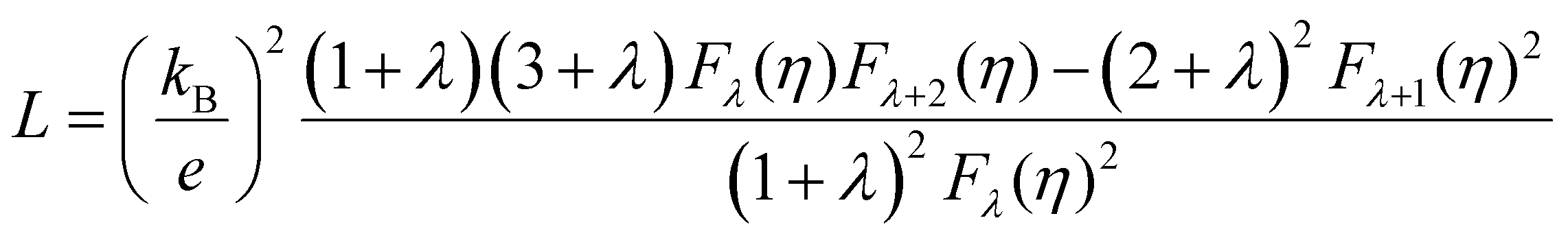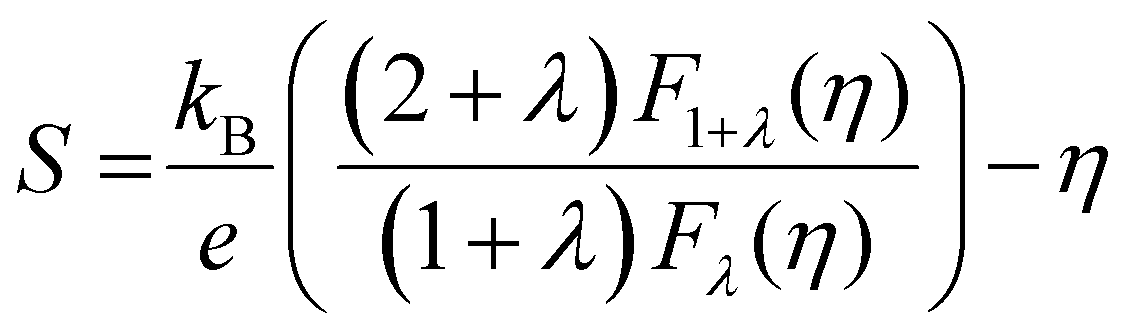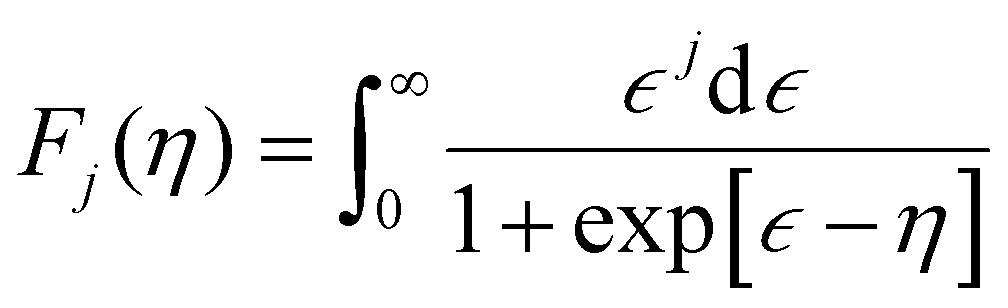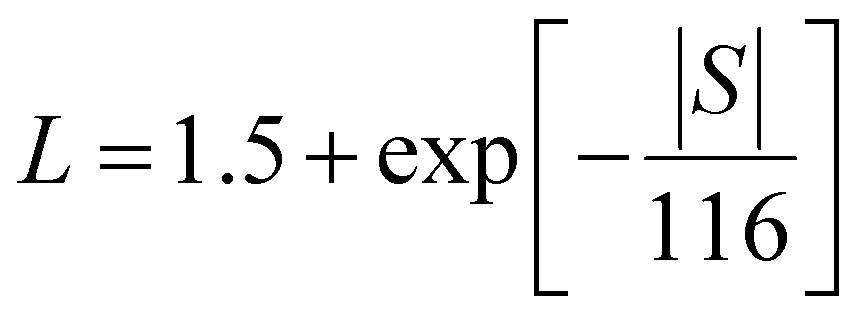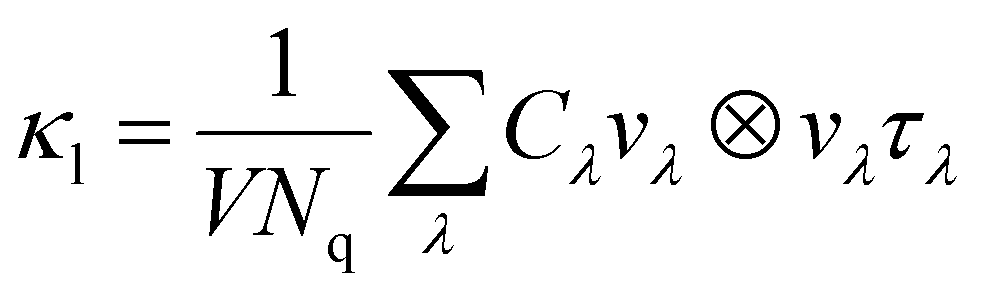Potential thermoelectric material Tl3XS4 (X = V, Nb, Ta) with ultralow lattice thermal conductivity†
Received
17th October 2024
, Accepted 4th December 2024
First published on 10th December 2024
Abstract
The demand for sustainable energy solutions has driven intensive research into advanced thermoelectric (TE) materials, to harness waste heat for efficient power generation. Recently, several studies have revealed that Tl3VS4 possesses an ultralow lattice thermal conductivity, despite its simple body-centered cubic lattice structure. This paper focuses on the TE properties of Tl3XS4 (X = V, Nb, Ta) compounds through a systematic exploration utilizing first-principles calculations and semiclassical Boltzmann transport theory. The results of the AIMD simulation and phonon calculation reveal the excellent dynamic stability and thermal stability of Tl3XS4 at 300, 500, and 700 K. Moreover, we find that the Tl3XS4 compounds present ultralow lattice thermal conductivity (<0.5 W m−1 K−1 at 300 K), and the nanostructure strategy is effective. Based on the outstanding Seebeck coefficient and ultralow lattice thermal conductivity, the optimal ZT values of Tl3VS4, Tl3NbS4, and Tl3TaS4 at 300 K are determined to be 1.17 (p-type), 0.84 (n-type), and 0.65 (p-type), respectively. Additionally, at each considered temperature, the maximum ZT values of p-type (n-type) Tl3XS4 follow the order: Tl3VS4 > Tl3NbS4 > Tl3TaS4 (Tl3NbS4 > Tl3VS4 > Tl3TaS4). Our results demonstrate that Tl3XS4 (X = V, Nb, Ta) compounds are promising thermoelectric materials. This exhaustive research enhanced our nuanced comprehension of the electronic, dynamic, and thermoelectric attributes of Tl3XS4 (X = V, Nb, Ta), thereby offering valuable insights into the TE field.
1. Introduction
The exploration of advanced thermoelectric (TE) materials has become paramount in the pursuit of sustainable energy technologies, as these materials offer the unique capability to convert waste heat into valuable electrical power. Thermoelectric materials are characterized by TE coefficients that play a crucial role in determining their efficiency. The thermoelectric power factor (PF), defined as PF = S2σ, where S is the Seebeck coefficient and σ is the electrical conductivity,1 represents a crucial metric. High power factors contribute to efficient TE energy conversion.2 Simultaneously, the dimensionless figure of merit (ZT) encapsulates the overall efficiency of TE materials and is given by ZT = S2σT/(κe + κl),3,4 where κe and κl are the electronic and lattice thermal conductivities,5 respectively. Achieving a high ZT value is paramount for the practical application of thermoelectric materials in energy conversion devices.
Traditionally, thermoelectric research has been guided by empirical models, such as the pioneering work of Slack,6 emphasizing the exploration of materials with large unit cells and complex structures to achieve ultralow lattice thermal conductivities. Several materials, predominantly exhibiting complex structures, have been suggested for utilization in TE applications, such as skutterudites,7–9 half-Heuslers,10–13 and clathrates.14–16 However, Tl3VSe4 challenges this paradigm by showcasing low lattice thermal conductivities despite its simple body-centered cubic lattice structure.17 This enigma prompts a reevaluation of existing models and calls for a deeper exploration of the underlying mechanisms governing thermoelectric behavior.
In 2018, Mukhopadhyay17 introduced a nuanced perspective to clarify the ultralow lattice thermal conductivity in Tl3VSe4. His work delves into the intricate interplay of heat transport mechanisms, involving lattice phonons and localized oscillators, thereby providing valuable insights into the TE phenomena exhibited by these compounds.18–20 The application of multichannel transport theory further enriches our understanding, highlighting the need for a comprehensive approach to decipher the TE behavior of the Tl3XSe4 family of compounds. In addition, theoretical studies suggested that Tl3TaSe4 and Tl3VS4 have ultralow thermal conductivity (0.1–0.4 W m−1 K−1 at room temperature).21 The high σ and ultralow κl contribute to the high ZT value of 0.85 and 0.84 at 300 K for Tl3TaSe4 and Tl3VS4, respectively. However, the relaxation time has been set at a constant of τ = 9 × 10−14 s in their calculations of PF and ZT. Recently, Wang et al. revealed that Tl3XSe4 (X = V, Nb, Ta) is a potential TE material with excellent performance, including large Seebeck coefficient, high electrical conductivity, high power factor, and low thermal conductivity.22 In conclusion, these compounds of the Tl3XSe417–20,22 family typically exhibit ultralow κl and excellent TE properties and are expected to be candidates for a new generation of TE applications.
The simplicity of Tl3XY4 (X = V, Nb, Ta; Y = S, Se) structures, marked by high lattice symmetry and minimal disorder, positions these compounds as promising alternatives for scalable and efficient TE applications. However, the intricacies of their TE behavior necessitate a comprehensive investigation. Notice that the Tl3XS4-compounds (X = V, Nb, Ta) with a cubic body-centered lattice have been prepared and synthesized successfully,23,24 which boosts the investigation of TE properties. However, it is important to highlight that there is a lack of systematic research addressing the electronic, phonon, and thermoelectric properties of Tl3XS4 (X = V, Nb, Ta) in the existing literature. This study employs first-principles calculations and semiclassical Boltzmann transport theory to understand the electronic structure, phonon dispersion, thermal conductivity, and thermoelectric transmission characteristics of Tl3XS4 (X = V, Nb, Ta). By unraveling the underlying mechanisms, we aim to contribute to the evolving field of thermoelectric materials and shed light on the unique properties of Tl3XS4 compounds.
2. Computational details
Density functional theory (DFT) calculations of Tl3XS4 (X = V, Nb, Ta) were comprehensively performed using the Vienna Ab initio Simulation Package (VASP).25 All calculations utilized a plane-wave basis set and the projector-augmented wave (PAW) method,26,27 employing a 500 eV plane-wave cutoff energy. The exchange–correlation functional was treated within the generalized gradient approximation (GGA), utilizing the Perdew–Burke–Ernzerhof (PBE) parameterization.28 A convergence criterion of energy and force were set to 10−6 eV and 10−4 eV Å−1 for computational efficiency and accuracy, respectively. To capture the dynamic behavior of Tl3XS4 at elevated temperatures, AIMD29 simulations were conducted using VASP within the NVT ensemble based on the machine-learned force fields (MLFF)30–32 feature. The Nosé–Hoover thermostat maintain temperatures of 300, 500, and 700 K.
The Ab initio scattering and transport (AMSET) package33 was employed to calculate the electron transport properties, including the Seebeck coefficient S, electrical conductivity σ, carrier scattering rates, and carrier mobilities. The AMSET code employs the momentum relaxation time approximation (MRTA) to solve the Boltzmann transport equation. It provides reliable estimates of electron relaxation times by incorporating multiple scattering mechanisms, including acoustic deformation potential scattering (ADP), polar optical phonon scattering (POP), ionized impurity scattering (IMP), and piezoelectric scattering (PIE).34 These estimates are in excellent agreement with experimental measurements and electron–phonon couplings computed through the Wannier function.33 The lattice thermal conductivities are obtained utilizing the ShengBTE code,35 in which a dense q-point mesh of 20 × 20 × 20 and the nearest neighbors of 8 (cutoff value) were adopted. Interatomic force constants (IFCs) including the second-order and third-order were extracted from DFT calculations using VASP, and a 2 × 2 × 2 supercell was employed for accurate force constant determination. The Dynaphopy code36 and Phonopy code37 were utilized to gain insights into anharmonic effects and calculate phonon dispersions at the finite temperatures of 300, 500, and 700 K.
3. Results and discussion
3.1. Crystal structures and electronic structures
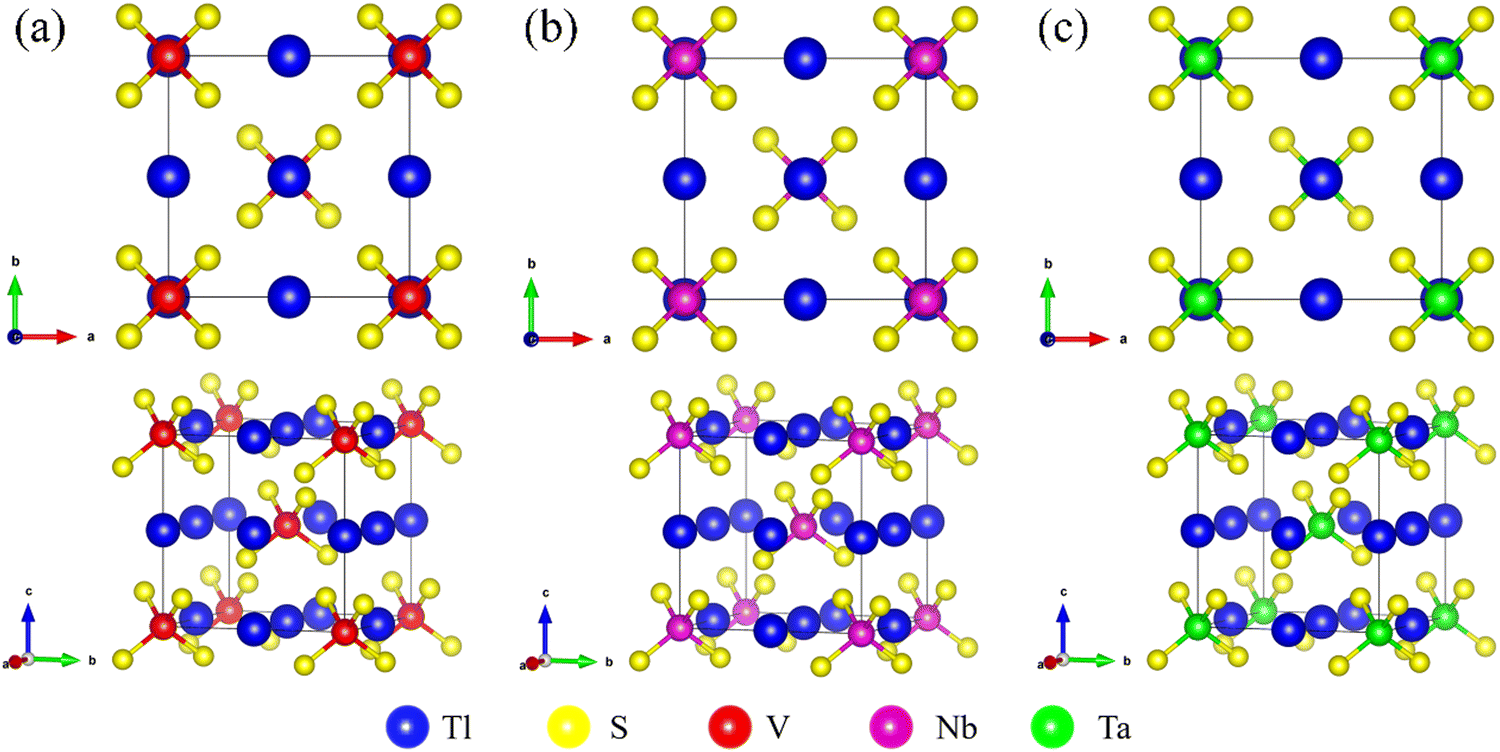
The Tl3XS4 (X = V, Nb, Ta) compounds exhibit a simple body-centered cubic phase with I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m (no. 217) space group. Fig. 1 demonstrate that the XS4− tetrahedra hold both the body center and vertices in the cubic lattice, while Tl atoms locate at the center of each edge and face of the cube. The relaxed lattice constants of Tl3XS4 (X = V, Nb, Ta) are given in Table 1, and the information of Tl3XSe4 (X = V, Nb, Ta) also been listed.
3m (no. 217) space group. Fig. 1 demonstrate that the XS4− tetrahedra hold both the body center and vertices in the cubic lattice, while Tl atoms locate at the center of each edge and face of the cube. The relaxed lattice constants of Tl3XS4 (X = V, Nb, Ta) are given in Table 1, and the information of Tl3XSe4 (X = V, Nb, Ta) also been listed.
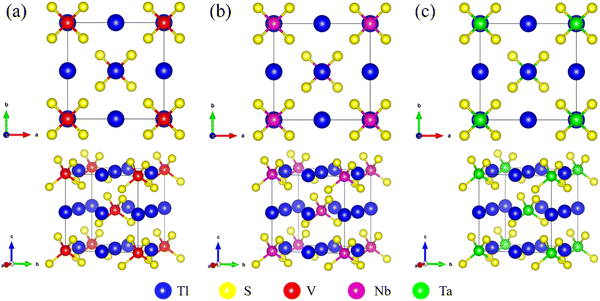 |
| | Fig. 1 The top views and side views of the crystal structure for Tl3VS4 (a), Tl3NbS4 (b), and Tl3TaS4 (c). | |
Table 1 Calculated lattice constants and band gap for Tl3XS4 (X = V, Nb, Ta). The results of Tl3XSe4 (X = V, Nb, Ta) are used for comparison
|
|
a = b = c (Å) |
E
g (eV) |
| Tl3VS4 |
7.67 |
2.88 (HSE06) 2.06 (PBE) |
| Tl3NbS4 |
7.81 |
3.24 (HSE06) 2.61 (PBE) |
| Tl3TaS4 |
7.84 |
3.31 (HSE06) 2.62 (PBE) |
| Tl3VSe422 |
7.89 |
2.33 (HSE06) 1.60 (PBE) |
| Tl3NbSe422 |
8.03 |
3.01 (HSE06) 2.18 (PBE) |
| Tl3TaSe422 |
8.05 |
2.89 (HSE06) 2.26 (PBE) |
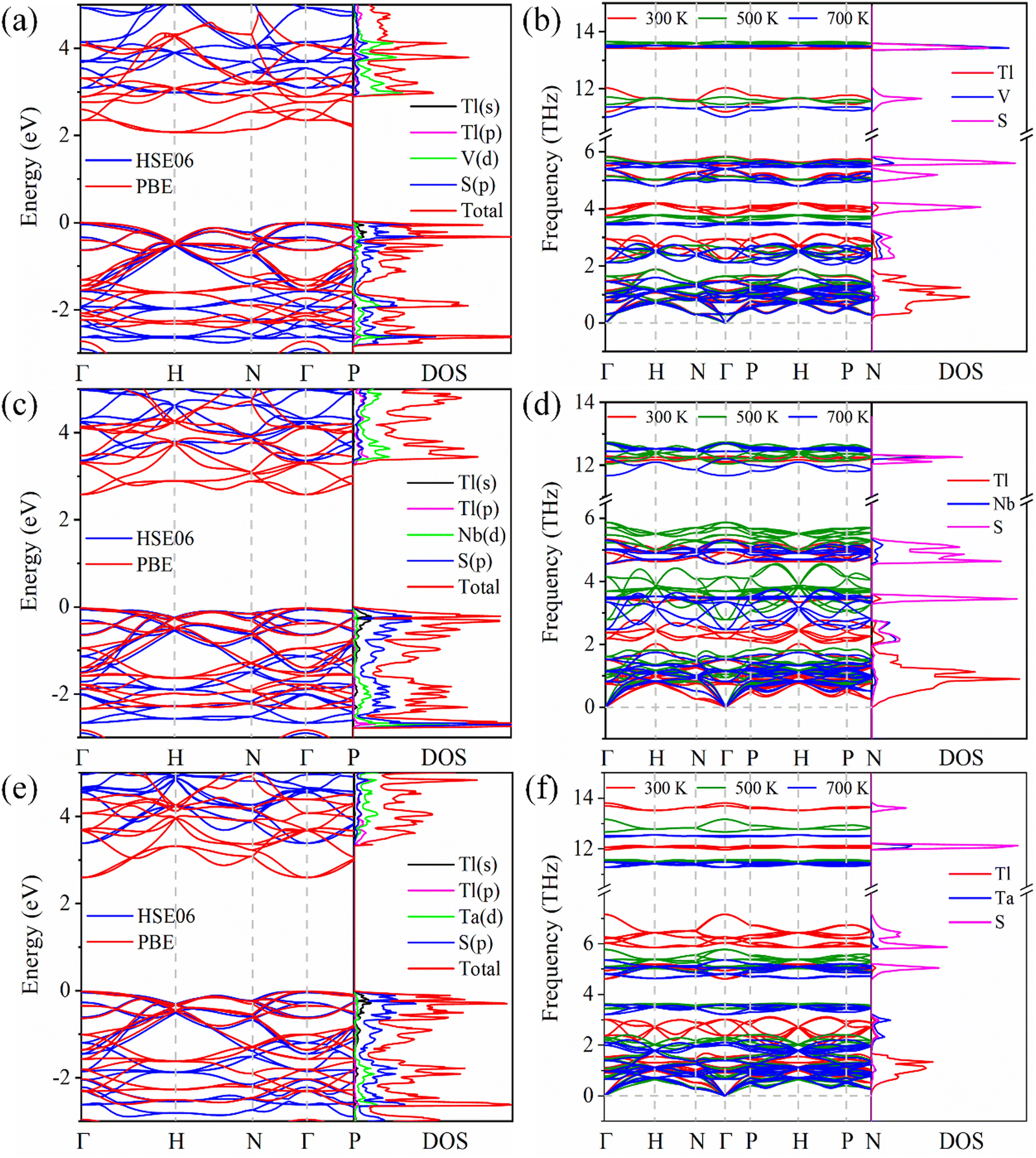
Using the PBE functional and HSE06 hybrid functional, we calculate the band structures of Tl3XS4 (X = V, Nb, Ta) (Fig. 2(a), (c) and (e)). The band gaps of Tl3VS4, Tl3NbS4, and Tl3TaS4 based on the PBE (HSE06) method are 2.06 (2.88), 2.61 (3.24), and 2.62 (3.31), respectively (the details are listed in Table 1). Additionally, the density of states (DOS) based on the HSE06 method revealed that p orbitals of S atoms largely contribute to the total DOS in the valence band maximum (VBM) with some contributions from s states of Tl atoms and V/Nb/Ta/-d orbitals, while the conduction band minimum (CBM) is predominately contributed by d orbitals of V/Nb/Ta atoms with some contributions from p states of Tl atoms. This is consistent with previously reported theoretical and experimental research findings.21,38,39
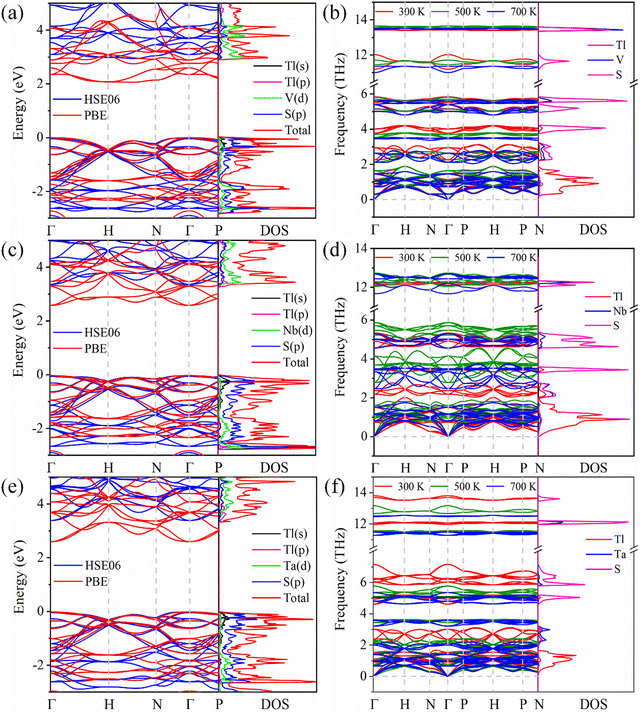 |
| | Fig. 2 Band structure and DOS, phonon structures and phonon DOS for Tl3VS4 (a) and (b), Tl3NbS4 (c) and (d), and Tl3TaS4 (e) and (f). | |
3.2. Stability and phonon structure
As is commonly understood, the establishment of a stable structure forms the foundation for the subsequent exploration of diverse material properties. To evaluate the dynamic stability of Tl3XS4 (X = V, Nb, Ta) and explore its phonon transport properties, we conduct calculations for the phonon band and DOS, as illustrated in Fig. 2(b), (d) and (f). Notably, imaginary frequency modes have not been observed in the phonon band of Tl3XS4 at 300, 500, and 700 K, indicating the extraordinary dynamical stabilities of Tl3XS4. The phonon DOS provides insights into the respective contributions of various atoms to the overall phonon spectrum. In the case of Tl3XS4, the predominant influence on the acoustic modes arises from the heavy Tl atoms. This observation can be attributed to the contribution of the atom to the phonon band, which is expressed as the ratio of atomic mass to unit cell mass.40 Besides, the optical modes primarily originate from the vibrations of lighter atoms S, with some additional contributions stemming from the vibrations of V/Nb/Ta. These findings are consistent with previously reported results.21 Therefore, for Tl3XS4 compounds, the contributions to lattice thermal conductivity from acoustic and optical modes predominantly arise from Tl and S atoms, respectively.
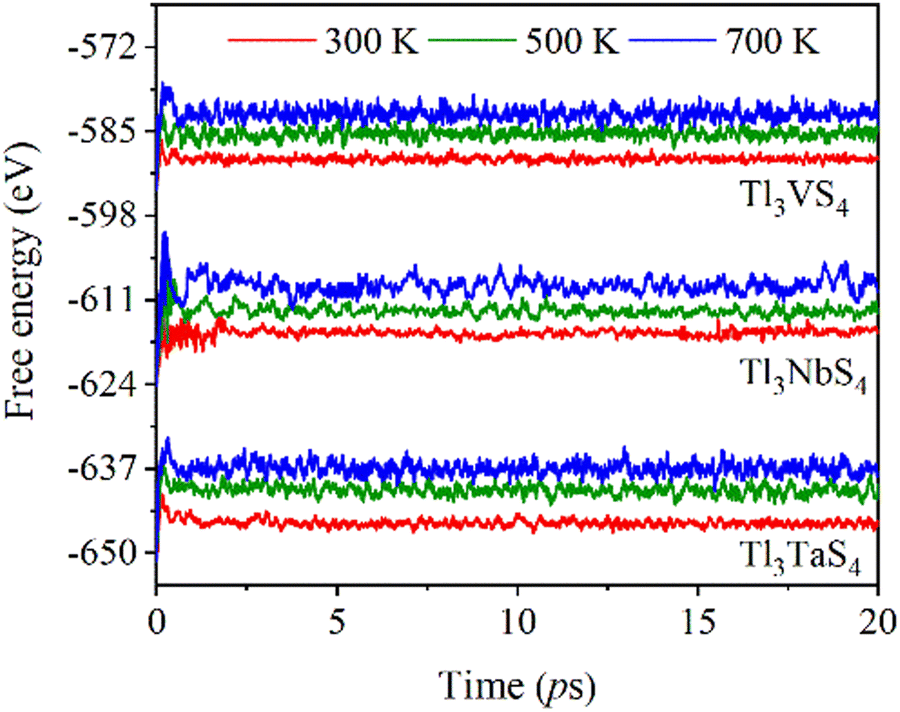
Furthermore, utilizing AIMD simulations, we obtain the free energy of Tl3XS4 (X = V, Nb, Ta) at temperatures of 300, 500, and 700 K (Fig. 3). A sufficiently long time of 20 ps was employed to guarantee the dependability of the simulation. These findings indicate the exceptional thermal stability of these materials across the three specified temperatures. After long enough simulation, the free energy of Tl3XS4 (X = V, Nb, Ta) remains stable, indicating the outstanding thermal stability of compounds at the specified temperatures.
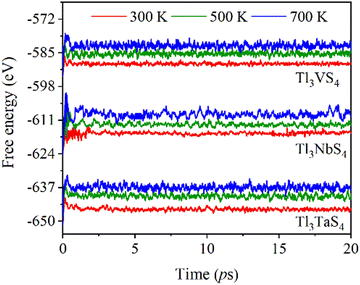 |
| | Fig. 3 Free energy as a function of simulation time in AIMD calculations for Tl3XS4 (X = V, Nb, Ta) at 300, 500, and 700 K. | |
3.3. Electrical transport properties
The absolute Seebeck coefficient of both p-type and n-type Tl3VS4 are presented in Fig. 4(a), (c) and (e). Generally, the Seebeck coefficient is defined as:41| | 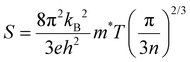 | (1) |
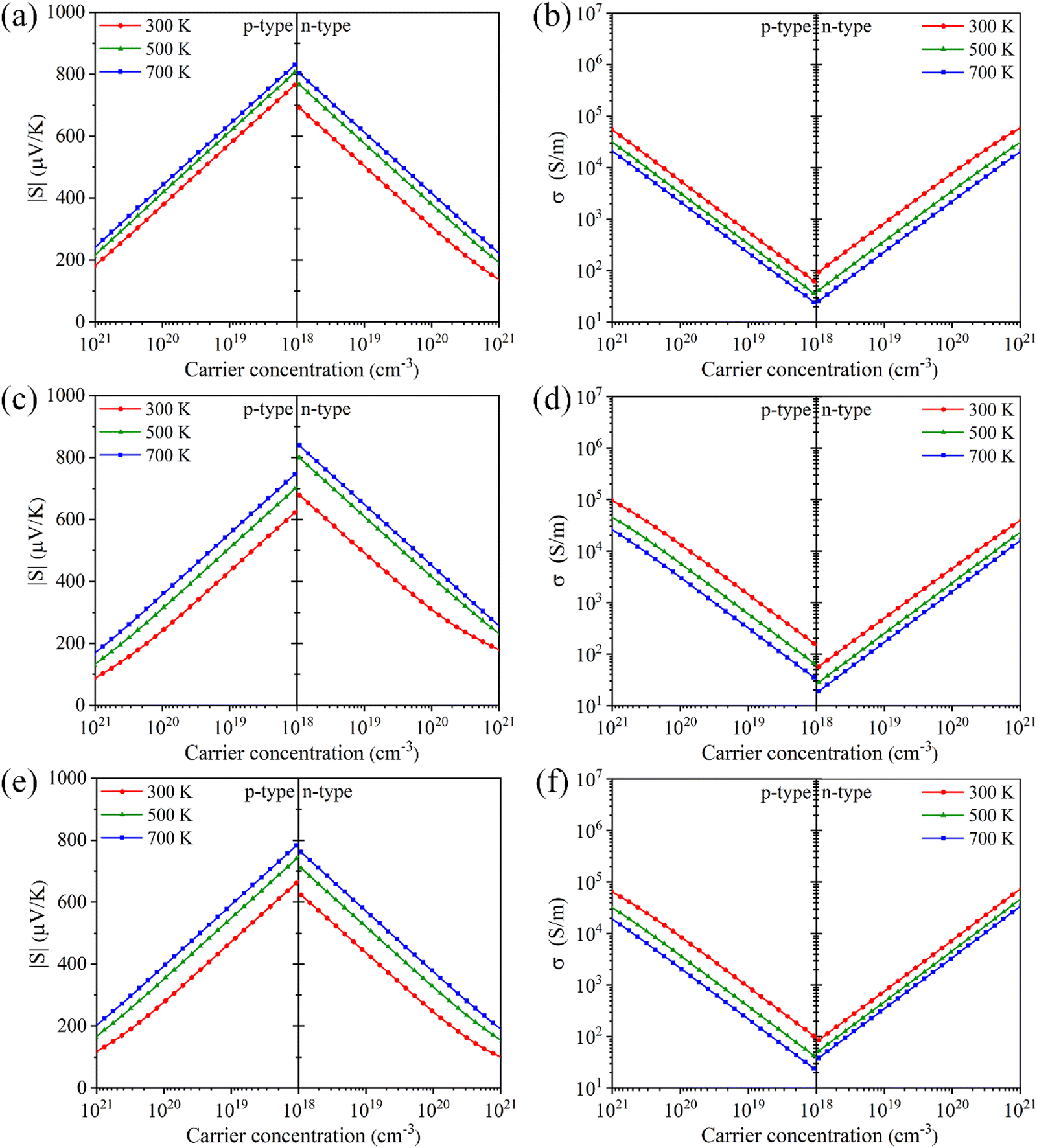
where e is the electron charge. It is observed that there is a minimal difference in the Seebeck coefficients of Tl3XS4 compounds under n-type and p-type doping. Specifically, for Tl3VS4 and Tl3TaS4, the Seebeck coefficient under p-type doping is slightly higher compared to n-type doping. However, Tl3NbS4 exhibits an opposite trend in its Seebeck coefficient compared to Tl3VS4 and Tl3TaS4, showing higher values under n-type doping. Furthermore, regardless of the doping type, the Seebeck coefficient of Tl3XS4 compounds increases with temperature and decreases with the increase in carrier concentration. These observations align with the trends predicted by eqn (1). It is noteworthy that Tl3XS4 compounds exhibit high Seebeck coefficients. Specifically, at 300 K, the Seebeck coefficients within the doping concentration range of 1018–1021 cm−3 are approximately 100–850 μV K−1, consistent with previous reports.21 Such outstanding Seebeck coefficients are comparable to excellent TE materials such as LaCuOSe/BiCuOSe.42
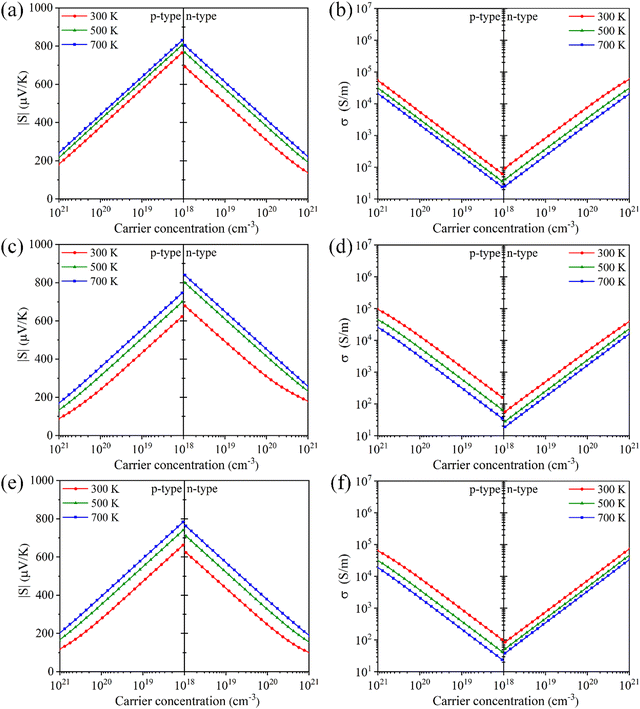 |
| | Fig. 4 The absolute value of Seebeck coefficient |S| and electrical conductivity σ for Tl3VS4 (a) and (b), Tl3NbS4 (c) and (d), and Tl3TaS4 (e) and (f) as a function of carrier concentration. | |
The results of the electrical conductivities with carrier concentrations are shown in Fig. 4(b), (d) and (f). The electrical conductivity of the material is closely related to the carrier concentration (n), as defined by:43
in which
μ is the carrier mobility. According to
eqn (2), the
σ increases with carrier concentration, which is consistent with our calculation results including p- and n-type Tl
3XS
4 (X = V, Nb, Ta). Moreover, there is a notable reduction in electrical conductivity as temperature increases, attributable to the increased carrier scattering and reduced carrier mobility at high temperatures. Based on the calculation results of AMSET, the carrier relaxation times
τtot and mobilities
μtot of Tl
3XS
4 (X = V, Nb, Ta) under ADP, IMP, and POP scattering mechanisms were calculated using the following equations:
44| | 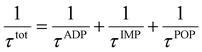 | (3) |
| | 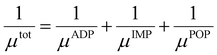 | (4) |
where
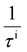
,
τi, and
μi (i = ADP, IMP, POP) are the carrier scattering rates, relaxation times, and carrier mobilities under the ADP, IMP, and POP scattering mechanisms, respectively. As shown in
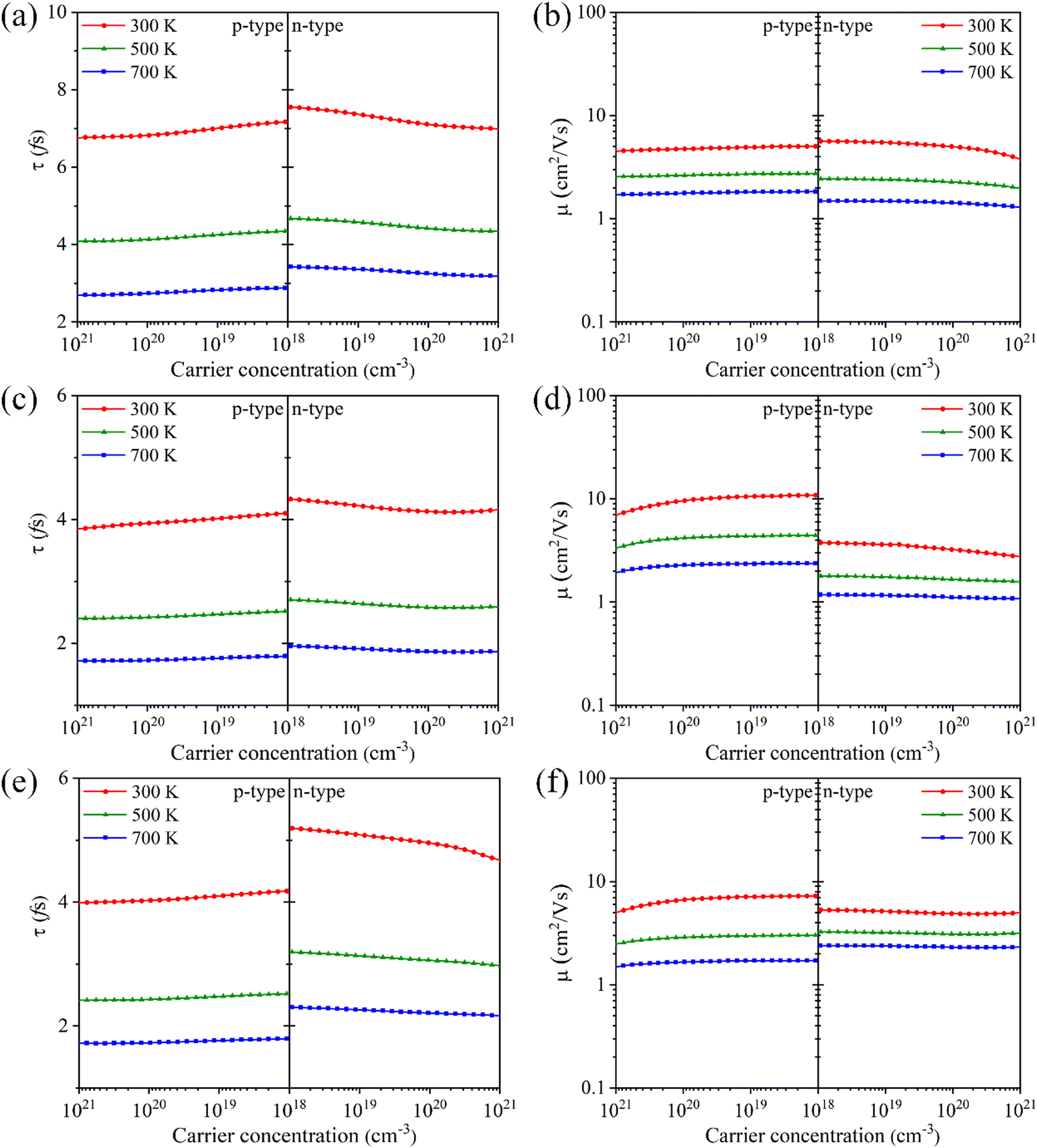
Fig. 5, the Tl
3XS
4 (X = V, Nb, Ta) system exhibits shorter carrier relaxation times (on the order of ∼10
−15 s) and smaller mobilities, which can be attributed to the strong carrier scattering. It is noteworthy that the carrier relaxation times of most thermoelectric semiconductor materials are typically on the order of ∼10
−15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
s.
42,45,46 For both p-type and n-type Tl
3XS
4 (X = V, Nb, Ta), the relaxation time decreases (
Fig. 5(a), (c) and (e)) and the mobility decreases (
Fig. 5(b), (d) and (f)) with the temperature from 300 K to 700 K, which is due to the enhanced carrier scattering with the increase of temperature. Additionally, as the carrier concentration increases, both relaxation time and mobility show a decreasing trend, which is consistent with their linear relationship
μ =
eτ/
m*. Since the effect of carrier concentration on mobility is not significant, the electrical conductivity exhibits a linear increasing trend with carrier concentration (as shown in
Fig. 4(b), (d), (f)), which is consistent with
eqn (2).
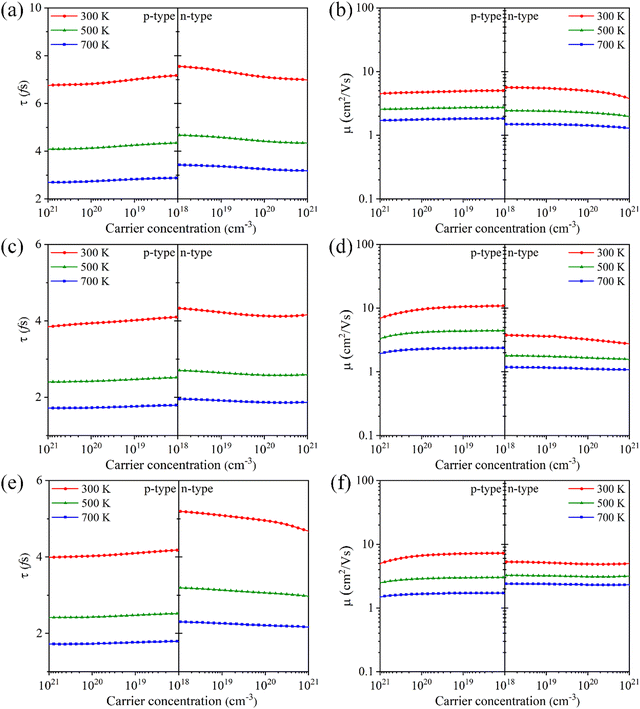 |
| | Fig. 5 The carrier relaxation time (τ) and carrier mobility (μ) for Tl3VS4 (a) and (b), Tl3NbS4 (c) and (d), and Tl3TaS4 (e) and (f) as a function of carrier concentration. | |
3.4. Thermal transport properties
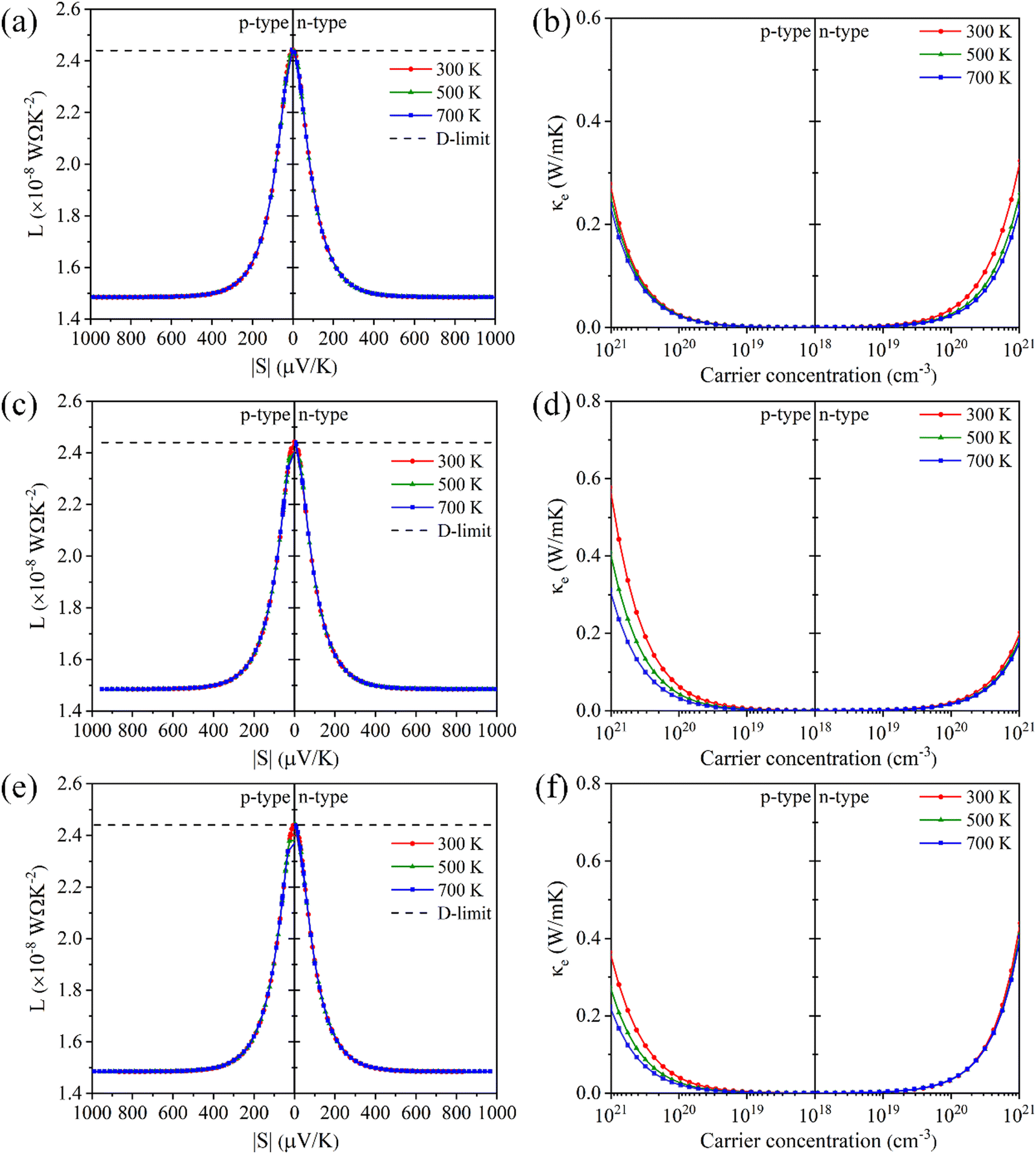
Electronic thermal conductivity (κe) exhibits a significant dependence on the Lorenz number (L), which is defined by the Wiedemann–Franz law as:47
For most metals, L converges to degenerate limit (D-limit) of ∼2.44 × 10−8 WΩ K−2.47 Although the Lorenz number for some heavily doped semiconductor thermoelectric materials is very close to the degenerate limit, its error can still reach up to ∼40%. This discrepancy arises because L is typically closely related to the scattering factor (λ), Seebeck coefficient (S), and chemical potential (η), according to the single parabolic band (SPB) model:47
| | 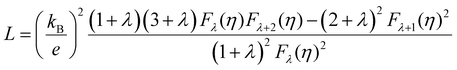 | (6) |
| | 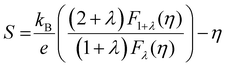 | (7) |
| | 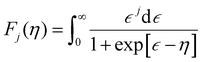 | (8) |
where
Fj(
η) represents the Fermi integral. Furthermore, Snyder
et al.47 proposed a satisfactory approximate equation for
L (referred to as Snyder's model in this paper):
| | 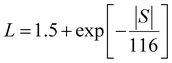 | (9) |
where
L is measured in 10
−8 WΩ K
−2, and
S is expressed in μV K
−1. Under the assumption of a single parabolic band and acoustic phonon scattering, Snyder's model achieves an accuracy within 5%. Based on the
eqn (6)–(8), we obtained the
L values corresponding the
S of p-type and n-type Tl
3XS
4 (X = V, Nb, Ta) at different temperatures, as shown in
Fig. 6(a), (c), (e). To verify the accuracy of the results, we further calculated the values of
L by using Snyder's model and compared with the calculation results of SPB model, as shown in Fig. S1–S3 (ESI
†). The
L values calculated by the two methods show close agreement, and the trend of
L as a function of
S is consistent with previous reports,
47 confirming the reliability of our calculations. Since
κe is directly proportional to
σ, its behavior closely follows that of
σ under varying carrier concentrations or temperatures (as depicted in
Fig. 6(b), (d), (f)).
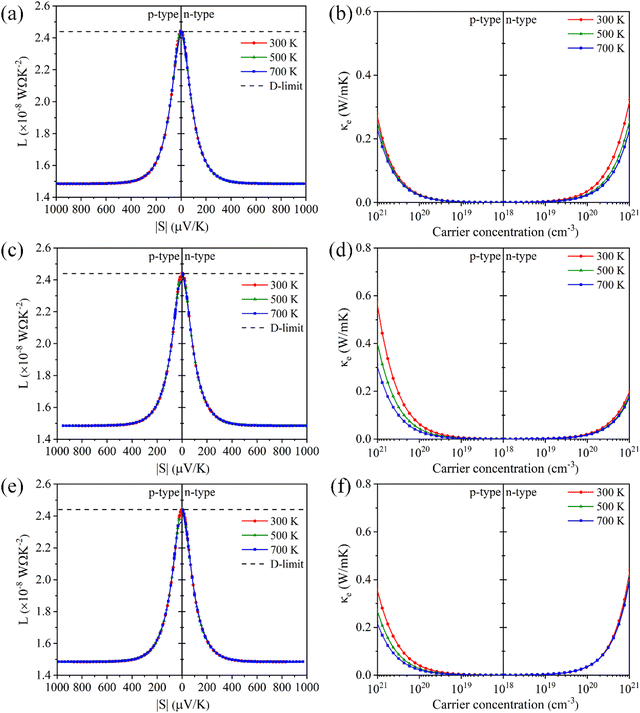 |
| | Fig. 6 The absolute Seebeck coefficient |S| dependent Lorenz number calculated by the SPB model of Tl3VS4 (a), Tl3NbS4 (c), and Tl3TaS4 (e). The electronic thermal conductivity κe for Tl3VS4 (b), Tl3NbS4 (d), and Tl3TaS4 (f) as a function of carrier concentration. | |
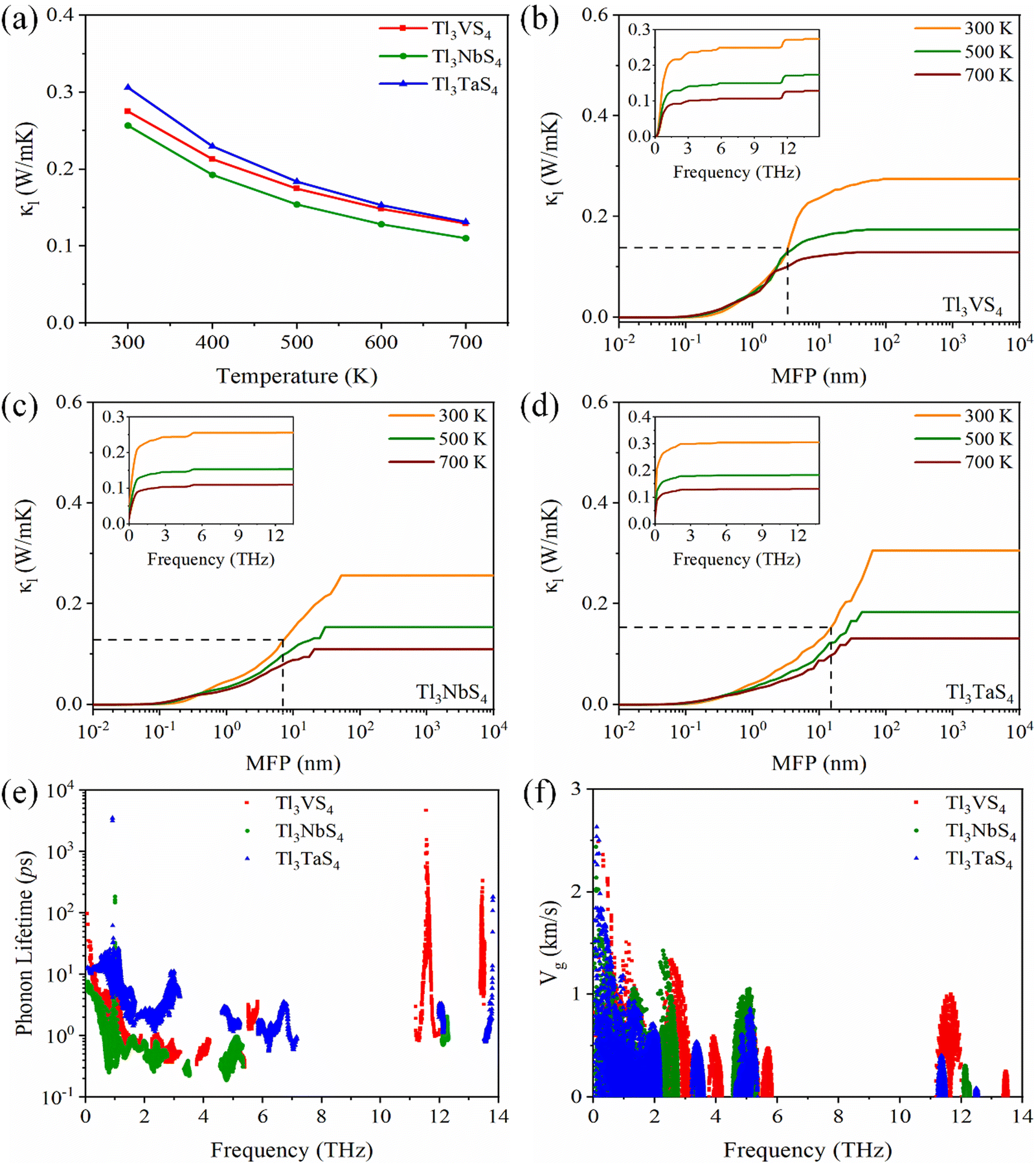
It is commonly acknowledged that the thermal conductivity in semiconductors is predominantly dominated by phonon part (κl).48 The lattice thermal conductivity of Tl3XS4 (X = V, Nb, Ta) at 300, 500, and 700 K is presented in Fig. 7(a). It is noteworthy that the entire compounds of the Tl3XS4 family exhibit exceptionally low lattice thermal conductivity. Specifically, the κl of the Tl3XS4 compounds is consistently below 0.5 W m−1 K−1 at room temperature. Furthermore, with increasing temperature, the scattering between phonons intensifies, leading to a decrease in κl as temperature rises. Moreover, we conducted calculations for the cumulative lattice thermal conductivity under different phonon mean free paths (MFP). The results indicate that, for Tl3XS4 compounds with a bulk structure, diminishing the material dimensions through a nanostructure strategy can effectively reduce its lattice thermal conductivity. For instance, at 300 K, when the size of Tl3VS4 samples is shortened to 3.34 nm, the κl can be halved, as plotted by the dashed line in Fig. 7(b). Besides, the cumulative κl of Tl3VS4, Tl3NbS4, and Tl3TaS4 are shown in the inset of Fig. 7(b), (c), and (d), respectively. Notably, at 300 K, nearly 90% of κl for Tl3XS4 originates from the acoustic modes and low-frequency optical modes (<2.5 THz). This means that the lattice thermal conductivity of Tl3XS4 is primarily contributed by acoustic phonons.
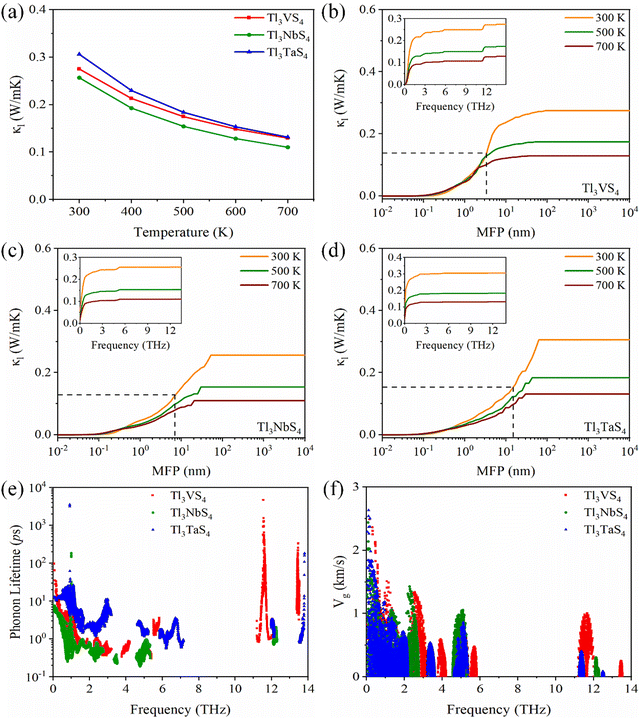 |
| | Fig. 7 (a) The lattice thermal conductivity κlvs. temperature of Tl3XS4 (X = V, Nb, Ta). (b)–(d) Cumulative lattice thermal conductivity as a function of phonon mean free path at 300, 500, and 700 K, inset of (b), (c) and (d) is cumulative lattice thermal conductivity as a function of frequency. The phonon lifetime (e) and phonon group velocity (f) vs. frequency at 300 K. | |
The κl is calculated as:
| | 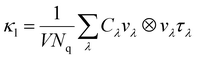 | (10) |
where
V and
Nq are the cell volume and the number of sampled
q points, respectively,
Cλ is the heat capacity,
vλ is the group velocity,
τλ is the phonon lifetime. To investigate the underlying factors of the low
κl in Tl
3XS
4, we calculated the phonon lifetime and group velocity at 300 K, as depicted in
Fig. 7(e) and (f). This analysis was pursued as
κl is predominantly influenced by the phonon lifetime and group velocity. Due to the significant band gap in the phonon dispersion of Tl
3XS
4, phonon lifetimes and phonon group velocities are predominantly distributed in the ranges of 0–6 THz and beyond 11 THz. This distribution aligns well with the frequency positions corresponding to the phonon spectrum. Since the
κl is mainly decided by the acoustic modes of the phonon band, we focus on the phonon lifetimes and phonon group velocities at the range of low frequencies. It is worth noting that the phonon lifetimes in the acoustic modes and low-frequency optical modes region follow the order: Tl
3TaS
4 > Tl
3VS
4 > Tl
3NbS
4 (as shown in
Fig. 7(e)). From
Fig. 7(f), the phonon group velocities also exhibit a similar trend, specifically, the group velocity of Tl
3TaS
4 is the largest and that of Tl
3NbS
4 is the smallest. These results indicate that the
κl of the Tl
3XS
4 (X = V, Nb, Ta) follows the order: Tl
3NbS
4 < Tl
3VS
4 < Tl
3TaS
4.
3.5. Power factor and figure of merit
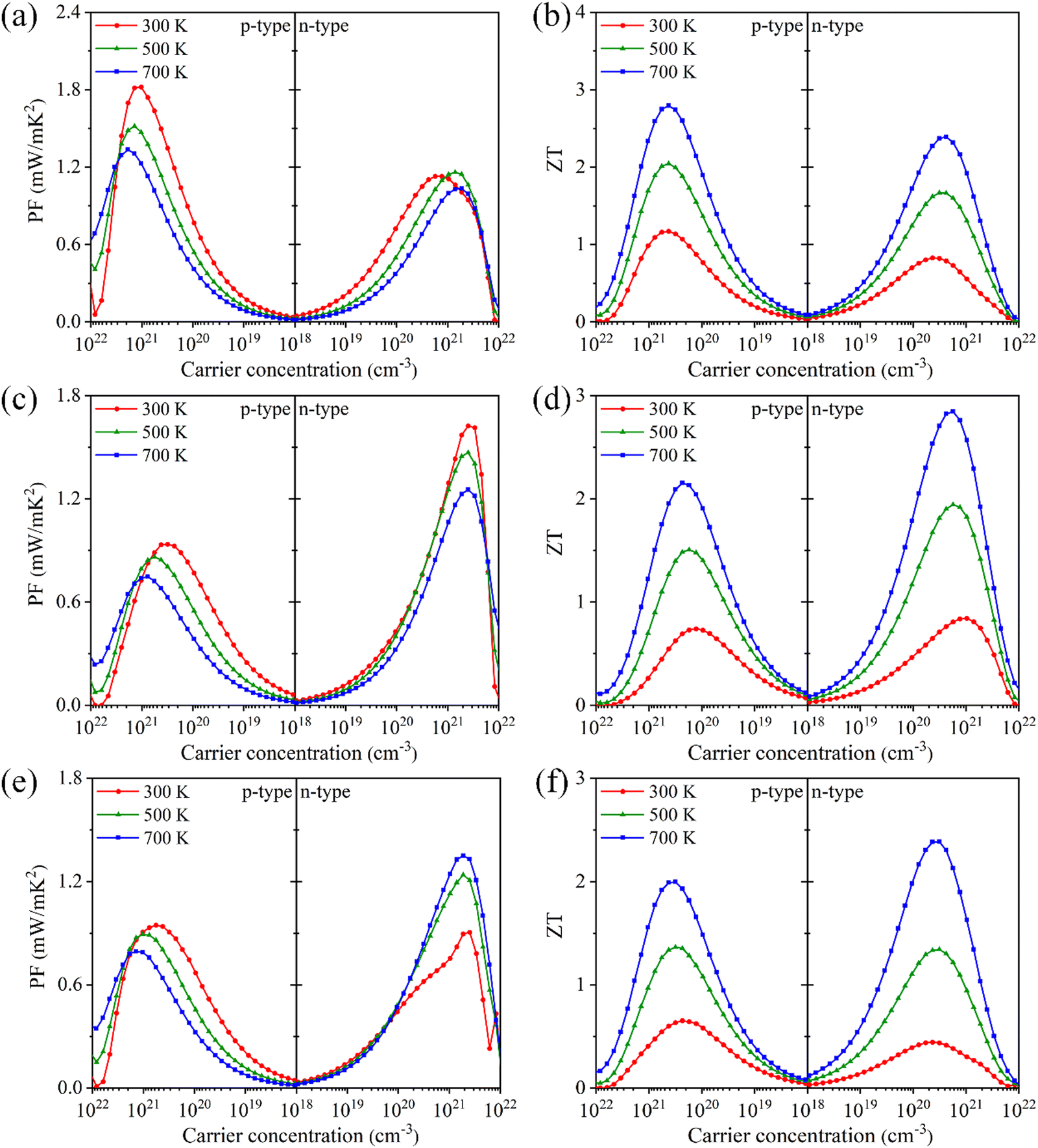
Based on PF = S2σ, the power factors of Tl3XS4 are given in Fig. 8(a), (c), (e). It is evident that the PF of Tl3XS4 significantly depends on the carrier type. Specifically, the p-type power factors of Tl3VS4 are higher than that of n-type, while Tl3NbS4 exhibits an opposite trend. This phenomenon is agreement with the dependence of the Seebeck coefficient on doping type, because the power factor is primarily determined by the Seebeck coefficient. Furthermore, the power factors exhibit a characteristic of initially increasing and then decreasing with the carrier concentration for both p- and n-type doping, reaching its maximum at a certain doping concentration. This is due to the increase of σ and the decrease of S with carrier concentration. Notably, at 300 K, the highest PF values of p-type (n-type) Tl3XS4 follow the order: Tl3VS4 > Tl3TaS4 > Tl3NbS4 (Tl3NbS4 > Tl3VS4 > Tl3TaS4). Specifically, at 300 K, the maximum PF values of p-type (n-type) Tl3VS4, Tl3NbS4, and Tl3TaS4 are 1.82 (1.13), 0.94 (1.62), and 0.95 (0.90) mW m−1 K−2, respectively.
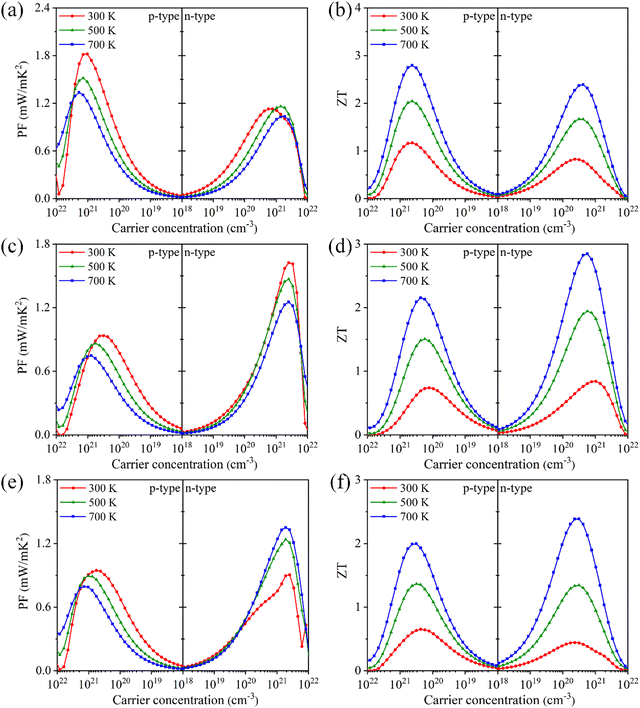 |
| | Fig. 8 The power factor PF and figure of merit ZT for Tl3VS4 (a) and (b), Tl3NbS4 (c) and (d), and Tl3TaS4 (e) and (f) under different carrier concentration at 300, 500, and 700 K. | |
Finally, the figure of merit with carrier concentrations of Tl3XS4 at 300, 500, and 700 K are presented in Fig. 8(b), (d) and (f). At all temperatures, the maximum ZT values of p-type Tl3XS4 follow the order: Tl3VS4 > Tl3NbS4 > Tl3TaS4, and the values of n-type follow the relationship: Tl3NbS4 > Tl3VS4 > Tl3TaS4. For example, at 300 K, the optimal ZT values of p-type (n-type) Tl3VS4, Tl3NbS4, and Tl3TaS4 are 1.17 (0.82), 0.74 (0.84), and 0.65 (0.44), respectively. The optimal ZT value of Tl3XS4 (X = V, Nb, Ta) at 300 K and corresponding parameters are listed in Table 2. Significantly, the peak ZT values are achieved at the carrier concentration of about 1020 cm−3, meeting the criteria for the optimal doping concentration in heavily doped semiconductors.49,50 The maximum ZT value for Tl3VS4, Tl3NbS4, and Tl3TaS4 is obtained at 700 K, for which the values are 2.79 (p-type), 2.85 (n-type), and 2.39 (n-type), respectively. Such outstanding ZT values can be on par with other excellent TE materials, such as LaCuOSe and BiCuOSe.42 Specifically, the optimal ZT values of p-type (n-type) LaCuOSe and BiCuOSe are reported to be about 0.32 (1.46) and 0.75 (1.20) at 900 K,42 respectively. In a word, these remarkable ZT values suggest that Tl3XS4 (X = V, Nb, Ta) compounds with a simple body-centered cubic structure are promising TE materials.
Table 2 Optimal ZT values and corresponding parameter conditions for Tl3XS4 (X = V, Nb, Ta) at 300 K
|
|
Carrier type |
n (1020 cm−3) |
|S| (μV K−1) |
σ (104 S m−1) |
PF (mW m−1 K−2) |
ZT
|
| Tl3VS4 |
Hole |
4.23 |
254 |
2.32 |
1.49 |
1.17 |
| Electron |
2.33 |
239 |
1.72 |
0.98 |
0.82 |
| Tl3NbS4 |
Hole |
1.28 |
222 |
1.69 |
0.83 |
0.74 |
| Electron |
10.4 |
178 |
4.08 |
1.29 |
0.84 |
| Tl3TaS4 |
Hole |
2.33 |
212 |
1.93 |
0.87 |
0.65 |
| Electron |
2.33 |
183 |
1.73 |
0.58 |
0.44 |
4. Conclusions
Collectively, utilizing the first-principles calculations, we investigated the electronic and thermal transport properties of Tl3XS4 (X = V, Nb, Ta) compounds with the simple body-centered cubic structure. First, the results of the AIMD simulation and phonon calculation reveal excellent dynamic stability and thermal stability of Tl3XS4 at 300, 500, and 700 K. Furthermore, the thermoelectric coefficients including the Seebeck coefficient S, electrical conductivity σ, power factor PF, and lattice thermal conductivity κl are further investigated at the considered temperatures. Our results demonstrate that the Tl3XS4 family exhibits ultralow κl (< 0.5 W m−1 K−1 at 300 K). The κl is primarily contributed by acoustic phonons, and the nanostructure strategy can effectively reduce κl. The investigations of phonon lifetimes and group velocities explain the lower κl of Tl3NbS4 compared to Tl3VS4 and Tl3TaS4. Finally, the combination of outstanding S and ultralow κl contributes to the remarkable ZT value. The optimal ZT values at 300 K for Tl3VS4, Tl3NbS4, and Tl3TaS4 are determined to be 1.17 (p-type), 0.84 (n-type), and 0.65 (p-type), respectively. For each considered temperature, the maximum ZT values of p-type (n-type) Tl3XS4 follow the order: Tl3VS4 > Tl3NbS4 > Tl3TaS4 (Tl3NbS4 > Tl3VS4 > Tl3TaS4). In a word, our results demonstrate that Tl3XS4 (X = V, Nb, Ta) compounds are promising thermoelectric materials, and the outstanding TE properties are comparable to excellent TE materials (i.e., LaCuOSe and BiCuOSe).
Data availability
Data available on request from the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 12365007, 12164050 and 12264022), the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities’ Association (Grant No. 202301BA070001-121, 202301BA070001-122, and 202301BA070001-094), and the Science Research Foundation of the Department of Education in Yunnan Province, China (Grant No. 2024J1056).
References
- L. Xie, C. Ming, Q. Song, C. Wang, J. Liao, L. Wang, C. Zhu, F. Xu, Y.-Y. Sun, S. Bai and L. Chen, Sci. Adv., 2023, 9, eadg7919 CrossRef CAS PubMed.
- X.-L. Shi, J. Zou and Z.-G. Chen, Chem. Rev., 2020, 120, 7399–7515 CrossRef CAS.
- D. Yang, Q. Gui, W. Yao, G. Wang and X. Zhou, Inorg. Chem., 2021, 60, 12331–12338 CrossRef CAS PubMed.
- J. Dong, D. Zhang, J. Liu, Y. Jiang, X. Y. Tan, N. Jia, J. Cao, A. Suwardi, Q. Zhu, J. Xu, J.-F. Li and Q. Yan, Inorg. Chem., 2023, 62, 17905–17912 CrossRef CAS PubMed.
- L.-D. Zhao, V. P. Dravid and M. G. Kanatzidis, Energy Environ. Sci., 2014, 7, 251–268 RSC.
- G. A. Slack, J. Phys. Chem. Solids, 1973, 34, 321–335 CrossRef CAS.
- R. Wei, G. Huiyuan, Z. Zihao and Z. Lixia, Phys. Rev. Lett., 2017, 118, 245901 CrossRef.
- B. C. Sales, D. Mandrus and R. K. Williams, Science, 1996, 272, 1325–1328 CrossRef CAS.
- G. Rogl, A. Grytsiv, P. Rogl, N. Peranio, E. Bauer, M. Zehetbauer and O. Eibl, Acta Mater., 2014, 63, 30–43 CrossRef CAS.
- J. Carrete, W. Li, N. Mingo, S. Wang and S. Curtarolo, Phys. Rev. X, 2014, 4, 011019 CAS.
- J. R. Sootsman, D. Y. Chung and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2009, 48, 8616–8639 CrossRef CAS.
- H. Zhu, R. He, J. Mao, Q. Zhu, C. Li, J. Sun, W. Ren, Y. Wang, Z. Liu, Z. Tang, A. Sotnikov, Z. Wang, D. Broido, D. J. Singh, G. Chen, K. Nielsch and Z. Ren, Nat. Commun., 2018, 9, 1–9 CrossRef.
- H. Zhu, J. Mao, Y. Li, J. Sun, Y. Wang, Q. Zhu, G. Li, Q. Song, J. Zhou, Y. Fu, R. He, T. Tong, Z. Liu, W. Ren, L. You, Z. Wang, J. Luo, A. Sotnikov, J. Bao, K. Nielsch, G. Chen, D. J. Singh and Z. Ren, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- T. Takabatake, K. Suekuni, T. Nakayama and E. Kaneshita, Rev. Mod. Phys., 2014, 86, 669–716 CrossRef CAS.
- G. S. Nolas, J. L. Cohn, G. A. Slack and S. B. Schujman, Appl. Phys. Lett., 1998, 73, 178–180 CrossRef CAS.
- Y. Saiga, K. Suekuni, S. K. Deng, T. Yamamoto, Y. Kono, N. Ohya and T. Takabatake, J. Alloys Compd., 2010, 507, 1–5 CrossRef CAS.
- S. Mukhopadhyay, D. S. Parker, B. C. Sales, A. A. Puretzky, M. A. McGuire and L. Lindsay, Science, 2018, 360, 1455–1458 CrossRef CAS PubMed.
- Z. Zeng, C. Zhang, Y. Xia, Z. Fan, C. Wolverton and Y. Chen, Phys. Rev. B, 2021, 103, 224307 CrossRef CAS.
- Y. Xia, K. Pal, J. He, V. Ozoliņš and C. Wolverton, Phys. Rev. Lett., 2020, 124, 065901 CrossRef CAS PubMed.
- A. Jain, Phys. Rev. B, 2020, 102, 201201 CrossRef CAS.
- S. Mukhopadhyay and T. L. Reinecke, J. Phys. Chem. Lett., 2019, 10, 4117–4122 CrossRef CAS.
- B. Li, C. Zhang, Z. Sun, T. Han, X. Zhang, J. Du, J. Wang, X. Xiao and N. Wang, Phys. Chem. Chem. Phys., 2022, 24, 24447–24456 RSC.
- C. Crevecoeur, Acta Cryst., 1964, 17, 757 CrossRef CAS.
- K. Wacker and P. Buck, Cryst. Res. Technol., 1985, 20, 1025–1030 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 3865 CrossRef.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef.
- R. Jinnouchi, J. Lahnsteiner, F. Karsai, G. Kresse and M. Bokdam, Phys. Rev. Lett., 2019, 122, 225701 CrossRef CAS PubMed.
- R. Jinnouchi, F. Karsai and G. Kresse, Phys. Rev. B, 2019, 100, 014105 CrossRef CAS.
- R. Jinnouchi, F. Karsai, C. Verdi, R. Asahi and G. Kresse, J. Chem. Phys., 2020, 152, 234102 CrossRef CAS PubMed.
- A. M. Ganose, J. Park, A. Faghaninia, R. Woods-Robinson, K. A. Persson and A. Jain, Nat. Commun., 2021, 12, 2222 CrossRef CAS PubMed.
- W. Dou, K. B. Spooner, S. R. Kavanagh, M. Zhou and D. O. Scanlon, J. Am. Chem. Soc., 2024, 146, 17679–17690 CrossRef CAS PubMed.
- W. Li, J. Carrete, N. A. Katcho and N. Mingo, Comput. Phys. Commun., 2014, 185, 1747–1758 CrossRef CAS.
- A. Carreras, A. Togo and I. Tanaka, Comput. Phys. Commun., 2017, 221, 221–234 CrossRef CAS.
- A. Togo and I. Tanaka, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- T. V. Vu, A. A. Lavrentyev, B. V. Gabrelian, K. F. Kalmykova, V. V. Sidorkin, D. M. Hoat, H. D. Tong, D. P. Tran, O. Y. Khyzhun and D. D. Vo, Opt. Mater., 2020, 99, 109601 CrossRef CAS.
- A. A. Lavrentyev, A. N. Gusatinskii, I. Y. Nikiforov, A. P. Popov and V. V. Ilyasov, J. Phys.: Condens. Matter, 1993, 5, 1447–1454 CrossRef CAS.
- E. Osei-Agyemang, C. E. Adu and G. Balasubramanian, npj Comput. Mater., 2019, 5, 116 CrossRef CAS.
- C. Lee, J.-H. Shim and M.-H. Whangbo, Inorg. Chem., 2018, 57, 11895–11900 CrossRef CAS.
- N. Wang, M. Li, H. Xiao, Z. Gao, Z. Liu, X. Zu, S. Li and L. Qiao, npj Comput. Mater., 2021, 7, 18 CrossRef CAS.
- G. Tan, L.-D. Zhao and M. G. Kanatzidis, Chem. Rev., 2016, 116, 12123–12149 CrossRef CAS.
- X. Song, Y. Zhao, J. Ni, S. Meng and Z. Dai, Mater. Today Phys., 2023, 31, 100990 CrossRef CAS.
- N. Wang, M. Li, H. Xiao, X. Zu and L. Qiao, Phys. Rev. Appl., 2020, 13, 024038 CrossRef CAS.
- X. Song, G. Wang, L. Zhou, H. Yang, X. Li, H. Yang, Y. Shen, G. Xu, Y. Luo and N. Wang, ACS Appl. Energy Mater., 2023, 6, 9756–9763 CrossRef CAS.
- H.-S. Kim, Z. M. Gibbs, Y. Tang, H. Wang and G. J. Snyder, APL Mater., 2015, 3, 041506 CrossRef.
- J. Li, W. Hu and J. Yang, J. Am. Chem. Soc., 2022, 144, 4448–4456 CrossRef CAS PubMed.
- G. Tan, L.-D. Zhao and M. G. Kanatzidis, Chem. Rev., 2016, 116, 12123–12149 CrossRef CAS PubMed.
- M. A. Haque, S. Kee, D. R. Villalva, W.-L. Ong and D. Baran, Adv. Sci., 2020, 7, 1903389 CrossRef CAS.
|
| This journal is © the Owner Societies 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Guangzhao
Wang
*a,
Guangzhao
Wang

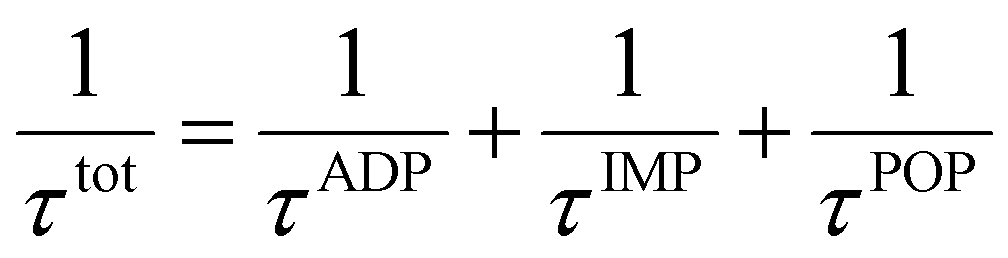
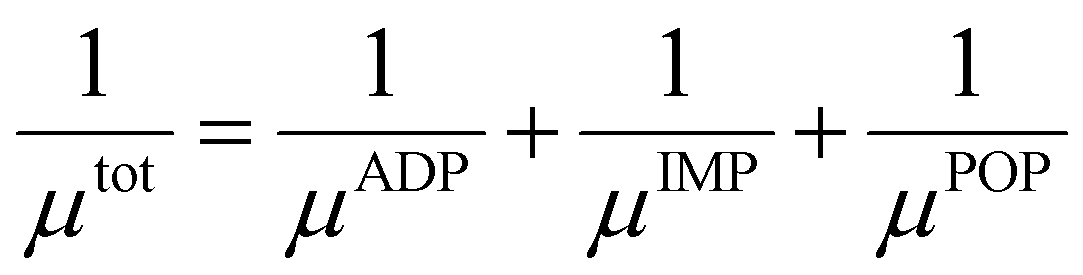
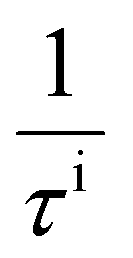 , τi, and μi (i = ADP, IMP, POP) are the carrier scattering rates, relaxation times, and carrier mobilities under the ADP, IMP, and POP scattering mechanisms, respectively. As shown in Fig. 5, the Tl3XS4 (X = V, Nb, Ta) system exhibits shorter carrier relaxation times (on the order of ∼10−15 s) and smaller mobilities, which can be attributed to the strong carrier scattering. It is noteworthy that the carrier relaxation times of most thermoelectric semiconductor materials are typically on the order of ∼10−15
, τi, and μi (i = ADP, IMP, POP) are the carrier scattering rates, relaxation times, and carrier mobilities under the ADP, IMP, and POP scattering mechanisms, respectively. As shown in Fig. 5, the Tl3XS4 (X = V, Nb, Ta) system exhibits shorter carrier relaxation times (on the order of ∼10−15 s) and smaller mobilities, which can be attributed to the strong carrier scattering. It is noteworthy that the carrier relaxation times of most thermoelectric semiconductor materials are typically on the order of ∼10−15