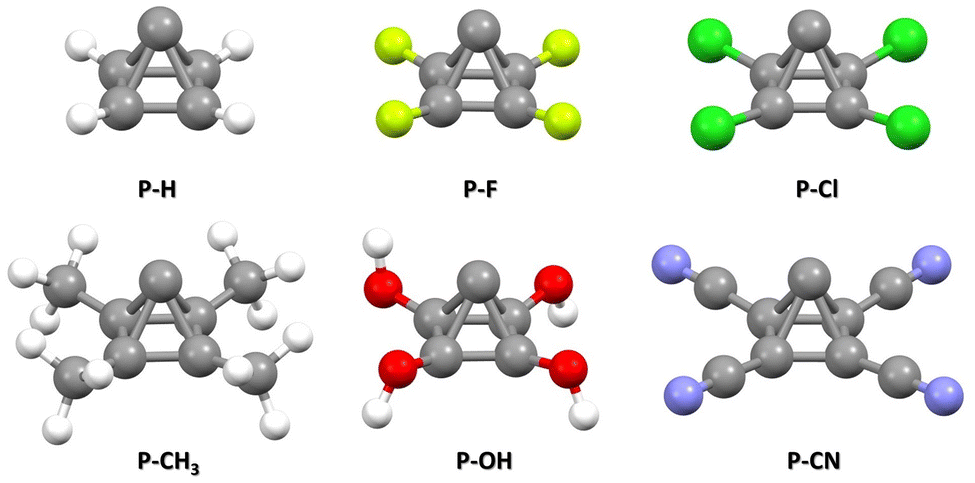 | ||
| Fig. 2 Optimized geometries of a pyramidane molecule and its derivatives with –F, –Cl, –CH3, –OH, and –CN substituents attached to four basal C atoms. | ||
Vibrational frequencies were calculated, and the results showed no negative frequencies for any of the studied molecules, indicating that the optimized geometries represent true minima (see Fig. S1–S6 in the ESI†). Optimized geometries were used for MEP calculations. Calculated MEPs with the electrostatic potential values at the critical points above the apical carbon atoms are given in Fig. 3.
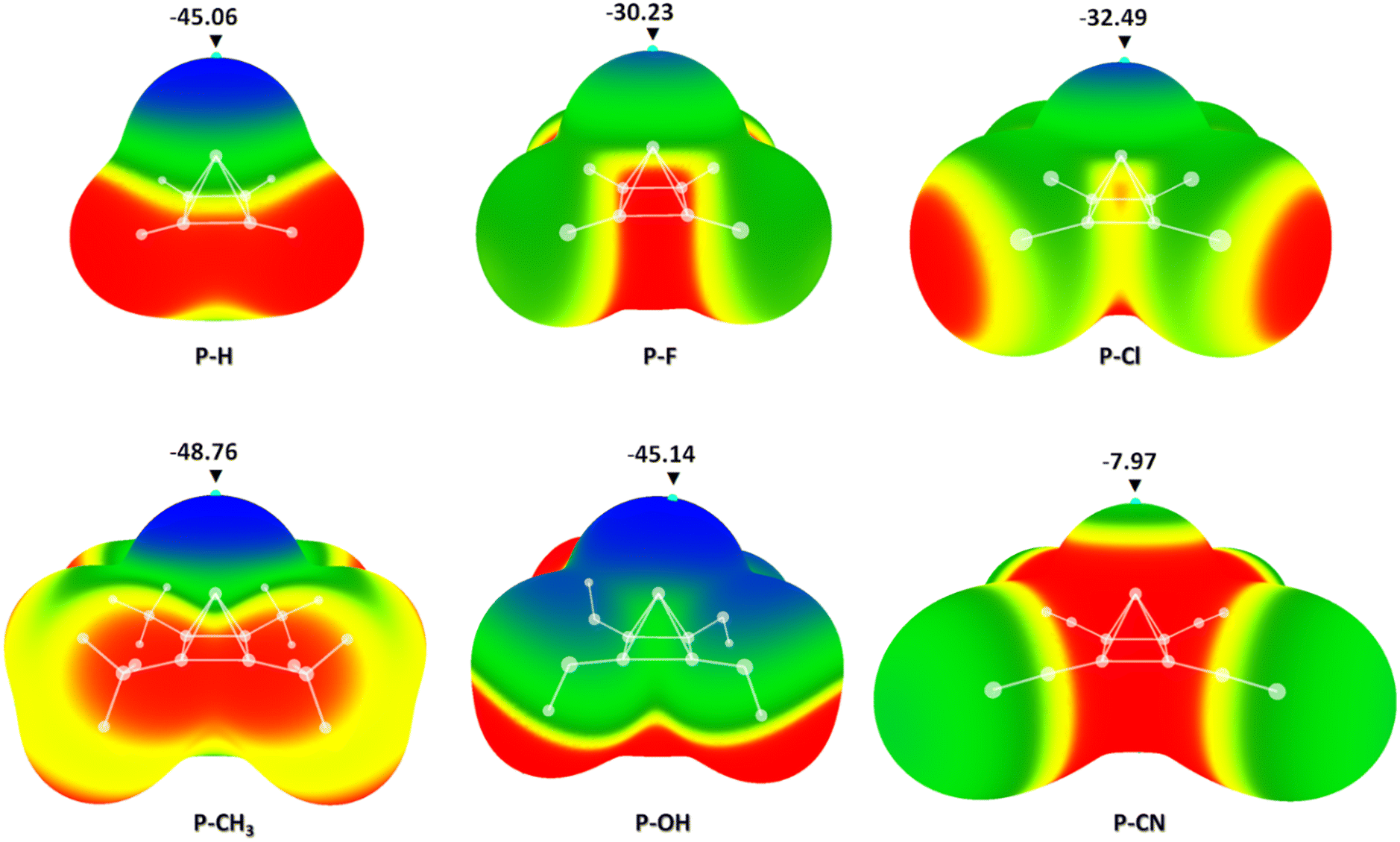 | ||
| Fig. 3 Calculated electrostatic potential maps of a pyramidane molecule and its derivatives with –F, –Cl, –CH3, –OH and –CN substituents attached to four basal C atoms. Values of energies in critical points are given in kcal mol−1. Colour ranges, in kcal mol−1, are: red, greater than 7.11; yellow, from 7.11 to 0.00; green, from −27.66 to 0.00; blue, more negative than −27.66. Blue dots refer to local minima on the molecular surfaces. | ||
The results of molecular electrostatic potential calculations showed that in the case of all six pyramidal molecules, there is a strong negative electrostatic potential above the apical carbon atom. The strongest negative potential was detected in the case of the pyramidane molecule with four –CH3 substituents (P-CH3) with the energy in the critical point of −48.76 kcal mol−1. Quite strong negative potentials were also detected in pyramidane molecules substituted with four –OH groups (P-OH, −45.14 kcal mol−1) and non-substituted pyramidane molecule (P-H, −45.06 kcal mol−1). The least negative potential was calculated in the case of the pyramidane molecule with four –CN substituents (P-CN, −7.97 kcal mol−1). For the structure containing the most negative critical point above the apical C atom (P-CH3) natural population analysis (NPA) charges were calculated. The results confirmed the existence of a negative charge on the apical carbon atom (NPA charge in the C atom: −0.21). This is in agreement with previous results indicating that the electrostatic potential on the surface above the apical carbon atom may be negative. In a recent study by Pendás and coworkers, it was shown through ELF and QTAIM analysis that the apical carbon atom possesses a lone pair of electrons which generates the negative electrostatic potential around it.43
Interaction energy calculations
Interaction energies were calculated on model systems consisting of pyramidane or substituted pyramidane molecules and water molecules. One O–H fragment of the water molecule was orientated to form linear O–H⋯C interaction with the apical carbon atom of pyramidane or substituted pyramidane. The distance between interacting carbon and hydrogen atoms was varied, and interaction energies were calculated. Interaction energies calculated at the MP2/aug-cc-pVTZ level are given in Fig. 4.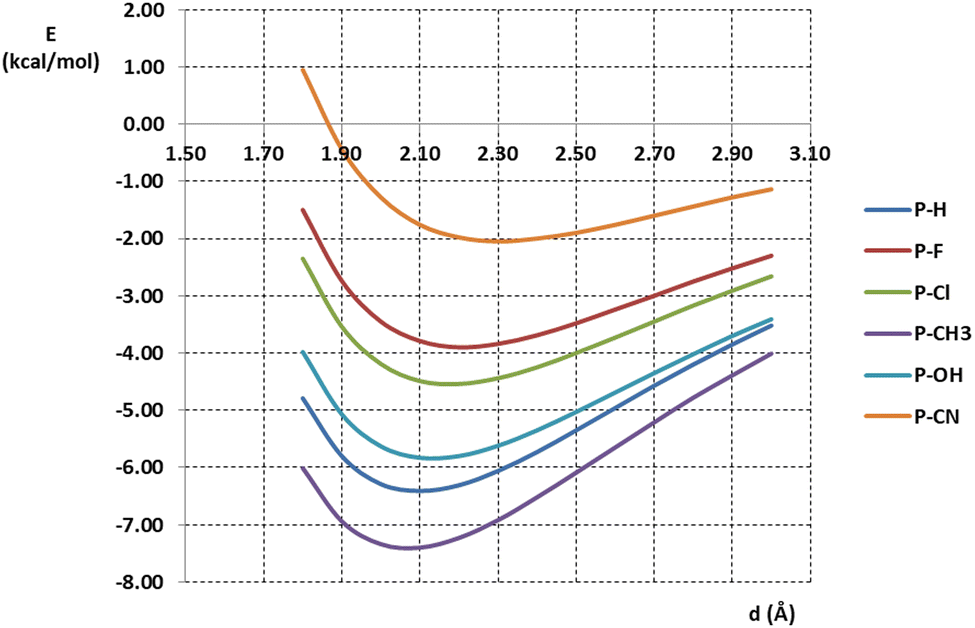 | ||
| Fig. 4 Energies of O–H⋯C interactions between pyramidane or pyramidane derivatives and water molecule calculated at the MP2/aug-cc-pVTZ level of theory. | ||
The calculated energies of O–H⋯C interaction were in the interval from −7.40 to −2.05 kcal mol−1. The strongest interaction (−7.40 kcal mol−1) was found in the model system containing the P-CH3 molecule. Very strong interaction was also found in the model system containing the unsubstituted pyramidane molecule, P-H (−6.41 kcal mol−1). On the other hand, the weakest interaction was found in model system containing pyramidane molecule with CN substituents (P-CN, −2.05 kcal mol−1). The geometries with optimal carbon–hydrogen distances are given in Fig. 5.
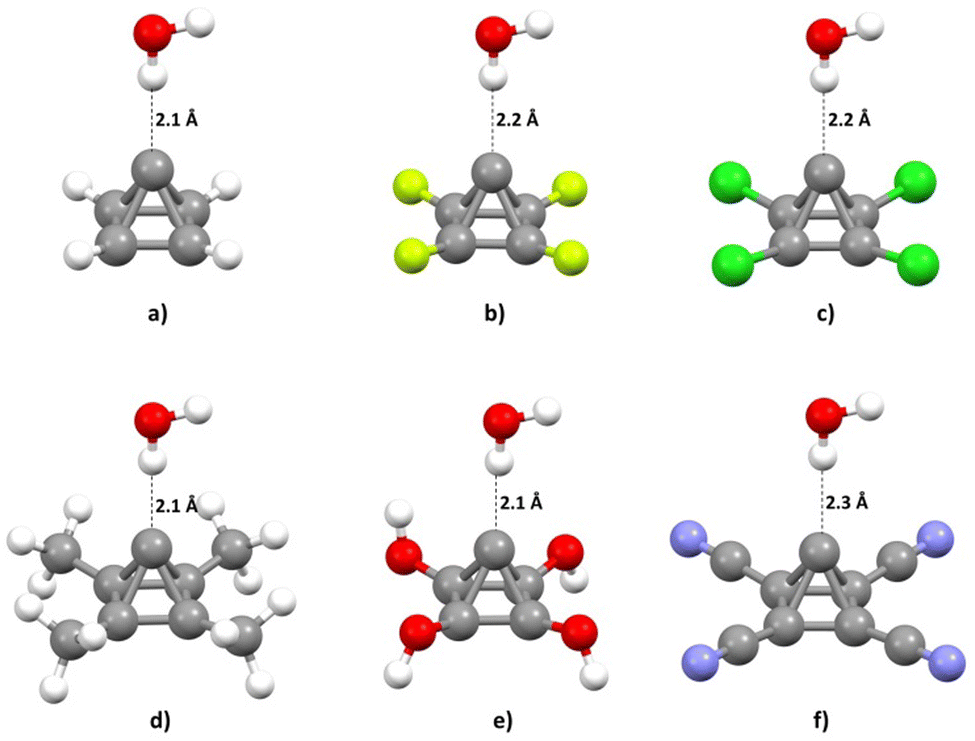 | ||
| Fig. 5 Calculated optimal geometries (a–f) for studied model systems. | ||
Analysis of the calculated hydrogen bond lengths is consistent with the results of interaction energies calculations and analysis of MEPs; the shortest hydrogen bonds (2.1 Å) are detected in the case of pyramidane molecules with the strongest interactions and the strongest negative electrostatic potential above the apical carbon atom (P-CH3, P-OH, P-H). On the other hand, the longest O–H⋯C interaction was detected in the case of a hydrogen bond involving the P-CN molecule (the weakest interaction and the lowest negative potential value over the apical carbon atom). These findings are also consistent with the calculated energies of O–H⋯C interactions (Table 1). For the optimal geometries calculated at the MP2/aug-cc-pVTZ level, interaction energies were re-calculated using a very accurate CCSD(T)/CBS level of theory (Table 1).
| Model system | O–H⋯C distance (Å) | ΔECCSD(T)/CBS (kcal mol−1) |
|---|---|---|
| P-H | 2.1 | −6.41 |
| P-F | 2.2 | −3.92 |
| P-Cl | 2.2 | −4.58 |
| P-CH3 | 2.1 | −7.43 |
| P-OH | 2.1 | −5.87 |
| P-CN | 2.3 | −2.00 |
The results showed that calculated interaction energies at the MP2/aug-cc-pVTZ level were in excellent agreement with the interaction energies calculated at the CCSD(T)/CBS level of theory. The strongest interaction calculated at the CCSD(T)/CBS level was found in the model system consisting of P-CH3 molecule and water (ΔECCSD(T)/CBS = −7.43 kcal mol−1). To better understand the nature of hydrogen bonding, the NBO analysis was performed on this model system. The results confirmed that electron transfer occurs from the lone pairs on the apical carbon atom to the antibonding orbital of the hydrogen atom in the water molecule. Very strong interactions were also calculated in the case of model systems containing P-OH (ΔECCSD(T)/CBS = −5.87 kcal mol−1) and P-H (ΔECCSD(T)/CBS = −6.41 kcal mol−1) molecules. Moderately strong interactions were calculated in the case of P-F and P-Cl molecules as hydrogen atom acceptors (−3.92 and −4.58 kcal mol−1, respectively). As expected, the weakest interaction was calculated for a model system containing the P-CN molecule (ΔECCSD(T)/CBS = −2.00 kcal mol−1). These trends are generally consistent with the trends in the calculated values of the negative electrostatic potential above the apical carbon atom (Fig. 3), except for P-OH and P-H molecules. Interaction energies were also calculated at the MP2/aug-cc-pVTZ level without taking into account the Basis Set Superposition Error (BSSE) and the trends in energy values were not changed (Table S13, ESI†).
Hydrogen bond critical points were calculated at the MP2/aug-cc-pVTZ level and visualized (Fig. S7, ESI†), since it was shown that analysis of electron densities in these points can give deeper insight into the properties of hydrogen bonds.44 Calculated values of electron density at bond critical points are in good correlation with calculated values of electrostatic potential at critical points above the apical carbon atom (Table S13, ESI†).
However, it is important to note that analysis of electrostatic potential maps could be used to explain only the contribution of electrostatics to the calculated interaction energies, while other contributions remain unknown. To reveal the role of other contributions to the total energy of the studied O–H⋯C interactions, we have performed an energy decomposition analysis.
Energy decomposition analysis
The energy decomposition analysis was performed using the SAPT2 method and the aug-cc-pVTZ level of theory. The results of the EDA calculations are given in Table 2.| P-H | P-F | P-Cl | P-CH3 | P-OH | P-CN | |
|---|---|---|---|---|---|---|
| Electrostatics | −9.65 | −5.47 | −6.01 | −10.64 | −8.72 | −2.66 |
| Exchange | 10.02 | 6.30 | 6.53 | 10.80 | 9.61 | 4.11 |
| Induction | −3.59 | −2.15 | −2.30 | −4.01 | −3.39 | −1.32 |
| Dispersion | −3.13 | −2.51 | −2.73 | −3.52 | −3.25 | −2.14 |
| Total SAPT | −6.35 | −3.84 | −4.50 | −7.37 | −5.75 | −2.00 |
| CCSD(T)/CBS | −6.41 | −3.92 | −4.58 | −7.43 | −5.87 | −2.00 |
The results of EDA calculations clearly indicate that the main energetic contribution to the O–H⋯C hydrogen bonds is the electrostatics. The strongest electrostatic contributions were calculated for model systems with the strongest interaction energies (P-CH3, −10.64 kcal mol−1, and P-H, −9.65 kcal mol−1). Not surprisingly, the lowest contribution of the electrostatics was calculated for interaction involving the P-CN molecule, −2.66 kcal mol−1. Besides electrostatics, contributions of induction and dispersion to the total energy of interaction are significant in all model systems. It could be noticed that the contribution of induction is stronger in the case of the P-H molecule than the P-OH molecule, which may be the reason why, for these two systems, values of electrostatic potential in critical points are not consistent with the trends in calculated interaction energies. It is important to note that interaction energies calculated at the SAPT2/aug-cc-pVTZ level are in excellent agreement with CCSD(T)/CBS energies.
Analysis of crystal structures
Although neither pyramidane nor its derivatives studied in the frame of this research were synthesized and crystalized yet, analysis of possible carbon–hydrogen contacts in similar structures could be helpful for the prediction of the potential of the apical carbon atom in pyramidal molecules to form hydrogen bonds as hydrogen atom acceptors. Using the criteria defined in the Methodology section, the CSD was searched, and only two structures in which short carbon–hydrogen contacts exist were extracted (refcodes VIZMUN and ZUQRUY). Three-dimensional representations of these structures are given in Fig. 6. Both structures contain Ru heteroatoms connected to an apical carbon atom.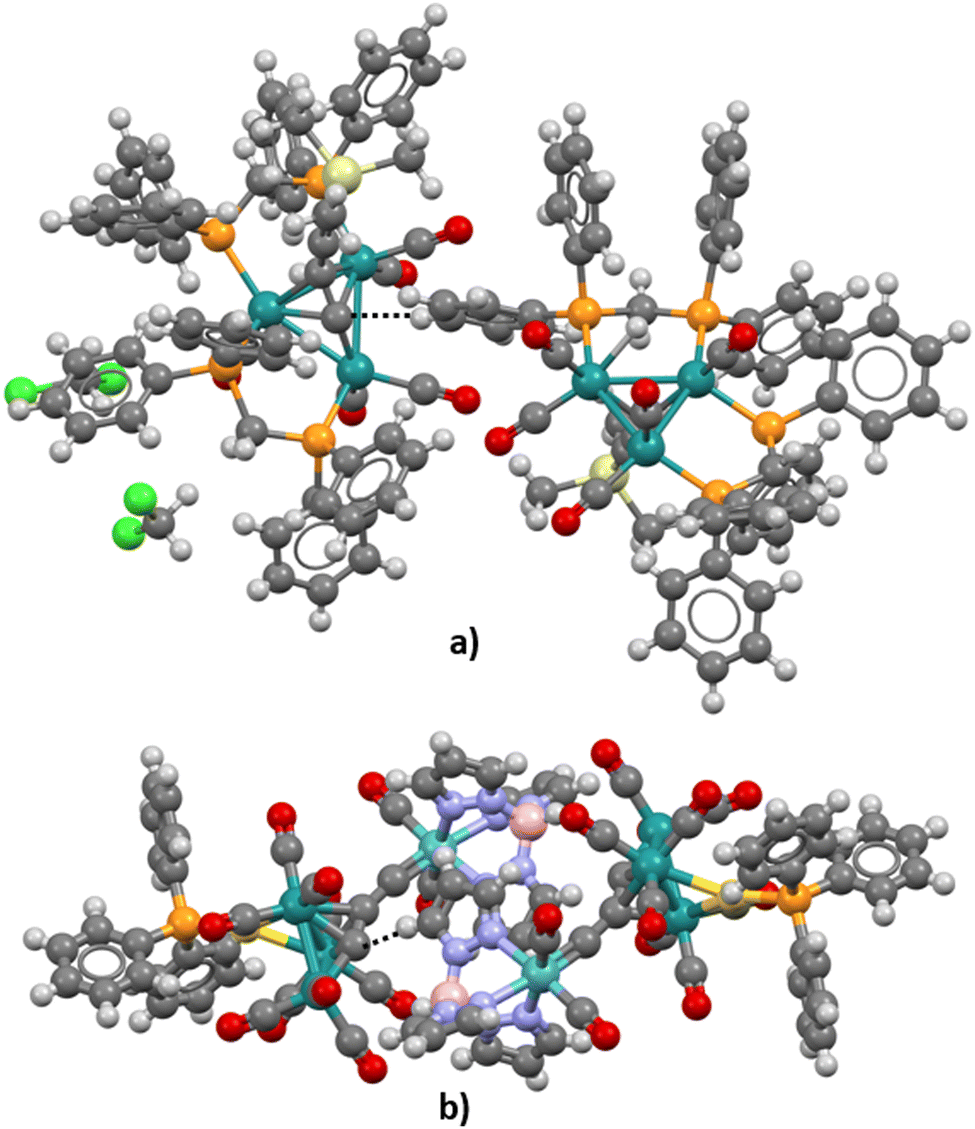 | ||
| Fig. 6 The fragment of (a) ZUQRUY and (b) VIZMUN structures with short contacts (dashed line) between apical carbon atoms and hydrogen atoms from another molecule. The carbon–hydrogen distances are d = 2.74 Å in ZUQRUY and d = 2.80 Å in the VIZMUN structure. | ||
In the case of the ZUQRUY structure, the distance between the apical carbon atom of the pyramidal fragment and the hydrogen atom of the C–H fragment is 2.74 Å, which is significantly shorter than the sum of van der Waals radii for C and H atoms (2.9 Å). Also, the angle defined by C–H⋯C atoms is 175.08°, indicating the presence of a directional interaction, which is usual in the case of strong hydrogen bonds. A similar situation was noticed in the VIZMUN structure (the distance between C and H atoms was measured to be 2.80 Å, while the angle defined by C–H⋯C atoms is 156.66°). Although the comparison between the measured distance between two atoms and the sum of van der Waals radii is not definite proof of the existence of non-covalent contacts, it is a good indicator of possible C–H⋯C interactions.
The tendency of apical carbon atoms to form non-covalent contacts with hydrogen atoms in these structures is evident from the analysis of Hirsfeld surfaces for ZUQRUY and VIZMUN structures (Fig. 7). Red regions above the apical carbon atoms on the Hirshfeld surfaces for both ZUQRUY and VIZMUN structures indicate the existence of C–H⋯C interactions between C–H fragments and apical carbon atoms.
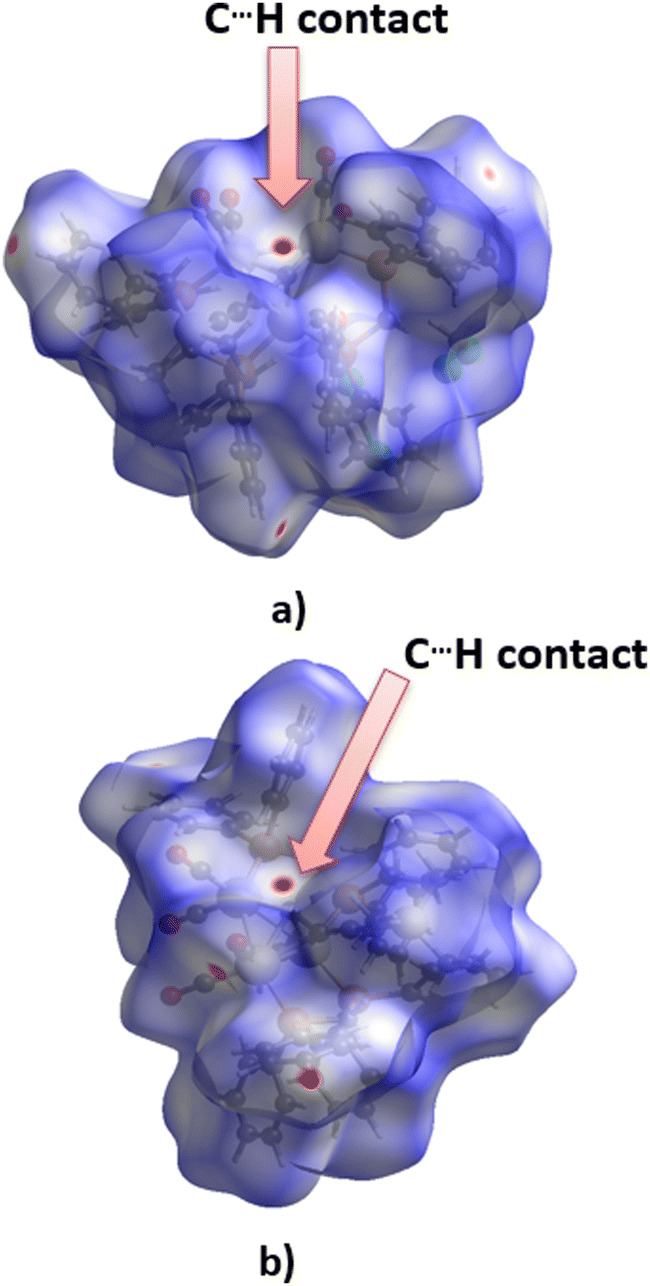 | ||
| Fig. 7 Hirshfeld surfaces (dnorm) calculated for fragments of (a) ZUQRUY and (b) VIZMUN structures. The red area on the surface above the apical carbon atoms indicates the existence of C⋯H contacts. | ||
To confirm the existence of C–H⋯C interactions in these crystal structures, non-covalent index analysis was performed. The calculated NCI plots are given in Fig. 8. Results of the analysis of NCI plots showed that in the case of the ZUQRUY structure (Fig. 8a), there is an area of non-covalent bonding between an apical carbon atom and a hydrogen atom from the neighbouring C–H fragment (green area). Similar results were obtained in the case of the VIZMUN structure (Fig. 8b). The green area between the apical carbon atom and a hydrogen atom of the C–H fragment in the VIZMUN structure also indicates the existence of an attractive non-covalent contact.
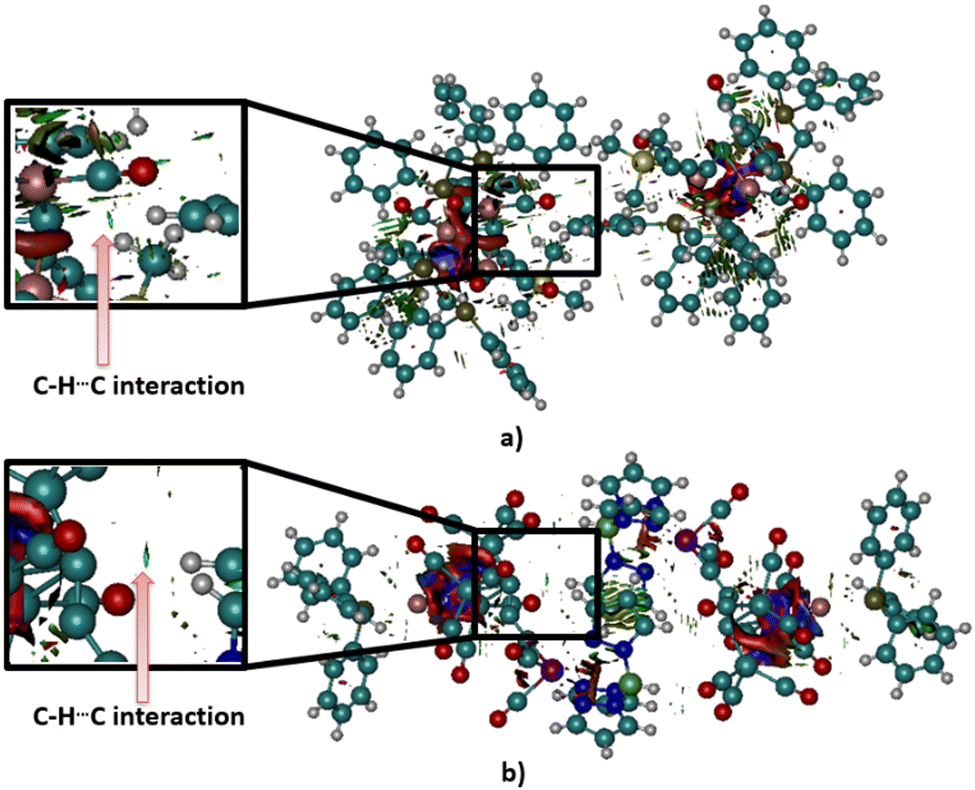 | ||
| Fig. 8 Calculated NCI plots for (a) ZUQRUY and (b) VIZMUN structures. Green surfaces correspond to the attractive regions of non-covalent interactions (the isosurface value was selected to be 0.5, while the interval of the color range is from −0.035 to 0.2). | ||
Conclusions
In this work, high-level quantum chemical calculations were combined with the analysis of available crystal structures to predict the properties of hydrogen bonds involving apical pyramidane carbon atoms as hydrogen acceptors. Six structures, including pyramidane (P-H) and its derivatives with four substituents on basal carbon atoms (P-F, P-Cl, P-CH3, P-OH, P-CN), were considered within this study. The results of the analysis of molecular electrostatic potentials showed that there is an area of negative electrostatic potential above the apical carbon atom in all six examined structures. The strongest negative potential was detected above the apical carbon atom of the P-CH3 structure (−48.76 kcal mol−1), while the very strong negative potential was detected in the case of P-OH (−45.14 kcal mol−1) and P-H (−45.06) molecules, too. The weakest negative potential was calculated above the apical carbon atom of the P-CN structure (−7.97 kcal mol−1). Since these results indicated that apical carbon atoms might act as hydrogen atom acceptors in hydrogen bonds, six model systems containing already mentioned pyramidal molecules and water molecules were made, and interaction energies were calculated. The results of interaction energy calculations showed that studied O–H⋯C interactions can be quite strong, up to ΔECCSD(T)/CBS = −7.43 kcal mol−1 in the case of P-CH3 molecule as a hydrogen atom acceptor. The weakest hydrogen bond was calculated in the case of a model system containing a P-CN molecule (−2.00 kcal mol−1). This agrees with the results of electrostatic potential calculations, indicating that these interactions are mainly electrostatic. Energy decomposition analysis was performed on all model systems, and the results confirmed that all studied interactions are indeed primarily electrostatic, with strong contributions from induction and dispersion. Although the studied molecules have not yet been synthesized, crystal structures of similar pyramidal molecules with apical carbon atoms involved in hydrogen bonds were found in the Cambridge Structural Database.The results presented in this study are in agreement with previous results showing that increased electron density is present at the apex of pyramidane-like molecules. To the best of our knowledge, this is the first study of the energies of hydrogen bonds involving apex pyramidane carbon as a hydrogen atom acceptor. The results obtained within this study may be of great importance for understanding the non-covalent bonding patterns in the case of hydrogen bonds involving pyramidal-shaped molecules.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported by the Ministry of Science, Technological Development and Innovation of Republic of Serbia (Contract numbers: 451-03-66/2024-03/200026 and 451-03-66/2024-03/200168).Notes and references
- T. Wang, Y. Zhang, B. Huang, B. Cai, R. R. Rao, L. Giordano, S.-G. Sun and Y. Shao-Horn, Nat Catal., 2021, 4, 753 CrossRef.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362 RSC.
- B. Dereka, Q. Yu, N. H. C. Lewis, W. B. Carpenter, J. M. Bowman and A. Tokmakoff, Science, 2021, 371, 160 CrossRef PubMed.
- P. A. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52 CrossRef PubMed.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399 CrossRef PubMed.
- M. Nishio, Y. Umezawa, J. Fantini, M. S. Weissd and P. Chakrabartie, Phys. Chem. Chem. Phys., 2014, 16, 12648 RSC.
- J. Ran and P. Hobza, J. Phys. Chem. B, 2009, 113(9), 2933 CrossRef PubMed.
- D. S. Kretić, D. I. Radovanović and D. Ž. Veljković, Phys. Chem. Chem. Phys., 2021, 23, 7472 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637 CrossRef.
- S. Grabowski, Chem. Commun., 2024, 60, 6239 RSC.
- A. J. Arduengo, III, S. F. Gamper, M. Tamm, J. C. Calabrese, F. Davidson and H. A. Craig, J. Am. Chem. Soc., 1995, 117, 572 CrossRef.
- M. Jabłoński and M. Palusiak, Phys. Chem. Chem. Phys., 2009, 11, 5711 RSC.
- S. J. Grabowski, CrystEngComm, 2023, 25, 4550 RSC.
- O. Loveday and J. Echeverría, Cryst. Growth Des., 2021, 21, 5961 CrossRef.
- V. Y. Lee, Y. Ito, A. Sekiguchi, H. Gornitzka, O. A. Gapurenko, V. I. Minkin and R. M. Minyaev, J. Am. Chem. Soc., 2013, 135, 8794 CrossRef PubMed.
- Q. Sun, C. G. Daniliuc, X. Yu, C. Mück-Lichtenfeld, G. Kehr and G. Erker, J. Am. Chem. Soc., 2022, 144, 7815 CrossRef PubMed.
- V. Y. Lee, H. Sugasawa, O. A. Gapurenko, R. M. Minyaev, V. I. Minkin, H. Gornitzka and A. Sekiguchi, Chem. – Eur. J., 2016, 22, 17585 CrossRef PubMed.
- V. Y. Lee, Y. Ito, O. A. Gapurenko, A. Sekiguchi, V. I. Minkin, R. M. Minyaev and H. Gornitzka, Angew. Chem., Int. Ed., 2015, 54, 5654 CrossRef PubMed.
- Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2003, 42, 3578 CrossRef PubMed.
- V. Y. Lee, O. A. Gapurenko, Y. Ito, T. Meguro, H. Sugasawa, A. Sekiguchi, R. M. Minyaev, V. I. Minkin, R. H. Herber and H. Gornitzka, Organometallics, 2016, 35, 346 CrossRef.
- F. Ren, D. Cao, W. Shi and H. Gao, J. Mol. Model., 2016, 22, 97 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- (a) R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef CAS; (b) T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. A. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479 CrossRef CAS.
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2004, 108, 10200 CrossRef CAS.
- (a) T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639 CrossRef CAS; (b) A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243 CrossRef CAS; (c) A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper and J. Olsen, Chem. Phys. Lett., 1999, 302, 437 CrossRef CAS; (d) D. Feller, J. Chem. Phys., 1992, 96, 6104 CrossRef CAS.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679 CrossRef.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. S. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185 CrossRef PubMed.
- E. G. Hohenstein and C. D. Sherrill, J. Chem. Phys., 2010, 133, 014101 CrossRef PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4, 17 CrossRef PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Cryst., 2008, 41, 466 CrossRef.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed; C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., 2016, 72, 171 CrossRef.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
- L. Vidal, D. Barrena-Espés, J. Echeverría, J. Munárriz and Á. M. Pendás, ChemPhysChem, 2024, 25, e202400329 CrossRef CAS PubMed.
- L. Soriano-Agueda, Int. J. Quantum Chem., 2024, 124, e27352 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03809f |
| This journal is © the Owner Societies 2025 |